
ABSTRACT
Chimeric antigen receptor (CAR)-engineered T cells have a proven efficacy for the treatment of refractory hematological B cell malignancies. While often accompanied by side effects, CAR-T technology is getting more mature and will become an important treatment option for various tumor indications. In this review, we summarize emerging approaches that aim to further evolve CAR-T cell therapy based on combinations of so-called universal or modular CAR-(modCAR-)T cells, and their respective adaptor molecules (CAR-adaptors), which mediate the crosslinking between target and effector cells. The activity of such modCAR-T cells is entirely dependent on binding of the respective CAR-adaptor to both a tumor antigen and to the CAR-expressing T cell. Contrary to conventional CAR-T cells, where the immunological synapse is established by direct interaction of CAR and membrane-bound target, modCAR-T cells provide a highly flexible and customizable development of the CAR-T cell concept and offer an additional possibility to control T cell activity.
Introduction
Beyond chemotherapy and targeted therapies such as kinase inhibitors, therapeutic antibodies, bispecific antibodies, and antibody-drug conjugates, engineered CAR-T cells have shown promising results in clinical trials, particularly in patients with B cell malignancies refractory to established therapies. Based on these data, Kymriah™ (tisagenlecleucel)Citation1 and Yescarta® (axicabtagene ciloleucel),Citation2 two anti-CD19 CAR-T cell-based therapies for patients with CD19+ hematological malignancies, including B cell acute lymphoblastic leukemia (ALL) and large B cell lymphoma, have been approved by the US Food and Drug Administration (FDA) and European Medicines Agency.Citation3–Citation5 These breakthrough therapies illustrate the progress in the development of these novel cell and gene therapies. Given the progress in the field, the focus of research is now shifting to approaches addressing the ease of use, robustness, and cost of manufacturing, safety, and achieving efficacy in solid tumors.
Table 1. Comprehensive overview of current CARs and their respective adaptor molecules.
One of the challenges associated with CAR-T cell therapies is cytokine release syndrome (CRS). CRS is a systemic inflammatory response caused by the rapid activation and expansion of CAR-T cells upon encountering the antigen, which initiates a vicious circle with accessory immune cells that can culminate in the life-threatening release of inflammatory cytokines.Citation6,Citation7 Another challenge of CAR-T cell therapy is the on-target off-tumor toxicity caused by targeting antigen expression on healthy tissue.Citation8,Citation9 As a consequence of the high potency of CAR-T cells, the tumor-antigen loss can be observed as an evasion strategy.Citation9 The majority of current CAR-T cell approaches target a single-specific tumor antigen, entailing the possibility of tumor escape caused by antigen loss due to mutations, downregulation or deletion of the target antigen. Additionally, in response to treatment, heterogeneous expression levels of tumor antigens may enable the selective survival of tumor subpopulations expressing low amounts of antigen, potentially favoring tumor relapse.Citation10–Citation12 To circumvent the relapse of such tumor variants, and to address the other hurdles mentioned above, novel approaches have been described that aim to build a controllable and diversified redirected anti-tumor immune response.
To address side effects of CAR-T therapy, methods that are very specific to cellular therapy, like the integration of a “suicide gene” to eliminate the CAR-T cells at a given time, have been developed. Obviously, these strategies result in the irreversible depletion of the engineered T cells, and so far have not solved the intrinsic inability to steer and control CAR-T cell activity.Citation13,Citation14 Furthermore, the high degree of flexibility required to target the right antigen at the right time – ultimately indispensable to provide durable antitumor effects and to fully benefit from targeted immunotherapy – is limited for most CAR-T cell approaches, as they require personalized preparation prior to treatment.
In this review, we summarize emerging approaches intended to further evolve CAR-T cell therapy based on combinations of so-called universal or modular CAR- (modCAR-)-T cells and their respective adaptor molecules, termed in this review CAR-adaptors, which mediate the crosslinking between target and CAR-T effector cells. While engineering and production of these CAR-adaptors has its own limitations, the combination of modCAR-T cells with such bi- or multi-specific CAR-adaptors provide the opportunity to control the activity of CAR-T cells and to target a variety of antigens simultaneously without the need for re-engineering T cells. Such a combinatorial approach promises to facilitate a modular platform for adoptive cell therapy. In the following, we describe various approaches that make use of orthogonal CAR-adaptors designed specifically to work with modCAR-T cells to tackle the issues mentioned above.
Concept and mechanism of modCAR-T cells
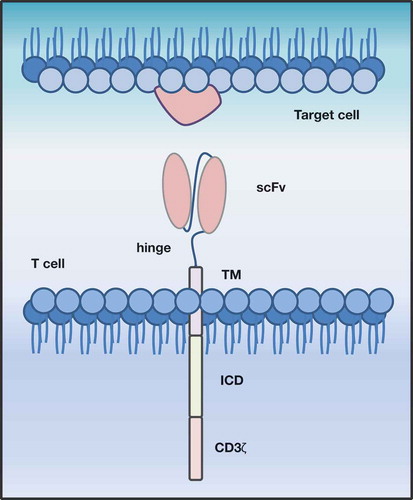
Typically, the term CAR-T cell is used as a synonym for second- or third-generation CAR-engineered T cells, and we refer to these as conventional CAR-T cells in this review. However, literature reveals that, due to rapid development and the constant engineering of chimeric receptors, a large “zoo” of CAR-T designs and different concepts exists that are summarized under the umbrella term “CAR”. The majority of these concepts follows the design of the conventional CARs and consists of three major domains: 1) an intracellular signaling domain (ICD) linked with 2) a transmembrane domain (TM) to 3) an extracellular antigen recognition domain (ECD) expressed on the cell surface (). Depending on the CAR concept, these domains can be fused or split into different parts; examples include inhibitory CARs, affinity tuned CARs, switch CARs and many more as recently described.Citation15–Citation17
Figure 1. Schematic representation of a second generation, conventional CAR-T cell. CAR-T cells target surface antigens directly in a major histocompatibility class-independent manner. They consist of an extracellular antigen recognition domain (ECD), conventionally a single chain variable fragment (scFv), which is linked via hinge region (hinge) to a transmembrane domain (TM), followed by an intracellular costimulatory domain (ICD) and a CD3ζ signaling domain.
Conceptually, alterations within the CAR design can further be made at the level of the ECD. Historically, the ECD in conventional CARs is a single-chain variable fragment (scFv) obtained by the fusion of the variable heavy and variable light chain sequences of monoclonal antibodies (mAbs) through a flexible linker. In most cases, the scFv is directed towards one particular (tumor) antigen. Recently, bispecific CARs, developed as a precaution to avoid antigenic escape, have been described, such as those targeting CD19 and CD20Citation18,Citation19 or CD19 and CD22,Citation20 and some of them are currently being tested in clinical trials (ClinicalTrials.gov Identifier: NCT03241940).Citation16 Recent approaches have illustrated the possibilities of substituting the scFv with other binding moieties. These include designed ankyrin repeat proteins (DARPins)Citation21 or nanobodiesCitation22 that can be selected to have IgG-like affinities, which have been described to possess advantages over immunoglobulin-based molecules in terms of expression and stability.Citation21,Citation23
Definition and properties of modCAR-T cells controllable through CAR-adaptors
Comprehensively, universal or modCARs describe chimeric receptors that do not directly recognize the target antigen, but instead make contact to adaptor molecules (CAR-adaptors), that in turn bind to the target antigen. Thus, the activity of modCAR-T cells is strictly dependent on the presence of the CAR-adaptors enabling the formation of an immunological synapse. Consequently, the formation of the synapse depends on the respective characteristics of the CAR-adaptors; the affinity of the CAR-adaptors for the tumor antigens, the affinity towards the chosen ECD of the modCAR, its half-life, as well as its bio-distribution, are all important.Citation24 ModCARs themselves follow the design of conventional CARs in terms of co-stimulatory and signaling domains. The ECD must not necessarily be an scFv-scaffold but can be interchangeable with any other structure capable of recognizing an antigen. Thereby, it is conceivable to choose from different ECDs and CAR-adaptors to facilitate a modular design that aims towards optimal antigen targeting.
By choosing suitable CAR-adaptors with respect to their half-life, bio-distribution, and target antigen, it is possible to tightly regulate and adapt modCAR-T cell activity. With regard to future clinical applications, the possibility for titration of CAR-adaptors allows the activity of modCAR-T cells to be “turned on or off” individually without the need to eliminate the T cells (). Thus, side effects could be mitigated more rapidly and therapy might be continued with adapted dosing at a later time point. Depending on the application, CAR-adaptors can be mAbs, antibody fragments, small molecules or any conceivable structure that is able to target at least one desired antigen, and that can simultaneously be recognized by the modCAR-T cells. Depending on its pharmacokinetic properties, the activity of modCAR-T cells in the organism is entirely dependent on the presence of the respective CAR-adaptors at sufficient levels.Citation25 The majority of CAR-adaptors reported in the literature are derived from the large repertoire of IgG antibodies or derivatives thereof, such as antigen-binding fragments (Fabs) or scFvs. These immunoglobulin-based CAR-adaptors can be customized by either chemically or genetically attaching a tag. The modification of CAR-adaptors can be either untargeted by coupling to primary amine residues and comprise commonly available tags like biotin, fluorescein isothiocyanate (FITC), or in a directed fashion via genetic fusion of peptide tags.Citation23,Citation25–Citation28 This allows for the creation of CAR-adaptors that vary in size and design, enabling the built-up of a flexible platform.
To date, more than 74 antibodies and Fc fusion proteins have been approved by the FDA for the treatment of diseases.Citation29 In addition, ca. 500 IgGs or antibody fragments are known to be in Phase 1 or 2 clinical studies,Citation29 prospectively providing a large pipeline for potential future CAR-adaptors. Recently, Townsend et al. identified antibodies of which several have shown promising results when converted into CAR formats targeting antigens such as CD38, CD33, CD123 or B cell maturation antigen (BCMA) for hematological malignancies or mucin (MUC1), Glypican-3 (GPC3) or prostate-specific membrane antigen (PSMA) for solid tumors.Citation30 As the availability of newly discovered antibodies and amenable targets expands, modCARs represent an approach to combine the worlds of therapeutic antibodies and CAR-T cells. A comprehensive review of modCAR approaches described in the literature is presented below.
Redirection of modCAR-T cells through small molecule-based CAR-adaptors
The redirection of modCAR-T cells by small molecules has been presented by two groups who made use of the small molecule folate conjugated with FITC as a CAR-adaptor.Citation31,Citation32 Small molecules like folate benefit from advantageous properties, like rapid diffusion out of blood vessels, deep tissue penetration, and the possibility for modifications to obtain the desired biophysical and pharmacokinetic properties while exhibiting short half-lives and rapid elimination.Citation31,Citation33 It has been shown that folate-FITC-based CAR-adaptors function in a highly selective manner and show dose-dependent cytotoxicity on target cells in vitro. Kim et al. demonstrated that the potency of the small molecule-based CAR-adaptors was not impaired by the presence of physiologically relevant folate concentrations. At the same time, the administration of high doses of free folate might be used to attenuate modCAR-T cell activity through competition.Citation31 Chu et al. showed that, besides redirecting modCAR-T cells against non-small-cell lung cancer (NSCLC) cells, it was also possible to target and efficiently lyse tumor-associated macrophages (TAMs) in samples from NSCLC patients in vitro.Citation32 TAMs play an important role in the development and progression of tumors and are often contributed to a tumor-favorable microenvironment.Citation34 Taking advantage of targeting both cancer cells as well as TAMs with one modCAR-T cell approach, it might be feasible to re-evaluate the immunosuppressive tumor microenvironment and achieve a faster clearance of the tumor. Finally, Lee et al. showed that small molecule-based adaptors based on FITC fusions to folate, DUPA, and AZA for the targeting of FOLR1+, PSMA+ or CAIX+ tumors, respectively, with FITC-specific modCAR-T cells, are able to control multiple antigenically different xenograft tumors.Citation35 Notably, they also showed that antigen escape could be prevented through the use of cocktails of bispecific small molecule-based CAR-adaptors.Citation35
Monovalent and bivalent nanobody-based CAR-adaptors
Using a single-domain camelid-derived nanobody-based CAR-adaptor directed against epidermal growth factor receptor (EGFR),Citation36 Albert et al. showed effective retargeting of modCAR-T cells towards EGFR-positive tumor cells both in vitro, as well as in a mouse-tumor xenograft model.Citation37 The modCAR-T cells were engineered to recognize the E5B9-tag,Citation38,Citation39 derived from La/SS-B, a nuclear protein that is not accessible on the surface of intact cells. The tag was fused C-terminal to nanobody-based CAR-adaptors () that had either been expressed in eukaryotic (Chinese hamster ovary) or prokaryotic (E. coli) cells. Interestingly, in this case, the CAR-adaptors produced in the prokaryotic system performed distinctly better in terms of killing than the similar CAR-adaptors produced in the eukaryotic system, which most likely reflects the stronger binding of the former that the authors had assessed by means of flow cytometry. This slight difference between CAR-adaptors being produced in pro- vs. eukaryotic expression systems can, however, not serve as a general rule, but is rather linked to that particular report. The group also addressed the question of bio-distribution of CAR-adaptors in high EGFR tumor-bearing mice monitoring nanobody-based CAR-adaptors labeled with 64Cu, and found enrichment at the tumor site, as well as a fast clearance of the molecules through the kidneys with a half-life of about 4–20 min.Citation37 As a basis for modCAR-T cell therapy, their findings suggested the requirement for continuous infusion of the CAR-adaptor, which would allow for a tight regulation of modCAR-T cell activity in case of undesired side effects.
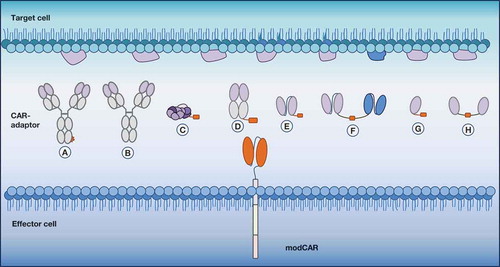
Figure 2. Indirect and flexible antigen targeting through CAR-adaptor molecules (CAR-adaptors). Depicted are the different designs of antigen-targeting CAR-adaptors that are used with a modular CAR (modCAR)-engineered effector cell. (a) IgG-tag-based CAR-adaptor | (b) IgG CAR-adaptor | (c) small molecule-based CAR-adaptor | (d) Fab-based CAR-adaptor | (e) scFv-based CAR-adaptor | (f) bispecific-based CAR-adaptor | (g) nanobody-based CAR-adaptor | (h) bivalent nanobody-based CAR-adaptor.
To further characterize the effectiveness of nanobody-based CAR-adaptors, Albert et al. designed a bivalent CAR-adaptor targeting EGFR. Two α-EGFR nanobodies were fused together via a linker including an E5B9-tag,Citation24 a short peptide-motif of 10 amino acids ((h)). Expressed in the eukaryotic system, the bivalent nanobody-based CAR-adaptors exhibited superior binding avidity on tumor cells, thus not only improving the enrichment of the CAR-adaptors at the tumor sites but also enhancing anti-tumor efficacy. Even in the presence of low-level target antigen, bivalent CAR-adaptors were able to redirect modCAR-T cells and induce killing successfully, which was not the case for monovalent CAR-adaptors. In general, the maximal anti-tumor activity of the bivalent CAR-adaptors was observed at concentrations of 0.05 nM, whereas for the monovalent CAR-adaptor, a 100-fold higher concentration was needed for equivalent efficacy. The clearance of the bivalent-CAR-adaptor occurred through glomerular filtration into the urine, as well as through the hepatobiliary system. As a side note, under the tested circumstances, neither the monovalent nor the bivalent CAR-adaptors interfered with EGFR-mediated signaling.Citation39 The benefit of avidity-gain and the use of CAR-adaptors with low affinity might provide a solution to achieve discrimination between tumor cells with high target expression and normal cells that only express low levels of the target.
scFv-based CAR-adaptors targeting one antigen or two variable antigens simultaneously
In another approach, T cells were equipped with modCARs, designed as above to recognize the E5B9-tag. The E5B9-tag was fused to a series of different scFv-based CAR-adaptors ((e)) targeting the Sialyl-Tn antigen, EGFR, PSMA or prostate stem cell antigen, respectively. All approaches resulted in effective recruitment of modCAR-T cells and potent anti-tumor activity in vitro as well as in vivo.Citation24,Citation40,Citation41 Subsequently, Cartellieri et al. designed a novel bispecific scFv-based CAR-adaptor ((f)), which consists of two different scFvs, one targeting CD123 and the other targeting CD33.Citation39 Both scFvs are connected via a linker-embedded 5B9-tag that can be targeted by modCAR-T cells engineered, respectively.Citation39 Since about 26% of AMLs are negative for either CD123 or CD33, monotherapies cannot cover all AML patients or deal with intratumoral heterogeneity, something that might be overcome by using this bispecific scFv-based CAR-adaptor targeting of both antigens simultaneously.Citation42 Redirected by the designed bispecific scFv-based CAR-adaptor, modCAR-T cells were able to effectively lyse CD33+ and CD123+ AML blasts. Notably, bispecific CAR-adaptors proved to be more efficient compared to the simultaneous application of the monospecific scFv-based CAR-adaptors supplemented at equal molar ratios.Citation39 Such bispecific scFv-based CAR-adaptors can be designed to be more selective for tumor cells expressing both antigens through avidity, thereby curtailing the risk of off-target toxicities. Moreover, bispecific CAR-adaptors of high affinity for both targets are able to redirect modCAR-T cells in the presence of only one displayed antigen, minimizing the chance of tumor escape upon antigen loss under treatment. To secure a safe treatment option and gain full potential of bispecific CAR-adaptors, careful selection of the right combination of tumor antigens also with regard to their expression level on healthy tissue is mandatory.
Fab-based CAR-adaptors effectively redirect modCAR-T cells
Rodgers et al. designed Fab-based CAR-adaptors ((d)) comprising a 14 amino acid peptide tag derived from yeast transcription factor GCN4 to secure the interaction of CAR-adaptors and anti-GCN4 modCAR-T cells.Citation27 Due to the smaller size and shorter half-life of Fabs compared to IgGs, the authors speculated that Fab-based CAR-adaptors may be more effective in tumor penetration and display a lower immunogenicity risk.Citation43 Furthermore, tumor uptake may be hindered as a consequence of the rapid elimination of Fabs from the circulation. However, the short half-life of the Fab-based CAR-adaptors might give an advantage to control modCAR-T cell activation more precisely compared to conventional CAR-T cells.
Rodgers et al. varied the stoichiometry and the location at which the tag was attached to the Fab-based CAR-adaptor. Hence, it was possible to systematically manipulate the orientation of the antigen-binding region of the CAR-adaptor relative to the modCAR-T cell. At all tested sites, the addition of the tag had minimal impact on protein stability, as well as on antigen binding. This finding might be attributed to the very high, single-digit picomolar affinity of the anti-GCN4 ECD for the CAR-adaptor GCN4-tag.Citation27 Dose-dependent cytotoxicity was observed for all tested Fab-based CAR-adaptors, whereby CAR-adaptors with tags proximal to the antigen binding site displayed the highest potency. This approach resulted in specific targeting and killing of CD19+ tumor cells with comparable efficacy to conventional CAR-T cell therapy in an in vivo Nalm-6 xenograft NSG mouse model. In a syngeneic murine model, Viaud et al. investigated modCAR-T cells possessing variable hinge domain or ICDs in combination with Fab-CAR-adaptors possessing the tag N-terminally attached.Citation44 Similar to the human system,Citation27 these Fab-based CAR-adaptors showed superior in vitro cytotoxicity, and, when combined with modCAR-T cells harboring an IgG4m hinge domain, an increased in vivo persistence.Citation44 Similarly to Fab-based CAR-adaptors with GCN4-tag, the study of Ma et al. confirmed the effectiveness of anti-CD19 and anti-CD22 FITC-labeled Fab-based CAR-adaptors as a combinatorial approach, demonstrating that successive targeting of two different antigens with a single modCAR approach enables the possibility to overcome tumor escape variants.Citation25 This approach of using Fab-based CAR-adaptors with engrafted GCN4- or FITC-tags was extended by Cao et al. to solid tumors. The group used trastuzumab Fab-based CAR-adaptors to target Her2+ breast cancer, and demonstrated complete eradication of the tumor in a xenograft NSG mouse model. No tumor relapse was observed until the end of the study (20-day post complete tumor clearance).Citation26
Taken together, for the mentioned approaches, nanobody-, scFv- or Fab-based CAR-adaptors, a short half-life has to be taken into account requiring frequent if not continuous dosing when safety is established.
ModCAR-T cells approaches with substituted ECD
More than two decades ago, Eshhar et al. designed CARs that comprised an scFv as ECD.Citation45 So far, most of the created CARs have followed that design, but some limitations, such as immunogenicity towards xenogenic regions of murine-derived scFv, poor expression or instability, are associated with scFv-ECDs.Citation46 Several approaches have illustrated the possibility to substitute the scFv with other binding moieties like DARPins,Citation21 nanobodies,Citation22 adenectins,Citation47 peptide ligands like T1E or receptor ligands like IL-13-zetakine, NKG2D or CD70.Citation48–Citation51 Preclinical experiments using scFv-substituted alternative CAR-ECDs show encouraging results, and, for example, IL-13zetakine CARs have already been tested in a first-in-human pilot safety and feasibility trial targeting IL13Rα2 for the treatment of recurrent glioblastoma.Citation52
Regarding the substitution of the scFv with an ECD targeted against an epitope suitable for the modCAR approach, ECDs like FcγRIII,Citation53–Citation57 modified avidinCitation58 or leucine zipperCitation59 have been described (). In the following, we highlight modCAR approaches with a focus on modCARs possessing ECDs different than scFv, but still making use of CAR-adaptors to establish T cell activation.
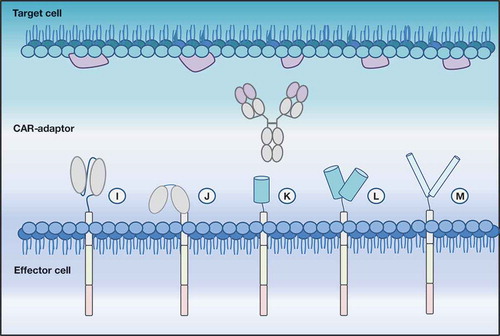
Figure 3. Depicted is a modular CAR (modCAR) engineered effector cell with diverse ECDs able to target a CAR adaptor molecule (CAR-adaptor), here represented by an IgG. (i) scFv-ECD | (j): FcR-ECD | (k) and (l) monomeric and dimeric avidin-ECD require a biotinylated CAR-AM to enable antigen targeting | (m) leucine zipper.
Redirection of modCAR-T cells through antibodies already used in the clinic
Besides using tagged CAR-adaptors, some modCAR approaches use therapeutic IgGs that are already clinically approved. For this purpose, the scFv-ECD of CARs can be substituted by the ECD of FcγRIIIa ((i)). For mAbs like rituximab, trastuzumab or mogamulizumab, it has been shown that the efficacy of cancer treatment can be impeded by chemotherapy-induced leukopenia and exhaustion of natural killer (NK) cells resulting from antibody-dependent cell-mediated cytotoxicity (ADCC).Citation60–Citation62 Several groups tried to overcome this drawback by engineering T cells to express a CAR in which the scFv-ECD is substituted by the ECD of the FcγRIII (CD16), resulting in a so-called “CD16-CAR”. For this approach, the clinically approved mAbs function as CAR-adaptors that can be bound by the high-affinity FcγRIIIa (with a 158V/V polymorphism), leading to T cell activation upon induced crosslinking in the presence of target cells.Citation53–Citation57,Citation63 D’Aloia et al. demonstrated cross-linking of FcγRIII-transduced murine hybridoma T cells leading to IL-2 secretion and FasL-mediated lysis of mAb-opsonized Fas+ tumor cells in vitro.Citation63 Along the same lines, the study conducted by Ochi et al. demonstrated proliferative potential and the differentiation of those FcγRIII modCAR-T cells into effector memory T cells. Furthermore, it has been shown that FcγRIII modCAR-T cells were capable of inhibiting the growth of CD20+ Raji cells in immunodeficient mice when concomitantly infused with rituximab.Citation55
Currently, Unum Therapeutics Inc. is evaluating FcγRIII modCAR-T cells in combination with rituximab to treat relapsed refractory B cell lymphoma (ClinicalTrials.gov Identifier: NCT03189836, NCT02776813), and so far, no serious adverse events related to the modCAR-T cells and no CRS have been observed (ClinicalTrials.gov Identifier. NCT02776813).Citation64 Furthermore, FcγRIII modCAR-T cells are being evaluated in Phase 1 clinical trials in combination with a humanized, afucosylated anti-BCMA antibody for a wide range of BCMA expressing myeloma cells (for example, see ClinicalTrials.gov Identifier: NCT03266692).Citation65 Further, another combination therapy using modCAR-T cells in combination with trastuzumab is currently in preclinical development, with an initiation of a Phase 1 trial in 2019 expected.Citation66
However, unrelated circulating antibodies in the blood are also recognized by the CD16 domain and, in the presence of autoimmune antibodies, could trigger an autoimmune response. Fc-engineered and/or glycoengineered antibodies with an enhanced affinity towards the CD16 modCAR may help to induce a preferential interaction of the CD16-CAR with the therapeutic antibody as CAR-adaptor. Rataj et al. tested activation and killing efficiency of modCAR-T cells in combination with glycoengineered antibodies compared to the respective unmodified antibodies with wildtype glycosylation, including the glycoengineered antibody obinutuzumab, which has about 10-fold enhanced affinity for the FcgRIII ECD.Citation67 Overall, the study showed the benefit of using glycoengineered antibodies in combination with high affinity CD16-modCAR-T cells, and proved that glycoengineering can help to better discriminate between endogenous and therapeutically applied antibodies for enhanced efficacy and safety.Citation57 In an analogous manner, glycoengineering (defucosylation) of mogamulizumab, a humanized anti-CCR4 IgG1 mAb, improved the binding to FcγRIIIa on activated effector NK cells in the presence of low antigen density.Citation68
Taken together, these studies demonstrate that FcγRIII modCAR-T cells are able to reliably induce killing in vitro as well as in vivo, in the presence of therapeutic mAbs directed towards the selected tumor antigens. Maintaining the biological and clinically relevant properties of the administered therapeutic antibodies, FcγRIII modCAR-T cells provide the additional benefit through enhanced ADCC.
Redirection of modCAR-T cells expressing a biotin-binding ECD
Avidin and streptavidin are both naturally occurring biotin-binding proteins with high affinities, but most of the monomeric or dimeric constructs reported so far are unstable, prone to aggregation, and reveal low biotin affinity. Several approaches have been undertaken to design monomeric or dimeric variants of avidin or streptavidin, respectively.Citation69,Citation70 Making use of modified avidin as ECD incorporated into a CAR, Urbanska et al. designed biotin-binding modCAR-T cells. The group showed that only modCAR-T cells expressing dimeric avidin were able to redirect biotinylated mAb- or scFv-CAR-adaptors. ModCAR-T cells expressing monomeric avidin did not show specific retargeting or activation of T cells. Since biotin is present in human plasma, the group demonstrated that, even at supra-physiological levels, soluble biotin did not cause antigen-independent activation of dimeric avidin engineered modCAR-T cells and did not inhibit the modCAR activity by acting as a competitor molecule.Citation23
Another approach making use of biotinylated mAb-CAR-adaptors has been described by Lohmueller et al.Citation58 The group engineered modCAR-T cells expressing an affinity-enhanced monomeric streptavidin 2 (mSA2) as CAR-ECD to bind to biotinylated mAb-CAR-adaptors. mSA2 has been shown to have a high affinity for biotin, comparable to dimeric avidin.Citation69 The authors showed mSA2 modCAR-T cell activation and lysis of target cells in vitro in a dose-dependent manner when using biotinylated rituximab as CAR-adaptor.Citation58 Although these variants of streptavidin and avidin represent proper tools to target biotinylated CAR-adaptors, there is the potential for immunogenicity towards those non-human proteins, which puts the therapeutic applications in human in question.
Redirection of modCAR-T cells expressing a leucine zipper as ECD
Another example of modCAR-T cells, in this case with the ECD of the CAR substituted through a leucine zipper, has been published by Cho et al. In this approach, modCARs are capable of binding to a cognate leucine zipper fused to scFv-based CAR-adaptors (zip-scFv-CAR-adaptor) ((l)). Making use of these modCAR-T cells, several questions could be addressed. Firstly, specific killing of target cells, including solid and hematological tumors (e.g., anti-Her2, anti-Axl, anti-mesothelin), could be demonstrated. Secondly, aiming to counteract potential side effects by preventing unwanted modCAR-T cell activation, Cho et al. developed antigen-targeting scFv-CAR-adaptors with different affinities complementary to zip-scFv-CAR-adaptors. The addition of such complementary CAR-adaptors exhibiting a high affinity towards the actual antigen-targeting CAR-adaptors showed a total impairment of target cell killing. Accordingly, adding competitive CAR-adaptors with weaker affinities led to tuned activation levels of modCAR-T cells. The approach was further developed to control different signaling domains (e.g., CD3ζ, CD28 or 4-1BB) within one modCAR-T cell, enabling independent control of different signaling pathways regulating modCAR-T cell potency. Pairing such modCAR-T cells with different zip-scFv-CAR-adaptors directed against one or multiple tumor antigens, with probably different leucine zipper-based CAR-adaptor affinities, enabled adjustable, combinatorial antigen detection. In an in vivo mouse model, zip-scFv-based CAR-adaptors dosage correlated with cytokine release in a dose-dependent manner, thus offering the possibility of adjusting cytokine release of modCAR-T cells. Furthermore, modCAR-T cell activity could be inhibited in vivo with complementary untargeted CAR-adaptors that could bind to the activating zip-scFv-CAR-adaptors and inactive it, reducing cytokine production to the level of the no-zip-scFv control. The unique feature of this modCAR approach is that the potency of the T cells can be regulated by using CAR-adaptors, not only with different affinities towards the tumor antigen but also towards the modCAR itself. This provides even more flexibility to modulate and adjust the level of modCAR-T cell activation. Taken together, it enables a more precise fine-tuning of the activation of the engineered T cells, thereby mitigating the potential for side effects.Citation59
Outlook and future perspectives
CAR-T cell therapies illustrate the progress being made in the field of cancer immunotherapy. However, conventional CAR-T cell therapy also suffers from drawbacks such as on-target off-tumor toxicity, severe side effects or tumor escape due to antigen loss. Here, we highlighted the development of modCAR-T cell designs that make use of adaptable CAR-adaptors to address some of the mentioned drawbacks. The different modCAR-approaches, with their various designs of modCAR-T cells and the corresponding CAR-adaptors, represent a flexible platform that allows selective and individually adaptable antigen targeting in a potentially safer manner than many conventional CAR-approaches, while preclinical studies have shown that modCAR-T cells and conventional CAR-T cells are able to kill target cells at comparable rates.Citation25,Citation41
The specific activation in the presence of CAR-adaptors only is a major advantage of modCAR-engineered cells over conventional CAR-T cell therapy. The titration of CAR-adaptors introduces the possibility of counteracting potential side effects by customized CAR-adaptor-dosing. Even unbound CAR-adaptors can be captured by administering complex-forming counter-CAR-adaptors that inhibit binding to the modCAR-T cells and prevent their activation.Citation59 This option, which is not feasible in conventional CAR-T approaches, could control CAR-T cell responses and minimize side effects without the loss of engineered T cells.
Interestingly, all of the highlighted modCAR-formats discussed here allow for an efficient formation of the immunological synapse and result in T cell activation (). This demonstrates the flexibility that comes with the modular design and implies that the distance and molecular geometry between modCAR-T-ECD and the target antigen can be more variable than anticipated. Considering this prerequisite in conjunction with the presented proof-of-concept studies, there is room for a virtually unlimited capacity of different combinations of modCARs and respective CAR-adaptors adjustable to individual needs.
While conventional CAR-T approaches often come with fixed affinities and CAR-expression levels that limit their effectivity, modCAR-T cells and the CAR-adaptors allow for the regulation of T-cell responses on mainly two levels. On one level, the activity of modCAR-T cells can be tuned through the affinity of the CAR-adaptor towards the modCAR, while on the other level, tuning can occur through the affinity of the CAR-adaptor to the tumor antigen. Use of CAR-adaptors with different affinities thus offers a degree of flexibility by, for example, allowing for the selection of tumor targets depending on their often heterogeneous expression levels.
Indispensable for the success of such modCAR-T approaches is the further development of CAR-adaptors specifically designed for use in CAR-T cell therapy aiming towards optimized antigen targeting, adapted half-life, as well as ideal recognition through modCARs. Conceptually, CAR-adaptors can be constructed to be multivalent, making it possible to target rather rarely presented antigens by making use of an avidity gain. Furthermore, variable glycosylation patterns improve effector function of IgG-based CAR-adaptors, as recently reviewed by Fonseca et al.Citation71 FcγRIII-based modCAR are the most developed concept that is already being tested in clinical trials and possess great potential to support mAb therapy by improving their clinical efficacy (ClinicalTrials.gov Identifier NCT02776813, NCT03266692).
Circumventing tumor escape and the outgrowth of tumor variants might be addressed by sequential or simultaneous targeting of multiple tumor antigens by using monovalent or multivalent CAR-adaptors. An example of this is the Fc-optimized anti-CD19 MOR208, which demonstrates increased binding to FcγIIIa and has been designed as a therapeutic drug for CLL, NHL and ALL.Citation72 In clinical trials, MOR208 showed promising efficacy in patients progressing with relapsed or refractory NHL that previously had received a rituximab-containing therapy (ClinicalTrials.gov Identifier NCT01685008).Citation72 Using the approach of FcγRIII-based modCAR-T cells, it is theoretically possible to administer rituximab or obinutuzumab as well as MOR208 simultaneously to make use of T cell-mediated killing and thereby prevent tumor escape, thus exploiting the benefits of both worlds.
While the standard procedure is to introduce CARs into a mixture of either isolated T cells or peripheral blood mononuclear cells, recent approaches to introduce CARs into precisely isolated subsets of immune cells such as CD4+, CD8+, regulatory T cells (Treg), or NK cells have been described.Citation59,Citation73,Citation74 Hypothetically, equipping different immune subsets with different receptors (e.g., inhibitory intercellular domains) raises the possibility of precisely silencing subsets of immune cells upon antigen encounter or even regulating different signaling pathways in different subsets of immune cells, allowing the intervention and fine-tuning of the immune response in a more advanced and probably less risky manner.
Currently approved CAR-T cell therapies are based on autologous T cells and must be generated patient-specifically. Dedicated facilities and a well-planned infrastructure are needed to deliver the customized therapy in a timely fashion. The combination of autologous universal CAR-T cells and CAR-adaptors, while providing advantages, for example, in terms of CAR-T cell control, will not necessarily provide benefit from a cost perspective. Generally, the production of clinical-grade CAR-adaptors can be based on established and cost-efficient processes for biologic agents such as therapeutic antibodies; however, the use of advanced and GMP compliant manufacturing facilities is required. While the cost of manufacturing biologics that serve as adaptors has decreased considerably during the past years, the combination of adoptive T cell therapy and CAR-adaptors will primarily be of interest when combined with allogenic universal CAR-T cells. This may allow the straightforward use of one single universal allogenic CAR-T cell line with diverse CAR-adaptors, for example, targeting different tumor targets, which in this case may decrease the burden of the production of diverse cell products. Recently, a novel promising approach to generate “off the shelf”, gene-modified cell products by knocking out MHCI and II and introducing CD47 has been described.Citation75 Thus, introducing modCARs into allogenic immune cells would enable the creation of a platform with high versatility. Such modCAR cells might persist in patients and could be reactivated if needed. In the future, it might be possible to combine allogenic modCAR-T cells from healthy donors with CAR-adaptors to guarantee an “off the shelf” approach. Taking allogenic modCAR-T cells and CAR-adaptor from storage would probably decrease the costs of CAR-T cell therapies. Individualized manufacturing processes, including safety testing and logistics, could be reduced drastically, and a readily available therapy for patients in urgent need would be possible. Depending on the application, the combination of different biologics might not only be restricted to cancer but could also function to address other diseases such as autoimmunity, infection or inflammation, paving the way towards highly personalized-treatment options.
In summary, modCAR-T cell approaches have the potential to advance the field of CAR-T therapy in the near future. The possibility of controlling the formation of the immunological synapse by simple titration of CAR-adaptors allows for rapid optimization of effector-cell-activity. Compared to conventional CAR-T approaches, modCAR-T cells possess improved safety without compromising the efficacy of CAR-T activity, allowing advanced treatment options and intelligent antigen targeting.
Disclosure of Potential Conflicts of Interest
DD, CJ, and CK declare employment and patents with Roche, CK declares ownership of Roche stock.
Additional information
Funding
References
- US Food and Drug Administration. KYMRIAH (tisagenlecleucel); 2017 Aug 30 and 2018 May 1. https://www.fda.gov/biologicsbloodvaccines/cellulargenetherapyproducts/approvedproducts/ucm573706.htm.
- US Food and Drug Administration. YESCARTA (axicabtagene ciloleucel); 2017 Oct 18. https://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/ucm581222.htm.
- First-ever CAR T-cell therapy approved in U.S. Cancer Discov. 2017 [cited 2018 Jul 28]accessed;7:OF1. http://cancerdiscovery.aacrjournals.org/content/7/10/OF1.
- Salmikangas P, Kinsella N, Chamberlain P. Chimeric antigen receptor T-cells (CAR T-cells) for cancer immunotherapy – moving target for industry? Pharm Res. 2018;35:152. doi:10.1007/s11095-018-2436-z.
- Zheng PP, Kros JM, Li J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov Today. 2018;23:1175–82. doi:10.1016/j.drudis.2018.02.012.
- Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, Mackall CL. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–95. doi:10.1182/blood-2014-05-552729.
- June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–65. doi:10.1126/science.aar6711.
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. doi:10.1038/mt.2010.24.
- Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol. 2013;31:999–1008. doi:10.1038/nbt.2725.
- Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber D, Schneck J, Gabrilovich D. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13:828–35. doi:10.1038/nm1609.
- Jackson HJ, Brentjens RJ. Overcoming antigen escape with CAR T-cell therapy. Cancer Discov. 2015;5:1238–40. doi:10.1158/2159-8290.CD-15-1275.
- Fisher J, Abramowski P, Wisidagamage Don ND, Flutter B, Capsomidis A, Cheung GW, Gustafsson K, Anderson J. Avoidance of on-target off-tumor activation using a co-stimulation-only chimeric antigen receptor. Mol Ther. 2017;25:1234–47. doi:10.1016/j.ymthe.2017.03.002.
- Thomis DC, Marktel S, Bonini C, Traversari C, Gilman M, Bordignon C, Clackson T. A Fas-based suicide switch in human T cells for the treatment of graft-versus-host disease. Blood. 2001;97:1249–57.
- Tey SK, Dotti G, Rooney CM, Heslop HE, Brenner MK. Inducible caspase 9 suicide gene to improve the safety of allodepleted T cells after haploidentical stem cell transplantation. Biol Blood Marrow Transplant. 2007;13:913–24. doi:10.1016/j.bbmt.2007.04.005.
- Maus MV, June CH. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin Cancer Res. 2016;22:1875–84. doi:10.1158/1078-0432.CCR-15-1433.
- Chu F, Cao J, Neelalpu SS. Versatile CAR T-cells for cancer immunotherapy. Contemp Oncol (Pozn). 2018;22:73–80. doi:10.5114/wo.2018.73892.
- Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer. 2019 Jan;120(1):26–37. doi:10.1038/s41416-018-0325-1.
- Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4:498–508. doi:10.1158/2326-6066.CIR-15-0231.
- Martyniszyn A, Krahl AC, Andre MC, Hombach AA, Abken H. CD20-CD19 bispecific CAR T cells for the treatment of B-cell malignancies. Hum Gene Ther. 2017;28:1147–57. doi:10.1089/hum.2017.126.
- Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, Wolters P, Martin S, Delbrook C, Yates B, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24:20–28. doi:10.1038/nm.4441.
- Hammill JA, VanSeggelen H, Helsen CW, Denisova GF, Evelegh C, Tantalo DG, Bassett JD, Bramson JL. Designed ankyrin repeat proteins are effective targeting elements for chimeric antigen receptors. J Immunother Cancer. 2015;3:55. doi:10.1186/s40425-015-0099-4.
- De Munter S, Ingels J, Goetgeluk G, Bonte S, Pille M, Weening K, Kerre T, Abken H, Vandekerckhove B. Nanobody based dual specific CARs. Int J Mol Sci. 2018;19:403. doi:10.3390/ijms19020403.
- Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, Powell DJ. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012;72:1844–52. doi:10.1158/0008-5472.CAN-11-3890.
- Albert S, Arndt C, Koristka S, Berndt N, Bergmann R, Feldmann A, Schmitz M, Pietzsch J, Steinbach J, Bachmann M. From mono- to bivalent: improving theranostic properties of target modules for redirection of UniCAR T cells against EGFR-expressing tumor cells in vitro and in vivo. Oncotarget. 2018;9:25597–616. doi:10.18632/oncotarget.25390.
- Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB, et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci U S A. 2016;113:E450–8. doi:10.1073/pnas.1524193113.
- Cao Y, Rodgers DT, Du J, Ahmad I, Hampton EN, Ma JS, Mazagova M, Choi S-H, Yun HY, Xiao H, et al. Design of switchable chimeric antigen receptor T cells targeting breast cancer. Angew Chem Int Ed Engl. 2016;55:7520–24. doi:10.1002/anie.201601902.
- Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A. 2016;113:E459–68. doi:10.1073/pnas.1524155113.
- Hermanson GT. Bioconjugate Techniques. Copyright © 2013, 1996, 2008. Elsevier Inc. All rights reserved; 2013.
- Strohl WR. Current progress in innovative engineered antibodies. Protein Cell. 2018;9:86–120. doi:10.1007/s13238-017-0457-8.
- Townsend MH, Shrestha G, Robison RA, O’Neill KL. The expansion of targetable biomarkers for CAR T cell therapy. J Exp Clin Cancer Res. 2018;37:163.
- Kim MS, Ma JS, Yun H, Cao Y, Kim JY, Chi V, Wang D, Woods A, Sherwood L, Caballero D, et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J Am Chem Soc. 2015;137:2832–35. doi:10.1021/jacs.5b00106.
- Chu W, Zhou Y, Tang Q, Wang M, Ji Y, Yan J, Yin D, Zhang S, Lu H, Shen J. Bi-specific ligand-controlled chimeric antigen receptor T-cell therapy for non-small cell lung cancer. Biosci Trends. 2018;12:298–308. doi:10.5582/bst.2018.01048.
- Krall N, Scheuermann J, Neri D. Small targeted cytotoxics: current state and promises from DNA-encoded chemical libraries. Angew Chem Int Ed Engl. 2013;52:1384–402. doi:10.1002/anie.201204631.
- Wang R, Zhang J, Chen S, Lu M, Luo X, Yao S, Liu S, Qin Y, Chen H. Tumor-associated macrophages provide a suitable microenvironment for non-small lung cancer invasion and progression. Lung Cancer. 2011;74:188–96. doi:10.1016/j.lungcan.2011.04.009.
- Lee YG, Marks I, Srinivasarao M, Kanduluru AK, Mahalingam SM, Liu X, Chu H, Low PS. Use of a single CAR T cell and several bispecific adapters facilitates eradication of multiple antigenically different solid tumors. Cancer Res. 2019;79:387–96. doi:10.1158/0008-5472.CAN-18-1834.
- Gainkam LO, Huang L, Caveliers V, Keyaerts M, Hernot S, Vaneycken I, Vanhove C, Revets H, De Baetselier P, Lahoutte T. Comparison of the biodistribution and tumor targeting of two 99mTc-labeled anti-EGFR nanobodies in mice, using pinhole SPECT/micro-CT. J Nucl Med. 2008;49:788–95. doi:10.2967/jnumed.107.048538.
- Albert S, Arndt C, Feldmann A, Bergmann R, Bachmann D, Koristka S, Ludwig F, Ziller-Walter P, Kegler A, Gärtner S, et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology. 2017;6:e1287246. doi:10.1080/2162402X.2017.1287246.
- Koristka S, Cartellieri M, Arndt C, Bippes CC, Feldmann A, Michalk I, Wiefel K, Stamova S, Schmitz M, Ehninger G, et al. Retargeting of regulatory T cells to surface-inducible autoantigen La/SS-B. J Autoimmun. 2013;42:105–16. doi:10.1016/j.jaut.2013.01.002.
- Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, von Bonin M, Bejestani EP, Ehninger G, Bachmann MP. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016;6:e458. doi:10.1038/bcj.2016.61.
- Loureiro LR, Feldmann A, Bergmann R, Koristka S, Berndt N, Arndt C, Pietzsch J, Novo C, Videira P, Bachmann M. Development of a novel target module redirecting UniCAR T cells to Sialyl Tn-expressing tumor cells. Blood Cancer J. 2018;8:81. doi:10.1038/s41408-018-0113-4.
- Feldmann A, Arndt C, Bergmann R, Loff S, Cartellieri M, Bachmann D, Aliperta R, Hetzenecker M, Ludwig F, Albert S, et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget. 2017;8:31368–85. doi:10.18632/oncotarget.15572.
- Ehninger A, Kramer M, Rollig C, Thiede C, Bornhauser M, von Bonin M, Wermke M, Feldmann A, Bachmann M, Ehninger G, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4:e218. doi:10.1038/bcj.2014.39.
- Herrington-Symes AP, Farys M, Khalili H, Brocchini S. Antibody fragments: prolonging circulation half-life special issue-antibody research. Adv Biosci Biotechnol. 2013;04:689–98. doi:10.4236/abb.2013.45090.
- Viaud S, Ma JSY, Hardy IR, Hampton EN, Benish B, Sherwood L, Nunez V, Ackerman CJ, Khialeeva E, Weglarz M, et al. Switchable control over in vivo CAR T expansion, B cell depletion, and induction of memory. Proc Natl Acad Sci U S A. 2018;115:E10898–E906. doi:10.1073/pnas.1810060115.
- Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720–24.
- Kong S, Sengupta S, Tyler B, Bais AJ, Ma Q, Doucette S, Zhou J, Sahin A, Carter BS, Brem H, et al. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin Cancer Res. 2012;18:5949–60. doi:10.1158/1078-0432.CCR-12-0319.
- Han X, Cinay GE, Zhao Y, Guo Y, Zhang X, Wang P. Adnectin-based design of chimeric antigen receptor for T cell engineering. Mol Ther. 2017;25:2466–76. doi:10.1016/j.ymthe.2017.07.009.
- Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64:9160–66. doi:10.1158/0008-5472.CAN-04-0454.
- Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D’Apuzzo M, Barish ME, Forman SJ, Jensen MC. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin Cancer Res. 2012;18:2199–209. doi:10.1158/1078-0432.CCR-11-1669.
- Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood. 2005;106:1544–51. doi:10.1182/blood-2004-11-4365.
- Shaffer DR, Savoldo B, Yi Z, Chow KK, Kakarla S, Spencer DM, Dotti G, Wu M-F, Liu H, Kenney S, et al. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood. 2011;117:4304–14. doi:10.1182/blood-2010-04-278218.
- Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, Naranjo A, Starr R, Wagner J, Wright C, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21:4062–72. doi:10.1158/1078-0432.CCR-15-0428.
- Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, Chng WJ, Campana D. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014;74:93–103. doi:10.1158/0008-5472.CAN-13-1365.
- Clémenceau B, Congy-Jolivet N, Gallot G, Vivien R, Gaschet J, Thibault G, Vié H. Antibody-dependent cellular cytotoxicity (ADCC) is mediated by genetically modified antigen-specific human T lymphocytes. Blood. 2006;107:4669–77. doi:10.1182/blood-2005-09-3775.
- Ochi F, Fujiwara H, Tanimoto K, Asai H, Miyazaki Y, Okamoto S, Mineno J, Kuzushima K, Shiku H, Barrett J, et al. Gene-modified human alpha/beta-T cells expressing a chimeric CD16-CD3zeta receptor as adoptively transferable effector cells for anticancer monoclonal antibody therapy. Cancer Immunol Res. 2014;2:249–62. doi:10.1158/2326-6066.CIR-13-0099-T.
- Tanaka H, Fujiwara H, Ochi F, Tanimoto K, Casey N, Okamoto S, Mineno J, Kuzushima K, Shiku H, Sugiyama T, et al. Development of engineered T cells expressing a chimeric CD16-CD3zeta receptor to improve the clinical efficacy of mogamulizumab therapy against adult T-cell leukemia. Clin Cancer Res. 2016;22:4405–16. doi:10.1158/1078-0432.CCR-15-2714.
- Rataj F, Jacobi SJ, Stoiber S, Asang F, Ogonek J, Tokarew N, Cadilha BL, van Puijenbroek E, Heise C, Duewell P, et al. High-affinity CD16-polymorphism and Fc-engineered antibodies enable activity of CD16-chimeric antigen receptor-modified T cells for cancer therapy. Br J Cancer. 2019 Jan;120(1):79–87. doi:10.1038/s41416-018-0341-1.
- Lohmueller JJ, Ham JD, Kvorjak M, Finn OJ. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology. 2017;7:e1368604. doi:10.1080/2162402X.2017.1368604.
- Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173:1426–1438. doi:10.1016/j.cell.2018.03.038.
- Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity. 2006;24:19–28. doi:10.1016/j.immuni.2005.11.010.
- Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–27. doi:10.1038/nri2744.
- Kohrt HE, Houot R, Marabelle A, Cho HJ, Osman K, Goldstein M, Levy R, Brody J. Combination strategies to enhance antitumor ADCC. Immunotherapy. 2012;4:511–27. doi:10.2217/imt.12.38.
- D’Aloia MM, Caratelli S, Palumbo C, Battella S, Arriga R, Lauro D, Palmieri G, Sconocchia G, Alimandi M. T lymphocytes engineered to express a CD16-chimeric antigen receptor redirect T-cell immune responses against immunoglobulin G-opsonized target cells. Cytotherapy. 2016;18:278–90. doi:10.1016/j.jcyt.2015.10.014.
- Michaels J. Unum Therapeutics Announces Presentation of Initial Clinical Data from the ATTCK-20-2 Trial and Pre-Clinical Data on its Antibody-Coupled T Cell Receptor (ACTR) Platform at the Upcoming 59th American Society of Hematology (ASH) Annual Meeting. BUSINESS WIRE, November 01, 2017 Report about oral presentation from Presentation Title: ACTR087, Autologous T Lymphocytes Expressing Antibody Coupled T-cell Receptors (ACTR), Induces Complete Responses in Patients with Relapsed or Refractory CD20-Positive B-cell Lymphoma, in Combination with Rituximab Authors (affiliation): L. Akard (IBMT), S. Jaglowski (OSU), S. Devine (OSU), M. McKinney (Duke), M. Vasconcelles, H. Huet, S. Ettenberg, A. Ranger, J. Abramson (MGH). https://www.businesswire.com/news/home/20171101005626/en/Unum-Therapeutics-Announces-Presentation-Initial-Clinical-Data.
- Cheema T, Hickman T, O’Callaghan K, Westendorf L, Manlove L, Gardai S, Nelson A, Boomer R, McGinness K, Schultes B, et al. Abstract 4605: efficient targeting of BCMA-positive multiple myeloma cells by antibody-coupled T-cell receptor (ACTR) engineered autologous T cells in combination with an anti-BCMA antibody. Cancer Res. 2017;77:4605. doi:10.1158/1538-7445.AM2017-4605.
- Inc. UT. Unum Therapeutics Inc.; 2018. https://www.unumrx.com/pipeline.
- Mossner E, Brunker P, Moser S, Puntener U, Schmidt C, Herter S, Grau R, Gerdes C, Nopora A, van Puijenbroek E, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115:4393–402. doi:10.1182/blood-2009-06-225979.
- Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, Uchida K, Anazawa H, Satoh M, Yamasaki M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278:3466–73. doi:10.1074/jbc.M210665200.
- Lim KH, Huang H, Pralle A, Park S. Stable, high-affinity streptavidin monomer for protein labeling and monovalent biotin detection. Biotechnol Bioeng. 2013;110:57–67. doi:10.1002/bit.24605.
- Qureshi MH, Yeung JC, Wu SC, Wong SL. Development and characterization of a series of soluble tetrameric and monomeric streptavidin muteins with differential biotin binding affinities. J Biol Chem. 2001;276:46422–28. doi:10.1074/jbc.M107398200.
- Fonseca MHG, Furtado GP, Bezerra MRL, Pontes LQ, Fernandes CFC. Boosting half-life and effector functions of therapeutic antibodies by Fc-engineering: an interaction-function review. Int J Biol Macromol. 2018;119:306–11. doi:10.1016/j.ijbiomac.2018.07.141.
- Jurczak W, Zinzani PL, Gaidano G, Goy A, Provencio M, Nagy Z, Robak T, Maddocks K, Buske C, Ambarkhane S, et al. Single-agent MOR208 in relapsed or refractory (R-R) Non-Hodgkin’s Lymphoma (NHL): results from Diffuse Large B-Cell Lymphoma (DLBCL) and indolent NHL subgroups of a Phase IIa Study. Blood. 2016;128:623. doi:10.1182/blood-2016-06-724161.
- Glienke W, Esser R, Priesner C, Suerth JD, Schambach A, Wels WS, Grez M, Kloess S, Arseniev L, Koehl U. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol. 2015;6:21. doi:10.3389/fphar.2015.00021.
- MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, Broady R, Levings MK. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. 2016;126:1413–24. doi:10.1172/JCI82771.
- Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, Thayer WO, Wahl A, Garcia JV, Reichenspurner H, et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat Biotechnol. 2019;37:252–58. doi:10.1038/s41587-019-0016-3.
- Cartellieri M, Koristka S, Arndt C, Feldmann A, Stamova S, von Bonin M, Töpfer K, Krüger T, Geib M, Michalk I, et al. A novel ex vivo isolation and expansion procedure for chimeric antigen receptor engrafted human T cells. PLoS One. 2014;9:e93745. doi:10.1371/journal.pone.0093745.
- Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, Davila E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. 2012;18:6436–45. doi:10.1158/1078-0432.CCR-12-1449.
- van Dam GM, Themelis G, Crane LM, Harlaar NJ, Pleijhuis RG, Kelder W, Sarantopoulos A, de Jong JS, Arts HJG, van der Zee AGJ, et al. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-alpha targeting: first in-human results. Nat Med. 2011;17:1315–19. doi:10.1038/nm.2472.