
ABSTRACT
Immune checkpoints are emerging as novel targets for cancer therapy, and antibodies against them have shown remarkable clinical efficacy with potential for combination treatments to achieve high therapeutic index. This work aims at providing a novel approach for the generation of several novel human immunomodulatory antibodies capable of binding their targets in their native conformation and useful for therapeutic applications.
We performed a massive parallel screening of phage libraries by using for the first time activated human lymphocytes to generate large collections of single-chain variable fragments (scFvs) against 10 different immune checkpoints: LAG-3, PD-L1, PD-1, TIM3, BTLA, TIGIT, OX40, 4-1BB, CD27 and ICOS. By next-generation sequencing and bioinformatics analysis we ranked individual scFvs in each collection and identified those with the highest level of enrichment.
As a proof of concept of the quality/potency of the binders identified by this approach, human IgGs from three of these collections (i.e., PD-1, PD-L1 and LAG-3) were generated and shown to have comparable or better binding affinity and biological activity than the clinically validated anti-PD-1 mAb nivolumab.
The repertoires generated in this work represent a convenient source of agonistic or antagonistic antibodies against the ‘Checkpoint Immunome’ for preclinical screening and clinical implementation of optimized treatments.
Introduction
Activation of immune cells involved in anti-tumor responses is regulated by multiple stimulatory and inhibitory pathways that can be targeted by monoclonal antibodies (mAbs). The immunomodulatory mAbs can either inhibit immunosuppressive receptors or activate co-stimulatory modulators expressed on the surface of T and B lymphocytes or natural killer (NK) cells (collectively named immune checkpoints) to enhance their tumor specific responses.Citation1
Translation of this concept into the clinic has led to the development of novel effective immunotherapies. To date, human or humanized mAbs targeting the immunosuppressive receptors CTLA-4 (ipilimumab), PD-1 (nivolumab and pembrolizumab), and PD-L1 (atezolizumab, durvalumab and avelumab) have been approved for the treatment of several tumors, including melanoma, non-small cell lung cancer, renal cell carcinoma, head and neck squamous cell carcinoma, Hodgkin lymphoma, urothelial carcinoma, liver carcinoma, microsatellite instable (MI) colorectal cancer and Merkel-cell carcinoma.Citation2-Citation5
Spurred by the remarkable success of these immunotherapies, many antibodies targeting other immune pathways have been brought to the clinic with the expectation to some may be more efficacious than the approved products. In fact, despite their success, the currently approved antibodies against immune checkpoints (hence collectively named checkpoint inhibitors; CI) are effective in only about 20–30% of the patients.Citation6-Citation8 Among the new targets are co-inhibitory receptors such as lymphocyte activation gene 3 (LAG-3), an immunosuppressive receptor expressed on activated T lymphocytes and T-regulatory lymphocytes, T cell immunoglobulin and mucin-3 (TIM3), T cell immunoglobulin and ITIM domain (TIGIT), which are expressed on exhausted CD8+ T cells in tumors.Citation9-Citation11 Besides improving CD8+ T cell function, LAG-3, TIM3, and TIGIT blockade is expected to affect tumor tissue Treg cells and interleukin (IL)-10-producing Tr1 cells, with TIM3 and TIGIT blockade also potentially improving dendritic cell phenotype. B- and T-lymphocyte attenuator (BTLA) is another co-inhibitory receptor whose expression is induced during activation of T cells, leading to inhibition of human CD8+ cancer-specific T cells.Citation12 Like PD-1 and CTLA-4, BTLA interacts with a B7 homolog, B7H4, but unlike PD-1 and CTLA-4, BTLA displays T cell inhibition via interaction with tumor necrosis family receptors (TNF-R), not just the B7 family of cell surface receptors.
Similarly, agonistic antibodies recognizing co-stimulatory receptors have reached the clinical stage such as OX40, a secondary co-stimulatory immune checkpoint molecule that prevents premature death of activated lymphocytes, and 4-1BB expressed on activated CD4+ and CD8+ T lymphocytes whose crosslinking enhances T cell proliferation, IL-2 secretion, survival and cytolytic activity. Other co-stimulatory proteins that are considered good targets for antibody-mediated immunotherapy are inducible T cell costimulator (ICOS), which is an immune checkpoint protein belonging to the CD28-superfamily that is expressed on activated T cells, and CD27, a member of the tumor necrosis factor receptor superfamily, which is required for generation and long-term maintenance of T cell immunity.Citation13-Citation15
Antibodies against different immune checkpoint receptors can also be combined to achieve additive or synergistic activity, potentially translating into a better efficacy. Proof of concept for this approach was provided by the finding of increased efficacy of the ipilimumab and nivolumab combination versus monotherapy in the treatment of metastatic melanoma.Citation16,Citation17
A numerous clinical trials with approved or novel antibodies against immunomodulatory receptors used in monotherapy or in combination with other biologics or small molecules are being carried out worldwide. Because large sets of antibodies against the different immune checkpoints are not available in a single lab, most of these trials are being developed without pre-clinical head-to-head comparison to allow for prediction of the most effective treatments.
Further progress in this field could be made by the generation of a complete repertoire of fully human antibodies specific for all these T cell checkpoint modulators. To this aim we tailored phage display technology to the rapid identification of large sets of antibodies recognizing several different immune checkpoints. The rationale of using a novel untested strategy for rapid, parallel screening of phage displayed antibody libraries by directly panning for the first time on activated human lymphocytes was also to ensure the selection of antibodies capable of recognizing the native receptor with the correct post-translational modification as displayed by live human lymphocytes. We demonstrated that we selected mAbs endowed with high binding affinity for their targets and functional activity on lymphocytes. Some of the antibodies identified by this approach compared very well in different functional assays with the clinically validated anti-PD-1/PDL-1 mAbs and were able to inhibit tumor growth also in vivo on a mouse model.
Results
Selection of scFvs on activated human PBMCs
Our goal was the generation of human antibody repertoires against immune checkpoints (IC). Since many IC are expressed on the surface of T lymphocytes and their expression is increased when T cells are exposed to antigen-dependent or independent stimuli, we sought to use unfractionated human peripheral blood mononuclear cells (hPBMCs) to screen libraries of human scFvs. To optimize the selection process, we first evaluated the kinetics and level of expression of 10 different IC () after in vitro activation with anti-CD3/CD28 beads. As shown in , peak expression measured by flow cytometric analysis varied between the different immunomodulators; however, the majority of them reached maximal levels of display after 96 hours of stimulation. After 96 hours of stimulation, all IC were well expressed in more than 50% of the gated population except BTLA, which reached a maximum of display in 40% of the population, and TIGIT, which only slightly increased its expression levels. Similar results were obtained by repeating the same treatment on the same hPBMC sample and on hPBMCs from different donors (not shown). In order to clarify the determination of expression (%) of each target on human lymphocytes, reported in , as well as gating strategy, representative fluorescence-activated cell sorting (FACS) analyses of the time course of activation for the indicated immune checkpoints on human lymphocytes have been reported in Figure S1. On the basis of this analysis, we chose 96 hours as the time point for the activation of lymphocytes to be used for the selection of the human scFv library.
Table 1. Expression profile (% of the positive cells) of each target on human lymphocytes. Percentage of expression was calculated as an average of the fraction of human lymphocytes expressing each target from 8 different donors untreated or activated at different time intervals. Standard Deviations ≤ 10%.
The strategy of using activated lymphocytes in the first panning was devised to ensure targeting of proteins with their native conformation, as that presented on the cell membrane, and to obtain a productive enrichment of several phage clones against multiple targets in one single shot, thus reducing also the time requested for multiple parallel selections.
To this aim about one million phage particles were selected by panning the library (1 × 1012 different phages) on activated hPBMCs as described in the Materials and Methods section (selection cycle 1). A negative panning on untreated lymphocytes was performed for some selections to remove from the repertoire all the non-specific phages that recognize common antigens on the cell surface (see Materials and Methods). For the isolation of scFvs against some targets also expressed at similar levels on untreated lymphocytes, such as CD27, BTLA and TIGIT, the negative panning was not carried out to avoid the loss of some specific phages in the subtractive selection.
The pool of phages obtained from cycle 1 potentially represented a large collection of binders to many different IC, henceforth referred to as the ‘Immunome Library’. To facilitate the identification of molecules recognizing specific receptors, we used a panel of 10 recombinant proteins to further affinity select the Immunome Library. With the aim of developing a universal protocol for the selection of antibodies to cell surface displayed proteins, we performed 10 different parallel selections with two subsequent cycles of selection on recombinant Fc-fusion proteins. This strategy represented a compromise between efficiency of selection and generation of IC-specific repertoires with significant diversity to allow for the identification of antibodies binding to different regions of the target IC and with different biological activities.
In parallel, a conventional selection performed by three panning cycles on the purified protein only was carried out for Lag 3-Fc, followed by enzyme-linked immunosorbent assays (ELISAs) in order to do a comparison with the novel proposed approach (data not shown).
Identification of scFv binders by next-generation sequencing
To identify individual phage clones selected by the combined ex vivo/in vitro approach, we sequenced the VH regions of the IC-specific Repertoires by massive parallel sequencing on the MiSeq Illumina platform (see Methods Section for details). To ensure efficient enrichment of target-specific phages, starting from the immunome library (cycle 1), we performed two subsequent cycles of selection on Fc-fusion recombinant proteins (cycles 2 and 3). Sequence analysis of the whole set of selections revealed that enrichments did occur already after cycle 2, but that significantly higher levels of enrichment (i.e., at least 10-fold) were obtained after the third cycle (). Analysis of parallel sequencing data allowed us to remove scFv clones, which were commonly enriched within each of the 10 selections. Since the Fc region was common to all the recombinant proteins used for selections, we interpreted the occurrence of these clones as unspecific Fc binders; accordingly, they were removed from further analysis. Similarly, the unspecific, biologically-enriched clones, bearing stop codons within the scFv coding sequence, were removed from the list of potential binders. Finally, in order to obtain the most relevant specific clones for each target, we set a threshold of 85 counts per million to the list of the different selection cycles 3. In this way, we captured a detailed snapshot of the best potential binders for 9 of 10 targets (). TIGIT selection was removed from analysis because it did not display significant enrichment of sequences, possibly due to its low expression on activated hPBMCs (see ). shows the detailed enrichment profiles and the heterogeneity for each of the top 10 binders within the nine successful selections, as assessed by phylogenetic analysis.
Figure 1. Immunome screening. The screening procedure started from a universal cycle, common to the different targets, performed by panning the unselected library on activated PBMCs (inner circle). Each divergent circle sector describes the enrichment profiles for the best 10 scFv clones of the indicated targets, scored according to their counts per million values within the cycle 3. The lines within each sector connect the individual enrichments, obtained after cycle 2 (small circles) and cycle 3 (large circles). Cycles 2 and 3 were both performed on the recombinant proteins. The asterisks indicate the clones, successfully converted into stable IgG4s, which were further characterized in this work.
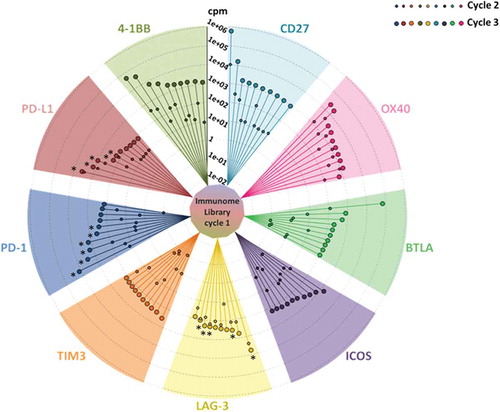
Figure 2. Trends of enrichments and phylogenetic correlations between the top 10 scFv clones, representative of each target. For each of the indicated targets, the left panel shows the representation of relative enrichments across the three selection cycles. The dendrograms shown at the right side report the phylogenetic clustering of the 10 most enriched scFv clones, according to the translated sequences of the corresponding hCDR3 regions.
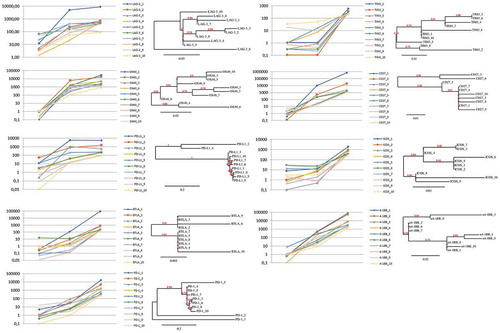
The scFv sequences that were specifically enriched after panning on three targets, i.e., LAG-3, PD-1 and PD-L1, were chosen for further studies. Antibodies specific for two of these targets, such as anti-PD-1 nivolumab and anti-PDL-1 atezolizumab, have been previously developed and widely used in the clinic, and thus they can be used for comparison in biological assays. Our goal was to identify, for further characterizations, at least 5 effective antibodies for each of the three targets. Starting from the most enriched sequences for each of the LAG-3, PD-1 and PD-L1 IC-specific repertoires, we accomplished the rescue of the corresponding scFvs and their conversion into human IgG4s. As described in Table S1, only 5 of 10 LAG-3 sequences (i.e., LAG-3_1, LAG-3_3, LAG-3_7, LAG-3_8, LAG-3_9) were able to produce stable IgG4s for further characterization (Table S1). In contrast, the initial sets of 6 and 5 sequences, respectively, for PD-1 and PD-L1, were all successfully converted into the corresponding, stable IgG4s (Table S1).
Human IgGs generated from selected binders show high binding affinity and receptor/ligand competitive activity
Human IgG4 antibodies generated from the best ranking LAG-3, PD-1 and PD-L1 binders were confirmed to recognize activated PBMCs and recombinant proteins with low nanomolar or sub-nanomolar affinity (see , Table therein, and Figure S2).
Figure 3. Binding of the selected antibodies to the purified recombinant proteins and tumor cells. Binding curves of the antibodies to each target protein/Fc (black curves) or Fc (grey curves) are reported in the left and central panels. The binding curves of the anti-PD-L1 mAbs to MDA-MB231 cells (black curves) or MCF-7 cells (grey curves) are reported in the right panel. The Kd values obtained from the binding curves of the antibodies to each target protein/Fc, activated hPBMCs and tumor cells are reported in the table. Binding values were reported as the mean of at least three determinations obtained in three independent experiments. Error bars depicted means ± SD. P values for the binding of the indicated mAbs to each target protein/Fc relative to their binding to Fc: ***P ≤ 0.001; **P < 0.01.
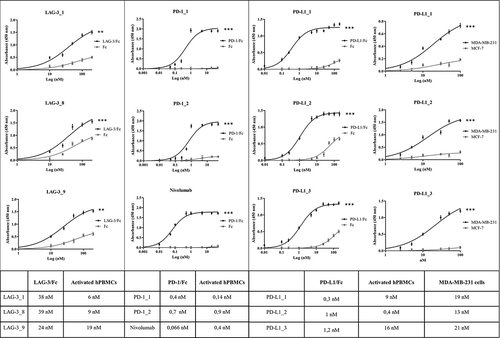
In particular, some anti-PD-1 mAbs (i.e., PD-1_1 and PD-1_2) showed comparable or higher apparent affinity compared to the clinically validated nivolumab. Most of the anti-PD-1 and anti-PD-L1 antibodies, including nivolumab, displayed similar binding to the recombinant protein compared to the activated hPBMCs, while the anti-LAG-3 antibodies showed higher affinity for the activated hPBMCs. Two or three of the original five antibodies chosen were used for further characterization because they were highly specific for both recombinant targets and activated lymphocytes, whereas they showed no or poor binding to Fc or untreated lymphocytes (see and Figure S2). The other IgG4 were discarded because they recognized the Fc or not activated lymphocytes (data not shown). As for the anti-Lag 3 scFvs selected by the conventional strategy on purified recombinant protein only and screened by ELISA assays, only a low percentage (15%) of positive clones contained a cDNA sequence encoding the whole scFv (with no stop codons or other rearrangements). The few clones expressing the whole scFvs were converted into human IgG4, and were found to be capable of binding to the purified protein, but they recognized the activated lymphocytes with lower affinity (data not shown). Furthermore, some of them were found to be unstable during their storage at 4°C for 2–4 days, thus they were not used for further characterization.
Because anti-PD-L1 antibodies have been shown to provide for clinical benefit by blocking interaction between PD-L1 expressed on cancer cells and PD-1 displayed by T cells,Citation4 we tested whether the anti-PD-L1 antibodies would also recognize their targets on the surface of cancer cells. All anti-PD-L1 antibodies showed high affinity for tumor cells expressing PD-L1, such as mammary MDA-MB-231 tumor cells, and the hierarchy of binding activity to tumor cells was similar to that observed with activated hPBMC ( and Table therein).
High affinity IgGs against immune checkpoint molecules display T cell immunostimulatory activity and effector functions
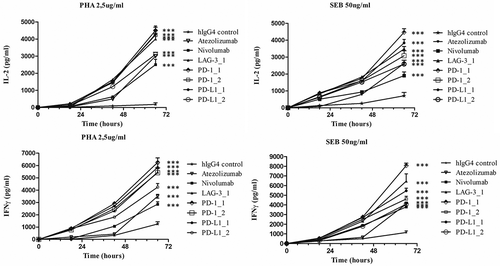
Previous reports showed that CD3-positive primary resting cells in unfractionated hPBMCs can be induced to proliferate in vitro by using Staphylococcal enterotoxin B (SEB) or phytohemagglutinin (PHA), and that this activity is modulated by CI.Citation18-Citation20 We therefore used this assay to test whether the selected antibodies against LAG-3, PD-1 or PD-L1 were able to increase cell division. To this end, some mAbs among those with the highest affinity and specificity were tested for their biological activity in a flow cytometry-based lymphocyte proliferation assay that allows identification of proliferating T cells by anti-CD3 staining. With this aim, carboxyfluorescein diacetate, succinimidyl ester (CFDA-SE)-stained lymphocytes were stimulated with PHA, and incubated in the absence or presence of the antibodies to induce antigen-specific T cells proliferation. In this assay, nivolumab consistently led to a 50% increase in proliferative activity and the anti-PD-1 antibodies PD-1_1 and PD-1_2 were also able to efficiently stimulate T cell proliferation with the former showing higher activity than nivolumab (). Similarly, all three antibodies against PD-L1 and one of the three LAG-3 antibodies also induced various degrees of proliferation. The ability to stimulate proliferation did not always correlate with the binding potency, suggesting that the different antibodies may have distinct modes of interactions with their targets.
Figure 4. Effects of the novel antibodies on lymphocyte proliferation. A. Proliferation of hPBMCs after stimulation with PHA at 2.5 μg/mL in the absence or in the presence of the immunomodulatory antibodies. Fold increase of CD3+ T cell proliferation determined by the indicated selected antibodies was measured by anti-CD3 staining by FACS with respect to activation of hPBMCs with PHA at 2.5 μg/mL in the absence of antibodies or in the presence of an unrelated IgG4. B. Effects of anti-PD-L1 antibodies on Lymphocyte proliferation as induced by tumor cells. Fold increase of hPBMCs proliferation as determined by normalized absorbance values obtained by ELISA with anti-BrdU antibodies in hPBMCs samples co-cultured with MDA-MB-231 (left panel) or MCF-7 (right panel) tumor cells in the absence (white bar) or in the presence of increasing concentrations (50 nM and 200 nM) of PD-L1_1 (light grey bars) or PD-1_1 (grey bars) antibodies for 72 hours at 37°C. Nivolumab was used, in both the experiments, as a positive control (black bars). Concentration values were reported as the mean at least three determinations in five independent experiments performed by using lymphocytes from 5–8 healthy donors. Error bars depicted means ± SD. P values for the indicated mAbs relative to unrelated IgG4 are: ***P ≤ 0.001; **P < 0.01; *P < 0.05.
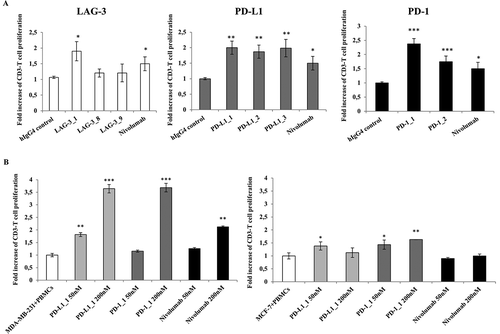
The antibodies PD-1_1 and PD-L1_1 that were the best activating antibodies on human lymphocytes confirmed this ability also on murine lymphocytes (Figure S3), thus suggesting that they are cross-reactive with mouse PD-1 and PD-L1.
We used MDA-MB-231 breast tumor cells, which express high levels of PD-L1 on their surface, to test the ability of the anti-PD-L1 and anti-PD-1 antibodies to suppress the inhibitory action of the PD-1/PD-L1 interaction on lymphocyte proliferation when the two cell types are co-cultured. As shown in , mAbs PD-L1_1 and PD-1_1 induced the proliferation of lymphocytes in a dose-dependent manner, whereas only a slight effect was detected on lymphocytes treated with mAbs PD-L1_2 and PD-1_2 (data not shown). Also in this assay mAbs PD-L1_1 and PD-1_1 displayed higher activity than nivolumab, while no effects on the proliferation of lymphocytes were observed when PD-L1-negative MCF-7 tumor cells were used. In competition ELISA assays, we showed that mAb PD-L1_1 and PD-L1_2 were able to interfere with the recognition of PD-L1 by its two receptors PD-1 and B7.1 (Figure S4). However, we suppose that these novel antibodies could act not only by interfering in PD-1 and PD-L1 interaction, but also by other antagonistic effects on their receptors or ligands. Indeed, it is likely that PD-1_1 has a different mechanism of action with respect to nivolumab, as it recognizes a different epitope, as evidenced from competitive ELISA assays performed by measuring the binding to PD-1 of biotinylated nivolumab in the absence or presence of saturating concentrations of unlabeled PD-1_1 mAb. Simultaneous binding of PD-1_1 and nivolumab revealed that the two mAbs did not interfere in their interactions with the receptor (data not shown).
Immunomodulatory antibodies, such as nivolumab, ipilimumab and pembrolizumab were shown to improve T cell effector functions, with this property potentially increasing their clinical benefit.Citation18-Citation20 We therefore tested five antibodies among those with the highest ability to induce T-cell proliferation (LAG-3_1, PD-1_1, PD-1_2, PD-L1_1, and PD-L1_2) in a cytokine secretion assay previously reported for the characterization of nivolumab.Citation19 In this assay, we included atezolizumab as an additional positive control.Citation3,Citation4 As shown in , all the tested antibodies were able to increase the secretion of both IL-2 and interferon (IFN)γ by hPBMCs stimulated with either PHA or SEB. Cytokine secretion increased over time upon addition of the different antibodies to the cell culture mixture. PD-1_1 mAb appeared to be consistently more potent than all other tested antibodies for its ability to stimulate secretion of both cytokines. Also, in this assay most of the newly identified antibodies compared well with nivolumab and atezolizumab, further supporting the conclusion that the Immunome Library that we generated is enriched in binders with good potential for being developed for clinical use.
Figure 5. Effects of the novel immunomodulatory antibodies on secretion of cytokines by stimulated T cells. IL-2 and IFNγ values obtained by ELISA assays on supernatants of hPBMCs stimulated with PHA (2.5 μg/mL) or SEB (50 ng/mL) in the absence or in the presence of the antibodies LAG-3_1, PD-1_1, PD-1_2, PD-L1_1, PD-L1_2 for 18–66 hours at 37°C. Nivolumab, atezolizumab and an unrelated antibody were used as a positive and negative control, respectively. Concentration values were reported as the mean at least three determinations in five independent experiments performed by using lymphocytes from 5–8 healthy donors. Error bars depicted means ± SD. P value for the indicated mAbs relative to unrelated IgG4 is: ***P ≤ 0.001.
In vivo antitumor activity
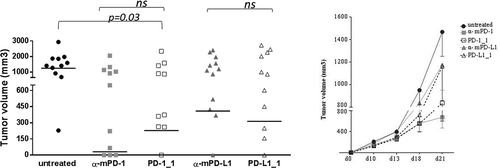
The two antibodies PD-1_1 and PD-L1_1, given their cross-reactivity with mouse PD-1 and PD-L1, were also tested in vivo on the CT26 colon cancer model. Mice were implanted with CT26 cells (day 0) and then treated with PD-1_1 and PD-L1_1 antibodies (day 3, 6, 10). Two commercially available antibodies reacting against murine PD-1 and PD-L1 (α-mPD-1 and α-mPD-L1), and previously validated in vivo, were used as positive controls.Citation21 While the growth rate of tumors in untreated mice was very fast and uncontrolled, with the majority of tumors reaching sizes of >650 mm3 at day 21, a reduction in tumor volume was found in mice treated with PD-1_1 (p = 0.03). The activity of the cross-reactive anti-PD-1 antibody is comparable to that of the commercial antibody against murine PD-1. A trend of comparable activity was also observed for the two anti-PD-L1 antibodies (). Treated mice consistently show a dichotomy in the response to PD-1 and PD-L1 blockade with two distinct treatment outcomes, responder and non-responder mice, as well described for this and other cancer models. During the period of treatment, the animals did not show signs of wasting or other visible signs of toxicity. Thus, the biological and functional activity of the two novel mAbs was also confirmed in a relevant in vivo model.
Figure 6. In vivo antitumor activities of PD-1_1 and PD-L1_1 antibodies. Tumor growth in groups of 10–12 mice inoculated with CT26 cells at day 0 and treated with PD-1_1, PD-L1_1 or positive control antibodies reacting against murine PD-1 and PD-L1 (α-mPD-1, α-mPD-L1) at day 3, 6 and 10. Shown is tumor volume for individual mice at day 21 (left) and mean of tumor volume for each group over time (right panel).
Discussion
Cancer immunotherapy based on immunomodulatory antibodies is becoming a mainstay of modern oncology. Checkpoint inhibitors (i.e., anti-PD-1, anti-PD-L1 and anti-CTLA-4 agents) have been shown to achieve unprecedented efficacy with long lasting clinical benefit. Perhaps most importantly, these antibodies have an extremely wide range of applications, with the currently approved ones used for the treatment of several tumors.Citation1-Citation5 This notwithstanding, treatment efficacy with CIs is still limited to 20–30% of the population, and could not be shown in highly prevalent cancer types such as colorectal, breast and prostate cancers.Citation6-Citation8 However, the recent enrichment of the armoury of mAbs against new targets with agonistic or antagonistic activity for co-stimulatory or inhibitory receptors, respectively, allow the design of new combination therapies to enhance the potential of the immune-based cancer therapy.
In this respect, a successful example is represented by the combination of anti-PD-1 and anti-CTLA-4 antibodies, which resulted in a longer progression-free survival and a higher rate of response than that observed with single mAb treatments in metastatic melanoma and MSI-associated colorectal cancer.Citation16,Citation17 Thus, further progress in the field could be made by the generation of a complete repertoire of fully human antibodies specific for many ICs to be tested preclinically alone and in appropriate combinations for rapid translation into the clinic. To this end we chose live activated hPBMCs as selectors for the generation of an unbiased library of IC binders, the Immunome Library, from which scFvs specifically recognizing a given receptor were identified by subsequent affinity selection cycles using recombinant proteins as specific baits.
Consistent with this working hypothesis, human IgG4 generated using the selected scFvs were able to recognize the cell-displayed receptors with apparent affinity in the low nanomolar range and down to 0.1–0.4 nM. This binding affinity is well within what has already been shown with clinically active checkpoint-specific antibodies, and compares well with that of the anti-PD-1 mAb nivolumab already approved for the treatment of many types of cancer.Citation19 To improve the efficiency of selection on hPBMCs, we increased the level of expression of the target IC by anti-CD3 crosslinking of the T cells. We have not tested the efficiency of selection on untreated hPBMCs; however, it is relevant to note that the only selection that did not yield enriched clones was the one on TIGIT, which displayed the lowest level of expression even after anti-CD3 activation, suggesting that there may be a threshold of receptor expression below which enrichment of specific clones is inefficient.
All the other selections achieved enrichment levels between 100 and 1000 counts per million, with four of them (PD-1, 4-1BB, CD27 and OX40) reaching even higher enrichment. Comparative sequence analysis of the different selection cycles confirmed that in most selections (i.e., seven of nine) there was a significant improvement in clones enrichment after the second cycle (i.e., at least 10 CPM), while selection on LAG-3 and OX40 did not seem to improve beyond the second cycle.
Our results support the conclusion that the combined ex vivo/in vitro selection with the next-generation sequencing approach used in this study is more rapid and efficient than the conventional selection carried out by panning on the purified protein only (on Lag 3-Fc as an example), followed by screening with ELISA assays. This is because, using the novel strategy, we can easily remove clones with stop codons and obtain more stable antibodies with respect to those selected by the conventional strategy. Furthermore, the novel strategy is even more convenient than that of combining selection on live cells and identification of specific binders by direct screening on the target of choice, which we previously adopted.Citation22
A major objective of this work was to generate repertoires of specific binders from which novel antibodies with biological activity could be rapidly identified. To prove that this was indeed the case, we generated human IgG4 from scFvs selected on PD-1, PD-L1 and LAG-3. The choice of these three targets was based on the observation that selections on them gave rise to median levels of enrichment and, most importantly, because there are well established in vitro assays to evaluate their functional activity.
From the list of the top 10 ranking scFv sequences, at least 5 effective antibodies for each of these three targets were converted in IgG4 and found to be expressed at satisfactory levels, stable in solution and capable of binding to their targets. Overall, of the sixteen IgGs with confirmed binding (five anti-LAG-3, five anti-PD-L1 and six anti-PD-1), eleven displayed low nanomolar apparent affinity for their cognate receptor expressed on hPBMCs, and three of them (PD-L1_2, PD-1_1 and PD-1_2) had sub-nanomolar apparent affinity and compared well with that of nivolumab. Among them, at least 2–3 of the 5 converted antibodies for each target were found to be highly specific for both recombinant purified targets and activated lymphocytes, with no significant binding to untreated lymphocytes or Fc, and thus they were chosen for further biological and functional assays.
When tested for their ability to stimulate T-cell proliferation, six of eight antibodies (LAG-3_1, PD-L1_1, PD-L1_2, PD-L1_3, PD-1_1 and PD-1_2) did show biological activity with the PD-1_1 mAb performing better than the clinically active nivolumab. PD-1_1 also displayed significant activity in promoting T cell proliferation in co-cultures with high PD-L1-expressing tumor cell line MDA-MB-231. In this assay also the PD-L1_1 mAb almost tripled T cell proliferation, consistently with its ability to interfere with PD-L1/PD-1 interaction.
We further demonstrated that some of the antibodies identified in this work can stimulate T cells to secrete IL-2 and IFNγ. PD-1_1 mAb consistently showed the highest activity in stimulating cytokine secretion and extended the observation that some of the antibodies identified in this work display binding affinities and biological properties similar to the clinically validated atezolizumab and nivolumab.Citation19-Citation25 In fact, some of the new antibodies showed even higher activities compared to nivolumab when tested in cell-based assays with co-cultures of lymphocytes with antigen presenting cells or tumor cells.
Due to the property of the two novel antibodies PD-1_1 and PD-L1_1 to cross-react with mouse lymphocytes differently from nivolumab, they were tested in vivo on mice bearing CT26 colon carcinoma and found to efficiently inhibit tumor growth without apparent toxic effects. Autoimmune reactions have been shown to be the major toxic effect of this class of therapeutic antibodies.Citation26 Further studies are needed to carefully monitor auto-immune side effects, potentially induced by these antibodies. However, the high affinity of the described novel immunomodulatory antibodies could allow for reducing the therapeutic doses and potentially the consequent eventual unwanted side effects.
Overall, the data presented in this work support the conclusion that our selection procedure allows the generation of antibodies specifically recognizing their target receptor in its native conformation and with high affinity without need for further affinity maturation. Furthermore, many of the novel antibodies displayed the ability to stimulate T cell proliferation and effector functions in a manner comparable to, or even superior than, the anti-PD-1 mAb nivolumab, which has shown clinical activity in different type of cancer diseases, suggesting that the Immunome Library that we generated is enriched in binders with good potential for being developed for clinical use.
Materials and methods
Antibodies and human recombinant proteins
The following antibodies were used: horseradish peroxidase (HRP)-conjugated anti-His mouse monoclonal antibody (Qiagen, 1,014,992); HRP-conjugated anti-M13 mouse monoclonal antibody (GE Healthcare, 27-9420-01); anti-human LAG-3 mouse monoclonal antibody (R&D Systems, MAB23193); anti-human PD-1 human monoclonal antibody nivolumab (Opdivo®, Bristol-Myers Squibb, 30,121,608); anti-human PD-L1 human monoclonal antibody (G&P Biosciences, MAB0199); HRP-conjugated anti-human IgG (Promega Madison, W4031); PE/anti-human CD2 mouse monoclonal antibody (BioLegend Inc., 300,208); APC/anti-human CD3 mouse antibody (557,597); PE/anti-human CD4 mouse monoclonal antibody (555,347); PerCP/anti-human CD8 mouse monoclonal antibody, 347,314 (all from BD Biosciences); HRP-conjugated anti-human IgG (Fab’)2 goat monoclonal antibody (Abcam, ab87422); APC/anti-human IgG Fc mouse antibody (409,306); APC/Cy7 anti-human CD366 (TIM3) mouse antibody (345,026); APC/anti-human CD272 (BTLA) mouse antibody (344,510); APC/anti-human CD137 (4-1BB) mouse antibody (309,810); APC/anti-human CD134 (OX40) mouse antibody (350,008); Brilliant Violet 510™/anti-mouse/rat/human CD27 hamster antibody (124,229); APC/anti-human/mouse/rat CD278 (ICOS) hamster antibody, 313,510 (all from BioLegend); FITC/anti-human TIGIT mouse antibody (Thermo Fisher Scientific, 11-9500-41).
The following recombinant chimeric proteins were used: human LAG-3/Fc; human PD-1/Fc (2319-L350); human PD-L1/Fc (156-B7-100); TIM3/Fc (2365-TM-050); BTLA/Fc (8385-BT-050); TIGIT/Fc (9464-TG-050); OX40/Fc (3388-OX-050); 4-1BB/Fc (838-4B-100); CD27/Fc (382-CD-10) and ICOS/Fc (169-CS-050); – human IgG1-Fc, 110-HG-100 (all from R&D Systems).
Cell cultures
MDA-MB-231 cells were cultured in Dulbecco’s Modified Eagle’s Medium (GibcoTM DMEM, Thermo Fisher Scientific, 11,965,092). MCF-7 cells were cultured in Modified Eagle’s Medium (GibcoTM MEM, Thermo Fisher Scientific, 11,095,080). Media were supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS, Sigma-Aldrich, MFCD00132239), 50 UI mL-1 penicillin, 50 µg mL-1 streptomycin (50 µg mL-1 streptomycin), 2 nM L-glutamine (A2916801) all from GibcoTM, Thermo Fisher Scientific. Cell lines were purchased from the American Type Culture Collection (ATCC) and cultured in a humidified atmosphere containing 5% CO2 at 37°C.
Isolation of human peripheral blood mononuclear cells
Human PBMCs were isolated from blood of healthy donors by using Greiner Leucosep® tube (Sigma-Aldrich, 227,288) following the manufacturer’s instructions and frozen in a solution containing 90% FBS and 10% dimethyl sulfoxide (DMSO) until use.
Cryopreserved cell vials were gently thawed by using RPMI 1640 medium (GibcoTM, Thermo Fisher Scientific, 11,875,093) supplemented with 1% L-glutamine, 1% CTL-Wash™ (Cellular Technology Limited, CTLW-010), and 100 U/mL Benzonase (Merck Millipore, 0500–3115).
The collected hPBMCs were then washed by centrifugation, plated and incubated overnight at 37°C in R10 medium consisting of RPMI 1640 supplemented with 10% inactivated FBS, 1% L-glutamine, 50 U mL−1 penicillin, 50 μg mL−1 streptomycin and 1% HEPES (GibcoTM, Thermo Fisher Scientific, 15,630,080).
After an overnight resting, the hPBMCs were collected in phosphate-buffered saline (PBS), counted by using the Muse® Cell Analyzer (Merck Millipore, 0500–3115) and resuspended at a density of 1 · 106 cells/mL.
Selection of scFv-phage clones
Phagemid particles were recovered from the library by using the M13-K07 helper phage (Invitrogen, Thermo Fisher Scientific, 18,311,019), as previously described.Citation27 For each round of selection, phages (1013 cfu) were blocked by incubating them with 5% (wt/vol) Skim Milk Powder (Fluka Analytical, Sigma-Aldrich, 70,166) for 30 mins in PBS before their use.
For the first round of selection, blocked phages (1013 cfu) were firstly submitted to one round of negative selection by incubation with untreated lymphocytes (5 × 106) in 2.5% (wt/vol) Skim Milk Powder (Fluka Analytical, Sigma-Aldrich, USA) in PBS for 2 hours at 4°C by gently rotation. The unbound phages were collected in the supernatant by centrifugation at 1200 rpm for 10 minutes and then used for the positive selection performed by incubating them with activated lymphocytes (1 × 106) overnight by gently rotation at 4°C in rotation in the presence of 2.5% skim milk powder.
For the selection of phages binding to CD27, BTLA and TIGIT the negative panning was not carried out to avoid the loss of some specific phages in the subtractive selection.
After extensive washes with PBS, the bound phages were eluted from activated lymphocytes with 76 mM citric acid (pH 2.5) in PBS for 5 minutes, and then neutralized with 1M Tris-HCl (pH 8.0). The recovered phages were amplified by infecting E. coli TG1 cells to prepare phages for the next rounds of selection on the purified chimeric protein.
To this aim, NuncTM polypropylene tubes (Fisher Scientific, Thermo Fisher Scientific, 151,232) were coated with the selected recombinant chimeric protein (previously described in ) at a concentration of 20 μg/mL in a 0,05 M NaHCO3 solution, for 72 hours at 4°C.
Blocked phages were submitted to two following rounds of negative selection by incubating them in the tubes coated with rhIgG1-Fc protein for 2 hours at 4°C in rotation. Unbound phages, recovered in the supernatant, were then incubated in the coated tubes, prepared as described above for positive selection, overnight at 4°C in rotation, and eluted as described above.
Alternatively, since a trypsin site is present between the scFv and the pIII protein of the phage coat, trypsin was used for a more efficient and selective elution of the binders. Briefly, after extensive washes with PBS, the bound phages were incubated with 50 mM Tris-HCl (pH 8.0) buffer containing 1 mM CaCl2 for 1 hour at 4°C in rotation, and then eluted from the chimeric proteins with trypsin (1 μg/mL) incubated for further 15 minutes at 25°C by gently shaking. The reaction was then blocked by using protease inhibitors (Complete™ EDTA-free Protease Inhibitor Cocktail, Sigma-Aldrich, 11,873,580,001). Phages were then collected and stored at 4°C until use.
DNA fragment preparation and high-throughput sequencing
For each sublibrary, obtained from each selection cycle, the phagemid double-strand DNAs containing the scFvs were purified from cultures of superinfected E. coli TG1 cells using Endo free Plasmid Maxi Kit (Qiagen, 12,362). The full-length scFvs were excised with restriction enzymes BamHI (R3136) and HindIII (R3104), all from New England Biolabs, and purified with Wizard® SV Gel and PCR Clean-Up System (Promega, A9281) from 1.2% agarose gel.Citation28 A second enzymatic excision was performed with NcoI (R3193) and XhoI (R0146), all from New England Biolabs, to isolate the VHs from the previously purified material, and then extracted from a 1.4% agarose gel. Library preparations for sequencing and preliminary bioinformatic analysis of the data were performed at the Center for Translational Genomics and Bioinformatics, Hospital San Raffaele, Milano, Italy. The VHs extracted from sub-libraries were bar-coded by TruSeq ChIP sample prep kit (Illumina, 15,023,092). A complementary scheme for bar-coding was implemented to obtain a deep and suitable sequencing of VH mixtures of several subcycles. Subcycles 2 and 3 for each target were mixed in a dedicated run. The first universal cycle 1 was sequenced separately to cover the larger complexity. The bar-coded samples were diluted to a final concentration of 10 pM and sequenced with 2 × 300 nt SBS kit v3 on an Illumina MiSeq apparatus.
scFv recovery
The clones of interest were isolated from the sublibrary at cycle 3 of the corresponding target. For high-ranking clones (relative representativeness ≥ 1% within cycle 3), the QuickChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies, 200,521) was used to perform copies of clones with overlapping primers, designed within the corresponding HCDR3 regions, according to the procedure previously described.Citation28 Briefly, the extension reactions were assembled as follows: 50–250 ng of template; 2/5 μL QuickSolution reagent; 1 μL Pfu Ultra High-Fidelity DNA polymerase (2.5 U/μL); 5 μL 10× reaction buffer; 1 μL dNTPmix; 125 ng forward primer; 125 ng reverse primer; H2O to a final volume of 50 μL. The template DNA was removed by restriction with 1 μL of DpnI enzyme, as suggested by the kit provider. An appropriate amount of reaction was used to transform XL10-GOLD ultracompetent cells (Agilent Technologies, 200,314) and then plated on LB/agar containing 100 μg/mL ampicillin. Some colonies were picked and evaluated by double digestion and sequencing.
For low ranking clones (relative representativeness < 1% within cycle 3), the DNA samples were isolated from cycle 3 by overlapping PCR. Briefly, Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific, F530S) was used to perform two PCR reactions to obtain separately VH and VL fragments, using primers designed within the corresponding HCDR3 regions and in constant region of plasmid upstream and downstream of VH and VL. In a second step, the PCR fragments corresponding to each clone were mixed and extended to get the full scFvs. The reactions were assembled as follows: 150 ng of template for VH and VL fragments amplification and 10 ng of template (VH and VL fragments) for full scFv amplification; 0,5 μL Phusion DNA Polymerase (0.02 U/μl); 10 μL 5× Phusion HF Buffer; 1 μL dNTP mix; 0,5 μM forward primer; 0,5 μM reverse primer; 1,5 μL DMSO; H2O to a final volume of 50 μL.
Antibody production and purification
The scFvs of interest were converted into whole IgG4 antibodies by cloning the corresponding VH and VL cDNAs in the pEU vectors 8.2VH and 4.2VL, expressing respectively, the constant antibody heavy and light chainsCitation29. Briefly, the VHs and VLs were amplified by CloneAmp HiFi PCR Premix in standard conditions with specific primers and purified with Wizard® SV Gel and PCR Clean-Up System (Promega, A9281) from 1,3% agarose gel. In-Fusion HD cloning kit (Clontech Laboratories, 639,692) was used to clone the VHs in BamHI (R3136) and BssHII (R0199) all from New England Biolabs, linearized pEU8.2VH vector, and the VLs in ApaLI (R0507) and AvrII (R0174) New England Biolabs, linearized pEU4.2VL vector. Stellar Competent Cells (Clontech Laboratories, 636,763) were transformed with the obtained vectors, and the colonies were screened by digestion and sequence analysis. The vectors containing the correct DNA, prepared with an endotoxin-free system (EndoFree Plasmid Maxi Kit, Qiagen, 12,362), were co-transfected in HEK293-EBNA by using Lipofectamine Transfection Reagent (Life Technologies, Inc. 11,668,019) and grown up for about 10 days at 37°C in chemical defined CD CHO medium (Gibco, Life Technologies, Inc. 10,743,029) supplemented with 5 mL of L-glutamine 200 mM (Gibco, Life Technologies, A2916801), 5 mL of Penicillin-Streptomicyn 10,000 U/mL-10 mg/mL (Sigma-Aldirch, P0781) in 6-well plates or in 150 mm Corning® tissue-culture treated culture dishes. The conditioned media were collected, and the antibodies were purified by using Protein A HP SpinTrapCitation30 (GE Healthcare Life Sciences, 28–9031–32). The purity of the final products was evaluated by SDS-PAGE NuPAGE™ 4–12% Bis-Tris Protein Gels, 1.0 mm, 10-well (Thermo Fisher Scientific, NP0321BOX) followed by the staining with Coomassie blue solution (Biorad, 1,610,786) for 20 minutes and de-stained with 7% of CH3COOH and 20% of Et-OH. All the antibody preparations were filtered with sterile 0.22 µm durapore hydrophilic filters (Millipore, MA, SLGS033SS) before their use.
Enzyme-linked immunosorbent assay
To confirm the binding specificity of the purified mAbs, ELISA assays were performed on chimeric proteins (coated at 5 μg/mL on microplates), tumor cells (PD-L1-positive breast cancer MDA-MB-231 cells or PD-L1-negative breast cancer MCF7 cells), and untreated or activated hPBMCs.
The ELISA assays on coated chimeric protein were performed by coating NuncTM flat-bottom 96-well plates (Thermo Fisher Scientific, 3596) with 5 μg/mL rhPD-1, rhPD-L1 and rhLAG-3 recombinant proteins in a solution of 0.05 M NaHCO3 for 72 hours at 37°C. After blocking of the coated 96-well plates with 5% nonfat dry milk in PBS for 1 hour at 37°C, the purified mAbs were added at increasing concentrations (10–200 nM) to the plates in 2.5% nonfat dry milk in PBS and incubated for 2 hours at room temperature by gently shaking. After extensive washes with PBS, the plates were incubated with HRP-conjugated anti-human IgG (Fab’)2 goat monoclonal antibody (Abcam, ab98535) for 1 hour, washed again and incubated with TMB reagent for 10 min before quenching with an equal volume of 1 N HCl.
Cell ELISA assays were performed by plating the cells in round-bottom 96-well plates (2 · 105 cells or 2 · 105 lymphocytes for each well) and incubating them with increasing concentrations (0,5–200 nM) of mAbs in 2.5% nonfat dry milk for 2 hours at room temperature with gentle agitation. Plates were then centrifuged; cell pellets were washed with PBS and incubated with HRP-conjugated anti-human IgG goat polyclonal antibody for 1 hour at room temperature. Following additional washes in PBS 1X, TMB reagent was added for 10 min before quenching with an equal volume of 1 N HCl. Absorbance at 450 nm was measured by the Envision plate reader (Perkin Elmer, 2102). Binding values were reported as the mean of at least three determinations obtained in three independent experiments. The Kd values were calculated by elaboration of ELISA binding curve analyses by Prism (Graphpad) tool according to the following model: Y = Bmax*X/(Kd+ X) + NS*X + Background. Bmax is the maximum specific binding in the same units as Y; Kd is the equilibrium binding constant, in the same units as X, and it is the ligand concentration needed to achieve a half-maximum binding at equilibrium; NS is the slope of non-specific binding in Y units divided by X units; background is the amount of nonspecific binding with no added ligand.
Lymphocyte proliferation ELISA assay based on the measurement of BrdU incorporation after co-culture with PD-L1 positive tumor cells
Since the MDA-MB-231 breast tumor cells express high levels of PD-L1 molecules on their surface, we tested the ability of the anti-PD-L1_1 and PD-Ll_2 antibodies to suppress the inhibitory action of the PD-1/PD-L1 interaction on lymphocyte proliferation when the two cell types are co-cultured. To this aim we performed a colorimetric immunoassay, by using Cell Proliferation ELISA BrdU kit (Roche® Applied Science GmbH, 11,644,793,001), according to the manufacturer’s recommendations, for the quantification of the lymphocyte proliferation.
Briefly, MDA-MB-231 (10,000 cells/well) were previously treated for 30 min at 37°C with mitomycin C (50 µg/ml), which inhibits the DNA synthesis of tumor cells, thus allowing the specific evaluation of lymphocyte proliferation to be followed. Then, the tumor cells were co-cultivated with hPBMCs in the absence or in the presence of increasing concentrations of anti-PD-L1 antibodies (50 nM, 100 nM and 200 nM) for 72 hours at 37°C. After an additional incubation of 24 hours with BrdU labelling solution, the cells were fixed and the DNA was denatured to improve the accessibility of the BrdU incorporated in newly synthesized DNA for detection with the HRP-conjugated anti-BrdU antibodies. The bound antibodies were detected by the addition of TMB and quantified spectrophotometrically by measuring the absorbance at 450 nm. The increase of the absorbance values correlate with DNA synthesis, and thus allow quantification of proliferating cells.
FACS analysis of expression levels of immune checkpoints on hPBMCs
Human PBMCs were isolated as described in the Materials and Methods section. After an overnight incubation they were activated with Dynabeads® Human T-Activator CD3/CD28 at a concentration of 1.103 beads/mL (GibcoTM, Thermo Fisher Scientific, 11131D). After 24–96 hours of activation, the cells were seeded in a round-bottom 96-well plate (1 · 106 cells/well) and then centrifuged at 1200 rpm for 5 minutes to remove the supernatant. Unlabeled anti-LAG-3, anti-PD-1 (nivolumab) or anti-PD-L1 primary antibodies were added to each well at a concentration of 10 μg/mL and incubated for 90 minutes at room temperature by gently shaking. After extensive washes, the cells were stained with 100 μL APC/anti-human IgG Fc antibody in FACS buffer (PBS, 1% FBS), and with 10 μg/mL PE/anti-human CD2 antibody (BioLegend, San Diego, 300,208) for 45 minutes at room temperature by gently shaking.
The labeled antibodies APC/Cy7 anti-human CD366 (TIM3), APC/anti-human CD272 (BTLA), APC/anti-human CD137 (4-1BB), PE/anti-human CD134 (OX40), Brilliant Violet 510™/anti-mouse/rat/human CD27, APC/anti-human/mouse/rat CD278 (ICOS), FITC/anti-human TIGIT antibodies were added to each well at a concentration of 10 μg/mL and incubated for 90 minutes at room temperature by gently shaking. After two washes with FACS buffer, the cells were resuspended in PBS and analyzed on CytoFLEX Flow Cytometer (Beckman Coulter).
Lymphocytes were identified by analyzing the forward (FSC) versus side (SSC) scatter and gated to exclude debris. Healthy and apoptotic cell populations were identified by using viability staining (LIVE/DEAD solution, Thermo Fisher Scientific, L3224). The live cells only were considered to measure the percentage of positive cells corresponding to the fraction of cell populations double positive for both the expression of CD2 (the T-Lymphocytes lineage marker) and for the specific target of interest (LAG-3, PD-L1, PD-1, TIM3, BTLA, TIGIT, OX40, 4-1BB, CD27 or ICOS). Unstained cells or cells treated with the appropriate isotype control were used as background control. Values were reported as the mean of at least three determinations (standard deviations ≤ 10%).
Lymphocyte proliferation assays
For the staining, pre-warmed 0.1% bovine serum albumin/PBS solution containing CFDA-SE (Vybrant® Cell Tracer Kit, InvitrogenTM, Thermo Fisher Scientific, V12883) at a concentration of 10 μM was used to resuspend hPBMCs at a density of 1 · 106 cells/mL, that were incubated at 37°C for 10 minutes. Ice-cold R10 medium was then used to permeabilize the cells by incubating on ice for an additional 5 min. The cells were then washed three times with PBS. Between the second and the third wash, the cells were incubated at 37°C for 5 minutes to allow the complete removal of excess of CFDA-SE. After the last wash and centrifugation at 1200 rpm for 10 minutes, the cells were resuspended at a density of 1 · 106 cells/mL in R10 and plated into a 48-well plate (1 · 106 cells/well). The lymphocyte proliferation assays were performed by incubating the plated lymphocytes with 2.5 µg/mL phytohemagglutinin-L (PHA-L, Roche, 11,249,738,001), in the absence or in the presence of the selected anti-LAG-3, anti-PD-1, anti-PD-L1 mAbs (10 µg/mL). The plates were then incubated at 37°C for 5 days. At the end of treatment, each sample was recovered, resuspended in 100 µL of PBS and transferred into a round-bottom 96-well plate. First, the cells were incubated with violet LIVE/DEAD solution (Thermo Fisher Scientific, L3224) for 30 minutes at 4°C. After several washes, the samples were further incubated with the anti-human CD3 (APC), anti-CD4 (PE) and anti-CD8 (PerCP) antibodies (10 μL/sample) for 1 hour at 4°C. Finally, the cells were extensively washed, resuspended in 200 µL of PBS and transferred into 5 mL polystyrene round-bottom tubes (BD Biosciences, 352,054) for acquisition at CytoFLEX Flow Cytometer (Beckman Coulter).
Effects of novel antibodies on the production of cytokines by stimulated hPBMCs
hPBMCs (1 · 106 cells) were cultured and stimulated with 2.5 μg/mL PHA-L or 50 ng/mL Staphylococcal enterotoxin B (SEB, Sigma-Aldrich, S4881) for 18, 42 and 66 hours, in the absence or in the presence of the selected anti-LAG-3, anti-PD-L1 and anti-PD-1 mAbs (20 μg/mL) or an isotype control antibody, used as negative control. Nivolumab and atezolizumab (InvivoGen, hpdl1-mab 9-1) were tested as a positive control in parallel assays, in the same conditions. The levels of IL-2 or IFNγ in cell culture supernatants were measured by ELISA assays (all from DuoSet ELISA, R&D Systems), according to the manufacturer’s recommendations. Concentration values were reported as the mean of at least three determinations in five independent experiments performed by using lymphocytes from 5 to 8 healthy donors.
In vivo studies
Six-week old female BalBC mice (Envigo) were used for in vivo studies. Mice were implanted subcutaneously on the right flank with 2 × 105 CT 26 tumor cells (day 0), and then treated intra-peritoneally with 200 μg of α-mPD-L1 (BioXcell, clone 10F.9G2, 50–562–314), α-mPD-1 (BioXcell, clone RMP114, BE0146), PD-1_1 or PD-L1_1 at day 3, 6, 10. Tumor growth was measured by caliper every 3–4 days using the formula LxW2/2 (L as the largest and W the smallest diameter of the tumor). Animals were sacrificed as soon as signs of distress or a tumor volume above 2000 mm3 occurred.
Statistical analyses
Error bars were calculated on the basis of the results obtained by at least three independent experiments. Analyses on activated lymphocytes were performed by using samples of hPBMCs obtained by at least three different donors. For these studies, differences between groups were assessed by Student’s t-test. Statistical significance was defined as p < 0.05.
Author contributions
C.D.L, A.N, N.Z., E.Scarselli conceived the project. C.D.A., E.Sasso, M.P., A.M.D.A., D.S., M.E., G.F. performed the experiments. C.D.L., C.D.A., E.S., M.P., A.M.D.A. analyzed data and prepared the figures. E.Scarselli, RC. gave project direction and important feedback on the paper. C.D.L., A.N., N.Z. wrote the paper. All authors edited and approved the manuscript.
Conflicts of interest
The authors declare that a patent has been recently filed by some authors of this manuscript. The authors A.M.D.A and E.Scarselli are employees of Nouscom srl; the author M.L.E. is an employee of ReiThera srl and A.N. owns shares in Keires AG.
Ethics approval and consent to participate
Experiments involving animals were approved by the Italian Ministry of Health (Authorizations 213/2016 PR) and have been done in accordance with the applicable Italian laws (D.L.vo 26/14 and following amendments), the Institutional Animal Care and Use Committee of CEINGE and Allevamenti Plaisant SRL.
Abbreviations
| APC | = | Allophycocyanin |
| BTLA | = | B- and T-lymphocyte attenuator |
| B.V.510 | = | Brilliant Violet 510 |
| CFDA-SE | = | Carboxyfluorescein Diacetate, Succinimidyl Ester; |
| CI | = | Checkpoint Inhibitors |
| FBS | = | fetal bovine serum |
| hPBMCs | = | human peripheral blood mononuclear cells |
| HRP | = | horseradish peroxidase |
| IC | = | Immune Checkpoints |
| ICOS | = | Inducible T cell Costimulator |
| LAG-3 | = | Lymphocyte Activation Gene 3 |
| PHA | = | Phytohemagglutinin |
| PD-1 | = | programmed cell death-1 |
| PD-L1 | = | programmed cell death ligand-1 |
| PE | = | Phycoerythrin |
| scFvs | = | single-chain antibody fragments |
| SEB | = | Staphylococcal Enterotoxin B |
| TIGIT | = | T cell Immunoglobulin and ITIM domain |
| TIM3 | = | T cell Immunoglobulin and Mucin-3; TNF-R, Tumor Necrosis Family Receptor |
Supplemental Material
Download MS Word (1.2 MB)Acknowledgments
The authors are grateful to Riccardo Cortese for his constant and remarkable support and guide, and they will always remember his high human qualities. The authors thank D. Lazarevic and D. Cittaro for optimization of sequencing and preliminary bioinformatic analysis.
Supplemental data
Supplemental data for this article can be accessed here.
Disclosure statement
The authors declare as potential conflicts of interest those reported in Conflicts of Interest.
References
- Peggs KS, Quezada SA, Allison JP. Cancer immunotherapy: co-stimulatory agonists and co-inhibitory antagonists. Clin Exp Immunol. 2009;157(1):9–19. doi:10.1111/j.1365-2249.2009.03912.x.
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi:10.1056/NEJMoa1200694.
- Patel R, Bock M, Polotti CF, Elsamra S. Pharmacokinetic drug evaluation of atezolizumab for the treatment of locally advanced or metastatic urothelial carcinoma. Expert Opin Drug Metab Toxicol. 2017;13(2):225–232. doi:10.1080/17425255.2017.1277204.
- Shultz D. Three Drugs Approved for Urothelial Carcinoma by FDA. Cancer Discov. 2017;7(7):659–660. doi:10.1158/2159-8290.CD-NB2017-071.
- Callahan MK, Flaherty CR, Postow MA. Checkpoint blockade for the treatment of advanced melanoma. Cancer Treat Res. 2016;167:231–250. doi:10.1007/978-3-319-22539-5_9.
- Lee CS, Cragg M, Glennie M, Johnson P. Novel antibodies targeting immune regulatory checkpoints for cancer therapy. Br J Clin Pharmacol. 2013;76(2):233–247. doi:10.1111/bcp.12164.
- Buqué A, Bloy N, Aranda F, Castoldi F, Eggermont A, Cremer I, Fridman WH, Fucikova J, Galon J, Marabelle A, et al. Trial watch: immunomodulatory monoclonal antibodies for oncological indications. Oncoimmunology. 2015; 4(4):e1008814. doi:10.1080/2162402X.2015.1008814.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi:10.1038/nrc3239.
- Anderson AC, Joller N, Kuchroo VK. LAG-3, TIM-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44(5):989–1004. doi:10.1016/j.immuni.2016.05.001.
- Goldberg MV, Drake CG. LAG-3 in Cancer Immunotherapy. Curr Top Microbiol Immunol. 2011;344::269–278. doi:10.1007/82_2010_114.
- Huang C-T, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, Ravi S, Kowalski J, Levitsky HI, et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21(4):503–513. doi:10.1016/j.immuni.2004.08.010.
- Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4(7):670–679. doi:10.1038/ni944.
- Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–242. doi:10.1038/nri3405.
- Sanmamed MF, Pastor F, Rodriguez A, Perez-Gracia JL, Rodriguez-Ruiz ME, Jure-Kunkel M. Agonists of costimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640–655. doi:10.1053/j.seminoncol.2015.05.014.
- Wen T, Bukczynski J, Watts TH. 4-1BB ligand-mediated costimulation of human T cells induces CD4 and CD8 T cell expansion, cytokine production, and the development of cytolytic effector function. J Immunol. 2002;168(10):4897–4906. doi:10.4049/jimmunol.168.10.4897.
- Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14(8):561–584. doi:10.1038/nrd4591.
- Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(13):1270–1271. doi:10.1056/NEJMc1509660.
- Maçon-Lemaître L, Triebel F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology. 2005;115(2):170–178. doi:10.1111/j.1365-2567.2005.02145.x.
- Wang C, Thudium KB, Han M, Wang X-T, Huang H, Feingersh D, Garcia C, Wu Y, Kuhne M, Srinivasan M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014;2(9):846–856. doi:10.1158/2326-6066.CIR-14-0040.
- Selby MJ, Engelhardt JJ, Johnston RJ, Lu L-S, Han M, Thudium K, Yao D, Quigley M, Valle J, Wang C, et al. Preclinical development of ipilimumab and nivolumab combination immunotherapy: mouse tumor models, in vitro functional studies, and cynomolgus macaque toxicology. PLoS One. 2016;11(11):e0161779. doi:10.1371/journal.pone.0161779.
- Wang S, Campos J, Gallotta M, Gong M, Crain C, Naik E, Coffman RL, Guiducci C. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc Natl Acad Sci USA. 2016;113(46):E7240–E7249. doi:10.1073/pnas.1608555113.
- Monaci P, Luzzago A, Santini C, De Pra A, Arcuri M, Magistri F, Bellini A, Ansuini H, Ambrosio M, Ammendola V, et al. Differential screening of phage-ab libraries by oligonucleotide microarray technology. PLoS One. 2008;3(1):e1508. doi:10.1371/journal.pone.0001508.
- Jiang T, Zhou C, Ren S. Role of IL-2 in cancer immunotherapy. Oncoimmunology. 2016;5(6):e1163462. doi:10.1080/2162402X.2016.1163462.
- Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16(3):131–144. doi:10.1038/nrc.2016.14.
- Pedicord VA, Montalvo W, Leiner IM, Allison JP. Single dose of anti-CTLA-4 enhances CD8+ T-cell memory formation, function, and maintenance. Proc Natl Acad Sci USA. 2011;108(1):266–271. doi:10.1073/pnas.1016791108.
- Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse events associated with immune checkpoint blockade in patients with cancer: a systematic review of case reports. PLoS One. 2016;11(7):e0160221. doi:10.1371/journal.pone.0160221.
- De Lorenzo C, Palmer DB, Piccoli R, Ritter MA, D’Alessio G. A new human antitumor immunoreagent specific for ErbB2. Clin Cancer Res. 2002;8(6):1710–1719. PMID: 12060608.
- Sasso E, Paciello R, D’Auria F, Riccio G, Froechlich G, Cortese R, Nicosia A, De Lorenzo C, Zambrano N. One-step recovery of scfv clones from high throughput sequencing-based screening of phage display libraries challenged to cells expressing native Claudin-1. BioMed Res Int. 2015;2015::703213. doi:10.1155/2015/703213.
- Paciello R, Urbanowicz RA, Riccio G, Sasso E, McClure CP, Zambrano N, Ball JK, Cortese R, Nicosia A, De Lorenzo C. Novel human anti-claudin 1 mAbs inhibit hepatitis C virus infection and may synergize with anti-SRB1 mAb. J Gen Virol. 2016;97(1):82–94. doi:10.1099/jgv.0.000330.
- Sasso E, Latino D, Froechlich G, Succoio M, Passariello M, De Lorenzo C, Nicosia A, Zambrano N. A long non-coding SINEUP RNA boosts semi-stable production of fully human monoclonal antibodies in HEK293E cells. mAbs. 2018;10(5):730–737. doi:10.1080/19420862.2018.1463945.