
ABSTRACT
Implementation of in vitro assays that correlate with in vivo human pharmacokinetics (PK) would provide desirable preclinical tools for the early selection of therapeutic monoclonal antibody (mAb) candidates with minimal non-target-related PK risk. Use of these tools minimizes the likelihood that mAbs with unfavorable PK would be advanced into costly preclinical and clinical development. In total, 42 mAbs varying in isotype and soluble versus membrane targets were tested in in vitro and in vivo studies. MAb physicochemical properties were assessed by measuring non-specific interactions (DNA- and insulin-binding ELISA), self-association (affinity-capture self-interaction nanoparticle spectroscopy) and binding to matrix-immobilized human FcRn (surface plasmon resonance and column chromatography). The range of scores obtained from each in vitro assay trended well with in vivo clearance (CL) using both human FcRn transgenic (Tg32) mouse allometrically projected human CL and observed human CL, where mAbs with high in vitro scores resulted in rapid CL in vivo. Establishing a threshold value for mAb CL in human of 0.32 mL/hr/kg enabled refinement of thresholds for each in vitro assay parameter, and using a combinatorial triage approach enabled the successful differentiation of mAbs at high risk for rapid CL (unfavorable PK) from those with low risk (favorable PK), which allowed mAbs requiring further characterization to be identified. Correlating in vitro parameters with in vivo human CL resulted in a set of in vitro tools for use in early testing that would enable selection of mAbs with the greatest likelihood of success in the clinic, allowing costly late-stage failures related to an inadequate exposure profile, toxicity or lack of efficacy to be avoided.
Introduction
During the development of therapeutic monoclonal antibodies (mAbs), a strategy for early identification of candidate mAbs with the greatest likelihood of success in the clinic is needed to avoid costly late-stage failures related to inadequate exposure, toxicity or lack of efficacy. Early screening and optimization of mAbs focus on characteristics such as affinity, potency and stability for selection of lead constructs, while pharmacokinetic (PK) properties, which can influence both efficacy and toxicity, are typically characterized later in development and on a small number of lead mAb constructs. Compared to the well-defined field of small molecule therapeutics, in vitro assays used as preclinical tools to predict human PK for mAbs have yet to be established for large molecule therapeutics. Despite the range of in vitro measures for characterizing physicochemical properties of biotherapeutics, in vitro in vivo correlations (IVIVC) have yet to be established. Implementing such tools for identifying mAbs at risk for poor PK during early phases of drug discovery will ultimately reduce the time needed for drug discovery and development by improving the lead mAb selection process.
The PK of mAbs is generally characterized by a slow systemic clearance (CL) and low volume of distribution, resulting in a long terminal elimination half-life (t1/2). Protection and recycling of mAbs mediated by the neonatal Fc receptor (FcRn) is well documented as one of the most important mechanisms in modulating mAb CL through its pH-dependent binding interactions in the endosomal compartment.Citation1–5 Observations of unexpectedly rapid CL of therapeutic mAbs in recent yearsCitation6–8 have led to interest in understanding CL mechanisms other than FcRn that may affect mAb PK. Multiple physicochemical attributes of a mAb, such as charge, target binding affinity, off-target binding (specific or non-specific), pH-dependent FcRn affinity, or degree and type of glycosylation, have been linked to CL.Citation9–20 Particularly striking are the associations between positive charge on the mAb variable domain and rapid CL. Several studies have reported poor PK profiles of mAbs with high positive charge, which can be dramatically improved through engineering efforts to remove or reposition positive charges or counterbalance them with negative charges.Citation6,Citation12,Citation13,Citation21,Citation22 The availability of high-throughput in vitro assays capable of identifying these properties in mAbs would facilitate the early identification and de-selection of potentially problematic mAbs before extensive research and development resources have been invested.
Several in vitro assays have been shown to correlate well with CL in humans, non-human primates (NHP), and mice.Citation23 Some assays indicate non-specific binding, such as an enzyme-linked immunosorbent assay (ELISA) showing mAb binding to baculovirus (BV) particlesCitation9 and a flow cytometric assay showing binding of IgG displayed on the surface of yeast to membrane preparations from Chinese hamster ovary (CHO) and other cells (termed PSR, or polyspecificity reagent),Citation24 while another assay measured retention of mAbs on a matrix-immobilized hFcRn affinity column.Citation13 Each of these assays is challenging to apply across large mAb screening campaigns for various reasons, such as use of complex reagents (BV particles) or non-standard antibody display formats (yeast display), and limited throughput (FcRn affinity chromatography). Recent work has also shown that high-throughput assays measuring undesired molecular interactions (e.g., aggregation, poor solubility, high viscosity, non-specificity) are capable of identifying problematic mAbs that may result in problems in later development, such as poor expression, difficulties with formulation and an increased risk of failure in the clinic that may be related to poor PK.Citation9,Citation20,Citation24 We therefore sought to identify in vitro assays that could be amenable to application with readily available reagents and have the ability to reproducibly identify mAbs with poor physicochemical properties.
Previously, we demonstrated the utility of the human FcRn (hFcRn) transgenic 32 (Tg32) homozygous mouse with allometric scaling as an in vivo tool for accurate prediction of mAb human CL, thus allowing early preclinical selection and de-selection of mAbs based on PK.Citation25 In this study, we compare the CL of a panel of mAbs in both hFcRn Tg32 mice and humans with their performance in a series of in vitro assays designed to measure non-specificity, self-association and hFcRn interaction. Our objective is to establish a panel of in vitro assays to identify mAbs with observed rapid and slow CL, and propose a screening paradigm for the early selection of mAbs with high probability of both unfavorable and favorable human PK.
Results
Antibodies
We studied two sets of mAbs (summarized in Table S1). One set contains 29 Pfizer research mAbs and 13 marketed therapeutic mAbs (total n = 42, designated mAb-01 through mAb-42), described as study mAbs. Study mAbs were selected to encompass a broad range of frameworks, isotypes, and target types (soluble or membrane-bound). Table S1 details mAb source (Pfizer vs Marketed), mAb numbering, availability of CL data in hFcRn Tg32 homozygous mouse and human, and in vitro assay score results. Of the 42 study mAbs, 22 had been administered to humans (linear CL range of 0.06 – 0.46 mL/hr/kg) and 40 were administered to hFcRn Tg32 homozygous mice (with allometrically projected human linear CL range of 0.07 – 1.86 mL/hr/kg ()) for comparison of PK to in vitro assay results. MAbs with non-linear CL values, presumably due to target-mediated drug disposition (TMDD) or anti-drug antibodies (ADAs), were excluded from the dataset in and any correlative analyses.
Table 1. mAb datasets and human linear clearance ranges.
The second set consists of a series of 23 mAbs generated using the variable region sequences from commercially available marketed mAbs that were grafted onto human IgG1/kappa or lambda constant regions (designated mAb-43 through mAb-65), described as clinical mAb analogs. Linear human CL values for the corresponding marketed mAbs were assigned to the clinical mAb analogs based on literature reports (linear CL range 0.05 – 0.24 mL/hr/kg, data sourced for 16/23 mAbs (). Non-linear CL values or mAbs with insufficient data in the literature to determine linearity of CL were excluded from the dataset in and any correlative analyses.
Strategy for in vitro assay selection
We sought to identify a set of in vitro assays examining various physicochemical properties of mAbs that may lead to unfavorable and favorable mAb PK. Our initial assessment focused on previously-described assays that were likely to be suitable for screening tens to hundreds of mAbs, consumed small amounts of mAb, and demonstrated precision, sensitivity and robustness. Assays that met these criteria measured non-specific binding (low-stringency DNA- and insulin-binding ELISAs), self-association (affinity-capture self-interaction nanoparticle spectroscopy (AC-SINS)), and interaction with matrix-immobilized human FcRn (surface plasmon resonance (SPR) and column chromatography assays), which are described in detail below. The performance of each in vitro assay was monitored using the same set of mAb controls () that included assay negative controls (mAb-01 and -02) and assay positive controls (mAb-03 and -04). MAb-05 is used as an assay positive control specifically for the AC-SINS assay. The study and clinical analog sets of mAbs were run in each assay to examine the relationship between in vitro assay results and in vivo CL in hFcRn Tg32 mice and humans.
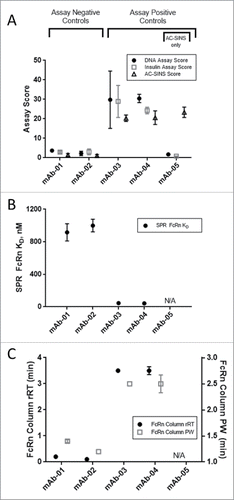
Figure 1. In vitro score range for assay controls. Assay negative control mAbs (mAb-01 and mAb-02) and positive control mAbs (mAb-03 and mAb-04) were tested in multiple replicates in the A) DNA binding assay (•), insulin binding assay (□), and AC-SINS assay (∆); B) hFcRn SPR assay (•); and C) immobilized hFcRn affinity column chromatography assay measuring relative retention time (rRT; •) and peak width (PW; □). Assay positive control mAb-05 (AC-SINS only) represents a distinct class of mAbs having high AC-SINS and low DNA and insulin scores. Error bars represent standard deviation.
Non-specific binding (DNA- and insulin-binding ELISAs)
Based on literature reports linking mAb CL and non-specific binding to charged or membrane-containing substrates,Citation6,Citation8,Citation9 we investigated published assays using a set of mAbs that had previously been observed to bind non-specifically to multiple targets (data not shown). In ELISAs, these mAbs showed similar binding to BV particles, bovine serum albumin (BSA)-blocked plastic, DNA and insulin. The assays for DNA and insulin binding, based on protocols designed to identify autoreactive antibodies from lupus patients,Citation17 gave similar results to the BV and BSA-binding ELISA and were more sensitive in detecting mAbs with low but above-background signal (Fig S1). Control mAbs showing nonspecific binding in these assays also bound to ELISA substrates, including lipopolysaccharide, heparin, serum proteins, extracellular matrix components, cellular nuclear extracts, fixed HepG2 cells, and to varying numbers of proteins in microarrays displaying thousands of human proteins (data not shown). However, these methods were unsuitable for screening implementation due to high assay variability, low throughput or high cost.
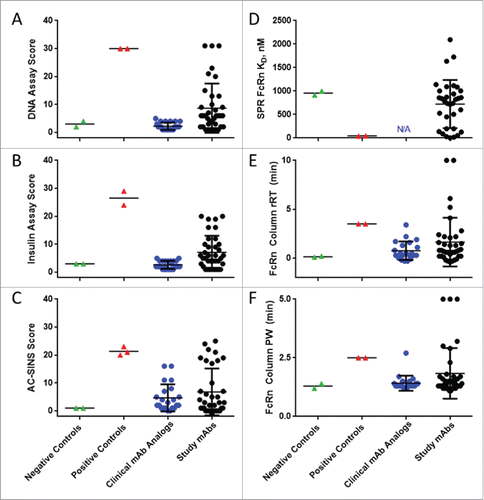
The DNA- and insulin-binding ELISAs proved to be the most sensitive assays to measure low-affinity charge-based interactions of mAbs. These two assays, which require less than 10 µg of protein, were established in 384-well format on automated liquid-handling platforms to achieve desired throughput and precision. Assay scores were calculated as the signal ratio of the mAb at 10 µg/ml binding to DNA or insulin relative to the ELISA signal in the absence of a primary antibody. For the DNA-binding assay, assay negative controls mAb-01 and -02 scored 4 ± 1 and 2 ± 1, respectively, and consistently differentiated from the assay positive controls mAb-03 and -04, which scored 30 ± 15 and 30 ± 2, respectively. Similarly, in the insulin-binding assay, assay negative controls scored 3 ± 0 and 3 ± 2 for mAb-01 and -02, respectively, and differentiated from assay positive controls mAb-03 and -04 with scores of 30 ± 8 and 24 ± 2, respectively ().
The set of study mAbs showed a broad range of scores from 1 – 31 for the DNA-binding assay, and from 1 – 29 for the insulin-binding assay (, ), reflecting the inclusion of mAbs covering the range of scores observed through antibody discovery and optimization (data not shown). By contrast, we observed that the set of clinical mAb analogs showed generally low scores in the DNA- and insulin-binding assays, ranging from 1 – 5 for each assay (, ). This observation raises the possibility that low scores in these assays may reflect favorable properties for clinical development, similar to the favorable profile suggested by Jain et al,Citation20 and these in vitro assays may allow early identification of problematic mAbs. This observation is also consistent with the below-threshold in vitro scores and CL observed for the 13 marketed mAbs within the study mAb dataset (identified in Table S1).
Figure 2. In vitro assay score range for mAb datasets. Assay control values are compared to the assay range observed for clinical mAb analog and study mAb datasets in assays A) DNA binding; B) insulin binding; C) AC-SINS; D) immobilized hFcRn SPR; and E-F) hFcRn column chromatography rRT and PW. Assay negative control mAbs (mAb-01 and mAb-02, green) and positive control mAbs (mAb-03 and mAb-04 for all assays + mAb-05 for AC-SINS, red) represent mean values of assay replicates. Clinical mAb analogs (blue) consist of the variable regions of commercially available marketed mAbs grafted onto human IgG1/kappa or lambda constant regions. Study mAbs (black) were chosen from research programs to represent a spectrum of in vitro scores or observed human CL.
Self-association (AC-SINS Assay)
We next examined the AC-SINS assay, which has been described as a high-throughput method for detection of mAbs with a propensity to self-associate.Citation14,Citation24,Citation26 This assay utilizes the optical properties of gold nanoparticles. Briefly, mAbs are captured by anti-human Fc antibodies coated on the gold nanoparticles. If a mAb tends to interact with itself, there is a clustering of the nanoparticles, which leads to a red shift in the absorbance wavelength. This assay has also been described in the literature as a potential screening tool for identifying developability issues related to solubility, viscosity, and aggregation.Citation14,Citation26 Within the panel of Pfizer mAbs, we observed a strong association of high AC-SINS scores with moderate to high viscosity levels, as defined by >20 cP at 150 mg/ml (data not shown). We hypothesized, however, that this assay may be useful in identifying mAbs with poor physicochemical properties, not solely viscosity but a more general property of non-specific interaction. Indeed, in the initial testing of control mAbs with high DNA- and insulin-binding scores, we observed high AC-SINS scores for the assay positive control mAbs-03 and -04 (20 ± 2 and 21 ± 3, respectively), while assay negative control mAbs-01 and -02 scored 1 ± 1 and 1 ± 1, respectively (). MAb-05, known to be highly viscous, was added as an additional positive control because it represented a distinct in vitro assay pattern, displaying high AC-SINS scores (23 ± 3) but low DNA- and insulin-binding scores (2 ± 0 and 1 ± 0, respectively) ().
Similar to their performance in the ELISAs, the study mAbs showed a broad range of AC-SINS scores, from -1–25 (). The clinical mAb analog set generally showed relatively low to moderate scores, ranging from 0–16. Approximately 10% of the mAbs included in the study scored above the assay threshold (1/13 of the marketed mAbs and 2/22 of the clinical mAb analog set), suggestive of potential developability issues for these mAbs. An AC-SINS assay was also included as one of the in vitro assays evaluated by Jain et al.,Citation20 who similarly utilized a panel of clinical mAb analogs, including 20 of the same marketed mAbs for which analogs were made for this study. Comparison of AC-SINS assay scores from these two independent datasets are in close agreement (ρ = 0.95, r2 = 0.83), demonstrating the robustness and general utility of this assay (Fig S2). MAb analogs basiliximab, golimumab and ipilimumab were used in both studies and scored similarly, at the high end of the range of AC-SINS scores.
Matrix-immobilized FcRn binding (SPR and FcRn column chromatography)
Measurements of mAb binding to immobilized hFcRn in affinity chromatography and SPR assays have been reported to show correlation between column retention time or binding affinity and CL in hFcRn Tg276 hemizygousCitation13 and Tg32 homozygous mice.Citation27 We evaluated the mAb sets in these two matrix-immobilized hFcRn binding formats, which require 50–200 μg of each mAb.
An SPR assay measured affinity of IgG to a complex of biotinylated FcRn immobilized on a streptavidin–carboxymethylated dextran Biacore chip. The assay negative controls mAb-01 and -02 displayed low affinity to immobilized biotin-FcRn (KD = 916.2 – 1000 nM), while assay positive controls mAb-03 and -04 had comparatively high affinity for FcRn (KD = 42.6 – 45.4 nM) (). KD values measured by SPR for the study mAb dataset ranged broadly (2.6 – 2167 nM, ) compared to assay controls, while KD values were not measured by SPR for the clinical mAb analog panel.
The matrix-immobilized human FcRn affinity chromatography assay was adapted from a literature methodCitation28 to determine the retention time (RT) and peak width (PW) of mAbs. Results are reported as a relative retention time (rRT) to that of an assay performance control, mAb-A, to normalize inherent assay variability relating to differing buffer lots and column preparations. Assay negative control mAbs-01 and -02 showed little deviation from the behavior of mAb-A, with rRT values of 0.2 and 0.1 min. and narrow PW values of 1.4 and 1.2 min., respectively (). Assay positive control mAbs-03 and -04, by contrast, both displayed delayed rRT of 3.5 min., with broader PW of 2.5 min.; . The study mAb dataset showed broad rRT ranges of -0.4 – 10.0 min and PW ranges of 1.1 – 5.0 ( E, ). The clinical mAb analogs showed generally low rRT (-0.3 – 2.2 min) and low PW (1.2 – 1.7 min) with the exception of obinutuzumab, which had rRT of 3.4 min. and PW of 2.7 min.
Correlation across in vitro assays
We compared the scores of study mAbs in the in vitro assay parameters and determined that the parameters clustered into three groups measuring non-specific interactions (DNA- and insulin-binding assays), self-association (AC-SINS assay) and matrix-immobilized FcRn binding (SPR and column chromatography assays). The scores for DNA and insulin binding, both charge-based measures of non-specific interactions, were highly correlated to each other (ρ = 0.93; Fig. S3). Likewise, the hFcRn column chromatography parameters (rRT and PW) showed good correlation with one another (ρ = 0.92). Affinity measurements of mAbs to immobilized FcRn by SPR were well correlated with all of the other in vitro assays (ρ = -0.84 with immobilized hFcRn column rRT and -0.71 to -0.79 for the other assays). AC-SINS showed little correlation with DNA and insulin (ρ = 0.49 and 0.36, respectively), and modest correlation with hFcRn column parameters (ρ = 0.58 for rRT and 0.62 for PW), suggesting that these in vitro assays measure distinct physicochemical properties and may each provide unique value (Fig S3).
Pharmacokinetics analysis and correlation to in vitro assay results
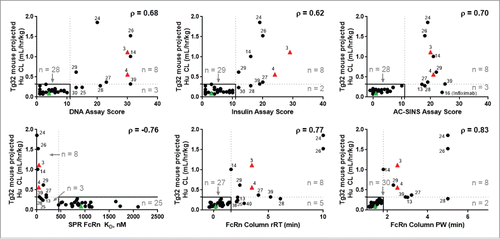
We next examined the relationship between the performance of study mAbs in the in vitro assays and their PK properties in vivo. We measured PK of each mAb via intravenous (IV) administration in the hFcRn Tg32 mouse, which we have shown previously to provide a strong correlation with human CL.Citation25 We allometrically projected human CL (using 0.93 exponent) based on the CL values measured in Tg32 mice, and we refer to this calculated value as Tg32-projected human CL. Plotting Tg32-projected human CL against each of the in vitro assay parameter results, we observed a strong trend for each parameter (). Overall, correlations were observed for mAbs in each in vitro assay with CL in hFcRn Tg32 mice: DNA assay (ρ = 0.68), insulin assay (ρ = 0.62), AC-SINS assay (ρ = 0.70), hFcRn column rRT/PW (ρ = 0.77/0.83) and hFcRn SPR KD (ρ = −0.76), with all correlations determined to be statistically significant (p < 0.05).
Figure 3. In vitro assay correlation with in vivo CL from mAbs administered to Tg32 mice and allometrically scaled to project human CL. The projected human CL is plotted against the measurements for each mAb from the in vitro assays: DNA binding, insulin binding, AC-SINS, hFcRn SPR, and hFcRn column chromatography rRT and PW. The threshold for identification of rapid CL is defined at ≥0.32 mL/hr/kg (dotted horizontal lines). The in vitro assay thresholds for the de-selection of mAbs based on CL is indicated by the dotted vertical lines, and defined as assay scores of ≥11 (DNA binding), ≥11 (insulin binding), ≥11 (AC-SINS), ≤451.7 nM (hFcRn SPR), ≥1.6 min (hFcRn column rRT), and ≥1.8 min (hFcRn column PW), based on ROC analysis (Fig. S4). Solid lines indicate the region where both in vitro assay scores and in vivo CL fall below threshold. Spearman correlation (ρ) is indicated for each in vitro assay with projected CL. All relationships show correlative significance at p<0.05. Circles and triangles, represent linear CL. Squares, apparent linear CL. •, study mAbs. Green ▴, negative control mAb-01. Red ▴, positive control mAbs-03 and -04. Control mAbs-02 and -05 do not have corresponding CL projected from Tg32 mice.
Using the IVIVC for mAb selection: Establishment of CL threshold
The correlation of in vitro data with CL suggests that these in vitro assays could be used for screening to identify mAbs at risk for poor PK. While scatter in the data may not allow robust prediction of the CL value from any one of the in vitro assays, the plots in show a cluster of mAbs in our dataset with low CL and low assay scores, suggesting that a threshold may exist for each in vitro assay parameter below which mAbs exhibit slow CL. To facilitate analysis, a threshold value for unacceptable (rapid) CL (both observed human and Tg32 mouse-projected human CL) was defined as ≥0.32 mL/hr/kg. This value is 2-fold the median CL (0.16 mL/hr/kg) of the observed human CL for both the study mAbs and the clinical mAb analogs (). The rationale for setting such a threshold value for CL builds in inter-subject variability and standard error for in vivo studies so that mAbs with likely reasonable PK properties stay in the selection pool for further development.
Using IVIVC for mAb selection: Establishment of in vitro assay thresholds
When the CL threshold of ≥0.32 mL/hr/kg is applied to the study mAb set, it is possible to define a threshold value for each in vitro assay that maximizes the utility of the assay to differentiate mAbs with CL above and below the CL threshold. Analysis of receiver operating characteristic (ROC) curves for each assay (Fig S4) provided the following assay parameter thresholds: ≥11 for the DNA-binding, insulin-binding and AC-SINS assays; ≥1.6 min. for hFcRn column rRT; ≥1.8 min. for hFcRn PW; and ≤451.7 nM for the FcRn SPR assay. Assay negative control mAb-01 consistently scores below in vitro and in vivo thresholds (where “below threshold” indicates scores < established threshold for DNA, insulin, AC-SINS and FcRn column assays, and > established threshold for FcRn SPR assay) in each assay compared to assay positive controls mAbs-03 and -04, which consistently score above threshold (). All mAbs in the dataset scoring below the assay thresholds have CL below 0.32 mL/hr/kg; i.e., using these thresholds, with the exception of mAb 13, there are no false negatives in our dataset. MAb 13 has a CL value = 0.32 mL/hr/kg, and scored above the thresholds in all assays except DNA- and insulin-binding ELISAs.
Although most mAbs scoring above the threshold for each in vitro assay have rapid CL, 20–38% of the total mAbs scoring above a given in vitro threshold have CL at or below 0.32 mL/hr/kg and are therefore defined as false positives. However, the majority of false positives showed a trend of CL approaching the 0.32 mL/hr/kg threshold, as well as a mixture of positive and negative results across the set of in vitro assays, suggesting that examination of a combined suite of assays could give a more nuanced understanding of PK risk rather than examination of each assay independently.
Retrospective application of thresholds assessing IVIVC of human CL
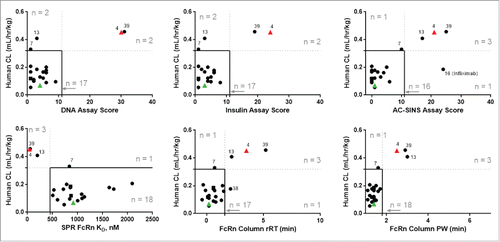
To understand how well the in vitro assay thresholds would identify mAbs with observed rapid CL in human, we conducted a retrospective analysis of the mAbs in our dataset for which historical human CL data were available. Overall, human CL and hFcRn Tg32-projected human CL showed a similar trend in comparison with in vitro results, where lower in vitro scores were associated with lower observed human CL for each assay (). These datasets contained only a small number of mAbs with observed rapid CL in humans (mAb-04, -07, -13 and -39), and of these, mAb-04 and -39 were above threshold in all six in vitro assay parameters and mAb-13 was above threshold in 4 of 6 assay parameters (AC-SINS, hFcRn column rRT and PW and SPR assay parameters). MAb-07 had Tg32-projected human CL below threshold (0.28 mL/hr/kg) and low-moderate in vitro scores in all assays, but its observed human CL was just above threshold (0.33 mL/hr/kg). When applying these CL and in vitro assay parameter thresholds, only two mAbs with observed human CL below threshold scored as above threshold in single assays, mAb-16 (infliximab) in the AC-SINS assay and mAb-38 in the hFcRn column assay. Above-threshold scores may also indicate other developability issues independent of PK. While the dataset contains too few rapidly-cleared mAbs for a robust measurement of correlation, the similar trends between hFcRn Tg32 mouse-projected human CL and observed human CL in and , respectively, confirm that this suite of in vitro assays represent a useful tool set for the risk assessment of unfavorable human PK for mAbs.
Figure 4. In vitro assay parameter correlation with in vivo CL from mAbs administered to humans. The observed CL of mAbs administered to humans is plotted against the measurements for each mAb from each of the in vitro assays: DNA binding, insulin binding, AC-SINS, hFcRn SPR, and hFcRn column chromatography rRT and PW. Threshold values reflect those shown in , where rapid CL is defined at >0.32 mL/hr/kg (dotted horizontal lines) and in vitro parameter thresholds are shown as vertical dotted lines. Solid lines indicate the region where both in vitro assay scores and in vivo CL fall below threshold. Spearman correlation (ρ) are not depicted as sparse data leads to values <0.6. Correlative significance (p < 0.05) is observed for human CL relationship to the AC-SINS assay, hFcRn column rRT and PW results. Circles and triangles, linear CL. Squares, apparent linear CL. •, study mAbs. Green ▴, negative control mAb-01. Red ▴, positive control mAb-04. Control mAbs-02, -03 and -05 do not have corresponding CL in human.
A similar trend was observed from examination of published human CL for the commercial mAb counterparts of the clinical analog mAbs tested in these in vitro assays. For the clinical mAbs that had published linear PK (16/23), human CL was below the 0.32 mL/hr/kg threshold, and in vitro assays scored below the thresholds for the DNA- and insulin-binding assays (Fig. S5). While the CL of clinical mAbs and the in vitro scores of their analogs are not directly comparable because the Fc regions of the analogs (all human IgG1) are not all identical to those of their clinical counterparts, the trends evident within this dataset further support the use of in vitro assay scores as an indicator of human CL risk.
Combining In vitro assay categories for favorable PK selection
Because individual in vitro assay parameters each had a number of false positive scoring mAbs, we examined the impact of combining the data from multiple parameters to more accurately identify mAbs with high CL in the study mAb set. lists each mAb in order of decreasing allometrically-projected human CL from the hFcRn Tg32 mouse, together with the corresponding result of each in vitro assay parameter noted as below (green) or above (red) the assay threshold. The majority of mAbs showed consistent in vitro scores that corresponded with their CL category, although mAb-07 did have CL just above threshold at 0.33 mL/hr/kg for observed human CL and was thus designated red for unfavorable CL. Interestingly, a small set of mAbs (mAbs-16, -40, -28, -30 and -25), scored above threshold for only one or two assay parameters.
Figure 5. Retrospective comparison of in vitro parameters and categories to human CL with triage assessment. Data from the set of study mAbs are rank-ordered by descending projected human CL from Tg32 mouse data. In vitro assay data and CL are scored as below threshold (green) or above threshold (red). Triage category: proposed decisions for each molecule based on the combined in vitro assay data are as follows: advance mAb candidate (green; accept mAbs scoring below threshold in 3/3 assay categories); characterize further (yellow; mAbs scoring above threshold in 1/3 or 2/3 assay categories); or do not advance mAb candidate (red; mAbs scoring above threshold in 3/3 assay categories). Key: Assay negative control (mAb-01) and positive controls (mAb-03 and -04) denoted in mAb listing. n/a indicates data not generated.
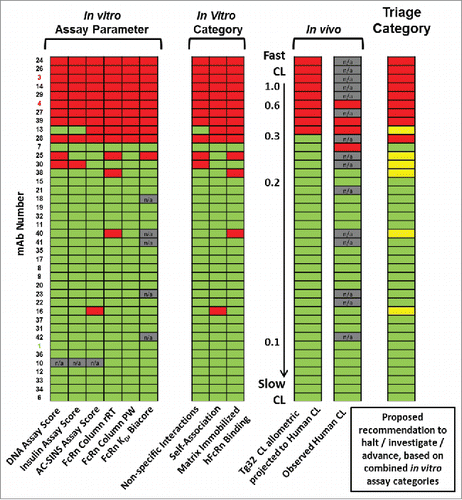
We assessed combining the results of in vitro assays that had high correlation with one another (Fig. S3). These assays fell into three categories, which measured: (1) non-specific interactions (DNA- and insulin-binding assays), (2) self-association (AC-SINS), and (3) matrix-immobilized hFcRn binding (SPR and hFcRn column chromatography). Using this categorization, the following guidelines are suggested for triaging mAbs assessed in in vitro assays for favorable PK: accept mAbs scoring below threshold in 3/3 assay categories; reject mAbs scoring above threshold in 3/3 assay categories, and investigate mAb PK in Tg32 mice for mAbs with scores above threshold in assays from 1/3 or 2/3 assay categories.
Applying these criteria, our dataset contains only one false positive, mAb-28, which would have been rejected based on in vitro categories, but had Tg32-projected human CL marginally below 0.32 mL/hr/kg and no comparable observed human CL; and one false negative, mAb-07, which had below-threshold in vitro scores and Tg32-projected CL at 0.28 mL/hr/kg, and observed human CL just above threshold at 0.33 mL/hr/kg. Therefore, the false positive rate combining assay parameters into categories is 11% (1/9 mAbs), improved over the 20–38% rates observed for single in vitro assay parameters. The false negative rate is 3%, with 1 of 31 mAbs having above-threshold CL and below-threshold in vitro scores. also illustrates that using categories allows the identification of mAbs that would need further characterization to be accepted.
Discussion
The high cost of advancing therapeutic mAbs to preclinical development and clinical testing has led to strong interest in identifying methods to predict late-stage failure, with regard to PK as well as manufacturing and formulation-related properties, during early construct design and screening stages. While the optimal physicochemical properties leading to favorable PK have been well defined for small molecules, embodied in Lipinski's Rule of Five,Citation29 a need exists to develop similar preclinical criteria and assays for the early identification of mAbs likely to result in favorable PK. As described here, our work has expanded upon previous studiesCitation9,Citation20,Citation24 that examined the relationship of individual in vitro assays and PK, by comparing the performance of a broad set of mAbs in a suite of in vitro assays against PK in an hFcRn Tg32 mouse model with an established correlation to human PK.Citation25 Our data have led us to a combinatorial triage approach that allows an informed decision on mAb PK risk at an early screening stage. Although these in vitro assays are not intended to directly predict the CL value of a given mAb, the implementation of this suite of in vitro assays can provide a tool for the selection of therapeutic mAb candidates with minimal non-target-related PK risk and minimize the likelihood that mAbs with poor PK would be advanced into costly preclinical and clinical development.
We chose a panel of 42 study mAbs, 22 of which have corresponding human PK data, to conduct both in vitro and in vivo measurements. The PK of 40 of these mAbs was assessed in the hFcRn Tg32 mouse, followed by allometrically projecting human CL and comparing CL with in vitro measures of non-specific interactions (DNA- and insulin-binding assays), self-association (AC-SINS assay), and binding to matrix-immobilized hFcRn (SPR and column chromatography assays). Each in vitro assay showed a general correlation with the Tg32 mouse-projected human CL (ρ range 0.62-0.83). The AC-SINS and immobilized hFcRn affinity column assays, as well as other measures of nonspecific binding to various substrates, have been described as assays of developability,Citation9,Citation20,Citation24,Citation30,Citation31 generally taken to mean that they are indicative of properties likely to lead to difficulties in manufacturing, high-concentration formulation, or stability, in addition to PK.
In assessing our study mAbs for which observed human PK is available, it was apparent that the number of mAbs with poor physicochemical properties was limited, likely because such mAbs were not selected for clinical studies. Only a small number of mAbs with poor physicochemical properties and human PK data have been reported in the literature.Citation9,Citation24 Thus, the scarcity of data at the high range of in vitro assay scores has limited the confidence with which to assign the boundary between acceptable and unacceptable assay behaviors using solely observed human CL. An important part of our study was the deliberate inclusion of a broad set of mAbs, not previously tested in vivo, and having above-threshold scores in the medium-to-high range in the DNA-binding, insulin-binding, or AC-SINS assays, to understand the in vivo impact of this molecular phenotype. The observation that these mAbs showed commensurate above-threshold CL was validation of the identification and differentiation power of the in vitro assays. As part of this study, we also sought to encompass a broad range of mAb frameworks, isotypes (IgG1, IgG2 and IgG4), and target types (soluble and/or membrane bound). We observed no trends or correlations to suggest that these factors had an effect on the reported correlations for this dataset (data not shown).
Consistent with published data, the results from this study demonstrate that in vitro assays measuring non-specificityCitation9,Citation11 and self-associationCitation14,Citation19,Citation24 can be utilized to de-select mAbs having unfavorable PK. Dostalek et al. recently summarized the literature availability of sequence-based in silico tools, in vitro assays (non-specific binding and FcRn binding assays) and in vivo tools (hFcRn Tg mice, cynomolgus monkeys), proposing a screening strategy for de-selection of mAbs with unfavorable properties. However, IVIVCs were not established to utilize or bridge any given tool towards establishing a human PK relationship.Citation23 Hotzel et al.Citation9 reported that BV particle binding in an ELISA format correlated with the CL of mAbs in humans and less well with NHP; Kelly et al.Citation24 have described a similar correlation with mAb CL and binding of CHO cell membrane preparations to IgGs displayed on the surface of yeast. Similarly, we have demonstrated that ELISAs measuring non-specific interactions to DNA and insulin show good correlation with mAb CL and are robust assays suitable for high-throughput use, with greater sensitivity and less variability across substrate batches than we observed with BV particles or cell extracts (data not shown). Non-specific interactions to negatively-charged substrates such as DNA and insulin (pI 5.7) is likely to result from localized positive charge patches on the mAb, consistent with previously reported findings.Citation6,Citation12,Citation13
We have also demonstrated a correlation between high self-interaction (measured by AC-SINS) and rapid CL, both projected from mAb hFcRn Tg32 mice and observed in humans. By contrast, a previous study measuring AC-SINS and mAb CL in wild-type C57Bl/6 mice did not result in correlation (ρ = 0.135),Citation24 likely due to the higher binding affinities of human IgG for mouse FcRn compared to human FcRn, which can significantly affect the resulting PK.Citation32 High mAb self-interaction has mostly been correlated with poor solubility and high viscosity,Citation14,Citation24 and is therefore a useful high-throughput screening assay for undesired physicochemical properties relating to formulation and development. Our study here further expanded the utility of this assay, and demonstrated for the first time the correlative value of this assay in terms of in vivo behavior of mAbs related to PK. Interestingly, most mAbs with above-threshold DNA and insulin binding also had above-threshold AC-SINS scores, although there are exceptions (mAb-05, -13 and -16) with above-threshold AC-SINS scores and below-threshold DNA/insulin-binding scores (Table S1). While the two mAbs tested in Tg32 mice (mAb-13, -16) had projected human CL below the threshold, this limited dataset precludes any interpretation of the relationship between the high AC-SINS/low binding to DNA and insulin pattern and CL.
Human FcRn binding and protection is one of the dominant factors governing mAb PK in vivo, but the literature around the use of in vitro hFcRn binding assays for selecting or deselecting mAbs has provided conflicting results that often depend on assay format.Citation33–37 Piche-Nicholas et alCitation27 have recently observed a correlation of positive charge in the mAb variable region with affinity to immobilized human hFcRn using the same SPR format for which we have observed a strong correlation with hFcRn Tg32-projected human CL. A combined interaction with both hFcRn and the supporting matrix is likely responsible for the behavior we and othersCitation13,Citation27 have observed in immobilized hFcRn affinity chromatography, as well as in SPR.
While mAb in vitro assay scores and CL showed statistically significant correlations, use of an individual assay alone is not an accurate indication of PK risk. The application of a threshold value of ≥0.32 mL/hr/kg for unfavorable CL, consistent with the rapid CL range reported in previous studies,Citation9,Citation25 allowed clear threshold designations in each in vitro assay for selection of low-risk mAbs for PK. For 20–38% of mAbs that scored above the in vitro threshold in a given assay, the CL was below threshold, and thus these assay results would be considered false positives. This false positive rate could be reduced to 11% (1/9, ) by classifying mAbs as high risk for rapid CL only if they scored above threshold in all three in vitro assay categories. Classifying mAbs based on the in vitro assay categories could thus identify nearly all of the molecules with moderate to rapid CL in our dataset, and thereby provides a basis for early triage during protein engineering campaigns. Examples of complementarity-determining region (CDR) sequence variants from campaigns to optimize four unrelated mAbs (families 1–4 in Fig S6) illustrate that measuring in vitro properties can often allow the identification of closely-related molecules with distinctly different CL.
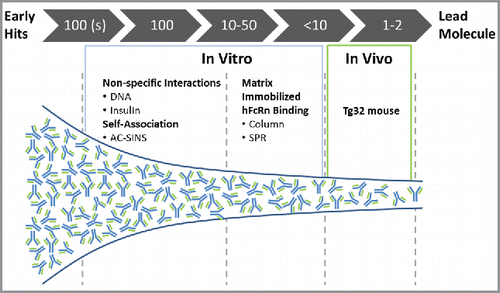
We have proposed a staged approach for de-risking PK of mAbs outlined in . Since the non-specific interactions (DNA- and insulin-binding assays) and self-association (AC-SINS assay) can be run in a high-throughput manner with only a few micrograms of mAb, hundreds of early-stage mAbs can be screened, and mAbs with unfavorable physicochemical properties can be rapidly rejected based on these two categories. An example collection of mAbs from three different therapeutic mAb screening campaigns, in which 82% of mAbs are below the thresholds (<11) for both DNA and AC-SINS scores, is shown in Fig. S7. In practice, users often apply more stringent criteria and deprioritize mAbs scoring close to the upper assay thresholds if alternative mAbs are available. For example, changing the threshold to a more stringent value of acceptable mAbs scoring <5 for both DNA and AC-SINS assays results in a change from 82% to 41% of mAbs in the acceptable range. While many of the mAbs rejected as a result of the more stringent thresholds may have had favorable PK, the cost of incorrectly eliminating such mAbs is often considered to be preferable to the cost associated with discovering mAbs with less favorable PK in later phases of development. MAb panels passing this initial filter are typically evaluated on the basis of functional criteria and additional physicochemical properties, such as aggregate formation and thermal stability,Citation38 and in parallel further evaluated for PK risk based on the matrix-immobilized hFcRn interaction assays. Only those lead mAbs with one or two above-threshold in vitro category scores would warrant further in vivo (Tg32) characterization. The strategy emerging from our studies correlating in vivo human CL (projected from Tg32 mice and observed) to in vitro categories enables an informed and contextual selection process, so that only those mAbs with desirable physicochemical properties advance to scale-up and preclinical testing with the highest likelihood of success for mAb product development and clinical PK outcome. Ultimately, development of in silico models that incorporate physiological parameters of the species, mechanistic understanding of PK and molecular considerations of the therapeutic mAb together with in vitro assay parameters would enable ideal selection processes, early PK risk assessments and human PK prediction of mAbs.
Figure 6. Screening paradigm for PK risk mitigation during mAb discovery and lead selection. High-throughput assays (DNA and insulin binding; AC-SINS) are implemented when hundreds of mAbs are available for screening. MAbs scoring below threshold in both categories would be accepted. Following additional screens including biological activity, expression, stability, etc., mAb panels of 10–50 would be screened by matrix-immobilized hFcRn binding interactions. MAbs with below-threshold scores in this category would be accepted and only those with 1/3 or 2/3 categories above threshold would require further characterization for de-risking PK.
Materials and methods
Antibodies
The 67 mAbs evaluated in this study included 42 study mAbs and 25 clinical analog mAbs. The 42 study mAb dataset comprised mAbs selected from Pfizer research programs on the basis of diverse properties, mAbs from Pfizer clinical development programs, and commercially available marketed mAbs (rituximab, infliximab, trastuzumab, adalimumab, bevacizumab, ustekinumab, basiliximab, cetuximab, eculizumab, omalizumab, tocilizumab, panitumumab, nivolumab). All marketed mAbs were obtained as US pharmaceutical-grade drug product via ProVen Pharmaceuticals, LLC.
MAbs were generated as recombinant proteins expressed in stably transfected CHO cells or in transiently-transfected HEK-293 cells. The clinical analog mAbs were generated by cloning the variable region sequences from the clinical mAbs onto a human IgG1 Fc and a human kappa or lambda constant region as appropriate. Analogs were transiently expressed in HEK-293 cells and purified with mAb Select SuRE resin (GE Healthcare), followed by buffer exchange into phosphate-buffered saline (PBS).
DNA and insulin ELISA
384-well ELISA plates (Nunc Maxisorp) were coated overnight at 4°C with DNA (10 µg/ml) (Sigma-Aldrich, D1626) and insulin (5 µg/ml) (Sigma-Aldrich, I9278-5mL) in PBS pH 7.5. The ELISA, adapted from assays described in Tiller et al,Citation17 was carried out on a PerkinElmer Janus Automated Workstation liquid handling robot. Wells were washed with water, blocked with 50 µl of Polyreactivity ELISA Buffer (PEB; PBS containing 0.05% Tween-20, 1 mM EDTA) for 1 hour at room temperature, and rinsed three times with water. Serially-diluted mAbs in 25 µl were added in quadruplicate to the wells and incubated for 1 h at room temperature. Plates were washed three times with water, and 25 µl of 10 ng/ml goat anti-human IgG (Fc specific) conjugated to horseradish peroxidase (Jackson ImmunoResearch, 109-035-008) were added to each well. Plates were incubated for 1 h at room temperature, washed three times with 80 µl of water, and 25 µl of TMB substrate (Sigma-Aldrich, T-0440) added to each well. Reactions were stopped after approximately 7 minutes by adding 25 µl of 0.18 M ortho-phosphoric acid to each well, and absorbance was read at 450 nm. DNA- and insulin-binding scores were calculated as the ratio of the ELISA signal of the antibody at 10 µg/ml to the signal of a well containing buffer instead of the primary antibody.
AC-SINS assay
The affinity capture self-interaction nanoparticle spectroscopy method was adapted as previously described.14 Briefly, the AC-SINS assay was standardized in a 384-well format on a Perkin-Elmer Janus liquid handling robot. 20 nm gold nanoparticles (Ted Pella, Inc., #15705) were coated with a mixture of 80% goat anti-human Fc (Jackson ImmunoResearch Laboratories, Inc. # 109-005-098) and 20% non-specific goat polyclonal antibodies (Jackson ImmunoResearch Laboratories, Inc. # 005-000-003) that were buffer exchanged into 20 mM sodium acetate pH 4.3 and diluted to 0.4 mg/ml. After one hour incubation at room temperature, sites unoccupied on the gold nanoparticles were blocked with thiolated polyethylene glycol (2 kD). The coated nanoparticles were then concentrated 10-fold using a syringe filter and 10 µl were added to 100 µl of mAb at 0.05 mg/ml in PBS pH 7.2. The coated nanoparticles were incubated with the antibody of interest for 2 hrs in a 96-well polypropylene plate and then transferred to a 384-well polystyrene plate and read on a Tecan M1000 spectrophotometer. The absorbance was read from 450–650 in 2 nm increments, and a Microsoft Excel macro was used to identify the max absorbance, smooth the data, and fit the data using a second-order polynomial. The smoothed max absorbance of the average blank (PBS alone) was subtracted from the smoothed max absorbance of the antibody sample to determine the antibody AC-SINS score.
FcRn column chromatography assay
A human FcRn affinity column was prepared as previously described.Citation28 To determine the human FcRn column retention time, 50 µg of mAb sample adjusted to pH 5.5 was injected onto a GE High Performance Streptavidin Sepharose column (cat# 17-5113-01) coated with in-house expressed and purified biotinylated hFcRn, and a linear pH gradient from pH 5.5 (MES) to 8.8 (Tris) in the presence of 150 mM NaCl was applied for elution of antibody sample. The FcRn column data are reported as a relative retention time where the elution time of sample is subtracted from that of an assay performance control, mAb-A, to normalize variability within assay.
FcRn binding affinity method using SPR
A Biacore™ T100 or T200 (GE Healthcare) was used to measure the steady-state binding affinities of the interaction between IgG molecules and matrix-immobilized FcRn as previously described.Citation27,Citation39 Briefly, 20–70 resonance units (RU) of biotinylated human FcRn-β2m complex were affinity captured on a carboxymethylated dextran chip pre-immobilized with streptavidin (SA) (GE Healthcare, #BR100531). A series of dilutions of IgG analytes (∼100 µg total mAb) were prepared in sample buffer (20 mM MES, 150 nM NaCl, 3 mM EDTA, 0.5% Polysorbate 20, pH 6.0) and sequentially flowed over the FcRn coated SA chip. Following the association and dissociation phases of the experiment, the SA chip was regenerated using 30 s injection of 10 mM HEPES with 150 nM NaCl, 3 mM EDTA, 0.05% P20, pH 7.4 (HBS-EP+ buffer, GE Healthcare). Assays were performed minimally in duplicate using at least two different FcRn-coated SA surfaces, and the average steady-state KD value and standard deviation for each IgG molecule were calculated. The resulting sensorgrams were double referenced and fit to steady-state affinity models using the Biacore T200 Evaluation Software (version: 1.0, GE Healthcare) or Biacore T100 Evaluation Software (version 2.0.1, GE Healthcare). The validity of the data was assessed using calculated chi-square to measure goodness of fit to the binding model and Rmax values.
PK analysis: hFcRn Tg32 mouse PK
PK studies in hFcRn Tg32 homozygous mice and mAb quantitation were performed as previously detailed.Citation25,Citation40 In brief, all mice (The Jackson Laboratory, #014565) were male, 6–10 weeks old and received a single IV dose (2, 5 or 10 mg/kg) with a dose volume of 4 mL/kg. Dose values (2 – 10 mg/kg) were selected to saturate target binding in the event of cross reactivity to mouse target, thus mitigating any TMDD to obtain linear PK parameter estimates. A total of 4–6 animal replicates were evaluated for each mAb with study durations ranging from 4 to 12 weeks depending on mAb properties. All PK studies were conducted at Pfizer Inc., and were designed and executed within accordance of the Animal Use Protocol and adherence to the Pfizer institutional animal care and use committee regulations. Quantitative analysis of plasma samples was conducted using either a radiolabeled approach (administration of 125I-labeled mAb) and measuring for radioactivity counts, or using a ligand binding assay (generic human IgG assay format) developed using a Gyros platform. Both methods were performed in accordance with described details.Citation25 PK parameter estimates were determined from individual animal data using non-compartmental analysis in WinNonlin (Pharsight Version 6.3.0.395) using the Plasma Data Module. Concentration values below the limit of quantitation (BLQ) were excluded from PK calculations. CL values are designated as linear or apparent linear as previously described.Citation25 Non-linear CL values (target-mediated CL (TMDD), or CL affected by presumed ADA) were excluded from all PK analysis. Human CL values were projected from Tg32 CL using a simple allometric scaling approach and empirically derived exponent of 0.93, as previously described.Citation25
PK analysis: Human PK
Of the 42 study mAbs, 22 had previously been administered to humans. For CL determination, a retrospective analysis of historical PK studies was conducted as previously described.Citation25 Briefly, PK parameters were obtained from single-dose IV PK studies conducted in healthy subjects or patients and assessed for linearity of CL (linear CL or apparent linear CL). MAbs were designated as having apparent linear CL when presumed linear though insufficient information was available against the criteria to confidently define as linear, as previously described.Citation25 All non-linear CL values were excluded from analysis. For 11 of 13 commercially available marketed mAbs, linear CL data were obtained from literature sources.Citation41–48
Statistical analyses
Analysis of in vitro to in vivo correlations was performed using GraphPad Prism (version 6.03) to determine Spearman correlation coefficient (ρ) where significance is denoted at p < 0.05. ROC analysis was carried out for each in vitro assay using a script in the Matlab R2013 software package (MathWorks. Matlab r2013a), setting mAbs with CL ≥0.32 mL/hr/kg threshold as positive. The True Positive Rate and False Positive Rate were calculated over a series of cut-off points for each assay, and the optimal cut-off point for maximal predictive power of each assay was determined by the point along the curve with the maximum distance to the diagonal.
Abbreviations
| ADA | = | anti-drug antibody |
| AC-SINS | = | affinity-capture self-interaction nanoparticle spectroscopy |
| BSA | = | bovine serum albumin |
| BV | = | baculovirus |
| CHO | = | Chinese hamster ovary |
| CL | = | clearance |
| FcRn | = | neonatal Fc-receptor |
| hFcRn | = | human FcRn |
| IVIVC | = | in vitro in vivo correlations |
| mAb | = | monoclonal antibody |
| NHP | = | non-human primates |
| PK | = | pharmacokinetics |
| PW | = | peak width |
| ROC | = | receiver operating characteristic |
| rRT | = | relative retention time |
| SPR | = | surface plasmon resonance |
| t1/2 | = | half-life |
| Tg | = | transgenic |
| TMDD | = | target-mediated drug disposition |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1417718_suppl_mat.zip
Download Zip (5 MB)Acknowledgments
All authors are employees of Pfizer Worldwide Research & Development. The authors are grateful to the following individuals for their contributions to this work: Rob Polzer, Will Somers, Matt McKenna, Trisha St-Fleur, Adam Root, Aaron D'Antona, Jane Guo, Arun Alphonse Ignatius, Lioudmila Tchistiakova, Martin Dowty, Jeff Salm, Joanne Brodfuehrer, Chris Shea, Laura Danner, Cyndi Filliettaz, Angela Nunez, Katie McGinn and Lisa Brideau.
References
- Roopenian DC, Akilesh, S. FcRn: the neonatal Fc receptor comes of age. Nat Rev. Immunol. 2007;7:715–725. doi:10.1038/nri2155. PMID: 17703228.
- Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, Petkova S, Avanessian L, Choi EY, Shaffer DJ, Eden PA, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J Immunol (Baltimore, Md. : 1950). 2003;170:3528–3533. doi:10.4049/jimmunol.170.7.3528. PMID:12646614.
- Challa DK, Velmurugan R, Ober RJ, Sally Ward E. FcRn: from molecular interactions to regulation of IgG pharmacokinetics and functions. Curr Top Microbiol Immunol. 2014;382:249–272. doi:10.1007/978-3-319-07911-0_12. PMID: 25116104.
- Oganesyan V, Damschroder MM, Cook KE, Li Q, Gao C, Wu H, Dall'Acqua WF. Structural insights into neonatal Fc receptor-based recycling mechanisms. J Biol Chem. 2014;289:7812–7824. doi:10.1074/jbc.M113.537563. PMID: 24469444.
- Ward ES, Zhou J, Ghetie V, Ober RJ. Evidence to support the cellular mechanism involved in serum IgG homeostasis in humans. Int Immunol. 2003;15:187–195. doi:10.1093/intimm/dxg018.
- Sampei Z, Igawa T, Soeda T, Okuyama-Nishida Y, Moriyama C, Wakabayashi T, Tanaka E, Muto A, Kojima T, Kitazawa T, Yoshihashi K, et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PloS one. 2013;8:e57479. doi:10.1371/journal.pone.0057479.
- Wu H., Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, White WI, Young JF, Kiener PA, et al. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol. 2007;368:652–665. doi:10.1016/j.jmb.2007.02.024. PMID: 17362988.
- Kelly RL, Yu Y, Sun T, Caffry I, Lynaugh H, Brown M, Jain T, Xu Y, Wittrup KD, et al. Target-independent variable region mediated effects on antibody clearance can be FcRn independent. mAbs. 2016;8:1269–1275. doi:10.1080/19420862.2016.1208330.
- Hotzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, Fielder P, et al. A strategy for risk mitigation of antibodies with fast clearance. mAbs. 2012;4:753–760. doi:10.4161/mabs.22189.
- Bumbaca Yadav D, Sharma VK, Boswell CA, Hotzel I, Tesar D, Shang Y, Ying Y, Fischer SK, Grogan JL, Chiang EY, Urban K, Ulufatu S, et al. Evaluating the Use of Antibody Variable Region (Fv) Charge as a Risk Assessment Tool for Predicting Typical Cynomolgus Monkey Pharmacokinetics. J Biol Chem. 2015;290:29732–29741. doi:10.1074/jbc.M115.692434.
- Datta-Mannan A, Lu J, Witcher DR, Leung D, Tang Y, Wroblewski VJ. The interplay of non-specific binding, target-mediated clearance and FcRn interactions on the pharmacokinetics of humanized antibodies. mAbs. 2015;7:1084–1093. doi:10.1080/19420862.2015.1075109.
- Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. mAbs. 2015;7:483–493. doi:10.1080/19420862.2015.1016696.
- Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc Natl Acad Sci U S A. 2015;112:5997–6002. doi:10.1073/pnas.1408766112.
- Liu Y, Caffry I, Wu J, Geng SB, Jain T, Sun T, Reid F, Cao Y, Estep P, Yu Y, Vásquez M, et al. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. mAbs. 2014;6:483–492. doi:10.4161/mabs.27431.
- Geng SB, Wittekind M, Vigil A, Tessier PM. Measurements of Monoclonal Antibody Self-Association Are Correlated with Complex Biophysical Properties. Mol Pharm. 2016;13:1636–1645. doi:10.1021/acs.molpharmaceut.6b00071. PMID: 27045771.
- Sigounas G, Harindranath N, Donadel G, Notkins AL. Half-life of polyreactive antibodies. J Clin Immunol. 1994;14:134–140. doi:10.1007/BF01541346.
- Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods. 2008;329:112–124. doi:10.1016/j.jim.2007.09.017. PMCID: PMC2243222.
- Notkins AL. Polyreactivity of antibody molecules. Trends Immunol. 2004;25:174–179. doi:10.1016/j.it.2004.02.004.
- Dobson CL, Devine PW, Phillips JJ, Higazi DR, Lloyd C, Popovic B, Arnold J, Buchanan A, Lewis A, Goodman J, van der Walle CF, et al. Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Scientific reports. 2016;6:38644. doi:10.1038/srep38644.
- Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, Cao Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci. 2017;114:944–949. doi:10.1073/pnas.1616408114.
- Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, Hattori K. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel: PEDS. 2010;23:385–392. doi:10.1093/protein/gzq009.
- Li B, Tesar D, Boswell CA, Cahaya HS, Wong A, Zhang J, Meng YG, Eigenbrot C, Pantua H, Diao J, Kapadia SB, et al. Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. mAbs. 2014;6:1255–1264. doi:10.4161/mabs.29809.
- Dostalek M, Prueksaritanont T, Kelley RF. Pharmacokinetic de-risking tools for selection of monoclonal antibody lead candidates. mAbs. 2017;9:756–766. doi:10.1080/19420862.2017.1323160.
- Kelly RL, Sun T, Jain T, Caffry I, Yu Y, Cao Y, Lynaugh H, Brown M, Vásquez M, Wittrup KD, Xu Y. High throughput cross-interaction measures for human IgG1 antibodies correlate with clearance rates in mice. mAbs. 2015;7:770–777. doi:10.1080/19420862.2015.1043503.
- Avery LB, Wang M, Kavosi MS, Joyce A, Kurz JC, Fan YY, Dowty ME, Zhang M, Zhang Y, Cheng A, Hua F, et al. Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. mAbs. 2016;8:1064–1078. doi:10.1080/19420862.2016.1193660.
- Wu J, Schultz JS, Weldon CL, Sule SV, Chai Q, Geng SB, Dickinson CD, Tessier PM, et al. Discovery of highly soluble antibodies prior to purification using affinity-capture self-interaction nanoparticle spectroscopy. Protein Eng Des Sel: PEDS. 2015;28:403–414. doi:10.1093/protein/gzv045.
- Piche-Nicholas NM, Avery LB, King AC, Kavosi M, Wang M, O'Hara DM, Tchistiakova L, Katragadda M. Changes in complementarity-determining regions significantly alter IgG binding to the neonatal Fc receptor (FcRn) and pharmacokinetics. MAbs. in press. doi:10.1080/19420862.2017.1389355.
- Schlothauer T, Rueger P, Stracke JO, Hertenberger H, Fingas F, Kling L, Emrich T, Drabner G, Seeber S, Auer J, Koch S, et al. Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies. mAbs. 2013;5:576–586. doi:10.4161/mabs.24981.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi:10.1016/S0169-409X(00)00129-0. PMID: 11259830
- Sule SV, Dickinson CD, Lu J, Chow CK, Tessier PM. Rapid analysis of antibody self-association in complex mixtures using immunogold conjugates. Mol Pharm. 2013;10:1322–1331. doi:10.1021/mp300524x.
- Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu TY, Torrey J, Thomas J, Bobrowicz P, Vásquez M, et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng Des Sel: PEDS. 2013;26:663–670. doi:10.1093/protein/gzt047.
- Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol. 2001;13:1551–1559. doi:10.1093/intimm/13.12.1551.
- Borrok MJ, Wu Y, Beyaz N, Yu XQ, Oganesyan V, Dall'Acqua WF, Tsui P. pH-dependent binding engineering reveals an FcRn affinity threshold that governs IgG recycling. J Biol Chem. 2015;290:4282–4290. doi:10.1074/jbc.M114.603712.
- Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. 2006;281:23514–23524. doi:10.1074/jbc.M604292200.
- Dall'Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol (Baltimore, Md. : 1950). 2002;169:5171–5180. doi:10.4049/jimmunol.169.9.5171.
- Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos: the biological fate of chemicals. 2011;39:1469–1477. doi:10.1124/dmd.111.039453. PMID: 21610128.
- Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28:157–159. doi:10.1038/nbt.1601.
- Jarasch A, Koll H, Regula JT, Bader M, Papadimitriou A, Kettenberger H. Developability assessment during the selection of novel therapeutic antibodies. J Pharm Sci. 2015;104:1885–1898. doi:10.1002/jps.24430.
- Thorn, M., Piche-Nicholas N, Stedman D, Davenport SW, Zhang N, Collinge M, Bowman CJ. Embryo-Fetal Transfer of Bevacizumab (Avastin) in the Rat Over the Course of Gestation and the Impact of Neonatal Fc Receptor (FcRn) Binding. Birth Defects Res B Dev Reprod Toxicol. 2012;95:363–375. doi:10.1002/bdrb.21026.
- Joyce AP, Wang M, Lawrence-Henderson R, Filliettaz C, Leung SS, Xu X, O'Hara DM. One mouse, one pharmacokinetic profile: quantitative whole blood serial sampling for biotherapeutics. Pharm Res. 2014;31:1823–1833. doi:10.1007/s11095-013-1286-y.
- Oitate M, Masubuchi N, Ito T, Yabe Y, Karibe T, Aoki T, Murayama N, Kurihara A, Okudaira N, Izumi T. Prediction of human pharmacokinetics of therapeutic monoclonal antibodies from simple allometry of monkey data. Drug Metab Pharmacokinetics. 2011;26:423–430. doi:10.2133/dmpk.DMPK-11-RG-011. PMID: 21606605.
- Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? mAbs. 2011;3:61–66. doi:10.4161/mabs.3.1.13799.
- Dong JQ, Salinger DH, Endres CJ, Gibbs JP, Hsu CP, Stouch BJ, Hurh E, Gibbs MA. Quantitative prediction of human pharmacokinetics for monoclonal antibodies: retrospective analysis of monkey as a single species for first-in-human prediction. Clinical pharmacokinetics. 2011;50:131–142. doi:10.2165/11537430-000000000-00000.
- Ling J, Zhou H, Jiao Q, Davis HM. Interspecies scaling of therapeutic monoclonal antibodies: initial look. J Clin Pharm. 2009;49:1382–1402. doi:10.1177/0091270009337134.
- Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clinical pharmacokinetics. 2010;49:493–507. doi:10.2165/11531280-000000000-00000.
- Bang LM, Plosker GL. Omalizumab: a review of its use in the management of allergic asthma. Treatments in respiratory medicine. 2004;3:183–199. doi:10.2165/00151829-200403030-00006.
- Oldfield V, Dhillon S, Plosker GL. Tocilizumab: a review of its use in the management of rheumatoid arthritis. Drugs. 2009;69:609–632. doi:10.2165/00003495-200969050-00007.
- Yang BB, Lum P, Chen A, Arends R, Roskos L, Smith B, Pérez Ruixo JJ. Pharmacokinetic and pharmacodynamic perspectives on the clinical drug development of panitumumab. Clinical pharmacokinetics. 2010;49:729–740. doi:10.2165/11535970-000000000-00000.