
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
HER2-ECD (human epidermal growth factor receptor 2 – extracellular domain) is a prominent therapeutic target validated for treating HER2-positive breast and gastric cancer, but HER2-specific therapeutic options for treating advanced gastric cancer remain limited. We have developed antibody-drug conjugates (ADCs), comprising IgG1 linked via valine-citrulline to monomethyl auristatin E, with potential to treat HER2-positive gastric cancer in humans. The antibodies optimally selected from the ADC discovery platform, which was developed to discover antibody candidates suitable for immunoconjugates from synthetic antibody libraries designed using antibody-antigen interaction principles, were demonstrated to be superior immunoconjugate targeting modules in terms of efficacy and off-target toxicity. In comparison with the two control humanized antibodies (trastuzumab and H32) derived from murine antibody repertoires, the antibodies derived from the synthetic antibody libraries had enhanced receptor-mediated internalization rate, which could result in ADCs with optimal efficacies. Along with the ADCs, two other forms of immunoconjugates (scFv-PE38KDEL and IgG1-AL1-PE38KDEL) were used to test the antibodies for delivering cytotoxic payloads to xenograft tumor models in vivo and to cultured cells in vitro. The in vivo experiments with the three forms of immunoconjugates revealed minimal off-target toxicities of the selected antibodies from the synthetic antibody libraries; the off-target toxicities of the control antibodies could have resulted from the antibodies’ propensity to target the liver in the animal models. Our ADC discovery platform and the knowledge gained from our in vivo tests on xenograft models with the three forms of immunoconjugates could be useful to anyone developing optimal ADC cancer therapeutics.
Introduction
HER2-ECD (human epidermal growth factor receptor 2 – extracellular domain) is a prominent therapeutic target validated for treating HER2-positive breast and gastric cancer. More than 600 clinical studies currently registered in ClinicalTrials.gov are related to HER2-positive cancers, and about a quarter of these studies are involved with antibody-based therapeutics/diagnostics targeting HER2-ECD. In addition to breast cancer and gastric cancer, HER2 over-expression has been found in other diverse cancers,Citation1,Citation2 suggesting that a substantial number of cancer patients could potentially benefit from antibody therapeutics targeting HER2-ECD. Two HER2-ECD-targeting monoclonal antibodies are already approved for the treatment of cancers in humans: 1) trastuzumab for HER2-positive breast, gastric and gastroesophageal junction cancer; and 2) pertuzumab for HER2-positive breast cancer in metastatic or neoadjuvant setting.Citation1,Citation3 In addition, trastuzumab emtansine, an antibody-drug conjugate (ADC) composed of trastuzumab conjugated through a non-reducible linker to the tubulin inhibitor mertansine, is approved for treating HER2-positive metastatic breast cancer and has shown overall survival benefit and favorable safety profile over long follow-up durations.Citation4 However, a recent report on the international randomized GATZBY Phase 2/3 trial indicates that trastuzumab emtansine is not superior to taxane in patients with previously treated HER2-positive advanced gastric cancer.Citation5 While HER2-ECD remains a validated target in several ongoing and planned clinical gastric cancer studies,Citation5,Citation6 HER2-specific therapeutic options for treating advanced gastric cancer are still limited.
The aim of our study was to test ADCs designed to treat human HER2-positive gastric cancer. The treatment efficacies and off-target toxicities due to the antibodies as targeting modules were measured with the N87 model system, which has been a standard system as a HER2 over-expressing human gastric cancer cell line, and xenograft model.Citation2 Three HER2-ECD-positive single-chain variable fragment (scFv) candidates (GH2-20, GH2-61 and GH2-75) from the synthetic antibody librariesCitation7,Citation8 were selected through the high throughput antibody-based immunoconjugate discovery platform.Citation9 Those with the highest potencies as targeting modules toward N87 cells were reformatted into human IgG1 antibodies and tested together with two positive control humanized IgG1 antibodies (trastuzumab and H32, which are humanized murine anti-HER2-ECD antibodiesCitation9) and one isotype negative control IgG antibody (nc). All these IgG1 antibodies were constructed with the same human IgG1 framework.
The efficacies of these IgG1 antibodies as the targeting modules for delivering cytotoxic payloads to N87 xenograft tumor models in vivo and to cultured N87 cells in vitro were scrutinized with three forms of immunoconjugates in this work: 1) ADC in the form of IgG1-vc-MMAE (monomethyl auristatin E linked to the IgG1 via valine-citrulline dipeptide cathepsin-cleavable linker);Citation10 2) immunotoxin of a single polypeptide chain in the form of scFv-PE38KDEL fusion protein, where PE38KDEL is a truncated form of Pseudomonas exotoxin (PE) A subunit toxin,Citation11,Citation12 as pioneered by Pastan and colleagues;Citation13 and 3) immunotoxin in the form of IgG1-AL1-PE38KDEL, where the IgG1 is non-covalently linked to PE38KDEL through the adaptor-toxin fusion protein AL1-PE38KDEL.Citation9 The AL1 fragment is composed of consecutive Protein A and Protein L separated by a polypeptide linker that enables Protein A and Protein L binding to the framework regions of the IgG1 simultaneously with nano-molar affinity.Citation9 Since the IgG1 candidates were reformatted from the scFvs derived from the synthetic antibody libraries, which were designed using antibody-protein interaction principles from computational and experimental analysesCitation8,Citation14–Citation17 and built on a single human variable domain antibody germline framework (IGKV1-NL1*01/IGHV3-23*04),Citation7–Citation9 the IgG1s bind to Protein A and Protein L through the variable domain framework heavy and light chain regions, respectively, without affecting the antigen binding sites of the antibodies.Citation7–Citation9
The differences in the cytotoxic payloads and linkers among the three forms of immunoconjugate were anticipated to result in different effects on the efficacies of the antibodies as the immunoconjugates’ targeting modules, and hence provide information for ADC candidate selection and further development. The results showed that the treatments with the selected ADC (IgG1-vc-MMAE) candidates eradicated the xenograft tumor at the endpoint of the treatment without signs of off-target toxicity even at the highest dosage used in the treatments. The immunotoxins (scFv-PE38KDEL), on the other hand, could be envisaged as a sensitive surrogate system for detecting potential off-target toxicity in vivo associated with the antibodies as the targeting modules in the immunoconjugates. The non-covalently linked immunotoxins (IgG1-AL1-PE38KDEL) were much easier to prepare in comparison with the corresponding ADCs, and were more tolerable in terms of off-target toxicity in comparison with the corresponding scFv-PE38KDELs, and hence could be used as an inexpensive, albeit qualitative, system to select IgG candidates for further ADC development in terms of efficacy and off-target toxicity. These findings supported the utility of the general ADC discovery platform on the basis of the synthetic antibody libraries. The in vitro and in vivo efficacy/toxicity comparisons among the antibodies and the three forms of immunoconjugates contributed to the ADC candidate selection with different prospects on the candidate antibodies’ potencies and off-target toxicities.
Results
The IgG1 candidates as targeting modules in immunoconjugates
The scFvs candidates were selected by their representative physicochemical properties. All the selected scFvs (GH2-20, GH2-61 and GH2-75) were among the most potent targeting modules in a large set of non-covalently linked immunotoxins, with IC50 for scFv-AL2-PE38KDEL < 0.01 nM and IC50 for scFv-AL1-PE38KDEL < 0.1 nM, as measured in previous work.Citation9 The epitopes of GH2-61 and GH2-75 overlap with that of the positive control antibody H32, for which the epitope has been identified on domain I of HER2-ECD as determined with negative stain electron microscopy.Citation9 The epitope of GH2-20 does not overlap with that of H32, and indirect evidence suggests that its epitope is situated on domain IV of HER2-ECD, but does not overlap with trastuzumab’s epitope,Citation8 which is also situated on domain IV of HER2-ECD as determined with x-ray crystallography (PDB code: 1N8Z).Citation18 GH2-61, GH2-20, H32 and trastuzumab IgG1 antibodies have similar on/off rates and nano-molar monovalent dissociation constants binding to HER2-ECD (measured with surface plasmon resonance(SPR) shown in Supplementary Table S1); the affinity of GH2-75 IgG1 to HER2-ECD is slightly inferior in terms of the monovalent dissociation constant and the off-rate binding to HER2-ECD (Supplementary Table S1).
Potent anti-tumor activities of the IgG1-vc-MMAEs in the xenograft models
The qualities of the ADCs in the form of IgG1-vc-MMAE were assured with in vitro and in vivo assays. The ADCs were prepared with purified IgG1(trastuzumab), IgG1(H32), IgG1(GH2-75), IgG1(GH2-61) and IgG1(GH2-20) conjugated with vc-MMAE to a drug-antibody ratio (DAR) of 2 (Supplementary Figure S1 (A) and (B)). The vc-MMAE conjugation to the IgG1s did not affect the binding affinity of the IgG1s to HER2-ECD, as measured with ELISA (EC50 measurements, Supplementary Figure S1 (C)), flow cytometry (mean fluorescence intensity (MFI) of cell surface HER2-binding, Supplementary Figure S1 (D)) and SPR (Supplementary Table S1). The EC50s determined with ELISA measurements for all the IgG1s and IgG1-vc-MMAEs were similar (Supplementary Figure S1 (C)), but, in contrast, the flow cytometry measurements indicated that IgG1(GH2-20) and IgG1(GH2-20)-vc-MMAE had lower binding efficiency to cell surface HER2-ECD in comparison with all the other IgG1s and ADCs (Supplementary Figure S1 (D)), most likely due to the close proximity of the epitope of GH2-20 to the cell membrane for which the steric effect was not involved in the ELISA and SPR assay systems. The IC50s measured with the in vitro cell-based assay for the IgG1-vc-MMAEs are shown in Supplementary Figure S1 (E).
Xenograft N87 tumors were almost completely eradicated by some of the ADCs at high dosage without signs of off-target toxicity. N87 tumors were implanted to NOD/SCID mice to the size of about 100 mm3 (day 0) and were then treated with ADCs at day 0, day 7 and day 14 at the dosage of 30 mg/kg ( (A-C) and Supplementary Figure S2). The efficacies of the treatments were ranked as IgG1(GH2-61)-vc-MMAE > IgG1(trastuzumab)-vc-MMAE > IgG1(GH2-20)-vc-MMAE > IgG1(H32)-vc-MMAE > IgG1(GH2-75)-vc-MMAE ( (A)). Photographic depictions of the endpoint tumors excised from the xenograft models are shown in Supplementary Figure S2. All the ADC-treated subjects except those treated with IgG1(GH2-75)-vc-MMAE survived to the endpoint (day 25) of the experiment ( (C)), and some of the ADC treatments almost completely eradicated the xenograft tumors at the endpoint ( (B) and Supplementary Figure S2). Serum biochemical analysis of all the subjects at the experiment endpoint indicated no substantial hepatic or renal toxicity due to the treatment (Supplementary Table S2), although the IgG1(trastuzumab)-vc-MMAE-treated mice had slightly elevated alkaline phosphatase (ALP), likely indicating drug-induced cholestatic hepatic toxicity. ADC treatments at 10 mg/kg dosage remained effective in preventing tumor progression, with the efficacy ranked as IgG1(H32)-vc-MMAE ≈ IgG1(GH2-20)-vc-MMAE > IgG1(GH2-61)-vc-MMAE > IgG1(trastuzumab)-vc-MMAE > IgG1(GH2-75)-vc-MMAE ( (D) and (E)); the survival curves were similar to those of the high-dosage ADC treatments ( (F)). However, the efficacy order of the ADCs, except that of IgG1(GH2-75)-vc-MMAE, were barely distinguishable at the 30 mg/kg dosage level ( (A) and (B)); therefore, precise differences in the efficacy order at the two dose levels could not be clearly defined.
Figure 1. Treatment of N87 xenograft mouse models with anti-HER2-ECD IgG1-vc-MMAEs. (A) Time course responses of the tumor size to the IgG1-vc-MMAE treatment for xenograft mouse models bearing N87 tumors were attained from the xenograft models randomly assigned into 8 groups and treated with 30 mg/kg of the respective IgG1-vc-MMAE at day 0, 7 and 14. Tumor size data for each of the mice of the treatment groups are shown in supplementary figure S1 (F). (B) endpoint tumor volume at day 21 for each of experimental subjects are plotted for each treatment group. (C) Kaplan-Meier survival curves depict the time courses of the fraction of the animal populations surviving the IgG1-vc-MMAE treatment (defined by tumor size below 400 mm3, see methods). (D)~(F) the description is the same as (A)~(C) for the 10 mg/kg IgG1-vc-MMAE treatments. Tumor size data for each of the mice of the treatment groups are shown in supplementary figure S1 (G).
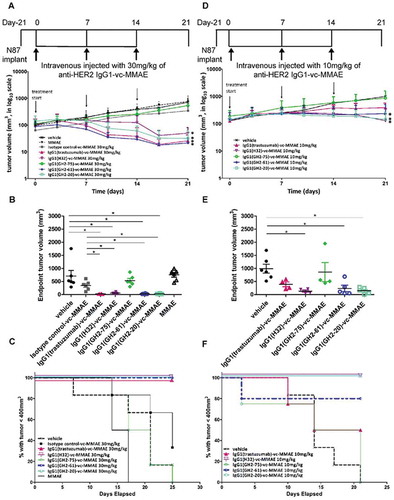
The in vitro efficacy measurements with cultured cells did not always reflect the in vivo efficacies on xenograft models for the ADCs tested. The highly effective in vivo treatments with IgG1(H32)-vc-MMAE, IgG1(trastuzumab)-vc-MMAE and IgG1(GH2-61)-vc-MMAE on xenograft N87 tumors were expected because of the high efficacies measured with cultured N87 cells ( (E)). Similarly, the poor performance of IgG1(GH2-75)-vc-MMAE in treating in vivo tumors was consistent with the low efficacy measured in vitro ( (E)). In contrast, it was difficult to reconcile the discrepancy between the in vivo and in vitro efficacies for the IgG1(GH2-20)-vc-MMAE, which was among the ADCs with the best in vivo efficacies and yet it had the worst in vitro efficacy among the tested ADCs.
Immunotoxins in the form scFv-PE38KDEL as surrogates for detecting antibody off-target toxicity in mouse models
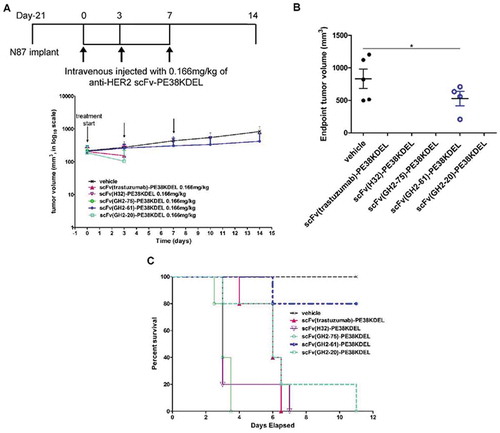
To compare the in vivo efficacies of the ADCs with the corresponding immunotoxins scFv-PE38KDEL, we carried out the in vivo tests of the corresponding scFv-PE38KDELs on the xenograft N87 tumor models. The scFv-PE38KDELs were prepared following the conventional immunotoxin construct (Supplementary Figure S3 (A) and (B)), for which the intoxication mechanism has been well-studied.Citation11,Citation13,Citation19 The EC50s and IC50s of the immunotoxins were measured to assure the qualities of the scFv-PE38KDELs (Supplementary Figure S3 (C) and (D), respectively). Except for the scFv(GH2-20)-PE38KDEL, all the other scFv-PE38KDELs have nM or sub-nM EC50, suggesting that the toxin module did not severely hinder the scFv module binding to HER2-ECD immobilized on ELISA wells (Supplementary Figure S3 (C)). Although the scFv(GH2-20)-PE38KDEL fusion protein had relatively lower affinity in comparison with the other scFv-PE38KDELs, likely due to the interference of the PE38KDEL module to the scFv(GH2-20)-antigen interaction, the scFv(GH2-20)-PE38KDEL cytotoxicity measured with the cell-based assay was only slightly inferior to those of the other scFv-PE38KDELs (Supplementary Figure S3 (D)). The correlation between EC50 and IC50 was qualitative at best (Supplementary Figure S3 (C) and (D), respectively), suggesting that, as expected, relationships between the molecular level affinity measurements and the cellular level cytotoxicity measurements are not straightforward.
The results of the in vivo tests indicated that the scFv-PE38KDEL immunotoxins were highly toxic to the xenograft models even at very low dosage level (0.166 mg/kg): 80% of the scFv(GH2-61)-PE38KDEL-treated xenograft mice remained alive at the endpoint of the in vivo experiment ( (B) and (C)), with slight benefit in preventing tumor progression ( (A) and (B)); all other scFv-PE38KDEL-treated mice did not survive the treatments ( (C)) at the endpoint. The deaths resulting from the immunotoxin treatments could have resulted from off-target toxicity, and, as such, the survival of the mice treated with scFv(GH2-61)-PE38KDEL indicated that the IgG1(GH2-61) could have the least off-target propensity among the antibodies tested as the targeting modules for ADCs. Since the off-target toxicity cannot be predicted with the in vitro assay systems, the immunotoxin form scFv-PE38KDEL could be used as a sensitive surrogate system for detecting the antibody’s propensity for off-target toxicity in vivo.
Figure 2. Treatment of N87 xenograft mouse models with anti-HER2-ECD scFv-PE38KDELs. (A) Data for the time course responses of the tumor size to the scFv-PE38KDEL treatment for xenograft mouse models bearing N87 tumors were attained from the xenograft models randomly assigned into 6 groups and treated with 0.166 mg/kg of the respective scFv-PE38KDEL at 0, 3 and 7 days as depicted in this panel. Note that the tumor size of the last one surviving mouse in the GH2-20 treatment group at day 7 and day 10 was not recorded and thus was not presented in the figure. Tumor size data for each of the mice of the treatment groups are shown in supplementary figure S3 (E). (B) Endpoint tumor volume at day 14 for each of experimental subjects are plotted for each treatment group. Data points for experimental subjects surviving the treatments are shown. (C) Kaplan-Meier survival curves depict the time courses of the fraction of the animal populations surviving (regardless of the tumor size) the scFv-PE38KDEL treatment.
The non-covalently linked IgG1-AL1-PE38KDEL immunotoxin treatments of xenograft models as an alternative test system for ADC development
Although the immunotoxin scFv-PE38KDEL system could provide information on the off-target toxicities of the antibodies as targeting modules in ADCs, the toxicities of the scFv-PE38KDELs were too potent to be measured properly, with few test subjects surviving the treatment (). Alternatively, we performed in vitro and in vivo tests of the non-covalently linked IgG1-AL1-PE38KDEL immunotoxins on the xenograft N87 tumor models to investigate the off-target toxicities and anti-tumor activities of the IgG1s as the targeting modules of the immunoconjugates.
The IC50s of the IgG1-AL1-PE38KDELs were measured in vitro to ensure the qualities of the prepared immunotoxins. All the immunotoxins had sub-nM IC50s; the cytotoxicity (IC50) of the IgG1(trastuzumab)-AL1-PE38KDEL was one order of magnitude less potent to those of the other IgG1-AL1-PE38KDELs (Supplementary Figure S4). The IC50s of IgG1-AL1-PE38KDELs had little correlation with those of corresponding scFv-PE38KDEL (Supplementary Figure S3 (D)), suggesting that, as expected, the cytotoxicity mechanism of IgG1-AL1-PE38KDEL is not identical to that of scFv-PE38KDELs because of non-covalently linked toxin and bi-valent antigen binding in IgG1-AL1-PE38KDEL versus covalently linked toxin and mono-valent antigen binding in scFv-PE38KDEL. The factors that could affect the cytotoxicity of the non-covalently linked immunotoxin include: IgG1 affinity to HER2-ECD, AL1-PE38KDEL affinity to the framework regions of the IgG1, interferences of the non-covalently linked AL1-PE38KDEL to the IgG1-HER2 interaction, cellular uptake efficiency of the IgG1-AL1-PE38KDEL complex and delivery efficiency of the cytotoxic payload to cytoplasm of the model cell. The latter two factors are more likely explanations for the inferior IC50 of the IgG1(trastuzumab)-AL1-PE38KDEL, as we had ruled out the former three factors based on our previous measurements at the molecular level.
The results of the in vivo tests indicated that the IgG1-AL1-PE38KDEL immunotoxins were less toxic in the xenograft models at moderate dosage level (0.75 mg/kg intraperitoneal injection; ) compared with the scFv-PE38KDEL immunotoxins at very low dosage level (0.166 mg/kg intravenous (i.v.) injection; ). Hence, the efficacies of the immunotoxins in the animals that survived the treatments could be compared ( (A) and (B)). All the test subjects treated with IgG1(GH2-61)-AL1-PE38KDEL and IgG1(GH2-20)-AL1-PE38KDEL survived the treatment with noticeable efficacies in tumor control ( (A) and (B)): IgG1(GH2-61)-AL1-PE38KDEL had the best relative efficacy in treating the xenograft models, followed by IgG1(GH2-20)-AL1-PE38KDEL. However, the efficacies shown in (A) and (B) were not as pronounced as those of the corresponding IgG1-vc-MMAEs ( (A) and (B)). Even so, the results showed that the non-covalently linked immunotoxins could be inexpensive substitutes for the corresponding IgG1-vc-MMAEs in preliminary in vivo efficacy/toxicity tests of the antibodies as targeting modules.
Figure 3. Treatment of N87 xenograft mouse models with anti-HER2-ECD IgG1-AL1-PE38KDELs. (A) Data for the time course responses of the tumor size to the IgG1-AL1-PE38KDEL treatment for xenograft mouse models bearing N87 tumors were attained from the xenograft models randomly assigned into 6 groups and treated (intraperitoneal injection) with 0.75 mg/kg of the respective IgG1-AL1-PE38KDEL at 0, 3 and 7 days as depicted in this panel. Tumor size data for each of the mice of the treatment groups are shown in supplementary figure S4 (B). (B) Endpoint tumor volume at day 10 for each of experimental subjects are plotted for each treatment group. Data points for experimental subjects surviving the treatments are shown. (C) Kaplan-Meier survival curves depict the time courses of the fraction of the xenograft mouse models bearing N87 tumors surviving (regardless of the tumor size) the IgG1-AL1-PE38KDEL treatment. (D) We repeated the same treatment protocol as shown in panel (C) on the mice without implanting xenograft tumors (n = 6 throughout). Kaplan-Meier survival curves depict the time courses of the fraction of the mice (n = 6 for each of the control/experimental groups) without implanting N87 tumors surviving the IgG1-AL1-PE38KDEL treatment.
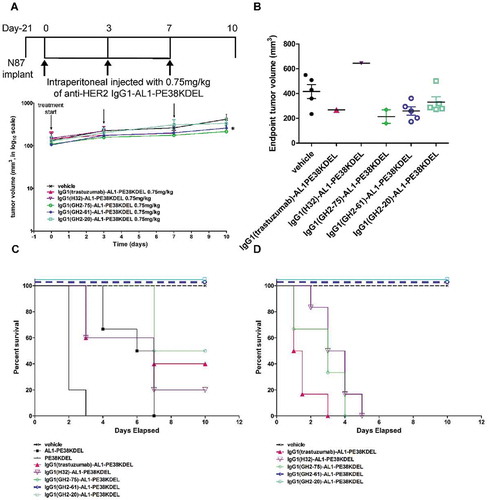
The mouse models were less tolerant to treatment of IgG1(H32)-AL1-PE38KDEL, IgG1(trastuzumab)-AL1-PE38KDEL and IgG1(GH2-75)-AL1-PE38KDEL ( (C)). The serum biochemical analysis (Supplementary Table S3) indicated that the immunotoxins IgG1(H32)-AL1-PE38KDEL, IgG1(trastuzumab)-AL1-PE38KDEL and IgG1(GH2-75)-AL1-PE38KDEL were more toxic than IgG1(GH2-61)-AL1-PE38KDEL and IgG1(GH2-20)-AL1-PE38KDEL, as shown by the substantially elevated serum alanine aminotransferase (ALT) and the R factor (ALT/ALP) levels (indicating drug-induced liver toxicity) in the mice treated with the former three immunotoxins.
To compare the off-target toxicities of the non-covalently linked IgG1-AL1-PE38KDEL immunotoxins in the mouse models in the presence and absence of the N87 xenograft tumors, we repeated the same treatment protocol shown in on the mice without implanting xenograft tumors ( (D)). The qualitative toxicities of the immunoconjugates indicated by the Kaplan-Meier survival curves attained from the treatment ( (D)) were similar to those from the treatment of the corresponding mouse models bearing the N87 tumors ( (C)), indicating that the off-target toxicities of the non-covalently linked IgG1-AL1-PE38KDEL immunotoxins originated from the intrinsic property of the IgG1 targeting module, rather than from unknown effects due to the xenograft tumors.
Together, the efficacy trend of the IgG1-AL1-PE38KDELs towards the xenograft models and the toxicity rank as shown in and Supplementary Table S3, respectively, could be used as a less costly surrogate system to provide information on antibody candidates for potent ADCs with less off-target toxicities in vivo.
Receptor-mediated endocytosis of the IgG1s as the targeting modules for the immunoconjugates
Cells with no HER2-ECD expressed on the cell surface are not sensitive to the immunotoxins with anti-HER-ECD antibodies as targeting modules,Citation9 suggesting that receptor-mediated endocytosis is essential to initiate the cytotoxicity mechanism of the corresponding immunotoxins. The receptor-mediated endocytosis of the 5 IgG1s was visualized with confocal microscopy before (30 minutes at 4°C after adding the antibodies) and after (16 hours at 37°C after adding the antibodies) the antibody endocytosis ( (A)). IgG1(trastuzumab), IgG1(H32), IgG1(GH2-75) and IgG1(GH2-61) bound to the N87 cell surface and then were internalized. IgG1(GH2-20) had low binding efficiency to the HER2-ECD on the cell surface ( (A)), in agreement with the measurement with flow cytometry (Supplementary Figure S1 (D)). Nevertheless, the internalization of IgG1(GH2-20) was clearly visible after 16 hours at 37°C ( (A)).
Figure 4. Internalization of anti-HER2 IgG1s in N87 cells. (A) Confocal microscopy images of N87 cells in the presence of anti-HER2-ECD IgG1s (5μg/mL concentration for each antibody) were taken 30 min after antibody addition at 4°C and 16 h at 37°C. HER2-antibody complexes were stained by Alexa Fluor 633-labeled anti-human antibody (red); lysosomes were stained by anti-human LAMP2 rabbit antibody followed by Alexa Fluor 488-labeled anti-rabbit antibody (green), and nuclei were stained by DAPI (blue). Scale bars: 10 μm. (B) Cellular uptake by N87 cells of the anti-HER2 DTPA-labeled IgG1 (Supplementary Figure S5 (B)) chelated with 111In are plotted versus incubation time with the 111In-labeled antibody; the mean values and standard deviations are calculated with three independent measurements. (C) Cell lysates from anti-HER2 IgG1-vc-MMAEs (10 μg/mL)-treated (18 h and 37°C) N87 cells were assessed by western blotting with β-tubulin as control. The details of the experimental procedures in this figure are described in Supplementary Methods.
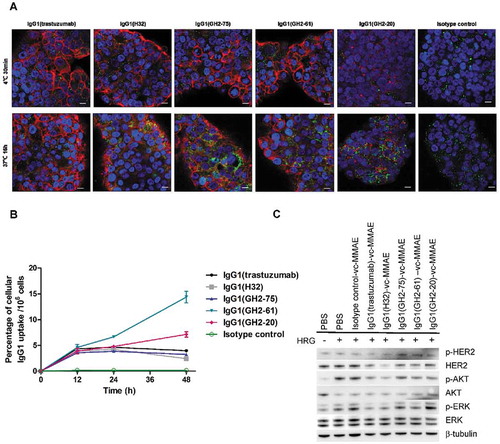
Monitoring of the receptor-mediated endocytosis of the IgG1s labelled with radio isotope 111In, which was in turn chelated to DTPA (diethylenetriaminepentaacetic acid) pre-conjugated on the IgG1 (EC50 measurements for the antibodies with/without the conjugated DTPA are shown in Supplementary Figure S5 (B)), further confirmed the internalization of IgG1(GH2-20) ( (B)). Two groups of IgG1 internalization patterns are evident in (B): 1) the internalization of IgG1(GH2-20) and IgG1(GH2-61) continued after 24 h incubation at 37°C, while 2) the internalization of IgG1(GH2-75), IgG1(trastuzumab) and IgG1(H32) stopped after 24 h incubation at 37°C ( (B)).
One mechanism for the cessation of the antibody internalization process could be the depletion of the HER2 receptors due to binding of the antibodyCitation8; depletion of HER2-ECD from N87 cell did occur after incubating IgG1(H32)-vc-MMAE and IgG1(trastuzumab)-vc-MMAE with the cells for 18 hours at 37°C ( (C)), partly explaining why the internalization of the IgG1s in the latter group stopped after 24 h incubation at 37°C ( (B)). However, the HER2 receptor depletion level due to IgG1(GH2-75)-vc-MMAE was largely indistinguishable from those due to IgG1(GH2-61)-vc-MMAE and IgG1(GH2-20)-vc-MMAE ( (C)), suggesting that other unknown mechanism(s) could be involved in prohibiting continuous internalization of IgG1(GH2-75)-vc-MMAE after 24 h incubation at 37°C. The continuous uptake of the IgG1(GH2-20) and IgG1(GH2-61) by the N87 cells ( (B)) could explain the high potency of the corresponding IgG1-vc-MMAEs and IgG1-AL1-PE38KDELs in treating xenograft N87 tumors ( and ).
Bio-distribution of the IgG1s as the targeting modules of the immunoconjugates
To gain further insights into the potencies and off-target toxicities of the immunoconjugates, we investigated the bio-distribution of the 5 IgG1s in xenograft models. The 5 IgG1s were conjugated with the fluorescence dye without substantial alteration of their affinities to HER2-ECD (Supplementary Figure S5 (A)). In vivo fluorescence imaging indicated that, qualitatively, the IgGs were locally concentrated in the N87 xenograft tumors one day after the administration of the antibodies to the mice ( (A)).
Figure 5. Bio-distributions of the anti-HER2 IgG1s in N87 xenograft models. (A) DyLight 680-labeled anti-HER2 IgG1s (3 mg/kg; Supplementary Figure S5 (A)) were injected into N87 tumor-bearing mice and imaged at 24 h post-injection. (B) Bio-distribution of DyLight 680-labeled anti-HER2 IgG1s in N87 tumor-bearing mice at 24 h post-injection (3 mg/kg) were determined ex vivo with IVIS (Supplementary Figure S6). The mean values and standard deviations are calculated with three independent measurements. (C) Anti-HER2 111In-labeled IgG1s were injected into N87 tumor-bearing mice and imaged at 24 h and 48 h after injection (4 mg/kg). The averaged values were calculated specifically for the images shown in the corresponding panels (n = 1). The details of the experimental procedures in this figure are described in supplementary methods.
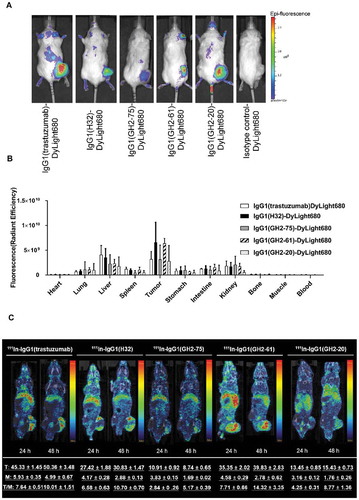
Quantitative ex vivo measurements of the bio-distribution indicated that IgG1(GH2-61) targeted the N87 tumor with high local concentration and low off-target propensity () and Supplementary Figure S6), explaining the high potencies of the corresponding IgG1-vc-MMAEs/IgG1-AL1-PE38KDEL ( and ) and the low off-target toxicities of the corresponding scFv-PE38KDEL/IgG1-AL1-PE38KDEL ( and ; Supplementary Table S3). In contrast, the high off-target distribution of IgG1(trastuzumab) could explain the toxicity of the scFv(trastuzumab)-PE38KDEL and IgG1(trastuzumab)-AL1-PE38KDEL in xenograft models ( and ; Supplementary Table S2 and S3). The ex vivo NIRF imaging of dissected organs of N87 tumor-bearing mice ( (B) and Supplementary Figure S6) 24 h after i.v. injection of fluorescence dye-conjugated IgG1 at about the same order of magnitude of the dosages used in IgG1-AL1-PE38KDEL/scFv-PE38KDEL treatments of N87 xenograft models suggested that the off-target distributions of the IgG1s to liver could explain the drug-induced liver toxicity of IgG1-AL1-PE38KDEL treatment. Specifically, the IgG1s’ liver bio-distribution level rank: trastuzumab > H32 > GH2-75 ≈ GH2-61 > GH2-20 ( (B)) is largely consistent with the ALT/ALP ratio (liver toxicity) trend (Supplementary Table S3 on serum biochemical parameters in N87 xenograft mice treated with IgG1-AL1-PE38KDEL): trastuzumab > GH2-75 > H32 > GH2-61 > GH2-20, suggesting that the off-target toxicities of the treatments with the 5 IgG1-AL1-PE38KDEL could be largely attributed to the off-target propensities of these IgG1s to liver.
MicroSPECT/CT imaging of the IgGs conjugated with radioisotopes 111In confirmed the specific targeting of the IgG1s to the xenograft N87 tumors. As indicated in (C), except for 111In-IgG1(GH2-75), the tumor (T) uptake increased with time in each group. The declined muscle (M) uptake leads to the elevated T/M ratio at 48 h post-injection, indicating a specific antibody targeting rather than passive diffusion. The difference in T/M ratio between 24 and 48 h post-injection is a noninvasive index of the antibody accumulation rate in tumor ( (C)), which is consistent with the high potency of IgG1(GH2-61) and the low potency of IgG1(GH2-75) as targeting modules in the corresponding ADCs ().
Results in (C) are compared to those in (B) and 4 (B) for experimental consistency. Although these in vivo/in vitro measurments were not meant for quantitative comparisons, the correlated measurements nevertheless indicated consistency in the measurements to an extent. The microSPECT/CT measurements (24 h tumor count = 45, 27, 11, 35, 13 for trastuzumab, H32, GH2-75, GH2-61, GH2-20, respectively, from (C)) and fluorescence measurements (24 h fluorescence x10−10 = 0.3, 0.7, 0.3, 0.7, 0.3 for the same set of antibodies, respectively, from (B)) are comparable in trend, except for trastuzumab as an outlier in the in vivo experiments. Interestingly, the increase rate of the T/M ratio from 24 h to 48 h related to the first 24 h: (T/M48h – T/M24h)/T/M24h for trastuzumab, H32, GH2-75, GH2-61, GH2-20 IgG1s were 0.3, 0.6, 0.8, 0.9, 1.0, respectively, which roughly followed (with GH2-20 as an outlier) the trend of cellular uptake rate of these IgG1s measured in the in vitro experiment shown in (B) ((uptake48h – uptake24h)/uptake24h = ~ 0, ~ 0, ~ 0, 0.8, 0.4, respectively), suggesting that the in vivo T/M ratio increase rate could be related to the in vitro cellular uptake rate, which had been anticipated (see above) as an indicator for potency of the IgG-based ADC in treating xenograft tumor. These comparable results suggest that cellular uptake of the ADCs tested in this work could be the dominant determinant for the ADCs’ efficacies.
Discussion
The IgG1(GH2-61)-vc-MMAE was comparably potent as the two control ADCs (IgG1(H32)-vc-MMAE and IgG1(trastuzumab)-vc-MMAE) in treating the xenograft tumor models at a 30 mg/kg dosage level, and out-performed the IgG1(trastuzumab)-vc-MMAE at a 10 mg/kg dosage level (). The immunotoxins IgG1(GH2-61)-AL1-PE38KDEL and scFv(GH2-61)-PE38KDEL were evidently superior to all other immunotoxins in terms of anti-tumor efficacy and off-target toxicity to the xenograft models ( and ). The superior efficacy and toxicity profile of the GH2-61-based immunoconjugates was attributed to efficient internalization of the antibody by the model cultured cells in vitro ( (B)) and the specific bio-distribution targeting mostly the tumors in the xenograft models in vivo ( (B)). In addition, the microSPECT/CT imaging of the IgG1 bio-distribution also supported that the IgG1(GH2-61) actively targeted the xenograft tumor with superior uptake rate over all the other IgG1 tested ( (C)).
In comparison, IgG1(GH2-20)-vc-MMAE was as potent as IgG1(GH2-61)-vc-MMAE in terms of eradicating xenograft tumors (). IgG1(GH2-20)-AL1-PE38KDEL was comparable to IgG1(GH2-61)-AL1-PE38KDEL in terms of anti-tumor efficacy and off-target toxicity (), but was more toxic in the form of scFv-PE38KDEL (). However, in contrast to the IgG1(GH2-61), the IgG1(GH2-20) bound to the cell surface HER2-ECD with substantially lower binding efficiency (Supplementary Figure S1 (D)), resulting in much higher IC50 measured with IgG1-vc-MMAE in vitro (Supplementary Figure S1 (E)), slower internalization rate ( (B)) and less efficient tumor uptake ( (C)). These results indicated that the in vitro and in vivo indirect assays on the antibodies as targeting modules did not necessarily reflect the in vivo efficacies on xenograft models for the immunoconjugates in general. This conclusion is in agreement with the observation that affinity of antibody measured in vitro does not necessarily suggest better tumor uptake of the antibody in vivo.Citation20 Since the NOD/SCID mice used in the xenograft models lacked matured B and T cells and deficient natural killer (NK) cell function, the in vivo and in vitro efficacy discrepancy of IgG1(GH2-20)-vc-MMAE is not likely to be attributed to the NK cell-based antibody-dependent cell-mediated cytotoxicity of the xenograft mice. Other in vivo attributes that could explain the in vivo and in vitro efficacy discrepancy remain unknown.
Although the antibody formats (IgG1 versus scFv), the linkers between the antibody and the toxic payload (covalent versus noncovalent) and the toxic payloads (MMAE versus PE38KDEL) are all different among the three forms of the immunoconjugates, our results regarding the off-target toxicities of the immunotoxins could nevertheless help to better select corresponding ADC candidates for further development. The in vivo mouse models were tolerant to all the HER2-ECD-targeting IgG1-vc-MMAEs at high dosage ( and Supplementary Table S2). Conversely, the mouse models were intolerant to all the HER2-ECD-targeting scFv-PE38KDELs except scFv(GH2-61)-PE38KDEL at very low dosage (). The intolerance of the immunotoxins could have resulted from off-target toxicity. Since the off-target toxicity cannot be measured or predicted with in vitro systems, the immunotoxin form scFv-PE38KDEL could be used as a sensitive surrogate system for detecting potential off-target toxicity in vivo associated with the antibody as the targeting module. This is particularly useful when the corresponding IgG1-vc-MMAE is not obviously toxic at elevated dosage levels in the experiments with xenograft models. However, the scFv-PE38KDEL could be a less relevant model mimicking the conventional IgG-based ADC because the monovalent scFv could result in substantially different effects in toxin delivery in comparison with that of the divalent IgG; therefore, the IgG1-AL1-PE38KDEL could be a more relevant model for the IgG-based ADC. The IgG1-AL1-PE38KDEL immunotoxins were less toxic to the mouse models than the scFv-PE38KDEL immunotoxins at medium dosage ( and Supplementary Table S3), and the potencies of the IgG1-AL1-PE38KDELs towards the xenograft models could be related qualitatively to those of the corresponding IgG1-vc-MMAEs. Hence, the IgG1-AL1-PE38KDELs could be used as a less costly replacement for the corresponding covalently linked ADCs to identify highly potent antibody candidates with low off-target toxicity. Although the targeting discrepancy between the IgG and scFv, the dissociation of the IgG and AL1-PE38KDEL and the formulation of the immunotoxins could hamper the utility of IgG1-AL1-PE38KDEL and scFv-PE38KDEL as surrogate systems to select corresponding ADC candidates in vivo for further development, both forms of immunotoxin could nevertheless be of use when the in vitro efficacy and toxicity measurements of the immunoconjugates with cultured cells do not reflect their in vivo efficacy and toxicity on xenograft models.
In conclusion, the potency of ADCs as anti-cancer therapeutics requires optimal delivery of the cytotoxic payloads by the ADCs’ antibodies as the targeting modules while maintaining low off-target toxicities. Antibodies from the synthetic antibody libraries underlying the high throughput immunoconjugate discoveries enabled a rapid process to develop potent ADCs capable of eradicating xenograft tumors in mice, as demonstrated in this work. In comparison with the two humanized control antibodies (trastuzumab and H32) derived from murine antibody repertoires, some of the antibodies (GH2-61 and GH2-20) derived from the synthetic antibody libraries have enhanced internalization rate, which could result in ADCs with optimal efficacies. Moreover, in vitro assays with cultured cells could narrow the antibody candidate population and gain insights into the optimization of the antibodies as the targeting modules in ADCs, but the in vivo efficacy and toxicity of the ADCs are frequently unpredictable from the in vitro measurements. We have found that in vivo experiments with the three forms of immunoconjugates on xenograft models further provide information for the ADC candidate selection in terms of potencies and off-target toxicities as anti-cancer therapeutics. These findings support the strategy to develop optimal ADCs against cancers with the utility of the general ADC discovery platform on the basis of the synthetic antibody libraries in combination with the in vivo tests on xenograft models with the three forms of immunoconjugates.
Material and methods
Cell culture and tumor xenograft studies
Human gastric carcinoma cell line NCI-N87 was acquired from American Type Culture Collection (ATCC). The cells were grown in RPMI 1640 medium (Gibco) containing 10% fetal bovine serum (Gibco) and Pen/Strep (10,000 U penicillin, 10 mg streptomycin, Gibco) at 37°C in a humidified incubator containing 5% CO2. All mouse experiments were conducted according to relevant guidelines and experimental protocols approved by the Institutional Animal Care and Utilization Committee (IACUC) of Academia Sinica (Protocol ID: 13–03-545). The N87 tumor-bearing mouse model was established by subcutaneously inoculating 1 × 106 cells into the right flank of 6–8 week old male NOD.CB17-Prkdcscid/NcrCrlBltw NOD/SCID mice (BioLASCO Taiwan Co. Ltd). When the tumors reached suitable tumor size of 80–100 mm3, the mice were randomly assigned into control and treatment groups and dosing was started. N87 tumor-bearing mice were treated once a week for a total of three doses with antibody-based anti-HER2 therapeutics through intravenous injection unless otherwise stated. The measurements of tumor size and weight change in mice were recorded twice a week. Tumor volume was calculated using the ellipsoid formula: length× width× height× 0.523. Survival probability over time was evaluated by the Kaplan–Meier method; mice with tumor of the size above 400 mm3 were considered as treatment failures and were removed from the surviving population in calculating the Kaplan-Meier curves; hence ‘survive’ means, in this work, ‘alive and with tumor burden less than 400 mm3ʹ unless otherwise indicated.
IgG1-vc-MMAE preparation
The IgG1s were conjugated with vcMMAE (MedChem Express) through the cysteine residues on the tris(2-carboxyethyl)phosphine hydrochloride (TCEP)-reduced IgG1s. Briefly, antibody was partially reduced for 1 h at room temperature with tris(2-carboxyethyl)phosphine (TECP, Sigma-Aldrich). We adjusted the concentration of the reductant to achieve equal DAR in the product ADCs: IgG1(trastuzumab), IgG1(H32), and isotype control antibodies were reduced with 28 μM TCEP (2 equivalent of reductant-to-IgG1 molar ratio), IgG1(GH2-75), IgG1(GH2-61), and IgG1(GH2-20) antibodies were reduced with 42 μM TCEP (3 equivalent of reductant-to-IgG1 molar ratio). The reduced antibodies were conjugated with drug by adding 5.25 equivalents of maleimide-vc-MMAE (10 mM DMSO solved stock). The conjugation reaction was kept at room temperature for 1 h. The N-acetylcysteine was used to quench the reaction at room temperature for 30 min. The quenched reaction mixture was desalted by gel filtration with a 5 mL desalting column (Thermo Scientific); the buffer was changed into phosphate-buffered saline (PBS) and the ADC product was concentrated by centrifugal ultrafiltration (Amicon Ultra 30K MWCO, MERCK Millipore). The ADC solutions were filtered through a 0.2 μm filter and stored at 4 °C. The ADC products were analyzed by sodium dodecyl sulfate-polyacrylamide electrophoresis (SDS-PAGE) (Supplementary Figure S1 (A)) and hydrophobic interaction chromatography (HIC) (Supplementary Figure S1 (B)).
ScFv-PE38KDEL preparation
The amino acid sequence of PE38KDEL domain has been documented;Citation21 the peptide linker (ASAAGGSGT) was inserted between scFv (with VL-linker-VH orientation) and PE38KDEL domain (Supplementary Figure S3 (A)). The codon optimized nucleotide sequences were chemically synthesized. The construction, expression and purification of scFv-PE38KDEL were similar to our previously described protocols.Citation17 In brief, the scFv-PE38KDEL coding region was sub-cloned into pET-32a expression vector with a thioredoxin fusion N-terminal to the scFv-PE38KDEL; the fusion protein contains a hexa-HIS tag followed by a TEV protease cutting site between the thioredoxin and the scFv-PE38KDEL. E. coli Rosetta-gami B (DE3) strain transformed with scFv-PE38KDEL-pET32a expression vector were grown in 2X YT medium (Tryptone 16 g/L, Yeast extract 10 g/L, NaCl 5 g/L) with ampicillin (200 μg/L), tetracycline (12.5 μg/L) and chloramphenicol (37.5 μg/L) at 37 °C until OD 600 reached 1.0, and then were shifted to 16 °C for additional 1 h incubation to reach temperature equilibrium between incubator and culture media. The protein expression was induced with final 0.2 mM IPTG overnight. The cell pellets were processed in lysis buffer (Tris-HCl, 20 mM, pH 8.0, 300 mM NaCl) with Microfluidizer (Microfluidics, MA). The recombinant thioredoxin fusion scFv-PE38KDEL immunotoxin was purified by nickel chelation chromatography with Ni2+ charged IMAC prepacked column (GE Healthcare Life Sciences). The fractions containing scFv-PE38KDEL immunotoxin were collected and digested by TEV protease (OD280 ratio 50:1) at 30 °C for at least 5 h, and then dialyzed against PBS buffer overnight. The hexa-HIS tag containing thioredoxin and TEV protease were removed by Ni2+ charged IMAC prepacked column. The tag-free scFv-PE38KDEL immunotoxin in the flow-through was collected and further purified with a Superdex 200 size-exclusion column (GE Healthcare Life Sciences) in PBS to around 95% purity (Supplementary Figure S3 (B)).
Cellular uptake and internalization of HER2-targeting IgG1s
For internalization assays, the procedure was based on the previous literature with some minor modifications.Citation22 N87 cells (1 × 106) were seeded in a 6-well plate and cultured in 3 mL of culture medium. After 24 h incubation, the culture medium was replaced by the serum-free medium (3 mL per well) containing 4 nM radiolabeled anti-HER2 antibodies. At designated time points (4, 12, 24, and 48 h post-incubation), the radioactive medium was aspirated, and the cells were washed with PBS (0.5 mL) to remove unbound radiolabeled antibodies. The medium and washing PBS were collected into a counting vial. 0.5% trypsin (0.5 mL) was added to detach the cells from the plate. Serum-containing culture medium (1.5 mL) was added to re-suspend the cells for avoiding possible damage from trypsin treatment. The number of cells in the cell suspension was counted for normalizing the cellular uptake of the radiolabeled antibody. The radioactivity of the collected cells was measured. After radioactivity measurement, the collected cells were treated with 1 mL buffer solution containing 200 mM sodium acetate and 500 mM sodium chloride (pH of the buffer was adjusted to pH = 2.5 with acetic acid) at 4°C for 5 mins and then centrifuged at 8000 g for 10 min.Citation22 The cells were washed twice with PBS. The supernatant of cell suspension and washing buffer were collected into a vial, and the cell pellets were added to another counting vial for radioactivity measurement. The cellular uptake was expressed as the percentage of treated dose accumulated in one million cells. The percentage of cellular IgG1 uptake per one million cells was calculated by the equation:
where,
Abbreviations
| ADC | = | antibody-drug conjugate |
| DTPA | = | diethylenetriaminepentaacetic acid |
| HER2-ECD | = | human epidermal growth factor receptor 2 - extracellular domain |
| MMAE | = | monomethyl auristatin E |
| NIRF | = | near-infrared fluorescence |
| scFv | = | single chain variable domain fragment |
| SPECT | = | single-photon emission computed tomography. |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author contributions
A.-S. Y. conceived the studies, and together with W.-Y. K. designed the in vivo experiments; W.-Y. K. carried out the xenograft model treatment experiments; H.-J. H. performed pharmacokinetics with W.-Y. K., synthesized ADCs, and performed Biacore analysis of antibodies; C.-Y. W. designed, carried out radionuclide imaging studies and analyzed data; Y.-L. T. performed in vitro cytotoxicity assay; H.-P. P. and J.-W. J were responsible for all bioinformatics analysis; H.-S. C. and C.-M. Y. prepared immunotoxins; C. -M. Y designed HIC analysis system and together with H.-J. H. carried it out; Y.-K. C., I.-C. C. and C.-P. T. perform other in vitro experiments and IgG production; M. H. supervised in vivo experiments; L.-C. L. was responsible for monitoring tumor growth; A. Y. W. interpreted experimental data; A.H.-J. W. oversaw this study; A.-S. Y., W.-Y. K., H.-J. H., C.-Y. W., H.-S. C., Y.-C. C., Y.-L. T. and H.-P. P. wrote the paper.
Competing interests
There is no competing interest.
Supplemental Material
Download Zip (20.8 MB)Acknowledgments
This research was supported by Academia Sinica and Ministry of Science and Technology [MOST104-0210-01-09-02, MOST105-0210-01-13-01, MOST106-0210-01-15-02] and by the Taiwan Protein Project [MOST105-0210-01-12-01 and MOST106-0210-01-15-04]. We would also like to thank the support from Program for Translational Innovation of Biopharmaceutical Development. We like to thank Dr. Yang, Chi Fan and National Center for Genome Medicine for experimental assistance. We like to thanks Taiwan mouse clinic in Academia Sinica for mouse serum biochemical analyses.
Supplementary Materials
Supplemental data for this article can be accessed on the publisher's website.
Additional information
Funding
Related Research Data
References
- Yan M, Parker BA, Schwab R, Kurzrock R. HER2 aberrations in cancer: implications for therapy. Cancer Treat Rev. 2014;40:770–780. doi:10.1016/j.ctrv.2014.02.008.
- Gravalos C, Jimeno A. HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Ann Oncology. 2008;19:1523–1529. doi:10.1093/annonc/mdn169.
- Gingras I, Gebhart G, de Azambuja E, Piccart-Gebhart M. HER2-positive breast cancer is lost in translation: time for patient-centered research. Nat Rev Clin Oncol. 2017;14:669. doi:10.1038/nrclinonc.2017.96.
- Metzger-Filho O, Winer EP. T-DM1 — an important agent in the history of breast cancer management. Nat Rev Clin Oncol. 2017;14:651. doi:10.1038/nrclinonc.2017.123.
- Thuss-Patience PC, Shah MA, Ohtsu A, Van Cutsem E, Ajani JA, Castro H, Mansoor W, Chung HC, Bodoky G, Shitara K, et al. Trastuzumab emtansine versus taxane use for previously treated HER2-positive locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma (GATSBY): an international randomised, open-label, adaptive, phase 2/3 study. Lancet Oncol. 2017;18:640–653. doi:10.1016/S1470-2045(17)30111-0.
- Doi T, Shitara K, Naito Y, Shimomura A, Fujiwara Y, Yonemori K, Shimizu C, Shimoi T, Kuboki Y, Matsubara N, et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody-drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: a phase 1 dose-escalation study. Lancet Oncol. 2017;18:1512–1522. doi:10.1016/S1470-2045(17)30604-6.
- Chen I-C, Chiu Y-K, Yu C-M, Lee -C-C, Tung C-P, Tsou Y-L, Huang Y-J, Lin C-L, Chen H-S, Wang AH-J, et al. High throughput discovery of influenza virus neutralizing antibodies from phage-displayed synthetic antibody libraries. Sci Rep. 2017;7:14455. doi:10.1038/s41598-017-14823-w.
- Chen HS, Hou SC, Jian JW, Goh KS, Shen ST, Lee YC, You -J-J, Peng H-P, Kuo W-C, Chen S-T, et al. Predominant structural configuration of natural antibody repertoires enables potent antibody responses against protein antigens. Sci Rep. 2015;5:12411. doi:10.1038/srep12411.
- Hou SC, Chen HS, Lin HW, Chao WT, Chen YS, Fu CY, Yu C-M, Huang K-F, Wang AH-J, Yang A-S. High throughput cytotoxicity screening of anti-HER2 immunotoxins conjugated with antibody fragments from phage-displayed synthetic antibody libraries. Sci Rep. 2016;6:31878. doi:10.1038/srep31878.
- Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, Rejniak SX, Gordon KA, DeBlanc R, Toki BE, et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102:1458–1465. doi:10.1182/blood-2003-01-0039.
- Weldon JE, Pastan I. A guide to taming a toxin–recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011;278:4683–4700. doi:10.1111/j.1742-4658.2011.08182.x.
- Chaudhary VK, Jinno Y, FitzGerald D, Pastan I. Pseudomonas exotoxin contains a specific sequence at the carboxyl terminus that is required for cytotoxicity. Proc Natl Acad Sci U S A. 1990;87:308–312.
- Alewine C, Hassan R, Pastan I. Advances in anticancer immunotoxin therapy. The Oncologist. 2015;20:176–185. doi:10.1634/theoncologist.2014-0358.
- Peng HP, Lee KH, Jian JW, Yang AS. Origins of specificity and affinity in antibody-protein interactions. Proc Natl Acad Sci U S A. 2014;111:E2656–65. doi:10.1073/pnas.1401131111.
- Hsu HJ, Lee KH, Jian JW, Chang HJ, Yu CM, Lee YC, Chen I-C, Peng H-P, Wu CY, Huang Y-F, et al. Antibody variable domain interface and framework sequence requirements for stability and function by high-throughput experiments. Structure. 2014;22:22–34. doi:10.1016/j.str.2013.10.006.
- Chang HJ, Jian JW, Hsu HJ, Lee YC, Chen HS, You JJ, Hou S-C, Shao C-Y, Chen Y-J, Chiu K-P, et al. Loop-sequence features and stability determinants in antibody variable domains by high-throughput experiments. Structure. 2014;22:9–21. doi:10.1016/j.str.2013.10.005.
- Yu CM, Peng HP, Chen IC, Lee YC, Chen JB, Tsai KC, Chen C-T, Chang J-Y, Yang E-W, Hsu P-C, et al. Rationalization and design of the complementarity determining region sequences in an antibody-antigen recognition interface. PLoS One. 2012;7:e33340. doi:10.1371/journal.pone.0033340.
- Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW Jr., Leahy DJ. Structure of the extracellular region of HER2 alone and in complex with the herceptin fab. Nature. 2003;421:756–760. doi:10.1038/nature01392.
- Spooner RA, Smith DC, Easton AJ, Roberts LM, Lord JM. Retrograde transport pathways utilised by viruses and protein toxins. Virol J. 2006;3:26. doi:10.1186/1743-422X-3-99.
- Rudnick SI, Lou J, Shaller CC, Tang Y, Klein-Szanto AJ, Weiner LM, Marks JD, Adams GP. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011;71:2250–2259. doi:10.1158/0008-5472.CAN-10-2277.
- Brinkmann U, Pai LH, FitzGerald DJ, Willingham M, Pastan I. B3(Fv)-PE38KDEL, a single-chain immunotoxin that causes complete regression of a human carcinoma in mice. Proc Natl Acad Sci U S A. 1991;88:8616–8620.
- Reilly RM, Kiarash R, Cameron RG, Porlier N, Sandhu J, Hill RP, Vallis K, Hendler A, Gariépy J. 111In-labeled EGF is selectively radiotoxic to human breast cancer cells overexpressing EGFR. J Nucl Med. 2000;41:429–438.