
ABSTRACT
Etanercept (ETN) (Enbrel®) is a soluble protein that binds to, and specifically inhibits, tumor necrosis factor (TNF), a proinflammatory cytokine. ETN is synthesized in Chinese hamster ovary cells by recombinant DNA technology as a fusion protein, with a fully human TNFRII ectodomain linked to the Fc portion of human IgG1. Successful manufacture of biologics, such as ETN, requires sophisticated process and product understanding, as well as meticulous control of operations to maintain product consistency. The objective of this evaluation was to show that the product profile of ETN drug substance (DS) has been consistent over the course of production. Multiple orthogonal biochemical analyses, which included evaluation of attributes indicative of product purity, potency, and quality, were assessed on >2,000 batches of ETN from three sites of DS manufacture, during the period 1998–2015. Based on the key quality attributes of product purity (assessed by hydrophobic interaction chromatography HPLC), binding activity (to TNF by ELISA), potency (inhibition of TNF-induced apoptosis by cell-based bioassay) and quality (N-linked oligosaccharide map), we show that the integrity of ETN DS has remained consistent over time. This consistency was maintained through three major enhancements to the initial process of manufacturing that were supported by detailed comparability assessments, and approved by the European Medicines Agency. Examination of results for all major quality attributes for ETN DS indicates a highly consistent process for over 18 years and throughout changes to the manufacturing process, without affecting safety and efficacy, as demonstrated across a wide range of clinical trials of ETN in multiple inflammatory diseases.
Abbreviations
| DMARD | = | disease-modifying anti-rheumatic drug |
| DP | = | drug product |
| DS | = | drug substance |
| ELISA | = | enzyme-linked immunosorbent assay |
| ETN | = | etanercept |
| Fc | = | crystallizable fragment |
| HIC | = | hydrophobic interaction chromatography |
| HCP | = | host cell protein |
| HPLC | = | high-performance liquid chromatography |
| IgG1 | = | human type 1 immunoglobulin G |
| JIA | = | juvenile idiopathic arthritis |
| mAbs | = | monoclonal antibodies |
| MAH | = | market authorization holder |
| RA | = | rheumatoid arthritis |
| SAE | = | serious adverse event |
| TNF | = | tumor necrosis factor |
Introduction
Targeting the pro-inflammatory cytokine tumor necrosis factor (TNF) has revolutionized the treatment of rheumatoid arthritis (RA) and other inflammatory diseases. With its approval by the European Medicines Agency (EMA) in 2000,Citation1 etanercept (ETN) (Enbrel®) was one of the first TNF inhibitors to be approved in the European Union (EU) for the treatment of RA. ETN has since been approved for the treatment of other autoimmune diseases,Citation2 including plaque psoriasis, psoriatic arthritis, ankylosing spondylitis,Citation3 and non-radiographic axial spondyloarthritis,Citation4 as well as polyarticular-course juvenile idiopathic arthritis (JIA), and the JIA categories extended oligoarthritis, enthesitis-related arthritis, and psoriasis arthritis.Citation5
ETN is a dimeric fusion protein consisting of the extracellular domain of human TNF receptor (TNFRII orp75), linked to the crystallizable fragment (Fc) of human type 1 immunoglobulin G (IgG1). ETN specifically inhibits TNF and binds to TNF (sTNF and tmTNF) reversibly in a 1:1 ratio.Citation6 The Fc component of ETN contains the CH2 and CH3 domains and the hinge region, but not the CH1 domain of IgG.
ETN is a complex protein with a total of 934 amino acid residues (Mr ≈ 150 kDa). It is heavily glycosylated, containing both N- and O-linked oligosaccharides, which can potentially influence the structure, activity, signaling, clearance, and immunogenicity of such glycosylated proteins. TNF blockade with ETN modulates several biologic responses that are induced or regulated by TNF, including expression of adhesion molecules responsible for leukocyte migration, serum levels of cytokines (e.g., IL-6), and serum levels of matrix metalloproteinase-3.Citation2
Incorporation of revisions in the manufacturing processes of a biologic after initial regulatory approval is part of the life cycle management of a drug.Citation7 These can range from relatively minor changes (e.g., a change in supplier of source materials) to more significant changes (e.g., introducing new purification steps),Citation7 and are governed by strict, regional-specific regulations.Citation8 A review of authorized manufacturing changes for 29 therapeutic monoclonal antibodies (mAbs) with European Public Assessment Report (EPAR) documents from 1998 and 2014Citation9 showed the annual average number of approved changes categorized by risk status (low, moderate, or high) was 1.8 (range 0–3.71). These findings suggest such changes are not unusual, and the EMA is highly experienced in assessing and assuring comparability of biologics pre- and post-manufacturing change.Citation8 The International Conference on Harmonisation (ICH) guidelines (ICH Q5E)Citation10 regulating this process reflect the complexity of the manufacture of biologics, in that demonstration of comparability does not necessarily mean the quality attributes of the pre- and post-change product are identical. Rather, they are required to be highly comparable, such that, based on the body of established knowledge and experience, it is possible to satisfactorily predict whether any differences in quality attributes will adversely affect the safety or efficacy of the drug product (DP).
Every batch of ETN drug substance (DS) is analyzed using multiple, orthogonal state of the art technologies, to evaluate the quality attributes that define the identity, strength, biological activity (potency and binding activity), purity (aggregated, misfolded, and clipped species), impurities (e.g., host cell proteins [HCP] and leached protein A), safety, physical characteristics, and overall quality profile. All batch-release test results must meet stringent specifications. Therefore, sophisticated controls are applied to the manufacturing process in order to ensure operation within tight operating ranges. The product quality parameters applied for batch-release testing have been selected following a development program, which includes extensive characterization of the process to facilitate understanding of the link between process performance and subsequent product quality profile. The product quality profile of the DS essentially defines the profile of the DP, as little change in quality attributes occurs through the DP manufacturing process.
The extent of the variance in quality attributes apparent for some marketed biologic products before and after a manufacturing change, illustrates the acceptable limits, within which, observed differences are considered by health authorities as not altering clinical profile.Citation11 Pfizer is the market authorization holder (MAH) for Enbrel in the EU and the rest of the world (excluding US and Canada, where Amgen is the MAH for Enbrel). Amgen and Pfizer both presently manufacture ETN, and have business systems in place to ensure product consistency is maintained across the globe. US- and EU-sourced product have been shown to be indistinguishable from one another, based on their physicochemical and in vitro functional biological activity, consistent with a single product.Citation12 Since 1998, the manufacture of ETN has been subject to various process changes, purposefully designed to enhance the efficiency of production, optimize process robustness, or adjust the raw material sources used to manufacture the DS. Information available from the EMA describes in the order of 20 process improvements that have been introduced in EU-manufactured ETN overall.Citation13 Of these improvements, only three have been major process revisions, with each change being supported by individual comparability exercises to demonstrate that the pre- and post-change products are comparable. Analytical comparability is demonstrated by evaluating all release test results against the pre-change historical normal values. Analytical comparability also includes a number of heightened characterization tests to confirm comparability with the reference standard. Heightened characterization includes analyses of primary, secondary, and tertiary structure, expected heterogeneity of the amino- and carboxi-termini, expected post-translational modifications, aggregation, fragmentation, molecular size, molecular charge distribution, and binding to multiple biological receptors, including the target molecule (TNF) and multiple Fcγ receptors.
The three major revisions to the original ETN DS manufacturing process were: 1) replacement of non-irradiated with γ-irradiated serum (2002); 2) addition of an extra purifi-cation step to decrease process impurities (protein A ligand) (2003); and 3) implementation of serum-free cell culture (2008). These changes were made proactively to improve the manufacturing process, and were not made as a consequence of any safety signals or concerns. Each of these changes in the manufacturing process was supported by an appropriate comparability package, including full product release and stability testing, together with a heightened characterization testing exercise and supportive clinical studies, where required, to meet the associated regulatory requirements.Citation8
To demonstrate consistency of product quality through each of these three major process revisions, data are presented for select analyses, which indicate the purity, levels of impurities, binding activity, potency, and overall product quality. The results span approximately 20 years of production across multiple sites and scales of manufacture, and demonstrate comparability before and after each process revision.
Results
Key quality attributes for ETN DS from >2,000 batches from three sites of commercial DS manufacture were assessed from 1998 to 2015. This assessment spanned implementation of the three major process revisions (Processes B, C, and D) to the original process (Process A), as well as manufacturing of ETN DP at three different sites (). The tests used for routine and heightened characterization of Enbrel quality attributes are listed in .
Table 1. Major revisions to ETN DP manufacturing process.
Table 2. Main Enbrel Quality Attributes and Characterization Tests.
Results from a number of release tests to assess key quality attributes over the ETN DS life cycle (1998–2015) according to the manufacturing process (A–D) are shown in . Overlap in the periods during which the different processes were used in the manufacture of Enbrel reflects the commissioning and time for licensure for additional manufacturing sites, while implementing the transition from one manufacturing process to another. These changes in process and manufacturing sites were performed in line with the relevant health authority requirements. The results for purity (, hydrophobic interaction chromatography [HIC] Peak 3), impurities (, HCP), binding activity (), cell-based potency (), and quality (, N-linked glycan profile) are highly comparable across each process revision.
Figure 1. Purity levels by HIC HPLC (Peak 3: levels of aggregated and misfolded species) (a); impurity levels as HCP by ELISA (b); binding activity to TNF by ELISA (c); and potency by inhibition of TNF-induced apoptosis in a cell-based bioassay (d), of ETN DP batches (1998–2015) across 3 manufacturing sites according to manufacturing process (A–D)* DP, drug product; DS, drug substance; ELISA, enzyme-linked immunosorbent assay; ETN, etanercept; HIC, hydrophobic interaction chromatography; HCP, host cell proteins; HPLC, high-performance liquid chromatography; TNF, tumor necrosis factor.
Minor variations in the number of batches included per process across each test method reflect minor differences in investigative or repeat testing. *Data (horizontal lines) are median values (95% confidence interval) and interquartile range; mean values are indicated by the square symbol. The dotted lines indicate the specification limits for ETN DS.
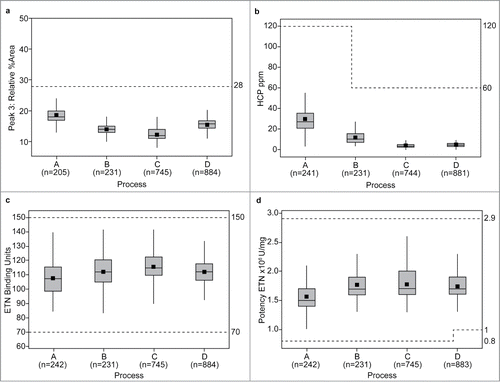
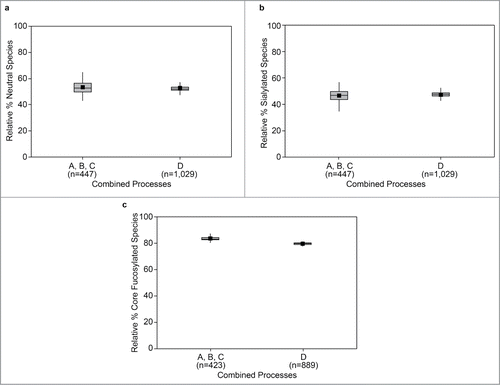
Figure 2. Relative abundance of neutral species (Peaks 1, 2/3, 4, and 5) (a); sialylated species (Peaks 6–9) (b); and core fucosylated (Peaks 1, 2/3, 5, 7, and 9) N-linked oligosaccharides (c), across process variations (A–D). Specifications for individual peaks were introduced following implementation of process D. Data available to create statistical representations therefore differ compared with those in . *Data (horizontal lines) are median values and interquartile range. The whiskers indicate the highest and lowest data values within the upper and lower limits, respectively. Mean values are indicated by the square symbol.
Results for the relative amount of misfolded and aggregated species, as measured by HIC Peak 3 (), are consistent across each process variation, with minor variations between each change, but the overall levels are comparable from one process to the next. The first major process change involved the transition from using bovine serum to γ-irradiated bovine serum in the production bioreactor and other minor process enhancements (i.e., change from Process A to Process B), resulting in reduction in the levels of HCP impurities detected by ELISA (). While there was no effect on the overall quality or safety following this process change, the specification limit for HCP was subsequently lowered (from 120 ppm to 60 ppm) to reflect the tighter process control (Processes B–D), and in line with regulatory practice. A further reduction in HCP levels as well as batch-to-batch variability can be seen in the transition to Process C, where an additional chromatographic step was added to production operations. For the key quality attributes of binding activity to TNF () and inhibition of TNF-induced apoptosis (), all batches were within the pre-specified limits for these product quality attributes both throughout the manufacturing history, and across the various process changes introduced.
To support the removal of serum from the cell culture (Process D), significant efforts were made from a process development perspective to ensure the overall carbohydrate profile, including the relative abundance of core-fucosylated structures and sialylated species, were not significantly altered. The relative abundance of neutral (), sialylated (), and core-fucosylated N-linked oligosaccharide species () across process variations showed that these attributes did not change significantly with the transition to Process D.
Results for analysis of all batches using the release and characterization tests met all specification, and acceptance criteria, and comparability of the ETN DS post-change was demonstrated in each case, following each process revision.
Discussion
Through over 18 years of clinical experience, ETN DP has been manufactured at three commercial sites, comprising >2,000 batches of DS. The manufacturing process has undergone three major revisions from that originally licensed, with a number of other more minor process changes incorporated.Citation13 These changes reflect plans either to enhance the product quality profile or reduce the already low risk of adventitious agent contamination.Citation13 A stringently controlled manufacturing environment, with the appropriately validated equipment and processes, has been designed to ensure consistency in the manufacture of the DS, and thus the DP, over time.Citation14 This is borne out by examination of results for all major quality attributes for ETN DS, which demonstrate that all product quality parameters, including process impurities, binding activity, charge distribution, N-linked oligosaccharide profile, and potency, have remained comparable between batches, across manufacturing sites, and through each planned process revision.
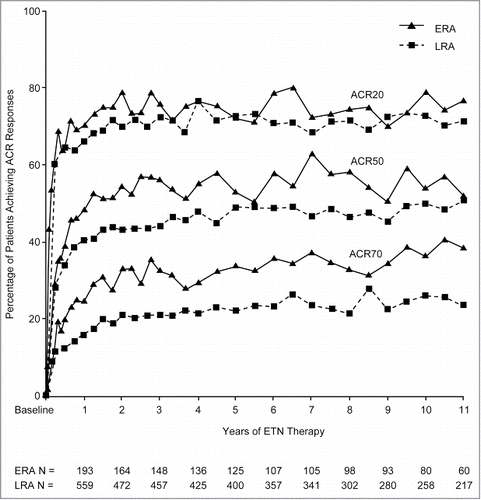
Maintenance of the structural consistency of ETN DS throughout the changes to the manufacturing process described here is reflected in a safety profile that has remained consistent with the approved product labeling,Citation15 across a wide range of clinical trials in a variety of inflammatory diseases.Citation3 For instance, in an open-label extension, where patients with disease-modifying anti-rheumatic drug (DMARD)-refractory RA who completed initial ETN trials were offered the opportunity to continue treatment, rates of serious adverse events (SAEs), serious infections, cancer, and deaths were stable each year, through 8 years of ETN exposure.Citation16 In addition, data from a long-term prospective study,Citation17 with some patients receiving >10 years of treatment, indicated that ETN was well-tolerated and effective as a long-term, continuous therapy for the treatment of both patients with early RA and long-standing RA (); with the risk/benefit ratio of continuous long-term treatment remaining favorable.Citation17 Consistency in activity is also evident from results from two large, prospective, 5-year, multicenter, observational registries (conducted from 2000 to 2008): RADIUS (Rheumatoid Arthritis DMARD Intervention and Utilization Study) 1, in which patients with RA required a change in treatment; and RADIUS 2, where patients commenced ETN treatment at study entry.Citation18 In total, over 6,000 patients initiated ETN treatment in the RADIUS registries, and data showed that rates of SAEs, serious infectious events, and events of medical interest in the ETN-treated patients were comparable to those seen in clinical trials, and did not increase with ETN exposure. Maintenance of efficacy of ETN has also been demonstrated in the treatment of patients with plaque psoriasis in long-term open-label extensions to placebo-controlled trials.Citation19,20 In patients receiving ETN (50 mg once-weekly), psoriasis activity and severity index (PASI) scores showed improvement from baseline of the extension study to 72 weeks.Citation19 In a separate analysis of data from the open-label extension phases of two Phase 3 trials, ETN demonstrated sustained effectiveness (based on Physician Global Assessment [PGA] and Dermatology Life Quality Index scores) and a favorable safety profile for up to 4 years.Citation20 This profile is also supported by 5-year data from an observational postmarketing safety surveillance registry, OBSERVE,Citation21 in which the proportion of patients rated as clear/almost clear based on PGA, increased from 12% at baseline to 51% at month 6, and remained relatively stable thereafter. No new safety signals were observed in this study of long-term real-world ETN use.
Figure 3. Sustained improvement in American College of Rheumatology (ACR) response (Reprinted with permission from Weinblatt et al., “Safety and Efficacy of Etanercept Beyond 10 Years of Therapy in North American Patients With Early and Longstanding Rheumatoid Arthritis” Arthritis Care & Research Vol. 63(3), 373–382, Wiley. © 2011, American College of RheumatologyCitation17)*, *Percentages of early rheumatoid arthritis (ERA) patients and longstanding rheumatoid arthritis (LRA) patients who achieved ACR criteria for 20%/50%/70% improvement in disease activity (ACR20/50/70) responses from baseline throughout year 11 are shown. ACR data are reported using C-reactive protein (CRP) values based on the enzyme immunoassay (EIA) of CRP (ERA: months 1–78, LRA: months 1–60), the average of the EIA and the new high-sensitivity method (ERA: months 84–90, LRA: months 66–90), and the high-sensitivity method (ERA and LRA: months ≥96). Numbers of ERA and LRA patients evaluated during each year are indicated. Values were not available for all patients at all time points.
Cumulatively, it is estimated that 28,123 subjects have participated in sponsor-initiated (Pfizer and Amgen) clinical trials, with 25,637 subjects exposed to ETN (unpublished data). There have been ∼5.6 million patient-years of collective clinical experience with ETN across the various indications (unpublished data). In all of these patient populations, ETN (with or without methotrexate) effectively reduced signs and symptoms, disease activity and disability, and improved health-related quality of life, with these benefits being sustained during long-term treatment.Citation3 During this time, changes to the manufacturing process (a familiarCitation7, 11 and carefully regulatedCitation8,10 aspect of the lifecycle of biologic products) have been implemented to ensure the efficacy, safety, and quality of the ETN DP currently available to patients is no different compared to when it was first introduced.
Material and methods
The test methods used to assess attributes reflective of the product purity, level of impurities, binding activity, potency, and quality are described below. During validation of planned manufacturing process changes, Enbrel batches were tested against the existing reference standard derived from the licensed process, historical product trends, and in side-by-side batch analysis, where applicable. Following licensure of the process change, a new reference standard was typically prepared, in line with the recommended approach for biological products.Citation22
Purity as measured by HIC high-performance liquid chromatography
HIC is performed to assess the purity of ETN DS and DP, and the extent of the related product species present. The method is used to resolve three variants of ETN, which differ in biological activity: Peak 1 is predominantly clipped species; Peak 2 is homogenous ETN; Peak 3 consists of misfolded species, aggregates, ETN fragments, and other process-related impurities. Results are expressed as relative percentage peak area.
Each sample (5 µL; 2 mg/mL) was injected onto a TSKgel Butyl-NPR (Tosoh) (2.5 µm) analytical column (4.6 mm × 35 mm) at 35°C, connected to an HPLC system. Product-related impurities were separated by gradient elution over 50 minutes using mobile phase A (ammonium sulfate [475.9 g] and anhydrous disodium hydrogen phosphate [28.4 g] in 1,950 mL water for chromatography, adjusted to pH 7.0 with phosphoric acid and diluted to 2,000 mL with water for chromatography), and mobile phase B (anhydrous disodium hydrogen phosphate [28.4 g] in water for chromatography and diluted to 1,950 mL with water, adjusted to pH 7.0 with phosphoric acid and diluted to 2,000 mL with water). The flow rate was 1.0 mL/min, and chromatography was monitored by fluorescence detection (278 nm for excitation; 350 nm for emission). Elution of the protein molecules occurred in order of increasing hydrophobicity, as the salt concentration decreased throughout the run, separating the sample into HIC Peak 1, HIC Peak 2, and HIC Peak 3, which were integrated and reported as relative %Peak 1, %Peak 2, and %Peak 3.
Impurities as measured by enzyme-linked immunosorbent assay (ELISA)
A standard sandwich ELISA using polyclonal antibodies was used to quantify the amount of HCP in DS samples from a calibration curve prepared from CHO cell HCP, using a null vector (the reagents used are proprietary to Pfizer). The units are specific to this assay, and sample results are reported as ppm of protein (ng HCP/mg protein).
Binding activity as measured by ELISA
The receptor binding assay used was a quantitative solid-phase ELISA. ETN (TNFR:Fc) in samples and standards binds to TNF adsorbed to the microplate wells and were detected using goat anti-human IgG antibody horseradish peroxidase conjugate (Sigma-Aldrich: #A0170). Binding activities are calculated based on the ratio of the ED50 values of the calibration curve relative to the control or sample curves.
Potency as measured by cell-based bioassay
The potency of ETN samples was quantified by measurement of their ability to neutralize TNF-mediated apoptosis in histiocytic lymphoma cell-line U937 (ATCC No. CRL-1593.2) via caspase activation. The U937 cells were incubated at 36.0–38.0°C for 30–60 min in a humidified incubator using 5% ± 2% CO2 with varying dilutions of test and reference preparations of ETN, in the presence of TNF. They were then incubated with Caspase-Glo 3/7 assay system (Promega: #G8092) at room temperature for 30–60 min, which resulted in caspase cleavage of a chemiluminescent substrate, subsequent release of a luciferase substrate, and generation of a luminescent signal. The luminescence produced was proportional to the amount of caspase activity present. Biologic activity of the sample was calculated based on the ratio of IC50 values of calibration curve, relative to each control and test sample curve. Sample-specific activity results are reported as U/mg.
Quality as measured by N-linked oligosaccharides map
N-linked oligosaccharides are a post-translational modification and each ETN monomer has three sites for the addition of N-linked oligosaccharide structures. Each of the three N-linked oligosaccharides on the ETN protein can be any one of multiple different species. The characteristic profile for the nine predominant species are routinely tested on every batch and must meet stringent acceptance ranges to ensure process consistency. N-linked glycans are chromatographically resolved into the various neutral (Peaks 1–5) and sialylated (Peaks 6–9) species after enzymatic cleavage from the protein. Each peak is controlled within a specified range.
0.25 M sodium phosphate, pH 7.5, (3 µL) and a 500,000 U/mL solution of peptide N-glycosidase F (2 µL) (New England Biolabs: #704L, Purified from Flavobacterium meningosepticum, free of proteases and Endo F activities) were added to the test solution (4 µL of 25 mg/mL solution). The mixture was incubated at 37°C for 20–24 h. The released N-glycans were labeled with 2-aminobenzamide. The labeled N-glycans were resuspended or diluted in water (100 µL) and separated by a reverse-phase HPLC (GlycoSepN HPLC column from Prozyme/ 5.0 µm) on an analytical column (4.6 mm × 250 mm) at 35°C. Components were separated by gradient elution of 80–20% of mobile phase B (mobile phase A, 0.5% formic acid in water [adjusted to pH with ammonia]; mobile phase B, acetonitrile) at a flow rate of 0.4 mL/minutes. Monitoring was by fluorescence detection (330 nm for excitation; 420 nm for emission). N-linked glycans are resolved into various neutral (Peaks 1–5) and sialylated (Peaks 6–9) species after enzymatic cleavage from the protein.
Disclosure of potential conflicts of interest
The authors are employees and shareholders of Pfizer.
Funding details
This work was funded by Pfizer.
Acknowledgments
Editorial/medical writing support was provided by Iain McDonald of Engage Scientific Solutions and was funded by Pfizer.
References
- European Medicines Agency. Enbrel. Authorisation details. EMEA/H/C/000262. European Medicines Agency; 2000 Feb 2 [accessed 2017 March 22]. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000262/human_med_000764.jsp&mid=WC0b01ac058001d124.
- Pfizer. Enbrel (etanercept) Summary of Product Characteristics. European Medicines Agency; 2010 Feb [accessed 2017 March 22]. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000262/WC500027361.pdf.
- Scott LJ. Etanercept: a review of its use in autoimmune inflammatory diseases. Drugs. 2014;74:1379–410. doi:10.1007/s40265-014-0258-9. PMID:25034360.
- Maksymowych WP, Dougados M, van der Heijde D, Sieper J, Braun J, Citera G, Van den Bosch F, Logeart I, Wajdula J, Jones H, et al. Clinical and MRI responses to etanercept in early non-radiographic axial spondyloarthritis: 48-week results from the EMBARK study. Ann Rheum Dis. 2016;75:1328–35. doi:10.1136/annrheumdis-2015-207596. PMID:26269397.
- Windschall D, Müller T, Becker I, Horneff G. Safety and efficacy of etanercept in children with the JIA categories extended oligoarthritis, enthesitis-related arthritis and psoriasis arthritis. Clin Rheumatol. 2015;34:61–9. doi:10.1007/s10067-014-2744-6. PMID:25034081.
- Scallon B, Cai A, Solowski N, Rosenberg A, Song XY, Shealy D, Wagner C. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301:418–26. doi:10.1124/jpet.301.2.418. PMID:11961039.
- Schneider CK. Biosimilars in rheumatology: the wind of change. Ann Rheum Dis. 2013;72:315–8. doi:10.1136/annrheumdis-2012-202941. PMID:23390018.
- European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Guideline on comparability of biotechnology-derived medicinal products after a change in the manufacturing process. Non-clinical and clinical issues. EMEA/CHMP/BMWP/101695/2006. London, UK: European Medicines Agency; 2007 July 19 [accessed 2017 March 22]. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003935.pdf..
- Vezér B, Buzás Z, Sebeszta M, Zrubka Z. Authorized manufacturing changes for therapeutic monoclonal antibodies (mAbs) in European Public Assessment Report (EPAR) documents. Curr Med Res Opin. 2016;32:829–34. doi:10.1185/03007995.2016.1145579. PMID:26808864.
- International Conference on Harmonisation (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonised tripartite guideline. Comparability of biotechnological/biological products subject to changes in their manufacturing process. Q5E. International Conference on Harmonisation; 2004 Nov 18 [accessed 2017 March 22]. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf..
- Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29:310–2. doi:10.1038/nbt.1839. PMID:21478841.
- Sandoz. Arthritis Advisory Committee Briefing Document. GP2015 (Proposed INN: etanercept) Biosimilar. US Food & Drug Administration. Silver Spring, MD.; 2016 June 6 [accessed 2017 March 22]. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM510494.pdf.
- European Medicines Agency. Enbrel: Procedural steps taken and scientific information after the authorisation. London (UK): European Medicines Agency; 2017. [accessed 2017 March 22]. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Procedural_steps_taken_and_scientific_information_after_authorisation/human/000262/WC500027366.pdf.
- Grampp G, Ramanan S. Managing unexpected events in the manufacturing of biologic medicines. Biodrugs. 2013;27:305–16. doi:10.1007/s40259-013-0018-5. PMID:23529766.
- US Food & Drug Administration. Enbrel® (etanercept) FDA approval letter. Rockville (MD) US: US Food & Drug Administration; 1998. Nov 2 [accessed 2017 March 22]. http://www.accessdata.fda.gov/drugsatfda_docs/appletter/1998/etanimm110298L.htm..
- Moreland LW, Weinblatt ME, Keystone EC, Kremer JM, Martin RW, Schiff MH, Whitmore JB, White BW. Etanercept treatment in adults with established rheumatoid arthritis: 7 years of clinical experience. J Rheumatol. 2006;33:854–61. PMID:16541481.
- Weinblatt ME, Bathon JM, Kremer JM, Fleischmann RM, Schiff MH, Martin RW, Baumgartner SW, Park GS, Mancini EL, Genovese MC. Safety and efficacy of etanercept beyond 10 years of therapy in North American patients with early and longstanding rheumatoid arthritis. Arthritis Care Res (Hoboken). 2011;63:373–82. PMID:20957659.
- Gibofsky A, Palmer WR, Keystone EC, Schiff MH, Feng J, McCroskery P, Baumgartner SW, Markenson JA. Rheumatoid arthritis disease-modifying antirheumatic drug intervention and utilization study: safety and etanercept utilization analyses from the RADIUS 1 and RADIUS 2 registries. J Rheumatol. 2011;38:21–8. doi:10.3899/jrheum.100347. PMID:20952478.
- Leonardi C, Strober B, Gottlieb AB, Elewski BE, Ortonne JP, van de Kerkhof P, Chiou CF, Dunn M, Jahreis A. Long-term safety and efficacy of etanercept in patients with psoriasis: an open-label study. J Drugs Dermatol. 2010;9:928–37. PMID:20684143.
- Papp KA, Poulin Y, Bissonnette R, Bourcier M, Toth D, Rosoph L, Poulin-Costello M, Setterfield M, Syrotuik J. Assessment of the long-term safety and effectiveness of etanercept for the treatment of psoriasis in an adult population. J Am Acad Dermatol. 2012;66:e33–45. doi:10.1016/j.jaad.2010.07.026. PMID:20850895.
- Kimball AB, Rothman KJ, Kricorian G, Pariser D, Yamauchi PS, Menter A, Teller CF, Aras G, Accortt NA, Hooper M, et al. OBSERVE-5: observational postmarketing safety surveillance registry of etanercept for the treatment of psoriasis final 5-year results. J Am Acad Dermatol. 2015;72:115–22. doi:10.1016/j.jaad.2014.08.050. PMID:25264239.
- International Conference on Harmonisation (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonised tripartite guideline. Specifications: test procedures and acceptance criteria for biotechnological/biological products. Q6B. International Conference on Harmonisation; 1999 March 10 [accessed 2017 Aug 16]. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q6B/Step4/Q6B_Guideline.pdf..