
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Biosimilars are products that are similar in terms of quality, safety, and efficacy to an already licensed reference/ innovator product and are expected to offer improved affordability. The most significant source of reduction in the cost of development of a biosimilar is the reduced clinical examination that it is expected to undergo as compared to the innovator product. However, this clinical relief is predicated on the assumption that there is analytical similarity between the biosimilar and the innovator product. As a result, establishing analytical similarity is arguably the most important step towards successful development of a biosimilar. Here, we present results from an analytical similarity exercise that was performed with five biosimilars of rituximab (Ristova®, Roche), a chimeric mouse/ human monoclonal antibody biotherapeutic, that are available on the Indian market. The results show that, while the biosimilars exhibited similarity with respect to protein structure and function, there were significant differences with respect to size heterogeneity, charge heterogeneity and glycosylation pattern.
Introduction
A biosimilar is defined as a biopharmaceutical drug designed to elicit clinical performance that is similar to that of an already licensed reference product.Citation1 Unlike their small molecule counterparts, monoclonal antibodies (mAbs) are more complex in nature due to their large size (150 kDa) and multi-chain structure (tetramer, IgG). Further, mAbs demonstrate significant micro-heterogeneity and batch-to-batch variability.Citation2
The European Medicines Agency (EMA) was the first regulatory agency to offer a regulatory framework for the development and approval of biosimilar products in 2006.Citation3 As per EMA's guidance, this approach expects the manufacturer to perform an extensive comparison with respect to quality, safety, and efficacy to show similarity between the reference, i.e., innovator product and the biosimilar. Since then, other countries have also introduced regulatory guidance for development of biosimilars. What remains common in all guidance documents thus far is the need for demonstration of similarity via extensive physicochemical and biological characterization as well as clinical studies.Citation4–5 In some jurisdictions, demand for extensive clinical trials have been challenged as being too cautious and hindering the development of biosimilars.Citation6
In a forward-looking step, the EMA has recently released a concept paper to revise the clinical requirements for granulocyte colony stimulating factor, thereby proposing criteria that would allow waiver of the clinical trial requirement for a biosimilar.Citation7 Recently, the US Food and Drug Administration (FDA) has released guidance on clinical pharmacological data to support a demonstration of biosimilarity to a reference product, indicating the possibilities to perform only selected clinical studies when comparative analytical characterization indicates a “highly similar proposed biosimilar with fingerprint-like similarity”.Citation8 At the 67th World Health Assembly, the World Health Organization (WHO) agreed that the next similar biotherapeutic product (SBP) guideline should also include affordability as a major consideration for biosimilars, while still ensuring their quality, safety, and efficacy.Citation9
Rituximab was the first mAb approved for treatment of cancer (B cell lymphoma), and it is also approved for immune-mediated and inflammatory diseases, e.g., rheumatoid arthritis, Wegener's granulomatosis.Citation10 An IgG1k chimeric mAb produced in Chinese hamster ovary (CHO) cells, rituximab targets the B-cell surface receptor CD20. Rituximab sequence is referenced as a 1328 amino acid protein in the International Immunogenetics Information system.Citation11 The heavy chain (HC) consists of 451 amino acids while the light chain (LC) comprises of 214 amino acids. HCs and LCs are linked by a single disulfide bond and the HCs by two S-S bridges located in a short hinge domain. Twelve additional cysteine bridges are intramolecular and delimit six different globular domains: one variable (VL) and one constant for the LC (CL); and, one variable (VH) and three constant for the HCs (CH1, CH2 and CH3).Citation12 Common post-translational modifications (PTMs) include a conserved N-glycosylation site within its Fc (Asn297) region, N-terminal glutamine to pyroglutamate (pyroGlu) cyclization, and partial C-terminal lysine loss during its synthesis in CHO cells.Citation13 The primary mechanism of action of rituximab comprises binding of its antigen-binding fragment (Fab) domains to CD20+ B-lymphocytes for induction of apoptosis by either antibody-dependent cell-mediated cytotoxicty (ADCC) and complement-dependent cytotoxicity (CDC).Citation14
In India, biosimilars are termed as “similar biologics” and are presently developed according to the guidelines issued by the Central Drugs Standard Control Organization (CDSCO) under the Ministry of Health and Family Welfare. Unlike those of EMA and US FDA, the Indian regulatory expectations do not include mandatory clinical testing for pharmacokinetics (PK)/ pharmacodynamics (PD) data, but do require a detailed in-depth physiochemical and functional characterization.Citation15 It is suggested that this be achieved by using an array of state-of-the-art analytical techniques, based on which a tailored non-clinical and clinical program can be designed. ICH Q5E and Q6B guidelines provide guidance on the physiochemical and structural features that should be considered for assessment of similarity.Citation16–17
Extensive literature exists on the topic of establishing structural and functional similarity of rituximab with respect to the innovator product, MabThera.Citation12,14,18–22 The physicochemical and biological characterization, followed by PD and immunogenicity studies on different batches of Kikuzubam from Probiomed S.A. and MabThera, has confirmed that similarity of critical quality attributes (CQA) results in comparable immunomodulatory activity.Citation14 A proposed biosimilar product, GP2013, produced in-house at Sandoz Biopharmaceuticals, has been a subject of several physicochemical and functional similarity exercises.Citation18 A stable expression in murine NS0 cells and biological evaluation of biosimilar 1B8 to rituximab from Roche has been shown to result in comparable biological properties of the molecules.Citation19 Another similarity exercise involving use of numerous mass spectrometric-based approaches for evaluation of primary structure and head-to-head similarity of functional properties has been performed on the biosimilar “Zytux” from AryoGen Pharmed (Iran) with respect to MabThera.Citation20 The functional similarity between the proposed biosimilar RTXM83 from PharmADN (Mabxience) and MabThera has been validated by calculating binding capacity and ADCC induction taking into account the different FcγRIIIa-158 polymorphisms.Citation21 In another study, an anti-CD20 mAb developed using RNA interference to decrease core fucosylation (RNAi-mediated) was characterized by LC-MS using dimethyl labeling and compared with MabThera to confirm the full sequence with the two mutation sites.Citation22 Determination and analysis of the relevant attributes for two proposed biosimilars (Reditux and Kikuzubam) showed differences in charge heterogeneity as related to Mabthera and demonstrated the capability of analytical methods with respect to identification of such physicochemical differences during similarity exercises.Citation12
Eight rituximab biosimilars have received marketing authorization in India thus far.Citation23 We evaluated five such marketed rituximab biosimilars (based on availability) against Ristova® using an extensive array of advanced analytical techniques to evaluate physiochemical and biological similarity. As shown here, our results demonstrate that, while the biosimilars exhibited similarity with respect to protein structure and function, there were measurable differences with respect to size heterogeneity, charge heterogeneity and glycosylation pattern.
Results
Structural and functional characterization was successfully performed using a wide array of state-of-the-art and orthogonal analytical techniques to compare rituximab biosimilars to Ristova®. Side-by-side comparison was performed to examine analytical similarity of rituximab biosimilars to Ristova® in accordance with the regulatory requirements.Citation24 The methodology was designed to identify any disparity in protein structure, mass, size, charge, hydrophobicity, and bioactivity as suggested in the Guidelines on Similar Biologics.Citation15 All of the biosimilars under consideration, as well as the innovator product (Ristova®), were found to be consistent with the manufacturer's specification on protein concentration (9.36–11.29 mg/ml) as detected by the UV/VIS spectrophotometer. presents an overview of extensive structural and functional characterization platform used for assessing similarity of rituximab biosimilars vs. Ristova®.
Table 1. Structural and functional characterization platform used for assessing similarity of Rituximab biosimilars vs. Ristova®.
The molecular mass and primary sequence were analyzed using reverse phase-high performance liquid chromatography (RP-HPLC) coupled with electrospray ionization-time of flight-mass spectrometry (ESI-TOF-MS). PTMs were identified by ESI-TOF-MS after tryptic digestion under reducing condition. N-glycan profiling was performed by hydrophilic interaction chromatography (HILIC) coupled with quadrupole time-of-flight-mass spectrometry (Q-TOF-MS). Higher order structure was evaluated by circular dichroism (CD), fluorescence, and Fourier transform infrared (FTIR) spectroscopy. Size heterogeneity was examined by size-exclusion-high performance liquid chromatography (SE-HPLC) and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Charge heterogeneity with or without carboxypeptidase B (CpB) digestion was assessed by cation exchange-high performance liquid chromatography (CEX-HPLC). Biological characterization was performed using in-vitro and receptor binding assays.
Structural characterization
Primary structure
MS was used to confirm the identity of rituximab through intact and reduced mass analysis, sequence verification using peptide mapping and N-glycan profiling.
Intact and reduced mass analysis
Intact mass analysis of rituximab revealed different molecular masses due to presence of glycoforms. ESI-TOF mass spectra extracted from total ion chromatogram (TIC) for the main peak with retention time (RT) at 11.3 min showed the occurrence of a typical charge envelope for high molecular weight proteins with the most abundant ion assigned to [M+35H]35+. This spectra was less resolved due to the presence of glycan heterogeneity, which, upon deconvolution, yielded masses corresponding to the most abundant glycoforms (G0F/G0F), (G1F/G0F), (G1F/G1F or G2F/G0F), (G2F/G1F), and (G2F/G2F), respectively, within mass accuracy of 10 ppm (). As given in Supplementary Table 1, Biosimilars 2, 4 and 5 correspond to the same glycoforms present in Ristova® except an unknown mass of 147726.2 Da. Reduced mass analysis of rituximab revealed the occurrence of light chain (LC) and heavy chain (HC), with the latter having different masses due to variability in glycoforms. ESI-TOF mass spectra extracted from the total ion chromatogram (TIC) for the LC (11 min) and HC (11.5 min) was highly resolved, which, upon deconvolution, yielded single mass for LC and masses corresponding to two glycoforms for HC, G0F/G0F and G1F/G0F, respectively (, Supplementary Table 1) within a mass accuracy of 10 ppm.
Figure 1. Comparison of deconvoluted ESI-TOF mass spectra for (A) intact and (B) reduced (LC – light chain, HC – heavy chain) Rituximab biosimilars and Ristova®, (C) Total compound chromatogram (TCC) mirror plots of peptide mapping for reduced Rituximab biosimilars (lower) and Ristova® (upper) obtained from ESI-TOF mass spectra upon trypsin digestion.
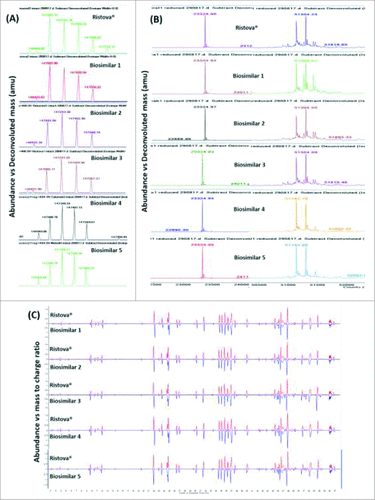
Based on the results, we concluded that the intact and reduced masses of rituximab biosimilars are near identical to Ristova®, with minor differences observed in the N-glycosylation pattern that require further characterization.
Peptide mapping and PTM identification
Trypsin cleavage at basic amino acid residues (lysine and arginine) resulted in generation of a moderate number of peptides from complete and incomplete digestion (miss cleavage of 2). The peak pattern (number and abundance of peaks) obtained in the total compound chromatogram using the Agilent MassHunter BioConfirm and Comparative Analysis software predicted 46 peptides for Ristova® and rituximab biosimilars, resulting in non-distinguishable chromatograms (). Minor differences in peak intensities were observed in samples, indicating that these peptides may have had small RT shifts. Though RP-HPLC is known to offer stable RT (typical variability ±0.02–0.05 min), when shallow gradients are used for protein separation, the variability can be of an order of magnitude or larger.Citation25 Protein sequence coverage of 100% for LC and 98% for HC were achieved within mass accuracy of 5 ppm. As given in Supplementary Table 2, the predicted sequences matched between Ristova® and rituximab biosimilars with occurrence of PTMs, such as N-terminal pyroGlu formation, deamidation, and oxidation at HC positions M20, M34, M256, M432 and LC position M21. The N-terminal sequences in HC corresponded to a peak at 34.95 min, matching the putative sequence QVQLQQPGAELVKPGASVKMSCK with mass of 2465.27 Da, and that of LC corresponded to a peak at 45.14 min, matching the putative sequence QIVLSQSPAILSASPGEKVTMTCR with mass of 2555.31 Da, with the presence of pyroGlu formation at position 1 in both. This result confirmed the conservation of the targeting region, which is also an important determinant of product half-life.Citation26 Minor differences were observed in N-terminal pyroGlu, wherein the modification was found in Ristova® and Biosimilar 2 only when the sequence was matched using predicted modification algorithm, but not using complete and incomplete digestion algorithm, indicating that N-terminal pyroGlu was not present in other rituximab biosimilars (Supplementary Table 2). Finally, the primary sequence of rituximab biosimilars compared to that of Ristova® was determined in accordance with earlier studies reported in the literature.Citation14,18
Table 2. N-glycan heterogeneity of Rituximab biosimilars and Ristova® as measured by HILIC QTOF MS after procainamide labelling (Number represents % glycan).
N-glycan profiling
N-linked glycosylation is widely considered as a CQA and its analysis is an important part of the characterization of therapeutic glycoproteins, especially mAbs. As given in , major glycoforms (G0F, G1F and G2F) were present in all samples with varying proportionsCitation27 and were in agreement with the estimated intact mass. More than 10 glycan species were identified and quantified individually (Man5, G0F-N, G0, Man6, G1F-N, G0F, G1, Man7, G1F, G2, Man8, G2F, G2FB, 1111 1A 0G, 1111 0A 1G, 2121 1A 0G, 2121 0A 1G) with differences in the number and abundance in all rituximab drug products, indicating the presence of high variability in the N-glycosylation pattern in rituximab biosimilars vs. Ristova®. While G0F (55.8% vs. 26.1%) was the most abundant glycoform present in Ristova® and most biosimilars, G2F (26.4% vs. 6.9%) was the most abundant glycoform in Biosimilar 3. Biosimilar 2 was found to be most similar to Ristova®, Biosimilar 3 possessed high % of galactosylated glycans (26.4% vs. 6.9%) and low % of sialylated glycans (3.1% vs. 0%) and Biosimilar 1 contained higher amount of mannose group (3.1% vs. 0%) and afucosylation (2.4% vs. 0.6%) as compared to Ristova®.
Higher order structure
CD, FTIR and fluorescence spectroscopy were employed to assess the higher order structure. CD spectroscopy in the far and near UV spectral regions provides an insight into the secondary structure (α-helix, β-sheet, random coil) and tertiary structure, respectively. The far UV CD spectra for all rituximab biosimilars were in compliance with Ristova®, determined by the presence of antiparallel β-pleated sheet and indicated by a characteristic negative dip at 216–218 nm (, ). The near UV CD spectra for all rituximab biosimilars did not overlay with that of Ristova® (), although the characteristic peaks of aromatic amino acids existed within the prescribed ranges, i.e., phenylalanine (b/w 255–270), tyrosine (Tyr) (b/w 275–282), and tryptophan (Trp) (b/w 290–305).
Figure 2. Comparison of higher order structure: overlay of (A) Far UV CD spectra, (B). Near UV CD spectra, (C) Secondary derivative FTIR spectra, (D) Intrinsic fluorescence spectra, and (E) Extrinsic fluorescence spectra for Rituximab biosimilars and Ristova®.
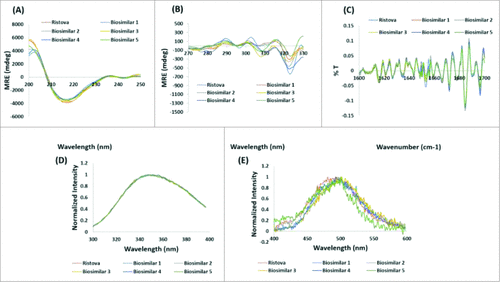
Table 3. Secondary structure analysis for Rituximab biosimilars and Ristova® as determined using Far UV CD, Fluorescence, FTIR spectroscopy.
FTIR provides information on the vibrational states of molecules. The protein backbone amide groups generated a number of characteristic IR bands that can be used to determine protein backbone conformation. The second derivative of FTIR spectra for all rituximab biosimilars and Ristova® showed indistinguishable overlap of the amide I and amide II bands (). summarizes results from analysis of the secondary structure for rituximab biosimilars and Ristova® as calculated from the percentage transmittance values obtained from FTIR spectra.Citation28 The presence of β-pleated sheet (57.7%), β-turn (22.3%), α-helix (9.4%), and random coil (10.4%) are observed. The results are in agreement with the observations made from analysis by CD spectroscopy. The percentage of β-sheets was lower (58% vs 70%) and the percentage of random coil was higher (11% vs 5%) than those previously published on mAbs.Citation29 This difference could be due to the presence of surfactants in the rituximab formulation buffer.
Fluorescence spectroscopy provides insight into the tertiary structure of the molecule. The changes in intrinsic fluorescence spectra for all rituximab biosimilars and Ristova® () indicated absorption of aromatic amino acids (Trp, Tyr) centered with λmax at 347 nm, revealing that Trp are exposed to the external environment. As given in , Biosimilar 3 and 4 showed λmax with a red-shift (moved to longer wavelength) by 2 nm, revealing that Trp had become more solvent exposed in these samples. The changes in extrinsic fluorescence spectra for all rituximab biosimilars and Ristova® () indicated continuous increase in fluorescence at 450 nm and decrease at 520 nm when excited at 280 nm, revealing that the hydrophobic patches and apolar sites have not changed in these samples.
Thus, it can be concluded that the higher order structures of the rituximab biosimilars were similar to that of Ristova®.
Size heterogeneity
Rituximab biosimilars and Ristova® were examined by SDS-PAGE under non-reducing condition for visual comparison of the various molecular forms based on size after SDS denaturation. Rituximab biosimilars were found to comply with Ristova®, and the principle band matched in position and intensity to the apparent molecular weight of rituximab (). Additionally, no high (HMW) and low molecular weight (LMW) variants with different molecular masses than rituximab were observed for all biosimilars.
Figure 3. Comparison of size and charge heterogeneity of Rituximab biosimilars and Ristova®: (A) Non reducing SDS-PAGE profile, (B) SEC chromatogram (insert shows a zoomed-in view), (C) Particle size distribution by DLS (insert shows a zoomed-in view), (D). CEX chromatogram before CpB digestion, (E) CEX chromatogram after CpB digestion.
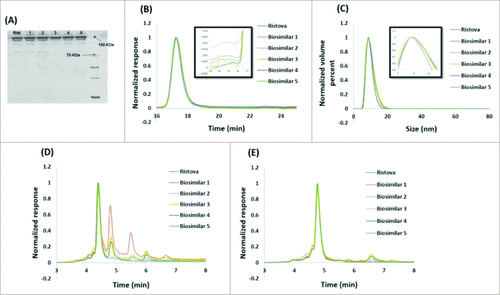
Purity of rituximab biosimilars and Ristova® were determined by SE-HPLC under native conditions. The chromatograms of rituximab biosimilars () showed a single peak with RT at 17.3 min corresponding to that of Ristova®, confirming low levels of HMW and LMW variants for four biosimilars. As given in , the HMW% of Biosimilar 3 was significantly higher (1.9% vs. 0.4%) than the other four biosimilars, although still within the specification of 2%.Citation30
Table 4. Table IV. Size and charge heterogeneity of Rituximab biosimilars and Ristova® as measured by SE-HPLC, DLS and CEX-HPLC.
Particle size distribution was examined with dynamic light scattering (DLS) by plotting the relative intensity of light scattered by particles in the various size classes as a function of hydrodynamic radii. It is evident from that a single peak was obtained corresponding to the rituximab monomer, and it had a diameter ranging from 10.5–11.1 nm (), in agreement with the values reported in literature.Citation31 Overall, we found all rituximab biosimilars except Biosimilar 3 to be comparable to Ristova® with respect to size heterogeneity.
Charge heterogeneity
Acid and basic variants can be formed by chemical and enzymatic modifications, with the acidic fractions typically consisting of deamidated, sialylated, and glycated forms and the basic fractions consisting of oxidated and C-terminal lysine variants.Citation18 Charge variants (acidic and basic) were examined by CEX-HPLC as per previously published protocol.Citation32 The specifications for charge variants are typically set based on equivalency of the summed percentage of acidic and basic variants vis-a-vis the innovator product. However, owing to difficulty associated with isolation of pure variants and the knowledge that not all affect the safety and efficacy of the product, a case-by-case investigation of individual variants is warranted.Citation33 CEX HPLC chromatograms of rituximab biosimilars exhibited a main peak with RT at 4.4 min (), corresponding to Ristova®. Two peaks with shorter retention time than the main were of acidic derivatives and four peaks with higher retention times than the main were of basic derivatives. As shown in , for all rituximab biosimilars, the sum of the acidic peaks was within the range of variability for Ristova® (9.5–15.2%), but the sum of the basic peaks was above the range of variability for Ristova®. While Biosimilar 3 contained more acidic species (13.7% vs. 8.9%), Biosimilar 1 contained more basic species (57.8% vs. 11.9%) than other drug products.
It should be noted that differences in charge heterogeneity are common among mAbs due to partial C-terminal lysine loss occurring in majority mAb species. This heterogeneity is widely believed not to have a significant effect on the safety and efficacy of biotherapeutics. Treatment of rituximab with carboxypeptidase B (CpB) was performed to remove the C-terminal lysine heterogeneity. The ubiquitous presence of this enzyme in almost all expression systems causes clipping of the C-terminal lysine.Citation34,35 CEX chromatograms of CpB-digested rituximab biosimilars showed main peak with RT at 4.8 min (), corresponding to that of Ristova®. As shown in , we observed that the sum of the acidic species remained the same for all rituximab biosimilars, whereas the sum of basic species reduced significantly, concluding the presence of C-terminal lysine variant. While Biosimilar 1 contained more acidic species (13.8% vs. 9.8%), Biosimilar 4 contained more basic species (8.2% vs. 1.2%) than other products.
Overall, we concluded that Biosimilars 1, 3, 4 and 5 are dissimilar to Ristova® with respect to charge heterogeneity.
Accelerated stability study
Accelerated stability studies are typically performed to investigate the effect of storage period, excipients, and environmental stress on drug product. They form an essential part of formulation development and similarity evaluations.Citation36 Guidelines for stability studies applied to similarity assessments are available as ICH Q5C.Citation37 Rituximab biosimilars were compared to Ristova® for multiple time periods under an elevated temperature stress condition at 50°C, as mAbs are more susceptible to degradation at 40°C and above. SE-HPLC profile showed a single peak with RT at 27 min corresponding to that of Ristova®, confirming low levels of HMW and LMW variants in rituximab biosimilars and Ristova®. This could be attributed to the occurrence of disulfide bonds, domain–domain interactions, and high content of β-sheets, providing stability to the mAb product.Citation38 After the third day onwards, differences were detected in levels of insoluble aggregation and the samples became visibly turbid. This was calculated by measuring concentration difference of the protein before and after centrifugation. As given in , Biosimilar 3 (29% vs. 15.8%) exhibited an increased rate of insoluble and soluble aggregate formation at 50°C compared to Ristova®. After 15 days, fragmentation was observed where Biosimilar 1 exhibited maximum fragmentation (4% vs. 2.4%) and Biosimilar 2 showed comparable fragmentation to Ristova®.
Biological characterization
A comprehensive set of bioanalytical methods were used to analyze functional integrity and similarity of rituximab biosimilars compared to Ristova®. Rituximab can induce death of CD20+ B cells by activating effector cells such as natural killer cells (NK), monocytes, and macrophages through the binding of the Fcγ receptors to its Fc domain.Citation39 These studies were designed taking into account the interactions of the Fc domains and their associated biological activities (affinity towards FcγR) that lead to B-cell depletion. FcγRIIIa, a pro-inflammatory receptor, expressed in human NK cells, is involved in the induction of ADCC.Citation40 Clinical studies have shown that affinity towards FcγRIIIa V158 is associated with a better response to rituximab in patients with follicular lymphoma.Citation41 The biological activity of rituximab biosimilars was compared to Ristova® using an ADCC assay. This assay involves the binding of a fully functional Fc domain to the target cell with binding to the Fcγ receptor (FcγRs) on the effector cells, triggering immune cell activation and death of the target cell.Citation42 The ability of the rituximab biosimilars to induce ADCC was assessed using Raji B cells as target cells and peripheral blood mononuclear cells (PBMCs) as effector cells measured as lactate dehydrogenase (LDH) activity.
In the CDC assay, C1q binds rituximab, and this triggers the complement cascade that leads to the formation of the membrane attack complex.Citation43 CDC activity was calculated using Raji B cells as target cells and human serum as the source of effector cells. It is well documented in literature that mAbs that display high ADCC and CDC are most suitable for therapeutic use against pathogens and cancer cells. Extensive efforts have been made to enhance these effector functions and successful approaches have been reported, wherein the binding activity of mAbs to FcγRIIIa or C1q is increased by introducing amino acid mutations into heavy chain constant regions or through glyco-modification of Fc-linked oligosaccharides.Citation44 and B shows the dose-response curve for the binding of rituximab biosimilars and Ristova® to FcγRs and C1q through ADCC and CDC, respectively. As given in , while the relative potency for rituximab biosimilars were comparable for Ristova® in ADCC assays, the relative potency for rituximab biosimilars were either lower or higher than for Ristova® in CDC assays. Biosimilars 1 and 3 particularly exhibited lower values in both ADCC and CDC, indicating lower activity with respect to Ristova®.
Figure 4. Comparison of in-vitro binding assays to receptors on CD20+ B cells through (A) ADCC, (B) CDC, (C) FACS for Rituximab biosimilars and Ristova® (Green line represents the range for Ristova®).
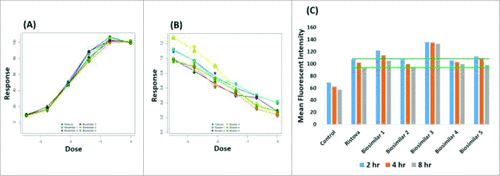
Table 5. Biological assays of Rituximab biosimilars and Ristova® as assessed by in-vitro target binding of CD20+ B cells using FACS, ADCC, CDC assays and binding to FcγRIIIa using Biacore X100TM.
Flow cytometry (fluorescence-activated cell sorting (FACS) and surface plasmon resonance (SPR) were used to measure antibody binding to cells that express specific Fab receptor and soluble Fc receptor, respectively. Binding affinity to CD20 receptor is the primary mechanism of action of rituximab, whose interaction is related to the structure of complementarity-determining regions (CDRs) in rituximab's Fabs.Citation45 This was assessed in a cell-based competitive binding assay, where FACS was performed to assess mean fluorescent intensity (MFI) of the drug product. As shown in , the binding of rituximab biosimilars and Ristova® to CD20+ Raji cells was compared through MFI values. As given in , Ristova® exhibited slightly lower binding than all except one of the rituximab biosimilars. Biosimilar 3 showed the highest binding, and Biosimilar 4 exhibited the lowest binding amongst the rituximab biosimilars. This difference between the mAbs was reproducibly observed with all the samples tested at different incubation times. Thus, rituximab biosimilars and Ristova® exhibited similar binding affinity towards CD20+ Raji cells (except Biosimilar 3). SPR was used to provide a comparison of the binding kinetics of rituximab biosimilars to human FcRγIIIa with respect to Ristova®. As given in , the equilibrium constant (KD) of rituximab biosimilars to FcγRIIIa was calculated to be within the same order of magnitude as Ristova®. KD values were compared, wherein Ristova® exhibited higher binding affinity than the rituximab biosimilars. Biosimilar 2 exhibited comparable affinity for binding to FcγRIIIa with respect to Ristova®, but others yielded lower values.
Biological characterization of the various rituximab biosimilars leads to the conclusion that, unlike the similarities seen during the characterization of protein structure, there are significant differences in the biological activities of the biosimilar products examined in this study.
Discussion
Patents for more than 20 mAb products have either expired or are on the verge of expiration.Citation46 While the respective biosimilar products are in the pipeline for regulatory approval, questions continue to arise with respect to their safety and effectiveness in comparison to the innovator product.Citation47 While all biosimilar guidelines continue to require clinical data, lack of awareness amongst patients still contributes to a low acceptance rate for biosimilars, which defeats the original motivation for development of the products.Citation48, 49 It is important to note that a biosimilar may potentially exhibit a range of differences compared to the innovator. In a study examining the similarity of rituximab biosimilars, variability in comparison to the innovator product was observed with respect to certain quality attributes.Citation50 Although biosimilarity is typically assessed using a panel of analytical tools and a number of physicochemical and biological methods are available for characterization of structure-function relationships, this examination is non-trivial.Citation51
The motivation of our study was to demonstrate the capability of the existing analytical similarity platforms for mAb products. Most of the previous studies have either presented the physicochemical properties of multiple batches of mAb product manufactured by a single biotech firmCitation52–55 or have focused on their biological properties.Citation56–58 Here, we utilize state-of-the-art structural and functional characterization methods and present a similarity exercise for rituximab biosimilars marketed in India, with respect to Ristova®, from five biotech firms. Indeed, the combination of several orthogonal methods, each having its own characteristic measuring principle, should be performed to study the physical, chemical and functional properties of biologics. As seen in , rituximab biosimilars were found to be comparable to Ristova® with respect to most of the criteria. However, significant differences were observed with respect to glycosylation profile, size heterogeneity, charge heterogeneity and biological properties.
Table 6. Summary of structural and functional similarity assessment of Rituximab biosimilars against Ristova®. Color code: Highly similar (green), moderately similar (yellow), not similar (red).
Since nearly all mAb biosimilars are produced in mammalian cells, they exhibit different PTMs, which results in heterogeneity. This could be attributed to differences in expression system, culture conditions, purification processes, formulations, or storage conditions of the product and need to be suitably addressed as per the “similar but not identical” paradigm.Citation59 Glycosylation greatly influences effector function by modulating binding to FcγR and has an impact on immunogenecity.Citation60 Fc sialylation has been known to lead to reduced ADCC activity and affect FcγRIIIa binding.Citation61 The presence of sialylated glycoforms in Biosimilar 3 () as compared to Ristova® resulted in increased acidic content of the protein, an observation supported by results obtained from CEX-HPLC (), resulting in reduced ADCC and CDC activity (). Increased galactosylation of CHO-derived antibodies enhances FcγRIIIa binding, ADCC and CDC activity.Citation62,63 The occurrence of a low amount of G0F and a high amount of G2F increased the level of galactosylation in Biosimilar 3 () as compared to Ristova®, but resulted in reduced ADCC and CDC activity (). Also, high mannose glycoforms exhibit higher FcγRIIIa binding, but show reduced CDC activity.Citation64,65 The presence of high mannose in Biosimilar 1 () as compared to Ristova® showed reduced CDC activity (). Antibodies lacking core fucosylation show a large increase in affinity for FcγRIIIa, leading to an improved receptor-mediated effector function.Citation66 Biosimilar 1 has more afucosylated glycoforms compared to Ristova®, and this perhaps explains the results from CD20 and FcγRIIIa binding (lowest KD), aggregation properties, ADCC and CDC assays. The presence of asialo-galactosylated biantennary and fucosylated monoantennary Fc glycans with varying level of sialic acid, N-acetylneuraminic acid (NANA) and N-glycolylneuraminic acid (NAGA) were observed in Biosimilar 1 and 3 (). However, the specific impact on biological activity requires further PK evaluation of these biosimilars.
It has been suggested that N-terminal pyroGlu formation in mAbs does not affect the safety and efficacy of the therapeutic, and rather it makes the protein more resistant to degradation.Citation67 Our results are in agreement with this observation. We observed that both Ristova® and Biosimilar 2 have this variant, exhibit similar biological activity and also better product stability compared to the other biosimilars under consideration (). Aggregates stimulate immune responses causing protein misfolding and can have deleterious and harmful effects. In a study, researchers have suggested that uptake and activation of immune response for aggregates are found to be more than that of its monomer.Citation68 This was in congruence with our results for Biosimilar 3, which contained higher level of HMW than Ristova®, showing higher binding affinity towards CD20+ B cells ().
In conclusion, biosimilarity was assessed at the quality level on the basis of a very comprehensive similarity exercise. Differences were detected mainly for Biosimilar 1 and 3 in the charged variants and glycan profiles, and found to have some effect on biological activity in the in vitro assays.
Materials and methods
Rituximab reference standard (Ristova® from Roche; 10 mg/ml) and five biosimilars manufactured by Indian companies were purchased from Sai Distributors, New Delhi, and Vardhman Health Specialities, Bangalore. The latter included Reditux (Biosimilar 1) from Dr. Reddy's Laboratories; 10 mg/ml, RituxiRel (Biosimilar 2) from Reliance Life Sciences; 10 mg/ml, Mabtas (Biosimilar 3) from Intas Biopharmaceuticals; 10 mg/ml, Maball (Biosimilar 4) from Hetero drugs; 10 mg/ml, and Cytomab (Biosimilar 5) from Alkem Laboratories; 10 mg/ml. All the products consisted of similar formulation buffer (sodium citrate dihydrate (7.35mg), sodium chloride (9mg), polysorbate 80 (0.7mg), pH-6.5).Citation69 Multiple 200 µl aliquots were made for each sample, stored at −80 °C, and used such that all samples underwent a single freeze/thaw cycle before analysis. The required concentration for different tools was achieved either by diluting or performing gel filtration chromatography-based buffer exchange using Sephadex G-25 resin (GE Healthcare) packed into a Tricon™ column (100 × 10 mm, GE Healthcare). Before every test, visual inspection was performed for the potential presence of visible particles. All products remained clear and colorless. In all cases, products were tested within their shelf lives and tests were conducted in triplicates.
Protein concentrations
The concentration of protein in the samples was determined by measuring optical density (OD) at 280 nm on ultraviolet-visible (UV-Vis) Spectra Max M2e Multimode Microplate Reader (Molecular Devices) in congruence to the Lambert-Beer's Law. Sample readings were recorded in triplicate and normalized by subtracting the readings from the blank buffer. Rituximab extinction coefficient ϵ280 = 1.7 mL/ (mg cm) was used for the calculation.
Intact and reduced mass analysis
RP-HPLC was performed to determine the intact mass of rituximab biosimilars on the AdvanceBio RP mAb C4 (4.6 × 100 mm, 5 μm, Agilent Technologies) column operated at 80°C using Agilent 1260 Infinity Bio-inert Quaternary LC system consisting of a quaternary pump with a degasser, an auto sampler with a cooling unit, and a diode array detector (DAD) coupled to an Agilent 6230 ESI-TOF-MS instrument. Prior to injection, the column was saturated with 90% mobile phase A (0.1% v/v formic acid (FA) in MilliQ) and 10% mobile phase B (0.1% v/v FA in acetonitrile). Rituximab samples were buffer exchanged to 0.1% v/v FA in MilliQ by filtration through 10 kDa MWCO Centricons (Pall Corporation, USA). 10 μg sample was loaded on the column and separated using a 30 min linear gradient from 10% – 65% B at a flow rate of 0.5 ml/min. Detection was performed by monitoring UV absorption at 280 nm and TIC was recorded for 1,000–7,000 m/z. MS spectra were calibrated in the positive ion mode prior to analysis. The capillary gas temperature/ voltage (Vcap) was set to 350°C and 5,500 V, respectively, and the fragmentor voltage (Vfrag) was 400 V. The MS spectra were deconvoluted using the maximum entropy (MaxEnt) and peak modeling (pMod) algorithm as part of the Agilent MassHunter Qualitative Analysis and BioConfirm software. For reduced mass analysis, 5 mM dithiothreitol (DTT) was added to 1 mL of sample and the mixture was incubated at 37°C for 30 min.
Peptide mapping and PTM identification
Peptide mapping was performed on reduced tryptic digest to determine the amino acid sequence and possible PTMs, including deamidation, oxidation, and glycosylation at Asn297 in rituximab biosimilars. Samples were solubilized in 6 M guanidine hydrochloride (Gn-HCl) + 100 mM Tris hydrochloride (Tris-HCl) at pH 7.8, reduced with 5 mM DTT, alkylated with 15 mM iodoacetamide (IAM) and diluted four folds with 100 mM Tris-HCl (pH 7.8). Digestion was performed using MS grade trypsin (1:20 w/w) from Agilent Technologies with overnight incubation at 37°C and acidified with FA. Samples were stored at −20°C while waiting for analysis. RP-HPLC was performed on AdvanceBio peptide mapping C18 (4.6 × 150 mm, 2.7 μm, Agilent Technologies) column operated at 55°C using Agilent 1260 Infinity Bio-inert Quaternary LC system coupled to an Agilent 6230 ESI-TOF-MS instrument operated in data-dependent mode. Prior to injection, the column was saturated with 95% mobile phase A (0.1% v/v FA in MilliQ) and 5% mobile phase B (0.1% v/v FA in acetonitrile). Digested samples were desalted to 0.1% v/v FA in 60% acetonitrile (ACN) using solid phase extraction by C18 ZipTip™ (Millipore Corporation), 10 μg digested sample loaded on the column and separated using a 75 min linear gradient from 5% – 65% B at a flow rate of 0.3 ml/min. Detection was performed by monitoring UV absorption at 215 nm and TIC recorded for m/z 100–3200. MS spectra was calibrated in positive ion mode prior to analysis. The capillary gas temperature/ Vcap was set to 300°C and 4500 V, respectively and the Vfrag was 300 V. MS spectrum was analyzed using the protein molecular feature extraction (MFE) algorithm in the Agilent MassHunter Qualitative Analysis and BioConfirm software with tolerance of 10.0 ppm to obtain list of probable peptides and matched with the masses of in silico digested Ristova® peptides to obtain sequence coverage. Mirror plot comparing the result of innovator to biosimilar was generated using Agilent MassHunter Comparative Analysis software. For disulfide bridging, samples were solubilized in 6 M Gn-HCl + 100 mM Tris-HCl and diluted four folds with 100 mM Tris-HCl (pH 7.8), skipping the reduction step and digested using MS grade trypsin (1:20 (w/w), Agilent Technologies) with overnight incubation at 37°C.
Released glycan profiling
Glycan profiling was performed to determine glycosylation pattern at Asn297 in Ristova® biosimilars. Samples were denatured by heating at 95°C for 10 min along with denaturation reagent (2% SDS, 1 M β-mercaptoethanol), followed by detergent solution (15% NP-40). Digestion was performed using MS grade PNGase F (4 mU, Promega; Cat# GKE-5006B) with 3 hour incubation at 37°C. The released glycans were purified using AdvanceBio N-glycan deglycosylation cleanup cartridges (Agilent Technologies) and dried overnight in vacuum concentrator. The dried glycan samples were mixed with active labelling reagent, InstantPC (10 mg of procainamide hydrochloride + 6 mg of sodium cyanoborohydride in 100 µL of 3:7 v/v acetic acid: dimethyl sulfoxide), and heated at 65°C for 3 hours. The labelled glycans were purified using AdvanceBio N-glycan labelling cleanup cartridges (Agilent Technologies) and reconstituted in 70:30 ACN: water. HILIC was performed to separate glycan from rituximab biosimilars on AdvanceBio glycan mapping column (2.1 × 100 mm, 1.8 μm, Agilent Technologies) using Agilent 1290 Infinity Bio-inert Quaternary LC system consisting of a quaternary pump with a degasser, an auto sampler with a cooling unit, and a DAD coupled to an Agilent 6550 iFunnel Q-TOF-MS with JetStream. Prior to injection, the column was saturated with 40% mobile phase A (100 mM ammonium formate, pH 4.5) and 60% mobile phase B (100% ACN). 10 μg samples were loaded on the column and separated using a 12 min linear gradient from 60%–75% B at a flow rate of 0.5 ml/min. Detection was performed by monitoring TIC at 300–3,200 m/z. MS spectra was calibrated in positive ion mode prior to analysis. The capillary gas temperature/ Vcap was set to 200°C/ 3,500 V, respectively, and the fragmentor voltage (Vfrag) was 175 V. The MS spectra was analyzed using the protein molecular feature extraction (MFE) algorithm in the Agilent MassHunter Qualitative Analysis and BioConfirm software with tolerance of 10.0 ppm to obtain list of probable glycans and matched with in-house PCDL (Personal Compound Database Library) database of PCA-labelled glycans (containing 20 compounds).
Circular dichroism
Far-UV CD spectra were recorded in the range of 200–250 nm at 25°C on a JASCO J-815 spectropolarimeter with a spectral band width of 5 nm using 0.1 cm path length quartz cell at a scan speed of 50 nm/min. Sample concentration was kept at 0.2 mg/ml and three spectra were scanned, averaged and finally plotted after subtracting the buffer baseline. Mean residue ellipticity (MRE, deg cm2/ dmole) was calculated, where Mo is mean residue weight (113 Da/ residue), θ is observed ellipticity (mdeg), l is light path (cm), and c is concentration (mg/ml).Citation70Near-UV CD spectra was recorded in the range of 250–340 nm at 25°C on a JASCO J-815 spectropolarimeter with a spectral band width of 5 nm using 1 cm path length quartz cell at a scan speed of 50 nm/min. Sample concentration was kept at 1 mg/ml and three spectra were scanned, averaged, and plotted after subtracting the buffer baseline. MRE (deg cm2/dmole) was calculated as described above.
Fluorescence spectroscopy
Intrinsic fluorescence spectroscopy was performed on a Spectra Max M2e Multimode Microplate Reader (Molecular Devices) spectrophotometer with a path length of 1 cm. Emission spectra of the samples were recorded at temperature of 25°C in the range of 300–400 nm with an excitation wavelength of 280 nm. The slits for excitation was 5 nm and emission was 10 nm. All samples were diluted to 0.2 mg/mL with sample buffer. Furthermore, extrinsic fluorescence spectroscopy was employed using fluorescent dye, 1-anilino-8-naphthalenesulfonate (ANS), to examine hydrophobic milieu such as protein interiors. Emission spectra of the samples were recorded in the range of 400–600 nm with an excitation wavelength of 280 nm.
Fourier transform infrared spectroscopy
FTIR spectroscopy measurements were made at 25°C using a Perkin Elmer 100 FTIR spectrometer equipped with a Universal Attenuated total reflectance (UATR) single reflection diamond accessory. The FTIR spectra of buffer blank and that of sample was recorded over a range of wave number from 4000–650 cm−1 and subtracted from the sample spectra. Each sample was analyzed in triplicate by using instrument specific software. The second derivative spectra was calculated using 11 point Savitsky-Golay smoothing of the original spectra. All samples were diluted to 3 mg/mL with formulation buffer.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Tris glycine SDS-PAGE was performed under non-reducing condition. Polyacrylamide gels were prepared from acrylamide and bisacrylamide stock solution (29:1), stacking buffer (1 M Tris-HCl, pH 6.8), resolving buffer (2 M Tris-HCl, pH 8.8), 10% SDS, APS, and TEMED as the catalyst. Stacking gel was used at 4% and resolving gel at 12% concentration. Samples were mixed with 5X loading buffer (1 M Tris-HCl, pH 6.8, 10% SDS, 50% glycerol, 2% w/v bromophenol blue) and heated for 10 min at 90°C before loading onto the gel. 5 ng of sample was loaded and run at 150 V until dye reached 80% gel length on a MiniPROTEAN® tetra cell (Bio-Rad Laboratories). Protein marker ladder (BioRad Laboratories) with molecular weights ranging between 10 and 250 kDa was used as standard. Later, the gel was fixed with a mixture of methanol: acetic acid solution (40%: 10%), stained with silver staining, and scanned on a GS-900™ Calibrated Densitometer (BioRad Laboratories).
Size-exclusion-high performance liquid chromatography
SE-HPLC was performed to quantify aggregates on TSKgel 3000SWXL (7.8 × 300 mm, 5 μm, Tosoh) column operated at 30°C using Dionex Ultimate 3000 UHPLC system (Thermo Scientific) consisting of a quaternary pump with a degasser, an auto sampler with a cooling unit, and a variable wavelength detector (VWD). 50 μg sample was loaded and isocratic elution was performed for 30 min at a flow rate of 0.5 ml/min with 50 mM sodium phosphate buffer, 300 mM NaCl, and 0.05% NaN3 at pH 6.8. All buffers were filtered with a 0.22 μm cutoff nylon membrane filter (Pall Corporation) and degassed prior to use. Detection was performed by monitoring UV absorbance at 280 nm. Chromeleon software (Thermo Scientific) was used for chromatogram integration and estimating monomer content through relative peak area percentage values.
Dynamic light scattering
The hydrodynamic radii and particle size distribution of samples were determined using Zetasizer Nano ZS 90 (Malvern Instruments) with temperature control fitted with a 633-nm He-Ne laser. The instrument uses dynamic light scattering to measure the diffusion coefficient, D, which is then converted to an average hydrodynamic size, RH, using the Stokes-Einstein equation, where kB is Boltzmann constant, T is absolute temperature and ηs is the solvent viscosity (taken as 0.8 mPa.S).Citation71The sample concentration was kept at 1 mg/ml and scattered intensities were recorded at a fixed scattering angle of 90°. The instrument has the ability to measure wide size range (0.3–5000 nm) and the diameter under consideration ranges from 10–210 nm, which is within the size range of the instrument.
Cation exchange-high performance liquid chromatography
CEX-HPLC was performed to quantify charge variants on MAbPac SCX-10 RS, BioLC™ (4.6 × 250 mm, 5 μm, Thermo Scientific) column operated at 25°C using Dionex Ultimate 3000 RSLC system (Thermo Scientific) consisting of a quaternary pump with a degasser, an auto sampler with a cooling unit, and a DAD. Prior to injection, the column was saturated with 95% mobile phase A (15 mM sodium phosphate buffer and 0.05% NaN3 at pH 6.2) and 5% mobile phase B (150 mM sodium phosphate buffer and 0.05% NaN3 at pH 6.2). All buffers were filtered with a 0.22 μm cutoff nylon membrane filter (Pall Corporation) and degassed prior to use. 20 μg sample was loaded and differently charged species were separated using a 13 min sigmoidal gradient from 5% – 35% B at a flow rate of 1 ml/min.Citation41 Detection was performed by monitoring UV absorbance at 280 nm. For C terminal lysine cleavage, 15 μL of 1 mg/mL carboxypeptidase B (CpB, Sigma-Aldrich; Cat# C9584) was added to 1 mL of sample and the mixture was incubated at 37°C for 30 min. Chromeleon software (Thermo Scientific) was used for chromatogram integration and estimating acidic, basic and main content through relative peak area percentage values.
Accelerated stability study
Thermal stability was examined at 50°C for 0, 3, 5, 10, 15 days with subsequent characterization of the degradation profile using SE-HPLC on Superdex 200 (7.8 × 300 mm, 5 μm, GE Healthcare) column using Dionex Ultimate 3000 UHPLC system (Thermo Scientific). 100 μg sample was loaded and isocratic elution was performed for 45 min at a flow rate of 0.5 ml/min w+ith 50 mM sodium phosphate buffer, 300 mM NaCl, and 0.05% NaN3 at pH 6.8. All buffers were filtered with a 0.22 μm cutoff nylon membrane filter (Pall Corporation) and degassed prior to use. Detection was performed by monitoring UV absorbance at 280 nm. Chromeleon software (Thermo Scientific) was used for chromatogram integration.
Cell lines and culture conditions
Raji cells lines were obtained from National Centre for Cell Science, Pune, India and maintained in RPMI 1640 media (Gibco) with 10% FBS (Gibco) and grown with 5% CO2 in an incubator at 37°C.
Antibody-dependent cell-mediated cytotoxicity assay
Cryopreserved standard Human PBMCs (Cellular Technology Limited; Cat# 70025.2) was thawed just before ADCC assay according to the manufacturer's protocol. PBMCs (2.5 × 105 cells/ well) and Raji cells (104 cells/ well) were suspended in RPMI 1640 media, seeded in a 96-well plate with serially diluted Ristova® and rituximab biosimilars. After incubation in 5% CO2 for 4 hours at 37°C, LDH activity of cell culture supernatants were measured by using LDH Cytotoxicity Assay Kit (Thermo Scientific) as described in the manufacturer's protocol. The percent cytotoxicity was calculated according to the following formula: %lysis = (experimental release – spontaneous release)/ (maximum release – spontaneous release) × 100. The absorbance at 490 nm was measured by Epoch microplate reader (BioTek Instruments). Relative potency was determined using parallel logistic (4-parameter fit) assay on PLA 3.0 bioassay software.
Complement-dependent cytotoxicity assay
Raji cells were incubated in RPMI 1640 media containing 10% FBS with different concentrations of rituximab biosimilars and complement human serum (Millipore Corporation; Cat# S1-100ml) in 5% CO2 overnight at 37°C. MTS substrate was added to each well with a further incubation of 3 hours. Relative potency was determined using parallel logistic (4-parameter fit) assay on PLA 3.0 bioassay software.
Binding to Raji cells
Binding of different concentrations of rituximab biosimilars to Raji cells was determined by a cell-based competitive binding assay in which test samples were used to displace a fluorescently labeled rituximab. One million cells per tube were incubated with rituximab biosimilars or with the corresponding conjugate in PBS containing 2% (w/v) bovine serum albumin for 2, 4 and 8 hours in 5% CO2 incubator at 37°C. After incubation, cells were washed twice with PBS and then incubated with FITC-labeled rat anti-rituximab polyclonal antibody (Bio-Rad Laboratories; Cat# MCA2260F) at 1:200 for 30 min on ice. After washing, cells were examined by using BD FACS Aria III (BD Biosciences) and results were analyzed using BD FACS Diva software (BD Biosciences).
Surface plasmon resonance
The kinetics of rituximab binding to human FcRγIIIa (CD16a) receptor (R&D systems; Cat# 4325-FC) was determined by Biacore X100TM (GE Healthcare). Samples were dialyzed against running buffer (HBS-EP buffer, pH 7.4), His-conjugated FcγRIIIa were immobilized on carboxymethyl dextran-coated CM5 sensor chips (GE Healthcare) by His coupling method (300 RU). Samples were injected for 120 s followed by 150 s dissociation phase. All measurements were performed at 25◦C with a flow rate of 30 μl/min according to the manufacturer's protocol. Five concentrations (0.5 to 16 nM) of Rituximab were then injected. Kinetic constants were calculated from the sensorgrams using the 1:1 fit model using BIA Evaluation 2.0.1 (GE Healthcare) software.
Statistical analysis
Data were analyzed using statistical analysis software (GraphPad Prism). All data represent the mean (± standard deviation, SD) of three independent experiments and each experiment was performed in triplicate. Student's t tests were performed to distinguish significantly different results (P < 0.05).
Abbreviations
| ACN | = | acetonitrile |
| ADCC | = | antibody-dependent cell-mediated cytotoxicity |
| ANS | = | 1-anilino-8-naphthalenesulfonate |
| APS | = | adenosine 5′-phosphosulfate |
| Asn | = | asparagine |
| CD | = | circular dichroism |
| CDC | = | complement-dependent cytotoxicity |
| CDR | = | complementarity-determining regions |
| CDSCO | = | Central Drugs Standard Control Organization |
| CEX-HPLC | = | cation exchange-high performance liquid chromatography |
| CHO | = | Chinese Hamster Ovary |
| CpB | = | carboxypeptidase B |
| CQA | = | critical quality attributes |
| D | = | diffusivity |
| DAD | = | diode array detector |
| DTT | = | dithiothreitol |
| EC50 | = | Half maximal effective concentration |
| EMA | = | European Medicines Agency |
| ESI-TOF-MS | = | electrospray ionization-Time-of-flight-mass spectrometry |
| FA | = | formic acid |
| FACS | = | fluorescence-activated cell sorting |
| FDA | = | Food and Drug Administration |
| FITC | = | fluorescein-5-isothiocyanate |
| FTIR | = | Fourier transform infrared |
| Gln | = | glutamine |
| Gn-HCl | = | guanidine hydrochloride |
| HC | = | heavy chain |
| HILIC | = | hydrophilic interaction chromatography |
| HMW | = | high molecular weight |
| IAM | = | iodoacetamide |
| ICH | = | International Conference on Harmonisation |
| IgG | = | Immunoglobulin G |
| IU | = | international unit |
| kDa | = | kilodalton |
| LC | = | light chain |
| LDH | = | lactate dehydrogenase |
| LMW | = | low molecular weight |
| m/z | = | mass-to-charge ratio |
| mAbs | = | monoclonal antibodies |
| mAU | = | milli absorbance unit |
| MaxEnt | = | maximum entropy |
| MCR | = | molecular coupling ratio |
| MFE | = | molecular feature extractor |
| MFI | = | mean fluorescent intensity |
| MRE | = | mean residue ellipticity |
| MWCO | = | molecular weight cut-off |
| NaCl | = | sodium chloride |
| NaN3 | = | sodium azide |
| NK | = | natural killer |
| OD | = | optical density |
| PBMC | = | peripheral blood mononuclear cells |
| PBS | = | phosphate-buffered saline |
| PD | = | pharmacodynamics |
| pyroGlu | = | pyroglutamate |
| PK | = | pharmacokinetic |
| pMod | = | peak modeling |
| ppm | = | parts per million |
| PTM | = | post-translational modification |
| Q-TOF | = | Quadrupole-Time of Flight |
| RP-HPLC | = | reversed-phase high-performance liquid chromatography |
| RT | = | Retention time |
| SD | = | standard deviation |
| SDS-PAGE | = | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SE-HPLC | = | size exclusion-high performance liquid chromatography |
| SPR | = | surface plasmon resonance |
| TEMED | = | N, N, N′, N′-tetramethylethylenediamine |
| TIC | = | total ion chromatogram |
| Tris-HCl | = | Tris (hydroxymethyl) methylamine hydrochloride |
| Try | = | typtophan |
| Tyr | = | tyrosine |
| UATR | = | Universal attenuated total reflectance accessory |
| UHPLC | = | ultra-high performance liquid chromatography |
| UV-VIS | = | ultraviolet-visible |
| Vcap | = | Capillary voltage |
| Vfrag | = | fragmentor voltage |
| VWD | = | variable wavelength detector |
| λmax | = | maximum wavelength |
Conflict of interests
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work and there is no conflict of interest regarding the publication of this paper.
SUPPLEMENTARY_MATERIAL.doc
Download MS Word (757.5 KB)Acknowledgments
This work was funded by the Center of Excellence for Biopharmaceutical Technology grant under Department of Biotechnology, Government of India (BT/COE/34/SP15097/2015). The authors would like to acknowledge Agilent Technologies India Pvt. Ltd. for providing technical support in this investigation.
References
- Rathore AS. Follow-on protein products: scientific issues, developments and challenges. Trends Biotechnol. 2009;27(12):698–705. doi:10.1016/j.tibtech.2009.09.004. PMID:19846228.
- Mazur MT, Seipert RS, Mahon D, Zhou Q, Liu T. A platform for characterizing therapeutic monoclonal antibody breakdown products by 2D chromatography and top-down mass spectrometry. AAPS J. 2012;14(3):530–541. doi:10.1208/s12248-012-9361-6. PMID:22581105.
- European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2014 (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf)
- Desanvicente-Celis Z, Caro-Moreno J, Enciso-Zuluaga M, Anaya JM. Similar biotherapeutic products in Latin America. Regulation and opportunities for patients with autoimmune diseases. Biosimilars. 2013;3:1–17.
- Nupur N, Singh SK, Narula G, Rathore AS. Assessing analytical comparability of biosimilars: GCSF as a case study. J Chromatography B. 2016;1032:165–171. doi:10.1016/j.jchromb.2016.05.027.
- Schellekens H, Moors E. Clinical comparability and European biosimilar regulations. Nat Biotechnol. 2010; 28(1):28. doi:10.1038/nbt0110-28. PMID:20062035.
- European Medicines Agency CHMP. Concept paper on the revision of the guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant granulocyte-colony stimulating factor. 2015 (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/07/WC500190635.pdf)
- U.S. FDA, Department of Health and Human Services. Guidance for Industry: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product (Draft). 2016 (https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm397017.pdf)
- World Health Organization. Draft guidelines on evaluation of monoclonal antibodies as similar biotherapeutic products (SBPs). 2016 (http://www.who.int/biologicals/mAb_1st_draft_KG_IK_1_March_2016_clean.pdf)
- Singh V, Gupta D, Almasan A. Development of novel anti-CD20 monoclonal antibodies and modulation in CD20 levels on cell surface: looking to improve immunotherapy response. J Cancer Sci Therapy. 2015;7(11):347–358. doi:10.4172/1948-5956.1000373.
- IMGT Repertoire (IG and TR). Monoclonal antibodies with clinical indications (http://www.imgt.org/IMGTrepertoire/GenesClinical/monoclonalantibodies/#rituximab)
- Flores-Ortiz LF, Campos-García VR, Perdomo-Abúndez FC, Pérez NO, Medina-Rivero E. Physicochemical properties of Rituximab. J Liquid Chromatography Related Technologies. 2014;37(10):1438–1452. doi:10.1080/10826076.2013.794738.
- Beck A, Diemer H, Ayoub D, Debaene F, Wagner-Rousset E, Carapito C, Van Dorsselaer A, Sanglier-Cianférani S. Analytical characterization of biosimilar antibodies and Fc-fusion proteins. Trends Analytical Chem. 2013;48:81–95. doi:10.1016/j.trac.2013.02.014.
- Miranda-Hernández MP, López-Morales CA, Ramírez-Ibáñez ND, Piña-Lara N, Pérez NO, Molina-Pérez A, Revilla-Beltri J, Flores-Ortiz LF, Medina-Rivero E. Assessment of physicochemical properties of rituximab related to its immunomodulatory activity. J Immunol Res. 2015;2015:910763. doi:10.1155/2015/910763. PMID:25973441.
- Guideline on similar biologics: Regulatory requirements for marketing authorization in India. Department Biotechnol Govt. India. 2012 (http://cdsco.nic.in/writereaddata/Bio%20Similar%20Guideline.pdf)
- Guideline IHT. Comparability of biotechnological/biological products subject to changes in their manufacturing process Q5E. 2004 (http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf)
- Guideline IHT. Specifications: Test procedures and acceptance criteria for Biological product Q6B. 1999 (http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q6B/Step4/Q6B_Guideline.pdf)
- Visser J, Feuerstein I, Stangler T, Schmiederer T, Fritsch C, Schiestl M. Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs. 2013;27(5):495–507. doi:10.1007/s40259-013-0036-3. PMID:23649935.
- Dorvignit D, Palacios JL, Merino M, Hernández T, Sosa K, Casacó A, López-Requena A, Mateo de Acosta C. Expression and biological characterization of an anti-CD20 biosimilar candidate antibody: a case study. MAbs. 2012;4(4):488–496. doi:10.4161/mabs.20761. PMID:22647435.
- Montacir O, Montacir H, Eravci M, Springer A, Hinderlich S, Saadati A, Parr MK. Comparability study of Rituximab originator and follow-on biopharmaceutical. J Pharmaceutical Biomed Analysis. 2017;140:239–251. doi:10.1016/j.jpba.2017.03.029.
- Cuello HA, Segatori VI, Alberto M, Pesce A, Alonso DF, Gabri MR. Comparability of antibody-mediated cell killing activity between a proposed biosimilar RTXM83 and the originator Rituximab. BioDrugs. 2016;30(3):225–231. doi:10.1007/s40259-016-0171-8. PMID:27053342.
- Li C, Rossomando A, Wu SL, Karger BL. Comparability analysis of anti-CD20 commercial (rituximab) and RNAi-mediated fucosylated antibodies by two LC-MS approaches. MAbs. 2013;5(4):565–575. doi:10.4161/mabs.24814. PMID:23751726.
- GaBI Online. Biosimilars of rituximab. Mol, Belgium: Pro Pharma Communications International. 2015 (http://www.gabionline.net/Biosimilars/General/Biosimilars-of-rituximab) [accessed on August 2017]
- Liao JJ, Darken PF. Comparability of critical quality attributes for establishing biosimilarity. Statistics Med. 2013;32(3):462–469. doi:10.1002/sim.5564.
- Dolan JW. How much retention time variation is normal? LCGC North Am. 2014;32(8):546–551.
- Varshavsky A. The N-end rule: functions, mysteries, uses. Proc Natl Acad Sci. 1996;93(22):12142–12149. doi:10.1073/pnas.93.22.12142. PMID:8901547.
- Raju TS, Jordan RE. Galactosylation variations in marketed therapeutic antibodies. MAbs. 2012;4(3):385–391. doi:10.4161/mabs.19868. PMID:22531450.
- Yang H, Yang S, Kong J, Dong A, Yu S. Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy. Nat Protocols. 2015;10(3):382–396. doi:10.1038/nprot.2015.024. PMID:25654756.
- Matheus S, Friess W, Mahler HC. FTIR and nDSC as analytical tools for high-concentration protein formulations. Pharmaceutical Res. 2006;23(6):1350–1363. doi:10.1007/s11095-006-0142-8.
- European Medicines Agency, CHMP. Guideline on development, production, characterization and specifications for monoclonal antibodies and related products. 2008 (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/08/WC500211640.pdf)
- Arosio P, Barolo G, Müller-Späth T, Wu H, Morbidelli M. Aggregation stability of a monoclonal antibody during downstream processing. Pharmaceutical Res. 2011; 28(8):1884–1894. doi:10.1007/s11095-011-0416-7.
- Joshi V, Kumar V, Rathore AS. Rapid analysis of charge variants of monoclonal antibodies using non-linear salt gradient in cation-exchange high performance liquid chromatography. J Chromatography A. 2015;1406:175–185. doi:10.1016/j.chroma.2015.06.015.
- Singh SK, Narula G, Rathore AS. Should charge variants of monoclonal antibody therapeutics be considered critical quality attributes? Electrophoresis. 2016;37(17–18):2338–2346. doi:10.1002/elps.201600078. PMID:27387433.
- Lyubarskaya Y, Houde D, Woodard J, Murphy D, Mhatre R. Analysis of recombinant monoclonal antibody isoforms by electrospray ionization mass spectrometry as a strategy for streamlining characterization of recombinant monoclonal antibody charge heterogeneity. Analytical Biochem. 2006;348(1):24–39. doi:10.1016/j.ab.2005.10.003.
- Du Y, Walsh A, Ehrick R, Xu W, May K, Liu H. Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. MAbs. 2012;4(5):578–585. doi:10.4161/mabs.21328. PMID:22820257.
- Alsenaidy MA, Jain NK, Kim JH, Middaugh CR, Volkin DB. Protein comparability assessments and potential applicability of high throughput biophysical methods and data visualization tools to compare physical stability profiles. Frontiers Pharmacol. 2014;5:39. doi:10.3389/fphar.2014.00039.
- Guideline IHT. Quality of biotechnological products: Stability testing of biotechnological/biological products Q5C. 1995 (http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5C/Step4/Q5C_Guideline.pdf)
- Liu H, Gaza-Bulseco G, Sun J. Characterization of the stability of a fully human monoclonal IgG after prolonged incubation at elevated temperature. J Chromatography B. 2006;837(1):35–43. doi:10.1016/j.jchromb.2006.03.053.
- Rafiq S, Butchar JP, Cheney C, Mo X, Trotta R, Caligiuri M, Jarjoura D, Tridandapani S, Muthusamy N, Byrd JC. Comparative assessment of clinically utilized CD20-directed antibodies in chronic lymphocytic leukemia cells reveals divergent NK cell, monocyte, and macrophage properties. J Immunol. 2013;190(6):2702–2711. doi:10.4049/jimmunol.1202588. PMID:23418626.
- Bowles JA, Wang SY, Link BK, Allan B, Beuerlein G, Campbell MA, Marquis D, Ondek B, Wooldridge JE, Smith BJ, et al. Anti-CD20 monoclonal antibody with enhanced affinity for CD16 activates NK cells at lower concentrations and more effectively than rituximab. Blood. 2006;108(8):2648–2654. doi:10.1182/blood-2006-04-020057. PMID:16825493.
- Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood. 2002;99(3):754–758. doi:10.1182/blood.V99.3.754. PMID:11806974.
- Weiner GJ. Rituximab: mechanism of action. Seminars Hematol. 2010;47(2):115–123. doi:10.1053/j.seminhematol.2010.01.011.
- Zhou X, Hu W, Qin X. The role of complement in the mechanism of action of rituximab for B-cell lymphoma: implications for therapy. Oncologist. 2008;13(9):954–966. doi:10.1634/theoncologist.2008-0089. PMID:18779537.
- Natsume A, Niwa R, Satoh M. Improving effector functions of antibodies for cancer treatment: Enhancing ADCC and CDC. Drug Design Dev Therapy. 2009;3:7.
- Du J, Wang H, Zhong C, Peng B, Zhang M, Li B, Huo S, Guo Y, Ding J. Structural basis for recognition of CD20 by therapeutic antibody Rituximab. J Biol Chem. 2007;282(20):15073–15080. doi:10.1074/jbc.M701654200. PMID:17395584.
- Tsiftsoglou AS, Ruiz S, Schneider CK. Development and regulation of biosimilars: current status and future challenges. BioDrugs. 2013;27(3):203–211. doi:10.1007/s40259-013-0020-y. PMID:23553340.
- Schellekens H. How similar do ‘biosimilars' need to be? Nat Biotechnol. 2004;22(11):1357–1359. doi:10.1038/nbt1104-1357. PMID:15529154.
- Schellekens H. Assessing the bioequivalence of biosimilars: the Retacrit® case. Drug Discovery Today. 2009;14(9):495–499. doi:10.1016/j.drudis.2009.02.003. PMID:19429509.
- Jacobs I, Singh E, Sewell KL, AL-Sabbagh A, Shane LG. Patient attitudes and understanding about biosimilars: an international cross-sectional survey. Patient Preference Adherence. 2016;10:937. doi:10.2147/PPA.S104891. PMID:27307714.
- Eon‐Duval A, Broly H, Gleixner R. Quality attributes of recombinant therapeutic proteins: an assessment of impact on safety and efficacy as part of a quality by design development approach. Biotechnol Progress. 2012;28(3):608–622. doi:10.1002/btpr.1548.
- Xie H, Chakraborty A, Ahn J, Yu YQ, Dakshinamoorthy DP, Gilar M, Chen W, Skilton SJ, Mazzeo JR. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. MAbs. 2010;2(4):379–394. doi:10.4161/mabs.11986. PMID:20458189.
- Park SS, Park J, Ko J, Chen L, Meriage D, Crouse‐Zeineddini J, Wong W, Kerwin BA. Biochemical assessment of erythropoietin products from Asia versus US Epoetin alfa manufactured by Amgen. J Pharmaceutical Sci. 2009; 98(5):1688–1699. doi:10.1002/jps.21546.
- Ayoub D, Jabs W, Resemann A, Evers W, Evans C, Main L, Baessmann C, Wagner-Rousset E, Suckau D, Beck A. Correct primary structure assessment and extensive glyco-profiling of cetuximab by a combination of intact, middle-up, middle-down and bottom-up ESI and MALDI mass spectrometry techniques. MAbs. 2013;5(5):699–710. doi:10.4161/mabs.25423. PMID:23924801.
- Liu J, Eris T, Li C, Cao S, Kuhns S. Assessing analytical similarity of proposed Amgen biosimilar ABP 501 to adalimumab. BioDrugs. 2016;30(4):321–338. doi:10.1007/s40259-016-0184-3. PMID:27461107.
- Jung SK, Lee KH, Jeon JW, Lee JW, Kwon BO, Kim YJ, Bae JS, Kim DI, Lee SY, Chang SJ. Physicochemical characterization of Remsima®. MAbs. 2014;6(5):1163–1177. doi:10.4161/mabs.32221. PMID:25517302.
- da Silva A, Kronthaler U, Koppenburg V, Fink M, Meyer I, Papandrikopoulou A, Hofmann M, Stangler T, Visser J. Target-directed development and preclinical characterization of the proposed biosimilar rituximab GP2013. Leukemia Lymphoma. 2014;55(7):1609–1617. doi:10.3109/10428194.2013.843090. PMID:24024472.
- Miranda-Hernández MP, López-Morales CA, Piña-Lara N, Perdomo-Abúndez FC, Pérez NO, Revilla-Beltri J, Molina-Pérez A, Estrada-Marín L, Flores-Ortiz LF, Ruiz-Argüelles A, et al. Pharmacokinetic comparability of a biosimilar trastuzumab anticipated from its physicochemical and biological characterization. BioMed Res Int. 2015;2015:874916. doi:10.1155/2015/874916. PMID:26682224.
- Velayudhan J, Chen YF, Rohrbach A, Pastula C, Maher G, Thomas H, Brown R, Born TL. Demonstration of functional similarity of proposed biosimilar ABP 501 to adalimumab. BioDrugs. 2016;30(4):339–351. doi:10.1007/s40259-016-0185-2. PMID:27422671.
- Schiestl M, Stangler T, Torella C, Čepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29(4):310–312. doi:10.1038/nbt.1839. PMID:21478841.
- Chelius D, Jing K, Lueras A, Rehder DS, Dillon TM, Vizel A, Rajan RS, Li T, Treuheit MJ, Bondarenko PV. Formation of pyroglutamic acid from N-terminal glutamic acid in immunoglobulin gamma antibodies. Analytical Chem. 2006;78(7):2370–2376. doi:10.1021/ac051827k.
- Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313(5787):670–673. doi:10.1126/science.1129594. PMID:16888140.
- Jiang XR, Song A, Bergelson S, Arroll T, Parekh B, May K, Chung S, Strouse R, Mire-Sluis A, Schenerman M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discovery. 2011;10(2):101. doi:10.1038/nrd3365. PMID:21283105.
- Okazaki A, Shoji-Hosaka E, Nakamura K, Wakitani M, Uchida K, Kakita S, Tsumoto K, Kumagai I, Shitara K. Fucose depletion from human IgG1 oligosaccharide enhances binding enthalpy and association rate between IgG1 and FcγRIIIa. J Mol Biol. 2004;336(5):1239–1249. doi:10.1016/j.jmb.2004.01.007. PMID:15037082.
- Yu M, Brown D, Reed C, Chung S, Lutman J, Stefanich E, Wong A, Stephan JP, Bayer R. Production, characterization and pharmacokinetic properties of antibodies with N-linked Mannose-5 glycans. MAbs. 2012;4(4):475–487. doi:10.4161/mabs.20737. PMID:22699308.
- Reusch D, Tejada ML. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology. 2015;25(12):1325–1334. doi:10.1093/glycob/cwv065. PMID:26263923.
- Ferrara C, Grau S, Jäger C, Sondermann P, Brünker P, Waldhauer I, Hennig M, Ruf A, Rufer AC, Stihle M, et al. Unique carbohydrate–carbohydrate interactions are required for high affinity binding between FcγRIII and antibodies lacking core fucose. Proc Natl Acad Sci. 2011;108(31):12669–74 doi:10.1073/pnas.1108455108. PMID:21768335.
- Yu L, Vizel A, Huff MB, Young M, Remmele RL, He B. Investigation of N-terminal glutamate cyclization of recombinant monoclonal antibody in formulation development. J Pharmaceutical Biomed Analysis. 2006;42(4):455–63. doi:10.1016/j.jpba.2006.05.008.
- Rosenberg AS. Effects of protein aggregates: an immunologic perspective. AAPS J. 2006;8(3):E501–E507. doi:10.1208/aapsj080359. PMID:17025268.
- Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability, and formulation. J Pharmaceutical Sci. 2007;96(1):1–26. doi:10.1002/jps.20727.
- Joshi V, Shivach T, Yadav N, Rathore AS. Circular dichroism spectroscopy as a tool for monitoring aggregation in monoclonal antibody therapeutics. Analytical Chem. 2014;86(23):11606–13. doi:10.1021/ac503140j.
- Rubinstein M, Colby RH. Polymer physics. New York: Oxford University Press; 2003.