
ABSTRACT
Selective inhibition of tumor necrosis factor (TNF) signaling through the proinflammatory axis of TNF-receptor 1 (TNFR1) while leaving pro-survival and regeneration-promoting signals via TNFR2 unaffected is a promising strategy to circumvent limitations of complete inhibition of TNF action by the approved anti-TNF drugs. A previously developed humanized antagonistic TNFR1-specific antibody, ATROSAB, showed potent inhibition of TNFR1-mediated cellular responses. Because the parental mouse antibody H398 possesses even stronger inhibitory potential, we scrutinized the specific binding parameters of the two molecules and revealed a faster dissociation of ATROSAB compared to H398. Applying affinity maturation and re-engineering of humanized variable domains, we generated a monovalent Fab derivative (13.7) of ATROSAB that exhibited increased binding to TNFR1 and superior inhibition of TNF-mediated TNFR1 activation, while lacking any agonistic activity even in the presence of cross-linking antibodies. In order to improve its pharmacokinetic properties, several Fab13.7-derived molecules were generated, including a PEGylated Fab, a mouse serum albumin fusion protein, a half-IgG with a dimerization-deficient Fc, and a newly designed Fv-Fc format, employing the knobs-into-holes technology. Among these derivatives, the Fv13.7-Fc displayed the best combination of improved pharmacokinetic properties and antagonistic activity, thus representing a promising candidate for further clinical development.
Introduction
Tumor necrosis factor (TNF) is a pro-inflammatory cytokine with pleiotropic activity on various cell types and a key factor in initiation and maintenance of chronic inflammatory diseases. Consequently, anti-TNF therapies, including anti-TNF antibodies (infliximab, adalimumab, golimumab, certolizumab pegol) and a soluble TNF receptor 2 (TNFR2) fusion protein (etanercept), have been developed as therapeutic entities and approved for various indications such as rheumatoid arthritis, juvenile idiopathic arthritis, psoriatric arthritis, plaque psoriasis, Crohn’s disease, and ulcerative colitis.Citation1,Citation2
TNF is expressed as a transmembrane type 2 protein (tmTNF) capable of binding and activating the two TNF receptors TNFR1 and TNFR2.Citation3–Citation5 TNFα converting enzyme-mediated cleavage converts TNF into a soluble protein (sTNF),Citation6 which can still bind both receptors; however, it is only able to efficiently activate TNFR1.Citation7,Citation8 Importantly, the two TNF receptors exhibit diametrically opposed (dichotomic) activities, with TNFR1 being primarily responsible for pro-inflammatory responses and TNFR2 primarily involved in tissue homeostasis, protection and regeneration.Citation9–Citation12 The approved anti-TNF agents neutralize both tmTNF and sTNF, thus globally blocking TNF activities.Citation13 Various adverse effects are related to this global inhibition, including common and opportunistic infections, reactivation of latent tuberculosis, lupus-like symptoms, demyelination, psoriasis, sarcoidosis, and an increase of malignancies, such as lymphomas.Citation14–Citation16
Selective inhibition of TNFR1 has emerged as novel approach for treatment of TNF-mediated inflammatory diseases.Citation16–Citation19 We previously developed a humanized anti-TNFR1 antibody (ATROSAB) that blocks receptor activation by TNF and lymphotoxin.Citation20,Citation21 ATROSAB, which is an IgG1 antibody deficient in Fcγ receptor and complement binding, demonstrated a protective role in a mouse model of N-methyl-D-aspartate-induced acute neurodegeneration by shifting the antithetic activity of TNF towards TNFR2.Citation22 Moreover, both H398 and ATROSAB ameliorated disease in a model of multiple sclerosis in TNFR1-humanized mice Citation9,Citation23 and ATROSAB was well tolerated in a preclinical safety study in cynomolgus monkeys (unpublished data). However, because in the absence of TNF ATROSAB showed in a narrow dose range a marginal agonistic activity in vitro, equivalent to about 1–3% of a TNF-induced response,Citation21 we sought to improve antagonistic activity and safety of this TNFR1-selective antibody by developing monovalent derivatives of ATROSAB. By applying affinity maturation by random and site-directed mutagenesis, as well as developing newly humanized variants based on alternative germline sequences, we developed a Fab (clone 13.7) with improved antagonistic activity. Furthermore, we generated and compared half-life-extended variants thereof applying modification with polyethylene glycol (PEGylation), fusion to serum albumin, an IgG half–antibody format, and a novel heterodimeric Fv-Fc format.
Results
Comparison of bioactivity of ATROSAB and its parental antibody H398
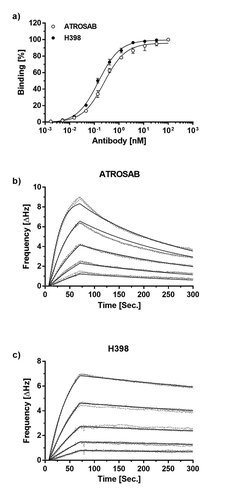
In accordance with previous findings,Citation20 ATROSAB and H398 bound to human TFNR1-Fc in an enzyme-linked immunosorbent assay (ELISA) with similar EC50 values of 0.25 nM and 0.15 nM, respectively (). Using quartz crystal microbalance (QCM) measurements, similar apparent affinities of ATROSAB and H398 were determined for binding to immobilized human and rhesus TNFR1-Fc. However, reducing antigen density on the chip to favor monovalent interactions revealed an approximately 3-fold lower apparent affinity of ATROSAB. This was caused mainly by faster dissociation of ATROSAB from the receptor, reflected by a higher koff value of 4.7 × 10−3 s−1 () compared to H398, with a koff value of 6.2 × 10−4 s−1 (, ). Consequently, a slower dissociation of monovalently bound H398 from TNFR1, and hence, a longer receptor occupation, could contribute to a superior blockade of TNFR1.
Table 1. Binding activities of ATROSAB and H398.
Figure 1. Receptor binding ATROSAB and H398.
a) Binding of ATROSAB and H398 to immobilized TNFR1-Fc in ELISA (n = 3, mean ±SD) and QCM (b, c) using a chip of 48 Hz ligand density and a concentration range from 62.5 nM to 3.9 nM in 2-fold dilution steps. Each concentration was analyzed in triplicates.
Affinity maturation and framework exchange of ATROSAB
In order to increase its affinity, and thus its antagonistic activity, the single-chain variable fragment (scFv) of ATROSAB (scFv IZI06.1)Citation20 was subjected to a first affinity maturation approach using site-directed mutagenesis of exposed residues within individual complementarity-determining regions (CDRs) (L1 and H2, CDR1 of the light chain and CDR2 of the heavy chain) or combinations of CDRs (H1/L2 and H2/L1) and selection by phage display against human TNFR1-Fc. For one of the selected clones, scFv IG11, which carries 6 mutations within CDRH2 (EIVPTQGEAKYNDKFKA, mutated residues are underlined), we observed a 3.9-fold slower receptor dissociation (, Fig. S1) and 3.3-fold improved equilibrium binding to human TNFR1-Fc in ELISA (), as well as a 1.6-fold higher bioactivity in terms of inhibition of TNF-induced TNFR1 activation in a HT1080-based interleukin-8 (IL-8) release assay (). This clone was subjected further to a random mutagenesis approach, resulting in another clone, scFv T12B, exhibiting 2 additional mutations in VH (Q1H, T53S, VH = variable domain of the heavy chain) and one in VL (S91G, VL = variable domain of the light chain). ScFv T12B revealed similar binding capacity compared to scFv IG11 in QCM and an ELISA (, Fig. S1, ). Of note, scFv T12B displayed reduced dissociation from immobilized TNFR1-Fc in QCM experiments revealing a rate constant of 2.6 × 10−3 s−1 as compared to koff values of 1.7 × 10−2 s−1 and 4.3 × 10−3 s−1 for scFv IZI06.1 and scFv IG11, respectively (Fig. S1, ). This translated into a further increased inhibitory activity of scFv T12B for TNF-induced IL-8 release from HT1080 cells by a factor of 3.4, compared to scFv IZI06.1 (, ).
Table 2. Characterization of scFv 13.7 in comparison to intermediate fragments scFv IG11 and scFv T12B and to the parental scFv IZI06.1.
Figure 2. Affinity maturation and framework replacement of scFv ATROSAB (scFv IZI06.1) results in improved activity and stability.
Binding of the parental scFv IZI06.1, the intermediate scFvs IG11 and T12B and the final version scFv 13.7, after affinity maturation and framework replacement was analyzed in ELISA (a, mean ± SD, n = 2). Inhibition of TNF-induced (0.1 nM TNF) IL-8 release from HT1080 cells (b) was analyzed for all scFvs (displayed are mean ±SD, n = 3). Thermal stability of scFv IZI06.1(c), scFv T12B (d) and scFv 13.7 (e) was analyzed using dynamic light scattering. Tm was determined upon visual interpretation of the displayed data points.
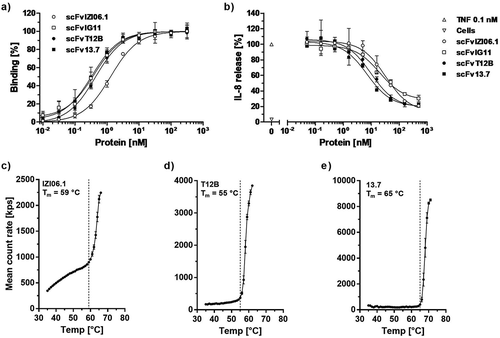
In a further approach, the humanization of H398 was revisited by an exchange of VH and VL framework regions of H398 with alternative germline genes in order to address the possibility of suboptimal CDR arrangement in IZI06.1 after the initial humanization.Citation24 Humanized versions with alternative framework regions (3–11*01 for VH and 1–39*02 for VL, using two variants for each germline differing in one amino acid position, VH: R or A at position 71, VL: I or V at position 2) were generated and combined with each other and with the VH and VL domains of scFv T12B, which was selected for this approach as the candidate with the most promising binding behavior (Fig. S2). All newly generated scFv molecules were expressed and, in case of sufficient producibility, analyzed for binding to TNFR1-Fc in ELISA, as well as inhibition and TNF-mediated TNFR1 activation in IL-8 release experiments (data not shown). Compared to scFv T12B, the most promising candidate, scFv 13.7 (composed of VH T12B and a newly humanized VL domain), revealed almost identical binding to human TNFR1-Fc in ELISA and QCM (, Fig. S1, ), as well as slightly improved inhibition of TNF-induced IL-8 release from HT1080 cells (). Furthermore, scFv 13.7 possessed an improved thermal stability with a melting temperature (Tm) of 65 °C compared to 55 °C for scFv T12B, as determined by dynamic light scattering (-).
Binding of IgG 13.7 and Fab 13.7 to human TNFR1
Based on the newly humanized and affinity-matured version 13.7, an IgG and a Fab with constant regions identical to ATROSAB were generated (IgG 13.7, Fab 13.7) and produced in transiently transfected HEK293T cells. Proteins were purified by affinity chromatography using protein A for IgG 13.7 and Kappa-Select for Fab 13.7. Correct assembly and integrity of IgG 13.7 and Fab 13.7 were confirmed by SDS-PAGE. In analytical size-exclusion chromatography (SEC), IgG 13.7 and Fab 13.7 as well as the reference proteins ATROSAB and its Fab (Fab ATR) revealed one single peak (-).
Figure 3. Expression, purification and receptor binding of 13.7 derived IgG and Fab.
Purity and correct assembly were analyzed by analytical size exclusion chromatography (a, Yarra SEC-2000 column, flow rate 0.5 ml/min) and SDS-PAGE (b, 12% separation gel, reducing conditions; c, 8% separation gel, non-reducing conditions). The indicated molecular weights were interpolated using a standard curve of proteins with known mass and retention time. Binding to human TNFR1 was evaluated in ELISA (d, n = 3, mean ± SD) and QCM experiments (e-h). In QCM measurements, concentrations between 64 nM and 4 nM (ATROSAB and IgG13.7) or 128 nM to 8 nM (FabATR and Fab13.7), were analyzed in triplicates. Measurements (grey, dotted line) and fit (black, solid) are shown.
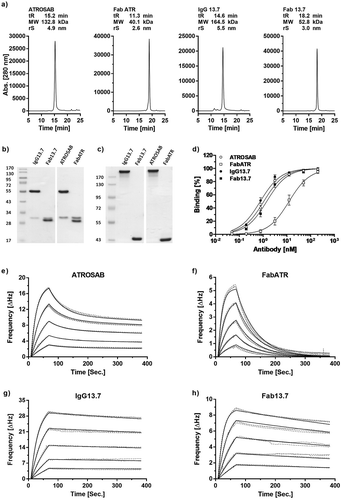
Binding of the proteins to human TNFR1-Fc was analyzed by ELISA, revealing EC50 values of 0.76 nM for IgG 13.7 and 1.4 nM for Fab 13.7. In comparison to ATROSAB, binding of IgG 13.7 to TNFR1 was improved 1.4-fold, while Fab 13.7 bound to human TNFR1 8.7-fold stronger than Fab ATR (). In addition, the proteins were analyzed by QCM for binding to immobilized human TNFR1-Fc using a moderate receptor density (86 Hz). Fab ATR showed a rapid dissociation from the chip, resulting in a KD value of 30 nM as analyzed by the “one to one” algorithm (, ). In contrast, Fab 13.7 revealed a reduced dissociation from immobilized TNFR1-Fc, substantiated by a KD value of 1.6 nM, i.e., a 25-fold increased affinity (, ). ATROSAB dissociated with biphasic kinetics from the receptor, indicating mono- and bivalent binding events with calculated koff values of 2.2 × 10−2 s−1 and 4.1 × 10−4 s−1 and KD values of 78 nM and 0.38 nM, respectively, in an analysis using a “one to two” binding algorithm (, ). In contrast, IgG 13.7 showed a very slow dissociation from the chip of moderate receptor density, rendering discrimination between mono- and bivalent binding impossible. Thus, based on Fab 13.7, the described approach of affinity maturation and framework replacement resulted in a strongly improved binding to TNFR1.
Table 3. Affinity determination of IgG13.7 and Fab13.7.
Selectivity and cross-reactivity of Fab and IgG 13.7 for TNFR1
Selectivity of Fab 13.7 and IgG 13.7 for binding to TNFR1 was confirmed by ELISA with immobilized TNFR1-Fc and TNFR2-Fc fusion proteins (Fig. S3a). Furthermore, species cross-reactivity of Fab 13.7 was analyzed using TNFR1 from different species (Fig. S3b). Fab 13.7 bound to human and rhesus TNFR1-Fc, while no binding could be detected for the mouse and rat TNFR1-Fc. ATROSAB showed a similar binding pattern. The mouse TNFR1-specific control antibody H55R-170 bound to mouse TNFR1-Fc as well as to rat TNFR1-Fc fusion proteins (Fig. S3c), confirming the integrity of the coated mouse and rat TNFR1-Fc fusion proteins.
In vitro bioactivity of IgG 13.7 and Fab 13.7
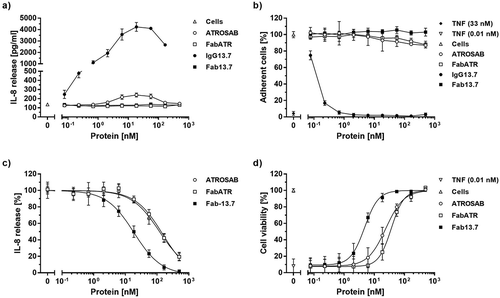
In a first set of in vitro experiments, we analyzed potential agonistic activities of IgG 13.7 and Fab 13.7. The monovalent Fabs (Fab ATR and Fab 13.7) did not show any signs of receptor activation as determined by the release of IL-8 () from HT1080 cells, compared with the cellular background (). In addition, no induction of TNFR1-mediated cytotoxicity in Kym-1 cells was detected for both proteins (, ). In accordance with previous experiments,Citation21 ATROSAB showed a marginal potential to activate TNFR1 in IL-8 release experiments using HT1080 cells (). The detected IL-8 level at the peak concentration was 1.8-fold higher in comparison to the IL-8 release from unstimulated cells, corresponding to around 1%, of the value obtained for TNF at a concentration of 33 nM (peak response in titration assay, data not shown) (). In cell death induction experiments using Kym-1 cells, however, no receptor activation could be detected (, ). Surprisingly, IgG 13.7 strongly activated TNFR1, resulting in released IL-8 amounts of ~ 87% compared to TNF (33 nM, , ). In a Kym-1 cytotoxicity assay, IgG 13.7 induced cell death in up to 100% of the cells as determined by crystal violet staining (, ). Consequently, only Fab 13.7 was employed to further test the inhibitory potential on TNF-induced cytokine release from HT1080 cells () and apoptosis of Kym-1 cells (). In these assays, TNF was used at a concentration of 0.1 nM to induce activation of TNFR1 in IL-8 release assays using HT1080 cells and at a concentration of 0.01 nM in Kym-1 cell death induction experiments. Interestingly, monovalent Fab ATR revealed similar activities as bivalent ATROSAB in IL-8 release experiments with IC50 values of 151 nM and 118 nM (). Furthermore, similar IC50 values of 37 nM and 24 nM were observed in the cytotoxicity assay using Kym-1 cells () (). Of note, Fab 13.7 revealed superior inhibitory potential towards TNFR1 compared to Fab ATR, but also to the bivalent IgG ATROSAB, resulting in an IC50 value of 19 nM using HT1080 cells (). An IC50 value of 4.7 nM was determined in the cell death induction assay using Kym-1 cells (, ). Taken together, these data demonstrate an improved inhibitory potential of Fab 13.7 compared to ATROSAB and Fab ATR, and a lack of agonistic activity.
Table 4. Bioactivity of ATROSAB and 13.7 antibodies.
Figure 4. Bioactivity of 13.7 antibodies.
The potential of IgG13.7 and Fab13.7 to stimulate TNFR1 on the surface of HT1080 and Kym-1 cells was analyzed by detection of IL-8 (a) release as well as by the induction of cell death (b). The inhibitory activity of Fab13.7 in the respective assays was analyzed (c and d) with respect to receptor stimulation by 0.1 nM TNF (c) or 0.01 nM TNF (d). ATROSAB and FabATR served as controls. Stimulatory experiments were performed twice (a and b, n = 2, mean ±SD), inhibitory experiments were performed three times (c and d, n = 3, mean ±SD).
Antibody-dependent cross-linking of anti-TNFR1 antibodies
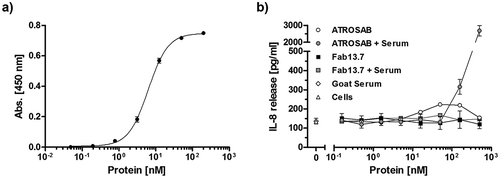
In order to rule out a potential agonistic activity in the presence of cross-linking anti-drug antibodies, IL-8 release assays were performed in the presence of an anti-human IgG serum preparation produced in goat and specific for the Fab. Binding of the goat-anti-human IgG serum to immobilized Fab 13.7 was demonstrated by ELISA revealing half-maximal binding at around 1 nM (). Incubation of increasing concentrations of Fab 13.7 with cross-linking goat anti-human Fab serum did not promote any TNFR1-mediated induction of IL-8 release (). In contrast, the anti-Fab serum induced increased levels of IL-8 in a concentration-dependent manner in the presence of ATROSAB ().
Figure 5. Fab13.7 lacks agonistic activity upon crosslinking.
Binding of a goat anti-human Fab antibody to Fab 13.7 in ELISA (a) and (b) the effect of this goat anti-human Fab antibody (64 µg/ml) on the stimulatory activity of Fab13.7 towards TNFR1 was analyzed in an IL-8 release assay using HT1080 cells and compared to ATROSAB (n = 2, mean ±SD).
Plasma half-life of Fab 13.7
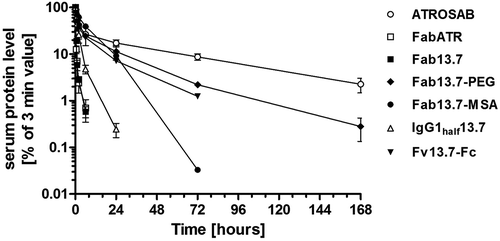
Aiming at therapeutic application, plasma half-life is a critical factor to be considered in order to maintain a prolonged drug exposure. The pharmacokinetic (PK) properties of Fab 13.7, which possesses a molecular mass of approximately 47 kDa, was analyzed in transgenic C57BL/6J mice expressing the extracellular domain of human TNFR1 instead of the respective mouse protein (TNFR1k/i mice). After a single intravenous (i.v.) injection of Fab 13.7, an initial half-life of 0.08 h, a terminal half-life of 1.4 h, and an area under the curve (AUC) of 4.2 µg/ml × h was determined. These values were similar to those of Fab ATR (t1/2,initial 0.17 h, t1/2,terminal 2.2 h, AUC 6.7 µg/ml × h) (). Thus, as expected, both proteins are more quickly cleared from circulation compared to ATROSAB, which exhibits an initial half-life of 0.44 h, a terminal half-life of 32.1 hours and an AUC of 181 µg/ml × h, respectively.
Figure 6. Pharmacokinetic study on Fab13.7 and half-life engineered variants.
PK profiles were obtained after i.v. bolus injection of 25 µg protein using C57BL/6J mice (n = 3) homozygously bearing the extracellular domain of human TNFR1 at the locus of the mouse gene. Initial and terminal half-lifes as well as the area under the curve were calculated after detection of remaining active protein in serum samples by ELISA.
Half-life extended monovalent 13.7 derivatives
In order to extend the half-life of antibody 13.7, we generated several monovalent derivatives exhibiting an increased molecular mass and/or utilizing neonatal Fc receptor (FcRn) recycling. In a first approach, a Fab′ fragment (Fab′ 13.7) was generated by introducing a free cysteine residue at the C-terminus of the first constant domain of the heavy chain (CH1), which was chemically coupled to a branched PEG40kDa moiety, resulting in Fab 13.7PEG (Fig S4). Furthermore, Fab 13.7 was fused through its Fd and a short flexible linker to the N-terminus of mouse serum albumin (MSA) (Fab13.7-MSA) (Fig. S5). A monovalent Fab-Fc fusion protein was generated by fusing Fab 13.7 to a modified Fc, lacking the cysteines in the hinge region as well as the ability to dimerize via the CH3 domain,Citation25,Citation26 resulting in a one-armed half-IgG molecule (IgG1half13.7) (Fig S6). Finally, a monovalent Fv-Fc molecule was generated by fusing the VH and VL domains to a heterodimerizing knob-into-hole (kih) Fc chainCitation27 lacking the cysteines in the hinge region (Fv13.7-Fckih, Fig. S7).
\These molecules were produced in HEK293-6E suspension cells after transient transfection and purified by affinity chromatography and subsequent preparative SEC in the cases of Fab13.7-MSA, IgG1half13.7 and Fv13.7-Fckih (SEC profiles and pooled fractions are displayed in Fig. S8). Molecular integrity was confirmed by SDS-PAGE analysis (Fig. S4c-S7c) and analytical SEC (Fig. S4d-S7d). Binding of all 13.7 derivatives to human TNFR1-Fc was demonstrated in ELISA. Here, compared to Fab 13.7, a slightly reduced binding was observed for Fab13.7PEG, Fab13.7-MSA and IgG1half13.7, while binding of Fv13.7-Fckih was not affected (, Fig. S4e-S7e). None of the newly generated Fab 13.7 derivatives showed detectable agonistic activity towards TNFR1 in an IL-8 release assay using HT1080 (Fig. S4f-S7f). Furthermore, inhibition of IL-8 release from HT1080 cells, induced by 0.1 nM TNF, was slightly reduced by factors ranging from 1.5 to 3.3 compared to Fab 13.7. Fv13.7-Fckih showed the lowest change in bioactivity (, Fig. S4g-S7g).
Table 5. Bioactivity of half-life engineered Fab13.7 Variants.
PK properties of the newly generated Fab 13.7-derived molecules were determined in TNFR1k/I mice after an i.v. injection of 25 µg protein. IgGhalf13.7 exhibited a similar terminal half-life of 1.5 hours as Fab 13.7 (1.4 h, , ). The AUC value of IgGhalf13.7 was increased by a factor of 7.1 compared to Fab 13.7 (, ). In contrast, Fab13.7PEG, Fab13.7-MSA, and Fv13.7-Fckih revealed strongly extended terminal half-lives with values of 14.4 h, 9.7 hours and 10.5 h, respectively, and strongly increased AUC values (, ).
Discussion
In this study, we identified a difference in TNFR1 interaction between two closely related TNFR1-specific antibodies, mAb H398 and its humanized derivative ATROSAB, the latter displaying a faster dissociation upon monovalent interaction with human TNFR1 (, ). To improve this parameter, the scFv IZI06.1, composed of the VH and VL of ATROSAB, was subjected to affinity maturation by phage display. After two consecutive rounds of site-directed and random mutagenesis, isolated clones revealed stronger binding to TNFR1 (scFv IG11; ; Fig S1; ) and improved inhibition of TNFR1 activation (scFv T12B, , ; Fig S1; ), respectively. The lead candidate was further combined with newly humanized variable domains based on VH and VL germline genes with the most human-like amino acid composition.Citation28 Functional scFvs were obtained in the combination of a newly humanized VL domain and the VH domain of the affinity matured scFv T12B, constituting the new scFv 13.7, which revealed similar binding to TNFR1-Fc and similar inhibition of TNF-induced activation of TNFR1 compared to its parental scFv T12B (, Fig. S1 and S2). However, scFv 13.7 showed higher thermal stability compared to scFv T12B and other newly humanized candidates, indicating an overall improved molecular structure, which was demonstrated to result in a higher product quality as revealed by a recent study showing a connection between thermal stability of antibodies, solubility and long-term stability (Fig. 2).Citation29
Compared to the initially humanized scFv IZI06.1, the combined engineering resulted in an ~ 3.5-fold higher apparent monovalent affinity than scFv IZI06.1 as determined by QCM (Fig. S1, ). However, QCM measurements might be affected by the presence of multimers, resulting in varying KD values as observed for scFv IZI06.1 when compared to previously published data.Citation20 Consequently, Fab 13.7 was produced, lacking aggregates and multimers, which revealed an 18.8-fold improved monovalent affinity when compared to Fab ATR (, ), comprising VH and VL of scFvIZI06.1.
Most importantly, the monovalent Fab 13.7 exhibited increased neutralizing activity compared to ATROSAB while lacking any agonistic activity, and thus represents a valuable alternative to the bivalent ATROSAB exhibiting marginally stimulatory activity (, ).Citation20,Citation21 Surprisingly, IgG 13.7 exhibited pronounced agonistic capacity when tested in vitro in different assays (, ). Taking into account that members of the TNFR superfamily (TNFRSF) typically require oligomeric receptor cluster formation, mediated by receptor-ligand/receptor-antibody binding and receptor-receptor interactions,Citation30–Citation32 this agonistic activity might be attributed to the improvement in affinity, especially to slower dissociation from TNFR1, which resulted in formation of stable signaling competent receptor-antibody complexes. These complexes must be formed also in the case of ATROSAB, but they seemed to be less stable, as indicated by its only marginal activation of TNFR1 on the cell surface, which might be due to the rather transient interaction between ATROSAB and TNFR1.
The agonistic power of IgG13.7 could also be the result of other effects. First, changes within the antibody’s paratope might have slightly altered the epitope or induced an epitope drift-like phenomenon. Such mechanisms have been described for in vivo matured antibodies,Citation33,Citation34 resulting in different binding behavior that fosters the formation of larger receptor clusters on the cell surface. Second, the changes within the CDRs could have enabled additional interactions between TNFR1 and the engineered antibody IgG 13.7, which could be involved in some kind of induced-fit mechanism as described for antibodies against protein or hapten antigens,Citation35,Citation36 leading to structural changes in the antigen, which have been described to possibly contribute to the activation of TNFR1.Citation37 Third, the changes during the process of maturation and exchange of framework regions could have altered the angle between TNFR1 and the Fab part of IgG13.7, rendering it more likely for the second Fab arm of the antibody to bind to another TNFR1 molecule, supporting the engagement of IgG 13.7 in larger receptor clusters that promote sustained receptor activation.Citation38 The latter argument would imply that ATROSAB, despite its two binding sites, represents a functionally monovalent molecule under most conditions. This reasoning is supported by the observation that ATROSAB shows no or only marginal receptor activation on its own and requires sustained secondary antibody-mediated receptor cross-linking to be converted into an antagonist. Moreover, ATROSAB and its corresponding Fab revealed identical bioactivities with respect to the inhibition of TNF-mediated activation of TNFR1, further supporting the view that bivalent binding of ATROSAB to cell surface TNFR1 is a rare event.
The fact that the monovalent interaction of 13.7 at the site of TNF binding resulted in sustained inhibition of TNFR1 activation underlines the potential of Fab 13.7 as a drug candidate amenable for further development. Aside from a clear improvement of bioactivity of Fab 13.7 (~ 5-fold compared to ATROSAB), the absence of any agonistic potential even in the presence of secondary crosslinking antibodies appears as a major advantage (). Indeed, anti-drug antibodies (ADAs) are known to represent a major hurdle in the application of protein therapeutics.Citation39 For example, in a previously published study, two of five patients treated with 2 to 10 µg/kg of an anti-human TNFR1 VH domain antibody showed mild to moderate signs of cytokine release syndrome.Citation40 This effect was attributed to pre-existing auto-antibodies directed against the VH, which were detected in 50% of the patients in a retrospective study. In the case of the anti-TNF drugs infliximab and adalimumab, a study revealed the development of ADAs in 50% and 31% of the patients, respectively, resulting in the termination of the applied therapy.Citation41 Of course, the apparent lack of conversion of the TNFR1 antagonist 13.7 into an agonist by secondary crosslinking antibodies, as suggested from the data obtained so far, requires additional studies to confirm this feature.
The short plasma half-life of the Fab 13.7 is a potential draw back for further drug development. Therefore, we generated several half-life extended derivatives of Fab 13.7, including PEGylated Fab, fusion of the Fab to MSA or a dimerization-impaired IgG Fc part (half-IgG), as well as an Fv-Fc fusion protein utilizing a knobs-into-holes Fc (Fig. S4-S7, ). All newly designed molecules revealed improved PK properties (, ), but all molecules also showed somewhat reduced binding to TNFR1-Fc and inhibitory potential of TNF-induced responses in vitro (Fig. S4-S7, ). This reduction in binding affinity after molecular modification is a well-described phenomenon, especially in the case of PEGylated Fabs, as demonstrated for Fabs directed against diverse targets.Citation42,Citation43 While Fab13.7PEG demonstrated superior PK behavior, which corresponded well with previously published PK data of an Alexa-Fluor-labeled anti-TNF Fab (certolizumab PEGol) in mice,Citation44 it also showed the strongest impairment of function. The molecule Fv13.7-Fckih, which was engineered for heterodimeric assembly of two peptide chains by use of the knobs-into-holes technology,Citation27 revealed a strongly improved PK profile, compared to the parental Fab, and showed the lowest impact on binding and antagonistic activity compared to all other generated Fab 13.7 derivatives.
Taken together, the novel monovalent TNFR1 inhibitor Fab 13.7 was generated by affinity maturation and framework exchange, resulting in stronger inhibition of TNF-induced cellular responses. The Fab was converted into the fusion protein Fv13.7-Fckih, improving its PK properties. The characteristics of the newly generated monovalent Fv13.7-Fckih are suitable to fulfill the previously proposed advantages of selective TNFR1 antagonism over systemic TNF blockade in TNFR1-mediated diseases due to unaffected TNFR2 signalingCitation15,Citation18,Citation45 and broader range of action due to simultaneous TNF and lymphotoxin α (LTα) inhibition. The latter is of special interest regarding the demonstrated pro-inflammatory role of LTα in different mouse models, e.g., collagen-induced arthritis and experimental autoimmune encephalomyelitis (EAE).Citation46–Citation48 LTα was furthermore reported to be elevated in the synovium as well as in the serum of rheumatoid arthritis (RA) patients.Citation49,Citation50 Of note, RA patients with resistance to infliximab treatment responded to etanercept, a TNFR1-Fc fusion protein that also neutralizes LTα.Citation51 Finally, Fv13.7-Fckih might also hold potential for the application in oncologic indications, as suggested by recent reports on the involvement of TNFR1 in tumor-associated lymphangiogenesisCitation52 or Helicobacter pylori infection-mediated development of gastric cancer in mouse models.Citation53 Thus, Fv13.7-Fckih represents a lead candidate for a drug development and for preclinical and clinical evaluation in diseases with a recognized pathogenic role of TNF and/or LT.
Materials and methods
Materials
ATROSAB and human TNFR1-Fc were provided by Baliopharm (Basel, Switzerland). H398 (HM2020SP-b) was purchased from Hycult biotech (Plymouth Meeting, PA, USA). Anti-His-HRP (HIS-6 His-Probe-HRP, sc-8036) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-human Fab (Goat, polyclonal, 2085–01) from SouthernBiotech, (Birmingham, AL, USA), anti-human IgG (Fab specific, A 0293) and anti-human IgG (Fc specific, A 0170) from Sigma-Aldrich (Taufkirchen, Germany) and anti-M13 monoclonal HRP-Conjugate (27,942,101) was purchased from GE Healthcare (Chalfont St Giles, GB).
Antibody production
All variants of ATROSAB and 13.7 were cloned into the expression vector pEE14.4 (IgG, Fab) or pSecTagAL1 (Invitrogen) (Fab’, Fv-Fckih, IgGhalf, Fab-MSA) and produced in stably or transiently transfected HEK293T cells using Lipofection (Lipofectamine™ 2000, 11,668–019, Invitrogen, Karlsruhe, Germany) for transfection. Proteins were purified from supernatant using either protein A affinity chromatography (TOYOPEARL®, AF-rProtein A-650F, 22,805, Tosoh, Stuttgart, Germany) or a HiTrap KappaSelect resin (17–5458-12, GE Healthcare, Chalfont St Giles, GB) followed by gel filtration if necessary (Fab13.7-MSA, IgG half13.7 and Fv13.7-Fckih, using an Äkta purifier with a Superdex 200 10/300 GL column at a flow rate of 0.5 ml/min using phosphate-buffered saline (PBS) as liquid phase. ScFv were produced in E. coli and purified from periplasmic extracts by IMAC (Ni-NTA Agarose, 64–17-5, Macherey-Nagel, Düren, Germany).
Generation of phage libraries
Individual CDR libraries were generated by amplification of the parental scFv gene, using PCR technology with degenerated primers containing randomized codons (NNK) at selected positions within the CDRs (library 1, 6 residues [H: 31, 32, 33; L:50, 53, 56]; library 2, 12 residues [H: 50, 52, 53, 54, 56, 58, 61; L: 27D, 27E, 28, 30, 32]; library 2a, 7 residues [H: 50, 52, 53, 54, 56, 58, 61]; library 2b, 5 residues [L: 27D, 27E, 28, 30, 32]).Citation54 Briefly, after digesting with NheI and BamHI (library 1), XmaI and PstI (library 2) or NcoI and PstI, the PCR products were ligated overnight at 16 °C into phagemid vector pHENIS, containing the parental scFv with a frameshift mutation and a stop codon in order to avoid wild-type phage in the libraries. DNA was precipitated and transformed by electroporation into E. coli TG1. Library EP03 was generated by error-prone PCR using the GeneMorph II Random Mutagenesis Kit (200,550, Agilent Technologies, Santa Clara, CA, USA) according to the manufacturers protocol.Citation55 Standard sequencing primers were used to amplify plasmid pHENIS-scFvLib2a_huBR6_IG11 while the conditions were adjusted to introduce a moderate number of mutations by using 0.1 µg template DNA in a 30-cycles PCR reaction. After digest with NcoI and NotI, the PCR product was ligated into phagemid vector pHENIS overnight at 16 °C. DNA was precipitated and transformed by electroporation into E. coli TG1.
Phage display
Phage display selections were performed in immunotubes essentially as described.Citation56 The concentrations of human TNFR1-Fc for coating of immunotubes were decreased from 1 and 0.1 µg/ml in round 1 to 0.1 and 0.01 µg/ml in the last round, respectively. Prior to the first round, a negative selection round was performed using immune tubes with immobilized human TNFR2-Fc (2 µg/ml). In some selections, soluble human TNFR1-Fc (5 µg/ml) was added to the tubes in order to capture quickly dissociating phages and hinder their re-binding to immobilized antigen. Additionally, an off-rate screening step was included in order to evaluate the reduced dissociation from the receptor by QCM technology. The supernatants of rescued monoclonal phages were diluted 1:2 in PBST (PBS, 0.1% Tween 20) and tested on a sensor chip with immobilized human TNFR1-Fc (48 Hz). For reasons of comparison, the running buffer was also mixed 1 to 1 with 2xTY media. The mean value of three measurements was analyzed using the Attaché office software (Attana, Stockholm, Sweden).
Pegylation of Fab13.7
Fab´13.7 was reduced by the addition of tris(2-carboxyethyl)phosphine (TCEP) to a final concentration of 5 mM and incubation for 2 hours at room temperature (RT). Remaining TCEP was removed in a dialysis step against nitrogen-saturated Nellis buffer (10 mM Na2HPO4/NaH2PO4 buffer, 0.2 mM EDTA, 30 mM NaCl, pH 6.7) overnight at 4 °C using a D-Tube™ Dialyzer Mini (MW cut-off 6 – 8 kDa). The reduced Fab´13.7 was mixed with methoxy-PEG2-Mal-40K (mPEG-Mal, NEKTAR 2D3Y0T01) at a molar ratio of 1:10, overlaid with nitrogen in order to avoid re-oxidation and incubated at RT for 1 hour. Finally, free and reactive maleimide groups were quenched by adding L-cysteine (final conc. 100 µM) for 10 minutes at RT. In a final step, PEGylated Fabs were purified using a HiTrap KappaSelect resin as described above.
Protein characterization
Proteins were analyzed by SDS-PAGE and stained with Coomassie-Brilliant Blue G-250. Purity and correct protein assembly was analyzed by SEC using a Waters 2695 HPLC in combination with a Phenomenex Yarra SEC-2000 column (300 × 7.8 mm, flow rate of 0.5 ml/min) or a TSKgel SuperSW mAb HR column (flow rate of 0.5 ml/min). Alternatively, an Äkta purifier with a Superdex 200 10/300 GL column was used at a flow rate of 0.5 ml/min using PBS as liquid phase. The HPLC mobile phase was 0.1 M Na2HPO4/NaH2PO4, 0.1 M Na2SO4, pH 6.7. Standard proteins used were: Thyroglobulin (669 kDa), Apoferritin (443 kDa), Alcohol dehydrogenase (150 kDa), BSA (66 kDa), carbonic anhydrase (29 kDa), FLAG peptide (1 kDa).
Thermal stability
Protein aggregation was analyzed by dynamic light scattering using the ZetaSizer Nano ZS (Malvern, Herrenberg, Germany) increasing temperature stepwise from 35 °C to 80 °C with 1 °C intervals and 2 minutes equilibration prior to each measurement. About 100 µg of protein in 1 ml PBS was analyzed and the melting point (Tm) retrieved upon visual analysis of the increasing signal (kcps).
Enzyme-linked immunosorbent assay
The indicated proteins were immobilized in 96-well microtiter plates at a concentration of 1 µg/ml in PBS overnight at 4 °C. Residual binding sites were blocked using 2% skim milk in PBS (MPBS, 200 µl/well) for 2 hours at RT. Samples were diluted in MPBS, applied to the plates and incubated for another hour at RT. For detection purposes, HRP-labeled or biotin-labeled detection antibodies were applied to the plate for another hour at RT. In the case of biotin-labeled detection antibodies, the plates were subsequently incubated at RT for 1 hour with an HRP-conjugated streptavidin reagent (Immunotools, IL kit component). Bound protein species were detected with 1 mg/ml TMB solution, 0.006% H2O2 in 100 mM Na-acetate buffer, pH 6 at RT, reaction was terminated using 50 µl 1 M H2SO4 and absorption was measured at a wavelength of 450 nm. The plates were washed three times with PBST (PBS + 0.005% Tween20) and twice with PBS in between each incubation step and in advance of the detection.
Quartz crystal microbalance
Binding kinetics were determined by quartz crystal microbalance measurements using a A-100 C-Fast or Cell-200 C-Fast (Attana, Stockholm, Sweden). Human TNFR1-Fc was immobilized on the sensor chip according to the manufacturer’s amine coupling protocol at the indicated densities. Studies were performed using PBST (0.1% Tween-20, pH 7.4) as running buffer at 37 °C, applying a flow rate of 25 µl/min. Reference measurements were performed after every second protein injection using PBST and subtracted from the obtained binding curves. Sensor chips were regenerated after each measurement and reference injection using 25 µl 5 mM NaOH or 20 mM glycine, pH 2.0. Data analysis was performed using Attaché Office Evaluation software (Attana, Stockholm, Sweden) and TraceDrawe (ridgview instruments, Vange, Sweden).
Interleukin release assay
HT1080 (IL-8) cells were seeded (2 × 104 cells/well) in a 96-well plate and incubated overnight. After exchange of the supernatant in order to remove constitutively secreted interleukin, the cells were incubated with the samples in RPMI 1640 + 5% fetal calf serum (FCS) at 37 °C, 5% CO2. For inhibition experiments, antibody samples and stimulating agent (TNF) were mixed prior to application to the cells. Unstimulated cells or cells treated with TNF alone served as controls. After 16–20 hours, plates were centrifuged at 500 × g for 5 minutes and diluted supernatants were analyzed by ELISA according to the manufacturers protocol (IL-8, 31,670,089, ImmunoTools, Friesoythe, Germany).
Cytotoxicity assay
Kym-1 cells (1 × 104 per well) were seeded into 96-well microtiter plates and incubated overnight at 37 °C and 5% CO2. Protein dilutions in 5% RPMI 1640 + 10% FCS were applied to the cells and incubated for another 24 hours at 37 °C and 5% CO2. In case of competition assays, both protein species were mixed together prior to application to the cells. The next day, supernatants were discarded and 50 µl/well crystal violet solution (0.5% crystal violet, 20% methanol in H2O) was added to the plates and incubated for 20 minutes at RT. Subsequently, plates were washed in H2O 20 times and air-dried. Remaining crystal violet dye was dissolved by the addition of 100 µl/well methanol. After 10 minutes shaking at RT, the absorbance at a wavelength of 595 nm was determined.
Pharmacokinetics
Animal care and experiments were in accordance with federal guidelines and have been approved by university and state authorities. In order to determine PK properties, 12 µg or 25 µg protein were injected i.v. into transgenic C57BL/6J mice bearing the gene of the extracellular domain of human TNFR-1 at the locus of the particular mouse gene (C57BL/6J-huTNFRSF1Aecdtm1UEG/izi).Citation22 Blood samples were collected from the tail vein after 3 min, 30 min, 1 h, 3 hours and 6 hours as well as after 3 days and 7 days and incubated on ice immediately. After centrifugation (13,000 × g, 4 °C, 10 minutes), serum was separated and stored at 20 °C until analysis. Remaining functional protein in the serum samples was analyzed by ELISA against immobilized human TNFR1-Fc. Data were calculated as percentage of the 3 minutes value.
Abbreviations
| ATROSAB | = | antagonistic TNF receptor one specific antibody |
| AUC | = | area under the curve |
| Fab | = | antigen-binding fragment |
| Fc | = | crystallizable fragment |
| Fv | = | variable fragment |
| PEG | = | polyethylene glycol |
| QCM | = | quartz crystal microbalance |
| TNF | = | tumor necrosis factor |
| TNFR1 | = | TNF-receptor 1 |
Supplemental Material
Download MS Word (1.8 MB)Acknowledgments
We would like to thank Nadine Heidel and Doris Göttsch for excellent technical assistance.
Disclosure statement
FR, KAZ, AH, PS, KP, and REK are named inventors on patents and patent applications covering the 13.7 technology.
Supplemental Material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Kotsovilis S, Andreakos E. Therapeutic human monoclonal antibodies in inflammatory diseases. Methods Mol Biol. 2014;1060:37–59. PMID:24037835. doi:10.1007/978-1-62703-586-6_3.
- Fischer R, Kontermann RE, Maier O. Targeting sTNF/TNFR1 signaling as a new therapeutic strategy. Antibodies. 2015;4:48–70. doi:10.3390/antib4010048.
- Brockhaus M, Schoenfeld HJ, Schlaeger EJ, Hunziker W, Lesslauer W, Loetscher H. Identification of two types of tumor necrosis factor receptors on human cell lines by monoclonal antibodies. Proc Natl Acad Sci U S A. 1990;87:3127–3131. PMID:2158104. doi:10.1073/pnas.87.8.3127.
- Loetscher H, Pan YC, Lahm HW, Gentz R, Brockhaus M, Tabuchi H, Lesslauer W. Molecular cloning and expression of the human 55 kd tumor necrosis factor receptor. Cell. 1990b;61:351–359. PMID:215886. doi:10.1016/0092-8674(90)90815-V.
- Loetscher H, Schlaeger EJ, Lahm HW, Pan YC, Lesslauer W, Brockhaus M. Purification and partial amino acid sequence analysis of two distinct tumor necrosis factor receptors from HL60 cells. J Biol Chem. 1990a;265:20131–20138. PMID:2173696.
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. PMID:9034190. doi:10.1038/385729a0.
- Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. PMID:8521496. doi:10.1016/0092-8674(95)90192-2.
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Diff. 2003;10:45–65. PMID:12655295. doi:10.1038/sj.cdd.4401189.
- Williams SK, Maier O, Fischer R, Fairless R, Hochmeister S, Stojic A, Pick L, Haar D, Musiol S, Storch MK, et al. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS One. 2014;9:e90117. PMID:24587232. doi:10.1371/journal.pone.0090117.
- Winsauer C, Kruglov AA, Chashchina AA, Drutskaya MS, Nedospasov SA. Cellular sources of pathogenic and protective TNF and experimental strategies based on utilization of TNF humanized mice. Cytokine Growth Factor Rev. 2014;25:115–123. PMID:25305470. doi:10.1016/j.trsl.2014.09.006.
- Probert L. TNF and its receptors in the CNS: the essential, the desirable and the deleterious effects. Neuroscience. 2015;302:2–22. PMID:26117714. doi:10.1016/j.neuroscience.2015.06.038.
- Tseng WY, Huang YS, Lin HH, Luo SF, McCann F, McNamee K, Clanchy F, Williams R. TNFR signalling and its clinical implications. Cytokine. 2016. doi:10.1016/j.cyto.2016.08.027.
- Mitoma H, Horiuchi T, Tsukamoto H, Ueda N. Molecular mechanisms of action of anti-TNF-α agents - Comparison among therapeutic TNF-α antagonists. Cytokine. 2018;101:56–63. PMID:27567553. doi:10.1016/j.cyto.2016.08.014.
- Desai SB, Furst DE. Problems encountered during anti-tumour necrosis factor therapy. Best Pract Res Clin Rheumatol. 2006;20:757–790. PMID:16979537. doi:10.1016/j.berh.2006.06.002.
- Willrich MA, Murray DL, Snyder MR. Tumor necrosis factor inhibitors: clinical utility in autoimmune diseases. Transl Res. 2015;165:270–282. doi:10.1016/j.trsl.2014.09.006.
- Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. 2016;12:49–62. PMID:26656660. doi:10.1038/nrrheum.2015.169.
- Kontermann RE, Scheurich P, Pfizenmaier K. Antagonists of TNF action - clinical experience and new developments. Expert Opin Drug Discov. 2009;4:279–292. PMID:23489126. doi:10.1517/17460440902785167.
- Van Hauwermeiren F, Vanderbroucke RE, Libert C. Treatment of TNF mediated diseases by selective inhibtion of soluble TNF or TNFR1. Cytokine Growth Factor Rev. 2011;22:311–319. PMID:21962830. doi:10.1016/j.cytogfr.2011.09.004.
- Fischer R, Maier O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Oxid Med Cell Longev. 2015;2015:610813. PMID:25834699. doi:10.1155/2015/610813.
- Zettlitz KA, Lorenz V, Landauer K, Münkel S, Herrmann A, Scheurich P, Pfizenmaier K, Kontermann R. ATROSAB, a humanized antagonistic anti-tumor necrosis factor receptor one-specific antibody. MAbs. 2010;2:639–647. PMID:20935477. doi:10.4161/mabs.2.6.13583.
- Richter F, Liebig T, Guenzi E, Herrmann A, Scheurich P, Pfizenmaier K, Kontermann RE. Antagonistic TNF receptor one-specific antibody (ATROSAB): receptor binding and in vitro bioactivity. PLoS One. 2013;8:e72156. PMID:23977237. doi:10.1371/journal.pone.0072156.
- Dong Y, Fischer R, Naudé PJ, Maier O, Nyakas C, Duffey M, Van der Zee EA, Dekens D, Douwenga W, Herrmann A, et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc Natl Acad Sci U S A. 2016;113:12304–12309. PMID:27791020. doi:10.1073/pnas.1605195113.
- Williams SK, Fairless R, Maier O, Liermann PC, Fischer R, Eisel OLM, Kontermann RE, Herrmann A, Weksler B, Romero IA, et al. Anti-TNFR1 targeting in humanized mice ameliorates disease in a model of multiple sclerosis. Sci Rep. 2018;8:13628. doi:10.1038/s41598-018-31957-7.
- Kontermann RE, Münkel S, Neumeyer J, Müller D, Branschädel M, Scheurich P, Pfizenmaier K. A humanized tumor necrosis factor receptor 1 (TNFR1)-specific antagonistic antibody for selective inhibition of tumor necrosis factor (TNF) action. J Immunother. 2008;31:225–234. PMID:18317365. doi:10.1097/CJI.0b013e31816a88f9.
- Jakob CG, Edalji R, Judge RA, DiGiammarino E, Li Y, Gu J, Ghayur T. Structure reveals function of the dual variable domain immunoglobulin (DVD-Ig™) molecule. MAbs. 2013;5:358–363. PMID:23549062. doi:10.4161/mabs.23977.
- Gu J, Yang J, Chang Q, Liu Z, Ghayur T, Gu J. Identification of anti-EGFR and anti-ErbB3 Dual Variable Domains Immunoglobulin (DVD-Ig) proteins with unique activities. PLoS One. 2015;10:e0124135. PMID:25997020. doi:10.1371/journal.pone.0124135.
- Ridgway JB, Presta LG, Carter P. “Knobs-into-holes” engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9:617–621. PMID:8844834. doi:10.1093/protein/9.7.617.
- Abhinandan KR, Martin AC. Analyzing the “degree of humanness” of antibody sequences. J Mol Biol. 2007;369:852–862. PMID:17442342. doi:10.1016/j.jmb.2007.02.100.
- Seeliger D, Schulz P, Litzenburger T, Spitz J, Hoerer S, Blech M, Enenkel B, Studts JM, Garidel P, Karow AR. Boosting antibody developability through rational sequence optimization. MAbs. 2015;7:505–515. PMID:25759214. doi:10.1080/19420862.2015.1017695.
- Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288:2351–2354. PMID:10875917. doi:10.1126/science.288.5475.2351.
- Branschädel M, Aird A, Zappe A, Tietz C, Krippner-Heidenreich A, Scheurich P. Dual function of cysteine rich domain (CRD) 1 of TNF receptor type 1: conformational stabilization of CRD2 and control of receptor responsiveness. Cell Signal. 2010;22:404–414. PMID:19879354. doi:10.1016/j.cellsig.2009.10.011.
- Chan FK. Three is better than one: pre-ligand receptor assembly in the regulation of TNF receptor signaling. Cytokine. 2007;37:101–107. PMID:17449269. doi:10.1016/j.cyto.2007.03.005.
- Bock K, Karlsson KA, Strömberg N, Teneberg S. Interaction of viruses, bacteria and bacterial toxins with host cell surface glycolipids. Aspects on receptor identification and dissection of binding epitopes. Adv Exp Med Biol. 1988;228:153–186. PMID:2459928.
- Root-Bernstein R. Rethinking molecular mimicry in rheumatic heart disease and autoimmune myocarditis: laminin, collagen IV, CAR, and B1AR as initial targets of disease. Front Pediatr. 2014;2:85. PMID:25191648. doi:10.3389/fped.2014.00085.
- Rini JM, Schulze-Gahmen U, Wilson IA. Structural evidence for induced fit as a mechanism for antibody-antigen recognition. Science. 1992;255:959–965. PMID:1546293. doi:10.1126/science.1546293.
- Wang W, Ye W, Yu Q, Jiang C, Zhang J, Luo R, Chen HF. Conformational selection and induced fit in specific antibody and antigen recognition: SPE7 as a case study. J Phys Chem B. 2013;117:4912–4923. PMID:23548180. doi:10.1021/jp4010967.
- Lewis AK, Valley CC, Sachs JN. TNFR1 signaling is associated with backbone conformational changes of receptor dimers consistent with overactivation in the R92Q TRAPS mutant. Biochemistry. 2012;51:6545–6555. PMID:22799488. doi:10.1021/bi3006626.
- Idriss HT, Naismith JH. TNF alpha and the TNF receptor superfamily: structure-function relationship(s). Microsc Res Tech. 2000;50:184–195. PMID:10891884. doi:10.1002/1097-0029(20000801)50:3<184::AID-JEMT2>3.0.CO;2-H.
- Lundkvist Ryner M, Farrell RA, Fogdell-Hahn A. The case for measuring anti-drug antibodies in people with multiple sclerosis. Expert Rev Clin Immunol. 2014;10:697–699. PMID:24780058. doi:10.1586/1744666X.2014.914852.
- Holland MC, Wurthner JU, Morley PJ, Birchler MA, Lambert J, Albayaty M, Serone AP, Wilson R, Chen Y, Forrest RM, et al. Autoantibodies to variable heavy (VH) chain Ig sequences in humans impact the safety and clinical pharmacology of a VH domain antibody antagonist of TNF-α receptor 1. J Clin Immunol. 2013;33:1192–1203. PMID:23832582. doi:10.1007/s10875-013-9915-0.
- Mok CC, van der Kleij D, Wolbink GJ. Drug levels, anti-drug antibodies, and clinical efficacy of the anti-TNFα biologics in rheumatic diseases. Clin Rheumatol. 2013;32:1429–1435. PMID:23887439. doi:10.1007/s10067-013-2336-x.
- Brocchini S, Godwin A, Balan S, Choi JW, Zloh M, Shaunak S. Disulfide bridge based PEGylation of proteins. Adv Drug Deliv Rev. 2008;60:3–12. PMID:17920720. doi:10.1016/j.addr.2007.06.014.
- Khalili H, Godwin A, Choi JW, Lever R, Brocchini S. Comparative binding of disulfide-bridged PEG-Fabs. Bioconjug Chem. 2012;23:2262–2277. PMID:22994419. doi:10.1021/bc300372r.
- Palframan R, Airey M, Moore A, Vugler A, Nesbitt A. Use of biofluorescence imaging to compare the distribution of certolizumab pegol, adalimumab, and infliximab in the inflamed paws of mice with collagen-induced arthritis. J Immunol Methods. 2009;348:36–41. PMID:19567252. doi:10.1016/j.jim.2009.06.009.
- Martin PL, Bugelski PJ. Concordance of preclinical and clinical pharmacology and toxicology of monoclonal antibodies and fusion proteins: soluble targets. Br J Pharmacol. 2012;166:806–822. PMID:22168335. doi:10.1111/j.1476-5381.2011.01812.x.
- Powell MB, Mitchell D, Lederman J, Buckmeier J, Zamvil SS, Graham M, Ruddle NH, Steinman L. Lymphotoxin and tumor necrosis factor-alpha production by myelin basic protein-specific T cell clones correlates with encephalitogenicity. Int Immunol. 1990;2:539–544. PMID:1707660. doi:10.1093/intimm/2.6.539.
- Suen WE, Bergman CM, Hjelmström P, Ruddle NH. A critical role for lymphotoxin in experimental allergic encephalomyelitis. J Exp Med. 1997;186:1233–1240. PMID:9334362. doi:10.1084/jem.186.8.1233.
- Chiang EY, Kolumam GA, Yu X, Francesco M, Ivelja S, Peng I, Gribling P, Shu J, Lee WP, Refino CJ, et al. Targeted depletion of lymphotoxin-alpha-expressing TH1 and TH17 cells inhibits autoimmune disease. Nat Med. 2009;15:766–773. PMID:19561618. doi:10.1038/nm.1984.
- Robak T1, Gladalska A, Stepień H. The tumour necrosis factor family of receptors/ligands in the serum of patients with rheumatoid arthritis. Eur Cytokine Netw. 1998;9:145–154. PMID:9681390.
- O’Rourke KP, O’Donoghue G, Adams C, Mulcahy H, Molloy C, Silke C, Molloy M, Shanahan F, O’Gara F. High levels of Lymphotoxin-Beta (LT-Beta) gene expression in rheumatoid arthritis synovium: clinical and cytokine correlations. Rheumatol Int. 2008;28:979–986. PMID:18379788. doi:10.1007/s00296-008-0574-z.
- Buch MH, Conaghan PG, Quinn MA, Bingham SJ, Veale D, Emery P. True infliximab resistance in rheumatoid arthritis: a role for lymphotoxin alpha? Ann Rheum Dis. 2004;63:1344–1346. PMID:15033655. doi:10.1136/ard.2003.014878.
- Ji H, Cao R, Yang Y, Zhang Y, Iwamoto H, Lim S, Nakamura M, Andersson P, Wang J, Sun Y, et al. TNFR1 mediates TNF-α-induced tumour lymphangiogenesis and metastasis by modulating VEGF-C-VEGFR3 signalling. Nat Commun. 2014;5:4944. PMID:25229256. doi:10.1038/ncomms5944.
- Oshima H, Ishikawa T, Yoshida GJ, Naoi K, Maeda Y, Naka K, Ju X, Yamada Y, Minamoto T, Mukaida N, et al. TNF-α/TNFR1 signaling promotes gastric tumorigenesis through induction of Noxo1 and Gna14 in tumor cells. Oncogene. 2014;33:3820–3829. PMID:23975421. doi:10.1038/onc.2013.356.
- Lou J, Marks JD. Affinity maturation by chain shuffling and site directed mutagenesis. In: Kontermann RE, Dübel S, editors. Antibody engineering. Berlin: Springer; 2010. p. 199. doi:10.1007/978-3-662-04605-0_9.
- Thie H. Affinity maturation by random mutagenesis and phage display. In: Kontermann RE, Dübel S, editors. Antibody engineering. Berlin: Springer; 2010. p. 199. doi:10.1007/978-3-662-04605-0_9.
- Kontermann RE. Immunotube selections. In: Kontermann RE, Dübel S, editors. Antibody engineering. Berlin: Springer; 2010. p. 199. doi:10.1007/978-3-662-04605-0_9.