
ABSTRACT
The development of alternative therapeutic strategies to tumor necrosis factor (TNF)-blocking antibodies for the treatment of inflammatory diseases has generated increasing interest. In particular, selective inhibition of TNF receptor 1 (TNFR1) promises a more precise intervention, tackling only the pro-inflammatory responses mediated by TNF while leaving regenerative and pro-survival signals transduced by TNFR2 untouched. We recently generated a monovalent anti-TNFR1 antibody fragment (Fab 13.7) as an efficient inhibitor of TNFR1. To improve the pharmacokinetic properties of Fab 13.7, the variable domains of the heavy and light chains were fused to the N-termini of newly generated heterodimerizing Fc chains. This novel Fc heterodimerization technology, designated “Fc-one/kappa” (Fc1κ) is based on interspersed constant Ig domains substituting the CH3 domains of a γ1 Fc. The interspersed immunoglobulin (Ig) domains originate from the per se heterodimerizing constant CH1 and CLκ domains and contain sequence stretches of an IgG1 CH3 domain, destined to enable interaction with the neonatal Fc receptor, and thus promote extended serum half-life. The resulting monovalent Fv-Fc1κ fusion protein (Atrosimab) retained strong binding to TNFR1 as determined by enzyme-linked immunosorbent assay and quartz crystal microbalance, and potently inhibited TNF-induced activation of TNFR1. Atrosimab lacks agonistic activity for TNFR1 on its own and in the presence of anti-human IgG antibodies and displays clearly improved pharmacokinetic properties.
Introduction
Tumor necrosis factor (TNF) plays a central role in the immune system, especially during inflammation and infection. TNF activates the immune system and is mainly expressed by macrophages, triggered, for example, by lipopolysaccharides (LPS) from microbial sources.Citation1–Citation5 However, prolonged exposure to endogenous levels of TNF during inflammatory reactions reduces the responsiveness of immune cells and thereby attenuates the immune system.Citation6–Citation8 Thus, TNF is an important regulator of the immune system that requires strict temporal and spatial regulation in order to maintain immunological responsiveness and to prevent autoimmunity. Dysregulated expression of TNF is associated with severe inflammatory conditions and has been reported to be involved in the development of diseases such as systemic lupus erythematosus,Citation9 type II diabetes,Citation10,Citation11 Crohn’s diseaseCitation12 and rheumatoid arthritis (RA). TNF overexpression in RA was demonstrated to induce the expression of further pro-inflammatory cytokines, to promote vascularization and to support the influx of immune cells, resulting in manifestation of chronic inflammation and tissue damage.Citation13–Citation16
Five TNF-targeting therapeutics (infliximab, adalimumab, certolizumab pegol, golimumab, etanercept) are presently approved and used for the treatment of different inflammatory diseases, resulting in dramatic improvement of the patients’ quality of life, for example, as assessed for inflammatory bowel disease.Citation17 However, the undisputable clinical success of anti-TNF therapeutics has limitations. Depending on the applied anti-TNF agent, 13–33% of treated patients do not respond to treatment and up 46% stop to respond to therapy, resulting in either discontinuation or dose increase.Citation18 Moreover, anti-TNF therapy can be associated with severe side-effects, including opportunistic infections, tuberculosis reactivation, development of malignancies (e.g., lymphomas) and neurological conditions (demyelinating disease), and the exacerbation of existing inflammatory symptoms (for reviews, see refs.Citation19,Citation20). Some side effects might be due to sustained and long-lasting blockade of TNF-mediated activation of its two distinct TNF receptors (TNFR1 and TNFR2), which have different or even opposing functions.Citation21,Citation22 While TNFR1 is mainly responsible for the induction of pro-inflammatory or, under certain conditions, apoptosis-inducing signals, TNFR2 is involved in cellular regeneration, tissue homeostasis, and immune attenuation.Citation23 Therefore, selective inhibition of TNFR1 under inflammatory conditions represents a more specific intervention in TNF biology and promises reduced side effects due to unaffected TNFR2 signaling.Citation20,Citation24–Citation26
In preclinical disease models, TNFR1-blocking agents have already proved to be effective. For example, a commercially available anti-mouse TNFR1 antibody reduced experimental autoimmune encephalomyelitis (EAE) symptomsCitation27 and a chemically modified TNFR1-specific TNF mutant (PEG-R1ant-TNF) showed therapeutic efficacy in murine models of hepatitis, collagen-induced arthritis, arterial inflammation and intimal hyperplasia.Citation28–Citation30 Furthermore, a domain antibody (dAb) directed against TNFR1 (GSK1995057) attenuated disease severity in a murine model of acute respiratory distress syndrome,Citation31 and a fusion protein composed of three different dAbs, two directed against different epitopes on TNFR1 and one dAb directed against serum albumin (intended to extend the serum circulation of the therapeutic), delayed the onset of EAE and reduced typical disease symptoms.Citation32 Importantly, cross-linking of TNFR1, which occurs, for example, by bivalent binding of the molecules or by secondary events like interactions with Fcγ receptors or anti-drug antibodies (ADA), must be avoided to maintain the antagonistic nature of the presently available TNFR1 inhibitors.
We recently converted a humanized anti-TNFR1 IgG1 antibody (Atrosab) into an affinity and stability improved monovalent antigen-binding fragment (Fab 13.7).Citation33 Fab13.7 efficiently inhibited TNF-mediated activation of TNFR1. Moreover, this Fab completely lacked intrinsic agonistic activity, which was observed to a limited extent for the IgG Atrosab at a narrow concentration range due to its bivalent molecular structure. In order to improve the half-life of the Fab, various half-life extension strategies were applied, including the fusion of the Fv domains to a knobs-into-holes Fc region.Citation33
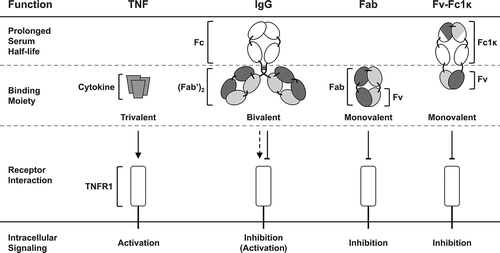
In this study, we applied a newly developed Fc heterodimerization strategy to generate a novel monovalent Fv-Fc fusion protein. The novel Fc heterodimerization technology, designated “Fc-one/kappa” (Fc1κ) is based on interspersed immunoglobulin (Ig) domains, derived from the per se heterodimerizing constant Ig domains CH1 and CLκ, containing CH3 sequence sections, intended to mediate neonatal Fc receptor (FcRn) binding. The concept of the monovalent, antibody-derived Fv-Fc1κ fusion protein and its molecular composition compared to the natural TNFR1 ligand TNF, bivalent IgG and the monovalent Fab is depicted in , highlighting the benefits of monovalency in combination with prolonged serum half-life.
Figure 1. Inhibition and activation of TNFR1. TNFR1 is strongly activated by its natural, trimeric ligand TNF. The bivalent IgG ATROSAB was shown to exert a dominant antagonistic activity in the presence of TNF, yet on its own exerts a marginal TNFR1 activation in a narrow dose range. Monovalent formats like the Fab or the newly developed Fv-Fc1k are effective antagonists of TNF mediated TNFR1 activation and lack intrinsic agonistic activity. In addition, the Fv-Fc1k comprises an Fc proportion, providing a prolonged serum half-life, comparable to an IgG.
Results
Development of a novel heterodimerization module
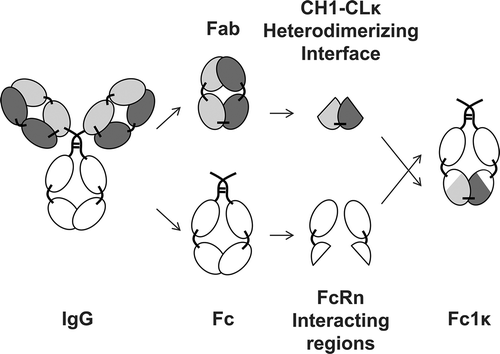
Novel heterodimerizing Ig domains were generated based on the first constant domain of the IgG1 heavy chain (CH1) and the constant domain of the κ light chain (CLκ, ). In order to enable binding to FcRn, amino acid sequence stretches of the third constant domain of the IgG1 heavy chain (CH3) that do not interfere with the CH3-CH3 interdomain interaction were transferred to the CH1 and CLκ domains in silico. This process resulted in two novel interspersed Ig domains. The first domain, CH31 contains amino acid sequence fragments of CH1 and CH3 and the second domain, CH3kappa (CH3κ) contains amino acid sequence fragments of CLκ and CH3. Furthermore, IgG1 CH2 domains were fused to the N-termini of CH31 and CH3κ, in order to provide the entire FcRn binding region of the IgG molecule.Citation34 Finally, by the addition of IgG1 hinge region to the N-termini of the CH2 domains, we generated a novel covalently linked heterodimerizing Fc moiety, designated Fc-one/kappa (Fc1κ, ).
Figure 2. Generation of a heterodimerizing Fc with FcRn-interacting regions. A novel heterodimerizing Fc was generated by replacing the IgG1 CH3 domains with interspersed constant Ig domains, based on the per se heterodimerizing constant Ig domains CH1 and CLκ, complemented with elements of IgG1 CH3 domains, that are responsible for the interaction with the FcRn. The newly generated heterodimerizing Fc was designated Fc-one/kappa (Fc1k).
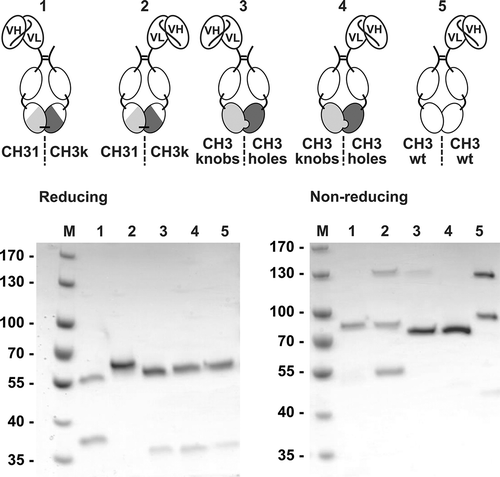
In order to evaluate the degree of heterodimerization, mediated by the newly generated Fc1κ chains, asymmetric single-chain variable fragment (scFv)-Fc1κ fusion proteins were generated by fusing an scFv moiety to the N-termini of either the CH31- or CH3κ-containing chains of Fc1k (). Moreover, similar molecules were cloned and produced, containing the state-of-the-art heterodimerizing “knobs-into-holes” Fc,Citation35 wherein amino acid residues of the CH3–CH3 interface were exchanged by amino acids with larger (“knobs”-chain) or smaller side chains (“holes”-chain). Finally, a construct containing a wild-type CH3 on both chains was included in the set of analyzed molecules. All proteins (see ) were produced in transiently transfected HEK293E cells and purified by protein A affinity chromatography. SDS-PAGE analysis under reducing conditions demonstrated successful expression of both chains of the heterodimeric fusion proteins, except for the Fc1κ fusion protein carrying the scFv at the CH3κ-containing chain, which expressed the scFv-CH3κ chain, but expression of the CH2-CH31 chain could not be detected. Under non-reducing conditions, the scFv-Fc1κ fusion protein containing the scFv moiety at the CH31 chain of the Fc1κ region and both proteins based on the knobs-into-holes technology revealed one clearly dominating band (89 kDa for scFv-CH31-CH3κ; 83 kDa for the two knobs-into-holes fusion proteins), demonstrating correct formation of the heterodimeric proteins with an expected molecular mass of 76 kDa and 77 kDa, respectively. Interestingly, the knobs-into-holes-based fusion protein, carrying the scFv moiety at the “knobs” chain showed an additional faint band around 130 kDa, indicating partial homodimerization of the heavy chain (scFv-Fcknobs). Furthermore, the Fc1κ fusion protein carrying the scFv at the CH3κ-containing chain, showed three bands, indicating the presence of covalently and non-covalently linked homodimers, as well as heterodimers, in the preparation. This observation indicates a residual propensity of the CH3κ-containing chain to form homodimers. Similarly, the fusion protein containing the wild-type Fc also showed three bands, indicating the formation of light chain and heavy chain homodimers and light chain/heavy chain heterodimers. Taken together, the generated interspersed Ig domains CH31 and CH3κ mediated heterodimerization of Fc chains in a configuration containing an scFv moiety connected to the N-terminus of the CH31-containing Fc chain in a manner that was similar or even superior to the established knobs-into-holes technology.
Figure 3. Proof of concept of Fc1k using asymmetric scFv-Fc1k fusion proteins. An scFv fragment was fused to the N-terminus of the hinge region of an Fc chain, containing either the newly generated CH31 or CH3κ domains, the heterodimerizing CH3knobs or CH3holes domains, or the wild type CH3 domains. Proteins were analyzed by SDS-PAGE (NuPAGETM 4–12% Bis-TRIS Midi Gel). Descriptions: M (Marker), 1: (scFv connected to Hinge-CH2-CH31); 2: (scFv connected to Hinge-CH2-CH3κ); 3: (scFv connected to Hinge-CH2-CH3knobs); 4: (scFv connected to Hinge-CH2-CH3holes); 5: (scFv connected to Hinge-CH2-CH3wt). Staining: Coomassie Brilliant Blue, de-staining: Water.
Generation of a monovalent TNFR1-specific antagonist
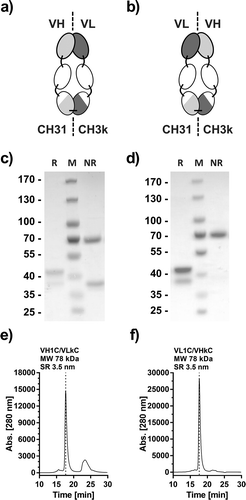
The variable domains of the recently developed TNFR1-specific Fab 13.7Citation33 were fused to the CH2 domain N-termini of the CH31- or CH3κ-containing Fc chains via a short peptide linker, either fusing the VH to the CH2-CH31 chain and VL to the CH2-CH3κ chain (VH13.7-CH2-CH31 and VL13.7-CH2-CH2k; VH1C/VLkC, )), or fusing VH to the CH-2CH3κ chain and VL to the CH2-CH31 chain (VL13.7-CH2-CH31 and VH13.7-CH2-CH3κ; VL1C/VHκC; )). After production in HEK293E cells by transient transfection and protein A affinity chromatography purification, both molecules revealed two bands of 38 kDa and 43 kDa in SDS-PAGE under reducing conditions (, d)). Moreover, under non-reducing conditions both proteins showed a band of 70 kDa, indicating correct assembly and covalent connection of the dimeric protein. However, in the VH1C/VLkC configuration, there was an additional band visible at a size of 38 kDa under non-reducing conditions, indicating the presence of non-covalently linked or monomeric chains ()). Finally, VH1C/VLkC and VL1C/VHκC both showed one major peak in size-exclusion chromatography (SEC) with an apparent molecular mass of 78 kDa (calculated 72 kDa) and a Stokes radius of 3.5 nm, corresponding to the assembled heterodimeric protein (,f)). Consistent with the SDS-PAGE analysis, the molecule with the VH1C/VLkC configuration showed additional peaks, confirming the existence of free unligated monomeric chains. A small fraction of aggregated or multimerized protein could also be discerned. In conclusion, the VL1C/VHκC configuration was selected for further development steps of this improved TNFR1-specific antagonistic antibody-derived molecule, which will be referred to below as Atrosimab.
Figure 4. Generation and optimization of Atrosimab. Two versions of a novel Fv-Fc1k fusion protein were generated, (a) composed of the chains VH-CH2-CH31 (VH1C)/VL-CH2-CH3κ (VLkC) or (b) composed of VL-CH2-CH31 (VL1C)/VH-CH2-CH3κ (VHkC). Both molecules were compared in SDS-PAGE (c and d, NuPAGETM 4–12% Bis-TRIS Midi Gel) under reducing (R) and non-reducing conditions (NR) and SEC (e and f, Phenomenex Yarra SEC-2000, 300 × 7.8 mm, flow rate of 0.5 ml/min, mobile phase Na2HPO4/NaH2PO4).
Biochemical characterization of Atrosimab
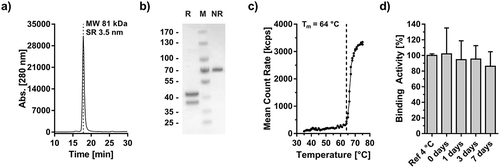
Atrosimab was produced in Chinese hamster ovary (CHO) cells after lentiviral transfection and purified by protein A affinity chromatography. The product was further purified by preparative SEC in order to remove oligomeric protein species. The final preparation revealed a single peak in SEC analysis with an apparent molecular mass of 81 kDa (72 kDa calculated) and a Stokes radius of 3.5 nm ()). Two bands in SDS-PAGE under reducing conditions of 38 and 43 kDa and one band under non-reducing conditions of ~70 kDa confirmed purity and correct assembly of Atrosimab ()). Mass spectrum analysis of deglycosylated Atrosimab further substantiated the correct formation of the heterodimer in the presence of negligible amounts of homodimerically assembled protein species (Fig. S1). Moreover, an aggregation point (melting temperature) of 64°C was determined by dynamic light scattering ()). Finally, Atrosimab maintained full binding activity after incubation in human plasma for up to seven days, indicating good plasma stability for the analyzed period of time ()).
Figure 5. Biochemical characterization of Atrosimab. Atrosimab was produced by Catalent (Catalent Pharma Solutions, Somerset, Ewing, NJ, US) from a CHO cell pool after stable lentiviral transduction and purified by protein A chromatography and subsequent gel filtration (performed at the University of Stuttgart). Characterization was performed by analytical SEC (a, TSKgel SuperSW mAb HR, Flow rate 0.5 ml/min, mobile phase Na2HPO4/NaH2PO4) and SDS-PAGE (b), NuPAGETM 4–12% Bis-TRIS Midi Gel under reducing (R) and non-reducing conditions (NR). M: Marker. (c) The melting temperature was determined by dynamic light scattering and visual interpretation of the obtained results. Plasma stability was analyzed after incubation in human plasma for the indicated time points followed by the determination of the EC50 values of residual binding protein by ELISA (D).
Binding of Atrosimab to TNFR1, C1q and Fcγ receptors
Atrosimab bound to human TNFR1-Fc in an enzyme-linked immunosorbent assay (ELISA) with an EC50 value of 0.37 nM, representing a 2.2-fold reduction in binding activity compared to the parental Fab 13.7 ()). The bivalent IgG antibody ATROSAB bound to TNFR1 with an EC50 value of 0.09 nM. Furthermore, in real-time binding analyses using the quartz crystal microbalance (QCM, )) technology, Atrosimab revealed a KD value of 2.66 nM with an association rate constant kon and a dissociation rate constant koff of 3.69 × 105 M−1s−1 and 9.83 × 10−4 s−1, respectively.
Figure 6. Antigen, Fc receptor and Complement binding of Atrosimab. Binding of Atrosimab to human TNFR1-Fc was analyzed in ELISA (a, n = 3, mean ± SD) and QCM (b). Five concentrations between 128 nM and 4 nM (1:2 dilution steps) were used to generate the kinetic data in b. A one-to-one binding algorithm was employed for fitting. C) Binding of human FcγRIa, IIb and IIIa and the complement protein C1q to immobilized Atrosimab was analyzed by ELISA. Rituximab (wild-type Fc part) and ATROSAB (silent Fc) were used as controls (n = 2, mean ± range).
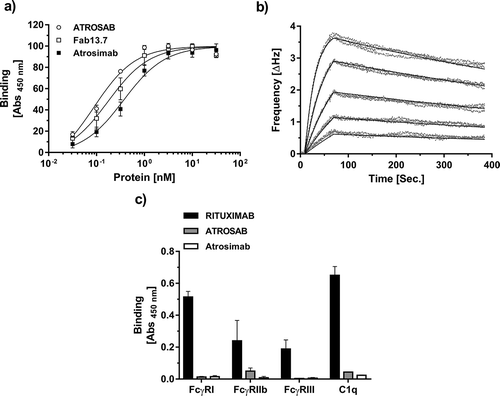
The inability of Atrosimab to mediate Fc effector functions due to the Δab mutations that were introduced into Fc1κCitation36 was demonstrated by analyzing the ability of the complement protein C1q and the Fcγ receptors Ia, IIb, and IIIa to bind to immobilized Atrosimab. Similar to the previously published antagonistic TNFR1-specific IgG ATROSAB,Citation37,Citation38 C1q and the Fcγ receptors Ia, IIb, and IIIa revealed strongly reduced or absent binding to Atrosimab compared to the control antibody Rituximab, which possesses a wild-type IgG1 Fc ()).
In vitro bioactivity of Atrosimab
Atrosimab demonstrated a complete lack of agonistic activity over a concentration range from 50 pM to 500 nM, as demonstrated in interleukin-6 (IL-6) and interleukin-8 (IL-8) release assays using HeLa and HT1080 cells, respectively, and in a cell death induction assay using Kym-1 cells (–c)). As expected, the parental Fab 13.7, used as monovalent control protein due to its previously demonstrated absence of agonism,Citation33 did not activate TNFR1. In contrast, the previously described presence of a marginal agonistic activity at a narrow dose range of the bivalent humanized antibody ATROSABCitation37,Citation38 was confirmed in IL-6 and IL-8 release experiments (,b)). However, this minor induction of TNFR1 activation was not detectable in the cell death induction assay using Kym-1 cells ()).
Figure 7. Antagonistic bioactivity of Atrosimab and lack of agonism. The inherent lack of agonistic activity of Atrosimab in terms of TNFR1 activation was demonstrated in three individual assays. a) IL-6 release from HeLa cells, b) IL-8 release from HT1080 cells and c) cell death induction assay using Kym-1 cells. The inhibitory potential of Atrosimab was shown in an IL-6 release assay using HeLa cells (d), in an IL-8 release assay using HT1080 cells (e) and in a cell death induction assay using Kym-1 cells (f), which were performed in the presence of a constant concentration of 0.1 nM TNF (d and e) or 0.01 nM TNF (f). ATROSAB (marginal activity) and TNF (strong activity) alone served as control molecules for the activation of TNFR1, Fab 13.7 served as negative control (a, b and c). Fab 13.7 and ATROSAB served as controls for the inhibition of TNF-induced TNFR1 activation (d, e and f). All graphs represent the mean of three individual experiments, error bars indicate SD.
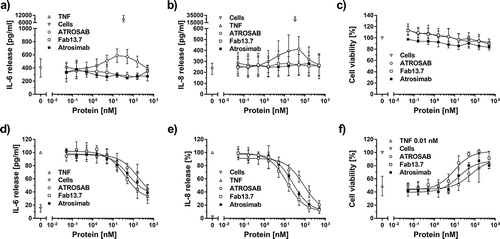
Atrosimab inhibited the activation of TNFR1 in the presence of a constant concentration of TNF in IL-6 (0.1 nM TNF) and IL-8 release (0.1 nM TNF), as well as in cell death induction assays (0.01 nM TNF) with IC50 values of 54.5 nM, 24.2 nM and 16.2 nM, respectively (), ). The corresponding IC50 values of the parental Fab 13.7 in IL-6 release, IL-8 release and in cell death induction assays were 37.1 nM, 12.7 nM, and 9.5 nM, respectively, indicating a slightly reduced bioactivity of Atrosimab (1.5- to 1.9-fold). However, in comparison to ATROSAB, the antagonistic activity of Atrosimab was three- to fourfold increased, depending on the assay system (IL-6: 3.0; IL-8: 3.5; cell death: 4.0) (–f), ).
Table 1. Bioactivity of Atrosimab.
Antibody-mediated cross-linking of Atrosimab
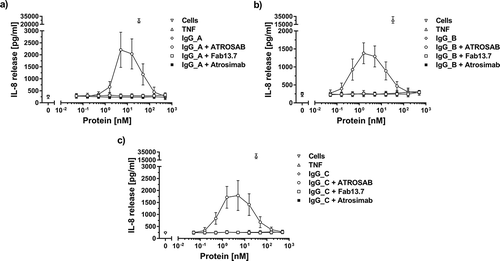
In order to assess the risk of drug-specific antibodies to turn Atrosimab into a potentially agonistic molecule by secondary cross-linking, three different goat anti-human IgG serum preparations were analyzed for their potential to induce TNFR1 activation in combination with Atrosimab. In IL-8 release experiments, a constant concentration of anti-human IgG serum was incubated together with increasing concentrations of Atrosimab and the control proteins Fab13.7 and ATROSAB (). Atrosimab and the monovalent control protein Fab13.7 did not induce release of IL-8 in combination with any of the tested sera, while the stimulation of HT1080 cells by the bivalent control protein ATROSAB in combination with all three anti-human IgG sera resulted in three- to fivefold increased induction of IL-8 release at the maximum of the respective dose response (–c); ATROSAB alone see )). Binding of all three used anti-human IgG sera to Atrosimab and the control proteins was confirmed by ELISA (Fig. S2a-c). Moreover, Atrosimab and Fab13.7 revealed only slightly reduced binding to TNFR1-Fc in presence of all three anti-human IgG sera, as determined by ELISA (Fig. S2 d-f). In contrast, TNFR1 binding of ATROSAB was strongly reduced in the presence of anti-human Ig, which, however, did not prevent crosslinking-mediated TNFR1 activation, as demonstrated above.
Figure 8. Complete lack of agonistic bioactivity of Atrosimab in the presence of anti-human IgG antibodies. The activation of TNFR1 on the surface of HT1080 cells by Atrosimab in the presence of a constant concentration (ca. 15.8 nM) of three different anti-human IgG serum preparations (a, b and c) was determined by the detection of IL-8 release into the culture supernatant. Unstimulated cells and 33 nM TNF were used as controls. The agonistic effect of potentially crosslinking antibodies was compared to Fab 13.7 and ATROSAB. All experiments show Mean ± SD of three individual experiments.
Pharmacokinetic analysis of Atrosimab
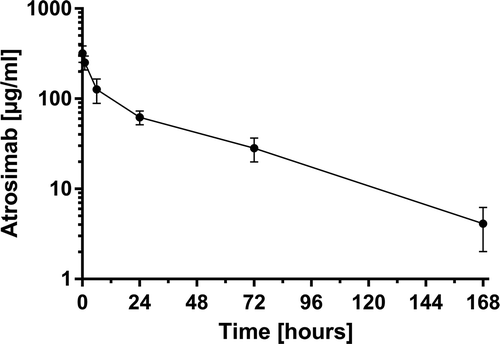
To evaluate the effect of the novel heterodimeric Fc on in vivo serum circulation, the pharmacokinetic (PK) properties of Atrosimab were analyzed after bolus injection of 400 µg using C57BL/6J-huTNFRSF1Aecdtm1UEG/izi mice carrying the gene encoding the extracellular domain of the human TNFR1 at the respective mouse locus (, ).Citation39 Atrosimab revealed initial and terminal half-lives of 2.2 ± 1.2 h and 41.7 ± 18.1 h, respectively, and an area under the curve of 5856.0 ± 1369.9 µg/ml × h.
Table 2. Pharmacokinetic analysis of Atrosimab.
Figure 9. Pharmacokinetic analysis of Atrosimab. Four hundred micrograms of Atrosimab were injected into C57BL/6J knock-in mice, carrying the gene of the human TNFR1 extracellular domain connected to the mouse transmembrane and intracellular domains instead of the wild-type mouse gene. Remaining intact protein in the serum was determined by ELISA for binding to TNFR1 at the indicated time points. Shown are the mean ± SD of five mice.
Discussion
The objective of this study was to develop a novel, monovalent inhibitor of TNFR1 activation with extended serum half-life based on the previously described antagonistic TNFR1-specific Fab 13.7.Citation33 To incorporate an IgG-like Fc into the molecule while retaining Fab-like heterodimerization of the polypeptide chains, the variable domains of Fab 13.7 were fused to the N-termini of newly generated heterodimerizing Fc chains, designated Fc-one/kappa (Fc1κ). Heterodimerization of this novel Fc moiety is mediated by interspersed Ig domains called CH31 or CH3κ. These interspersed Ig domains are composed of sequences originating mainly from the per se heterodimerizing IgG1 CH1 and CLκ domains, and also sequence stretches originating from CH3 domains in order to retain FcRn binding and thus enable FcRn-mediated drug recycling in vivo. In contrast to most of the described Fc heterodimerization technologies (for reviews, see refs.Citation40,Citation41), which rely on the replacement of single or multiple amino acids located at the CH3–CH3 interface, the herein presented Fc heterodimerization technology is based on the exchange of larger amino acid sequence stretches obtained from human antibody sequences. This approach hypothetically has a lower probability to create de novo T cell epitopes, although this needs to be verified in further studies.
The degree of heterodimerization, mediated by the newly generated Fc1κ, was demonstrated on the basis of asymmetric scFv-Fc1κ fusion proteins and compared to the knobs-into-holes technology,Citation35 a state-of-the-art heterodimerization platform. In the composition carrying the scFv moiety on the N-terminus of the CH31-containing Fc chain, heterodimer formation was similar compared to knobs-into-holes with the scFv fused to the “holes” Fc chain or even superior compared to knobs-into-holes with the scFv fused to the N-terminus of the “knobs” Fc chain. Notably, in a recently published study on improved versions of the BEAT-Fc part, both asymmetric dAb Fc fusion proteins based on the classic BEAT technology and those based on the SEEDbody technology revealed the residual formation of homodimers.Citation42 This observation further supports the exceptional heterodimerization quality of the newly generated Fc1κ.
The isolated Fc chain, comprising the CH31 domain, was expressed poorly, even in the presence of the Fc chain containing scFv and CH3κ moieties. This inefficient protein expression might be due to an altered interaction with the luminal endoplasmic reticulum chaperone BiP, which is involved in the processing of the CH1 domain, ensuring that IgG heavy chains are only secreted in combination with the light chain.Citation43 Similarly, also the AGSb chain of the SEEDbody technology could not be expressed individually; however, this chain does not contain elements of the CH1 domain and was expressed successfully in combination with all other relevant chains of the analyzed Fc-IL-2 fusion protein.Citation44 Productivity of the Fc1κ chains, and especially of the CH31 comprising chain, will be addressed in detail in the future, such as with respect to cysteine residues, which were demonstrated to control the interaction with the BiP protein and thereby the secretion of IgG heavy chains.Citation45
The Fv-Fc1κ fusion protein Atrosimab in the composition VL-CH2-CH31/VH-CH2-CH3κ revealed correct heterodimeric assembly without any detectable aggregation or free monomeric polypeptide chains after one-step purification by protein A affinity chromatography, indicating good processability with respect to further drug development. Additional purification steps, such as cation exchange chromatography, recently reported to be required to remove unwanted homodimers during the purification process of a bispecific molecule based on a charge-directed Fc heterodimerization technology,Citation46 might be included to remove residual side products (for review, see Refs.Citation47,Citation48).
Atrosimab revealed an aggregation temperature of 64°C, which is similar to that of intact IgG molecules when determined by dynamic light scattering.Citation49,Citation50 However, there might still be space for optimization, especially considering newly generated interfaces between individual constant Ig domains, which have been demonstrated to represent an efficient target for stability engineering in the case of an scFv to Fab conversionCitation51 or the fusion of VH or VL to the N-terminus of a CH3 domain.Citation52
Atrosimab bound to TNFR1 with high affinity (KD 2.7 nM) and strongly inhibited TNF-induced receptor activation with IC50 values ranging from 16 nM to 55 nM in different in vitro assays. Compared to the parental Fab 13.7 molecule, TNFR1 binding and inhibition of activation was slightly reduced. Since correct assembly of the heterodimeric protein was confirmed by mass spectrometry analysis, the reduced binding and bioactivity might rather be attributed to alterations in the VH and VL pairing after fusion to the CH2 domain. Similar effects are described in the literature, such as for the conversion of an anti-scorpion toxin scFv into a Fab moleculeCitation53 or an anti-transforming growth factor β1 scFv into an IgG4,Citation54 and has been studied intensively in a comprehensive computational and experimental approach of variable domain exchange in the case of an anti-CD20 scFv.Citation55
In order to disable the induction of antibody-mediated effector functions via the complement system or natural killer cells, Atrosimab was equipped with a “silent” FcCitation36 unable to interact with FcγRI, FcγIIb and FcγIII, as well as with the complement protein C1q. Furthermore, this lack of binding to effector molecules of the immune system should also prevent the activation of TNFR1 due to secondary crosslinking of Atrosimab bound to cells expressing FcγRs, as described to be indispensable for antibody-mediated activation of different members of the TNFR superfamily, e.g., CD134.Citation56,Citation57 In addition, secondary crosslinking could be mediated by natural or induced ADAs, which have been reported to be a general phenomenon in patients treated with biopharmaceuticals.Citation58 More particularly, ADAs were observed in the case of patients treated with either infliximab or adalimumab at rates of 50% or 31%, respectively.Citation59 The detrimental effects of ADAs on anti-TNFR1 agents became obvious in a clinical Phase 1 study concerning a VH dAb (GSK1995057).Citation60 Moderate symptoms of a cytokine release syndrome (CRS) at doses of 2 to 10 µg/kg were observed due to pre-existing ADAs as determined to be present in 50% of drug naïve patients in subsequent analyses. Atrosimab demonstrated a lack of activation of TNFR1, even in the presence of three different goat anti-human IgG sera. Together with the lack of binding to FcγRs and the complement protein C1q, these data suggest an absence or reduced propensity to cause systemic activation of TNFR1, even in patients with ADAs in their circulation.
Furthermore, Atrosimab revealed a terminal half-life of 41.8 h in C57BL/6J-huTNFRSF1Aecdtm1UEG/izi mice after injection of 20 mg/kg body weight, representing an almost 40-fold extension compared to the terminal half-life of Fab 13.7 (1.4 h).Citation33 Surprisingly, even when compared to ATROSAB (32.1 h),Citation33 Atrosimab had a 1.3-fold elongated terminal half-life. However, the data concerning Fab 13.7 and ATROSAB were collected after injection of a lower dose (1.25 mg/kg), which might have an impact on the PK characteristics, such as on target-mediated clearance as shown for other antibodies directed against broadly expressed targets.Citation61,Citation62 This effect depends on the applied dose, and is detectable only at doses below a drug-specific threshold of target saturation.Citation63 Conclusively, additional experiments using identical doses of the different agents must be performed to identify the exact PK properties of Atrosimab in comparison to Fab 13.7 and ATROSAB.
In summary, Atrosimab revealed strong binding to TNFR1 and sustained inhibition of TNF-mediated TNFR1 activation in combination with a complete lack of agonistic activity, even in the presence of cross-linking antibodies. Atrosimab furthermore showed a clearly improved in vivo serum circulation time. Regarding the therapeutic application of Atrosimab, it is important to mention that blockade of TNFR1 also inhibits the activity of Lymphotoxin-α, which contributes to the inflammatory condition in experimental allergic encephalomyelitis in miceCitation64 and was further shown to be elevated in the synovium of RA patients.Citation65 Lymphotoxin as a relevant therapeutic target is apparent from a case report on the successful treatment of an infliximab-resistant RA patient with etanercept, the only approved anti-TNF drug that also inhibits Lymphotoxin-α activity.Citation66 Furthermore, long-lasting blockade of complete TNF activity by treatment with classic TNF inhibitors are accompanied by various side effects like susceptibility to infections or, more rarely, the exacerbation of inflammatory symptoms and the development of malignancies.Citation20,Citation26 In contrast, selective inhibition of TNFR1, leaving TNFR2 signaling unaffected, represents a more specific intervention in TNF’s pathophysiologic actions, both in diseases where anti TNFs show therapeutic activity and those where anti-TNF blockade failed. Moreover, recent publications revealed the involvement of TNFR1 in mouse models concerning tumor lymphangiogenesis and metastasisCitation67 and in gastric tumorigenesis,Citation68 which further underlines the potential of Atrosimab as a candidate for treatment not only of inflammatory diseases but also of certain oncological indications.
Materials and methods
Materials
ATROSAB and human TNFR1-Fc were provided by Baliopharm (Basel, Switzerland). The final Atrosimab batch was produced by Catalent (Catalent Pharma Solutions, Somerset, Ewing, NJ, US) in CHO cells after lentiviral transfection and purified by protein A affinity chromatography. Anti-His-horseradish peroxidase (HRP) (HIS-6 His-Probe-HRP, sc-8036) was acquired from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-human IgG A (Goat, polyclonal, 2010–01) from SouthernBiotech, anti-human IgG B (Goat, polyclonal, MBS571163) and anti-human IgG B (Goat, polyclonal, MBS571678) from MyBioSource, (San Diego, CA, USA), anti-human IgG (Fab specific, A 0293) and anti-human IgG (Fc specific, A 0170) from Sigma-Aldrich (Taufkirchen, Germany).
Protein production
DNA encoding the interspersed constant Ig domains connected to the C-terminus of an IgG CH2 domain were ordered from GeneArt® (Regensburg, Germany) and cloned into the expression vector pSecTagAL1 (modified from pSecTag-FcHis),Citation69 which already contained the variable domains of the heavy or light chain of Fab 13.7 using the restriction enzymes KpnI and EcoRI. All Atrosimab variants and the Fab 13.7 protein were produced in HEK293E cells after transient transfection of a mixture of two vectors, encoding for the different chains of the dimeric molecules using polyethylenimine (linear, 25 kDa, Sigma-Aldrich, Taufkirchen, Germany). Proteins were purified by protein A affinity chromatography according to the manufacturers' protocol (TOYOPEARL®, AF-rProtein A-650F, 22805, Tosoh, Stuttgart, Germany). In the case of the Atrosimab batch produced by Catalent (Catalent Pharma Solutions, Somerset, Ewing, NJ, US), a preparative gel-filtration step was performed (Äkta purifier, Superdex 200 10/300 GL column, flow rate of 0.5 ml/min, phosphate-buffered saline (PBS) as liquid phase).
Protein characterization
Purified protein samples were analyzed by SDS-PAGE using 4 µg of each sample under reducing and non-reducing conditions. Proteins were stained using Coomassie-Brilliant Blue G-250 and acrylamide gels were de-stained with water. Correct assembly under native conditions was visualized by SEC using a Waters 2695 HPLC and a Phenomenex Yarra SEC-2000 column (300 x 7.8 mm) or a TSKgel SuperSW mAb HR column (flow rate of 0.5 ml/min, 0.1 M Na2HPO4/NaH2PO4, pH 6.7 as mobile phase). Standard proteins (MW, rS): Thyroglobulin (669 kDa, 8.50 nm), Apoferritin (443 kDa, 6.10 nm), beta Amylase (200 kDa, 5.4 nm), bovine serum albumin (67 kDa, 3.55 nm), Carbonic anhydrase (29 kDa, 2.35). Mass spectrometry analysis was performed by Xin Chen at Catalent Pharma Solutions (New Jersey, US) after deglycosylation using PNGase F (1 unit/µg protein) for 18 h at 37°C. Separation was performed on an ACQUITY UPLC® H-Class System (Waters; Milford, MA). Measurements were implemented on a Xevo G2-XS system with an electrospray ionization (ESI) source (Waters; Milford, MA). The system was controlled by UNIFI 1.8 (Waters; Milford, MA). The prepared samples were injected into a Waters BEH C4 column (2.1 × 100 mm, 1.7 µm) at an injection volume of 5 µL. Mobile Phase A was water with 0.1% formic acid (FA) and Mobile Phase B was acetonitrile with 0.1% FA. A linear gradient was used.
Thermal stability
Temperature-dependent aggregation of Atrosimab was analyzed using the ZetaSizer Nano ZS (Malvern, Herrenberg, Germany). Increasing temperatures were applied stepwise from 35°C to 80°C with intervals of 1°C and equilibration times of 2 min prior to each measurement. One hundred micrograms of protein were diluted in 1 ml PBS and the melting/aggregation temperature (Tm) was determined by visual interpretation of the increasing signal of the particle size (kcps).
Plasma stability
Atrosimab samples were diluted to 100 nM in human plasma, incubated at 37°C for 1, 3 and 7 days and subsequently analyzed by ELISA for binding to human TNFR1 (as described below) after serial dilution in 2% skim milk in PBS (2% MPBS) by steps of 1 to 3.16 (square root of 10). A protein sample, stored at 4°C and a protein sample, frozen immediately after dilution in human plasma, were used as controls.
Enzyme-linked immunosorbent assay
Indicated proteins were diluted to 1 µg/ml in PBS, transferred to a 96-well microtiter plate and incubated overnight at 4°C. Skim milk in PBS (2% MPBS) was used to block residual binding sites (200 µl/well for 1–2 h at room temperature (RT)). All samples were diluted in 2% MPBS to the indicated maximal concentrations and diluted in steps of 1 to 3.16 (square root of 10). Each sample was transferred to the microtiter plates and incubated at RT for another hour. HRP-labeled detection antibodies were diluted in 2% MPBS as recommended by the manufacturer and applied to the plates for 1 h at RT. Binding of the analyzed proteins was detected using 100 µl substrate solution (1 mg/ml 3,3ʹ,5,5ʹ-Tetramethylbenzidine[TMB], 0.006% H2O2 in 100 mM Na-acetate buffer, pH 6 at RT) and the detection reaction was stopped using 50 µl 1 M H2SO4. Absorption was determined at a wavelength of 450 nm. In each step, a working volume of 100 µl was used and between each step, the plates were washed twice with PBS containing 0.005% Tween20 (PBST) and twice with PBS.
Quartz crystal microbalance
Real-time binding kinetics were analyzed using the A-100 C-Fast or Cell-200 C-Fast biosensors (Attana, Stockholm, Sweden). A human TNFR1-Fc fusion protein was covalently immobilized to the Attana LNB Carboxyl Sensor Chip (3623–3103, Attana, Stockholm, Sweden) at the indicated ligand density, using the Amine Coupling Kit (3501–3001, Attana, Stockholm, Sweden) as recommended by the manufacturer. Binding of analytes was performed at 37°C, using PBST (0.1% Tween-20, pH 7.4) as running buffer at a flow rate of 25 µl/min. Reference injection of running buffer was performed after every other measurement and subtracted from the binding curves during data evaluation using the Attaché Office Evaluation software (Attana, Stockholm, Sweden) and TraceDrawe (Ridgeview instruments, Vange, Sweden). Sensor chip regeneration was accomplished by injecting 20 mM glycine, pH 2.0 twice for 12 s.
Interleukin release assay
HeLa (IL-6) or HT1080 (IL-8) cells were adjusted to a concentration of 2 × 105 cells/ml and 100 µl were distributed in 96-well plates and incubated at 37°C, 5% CO2 overnight. The used medium (RPMI 1640 + 5% FCS) was discarded subsequently to remove present IL-6 or IL-8, and the protein samples were diluted in culture medium and applied to the cells. In the case of inhibition experiments, antibody samples were added to the cells immediately following to the addition of the stimulant (TNF). After 16–20 h of incubation at 37°C, 5% CO2, the supernatants were harvested subsequent to 5 min of centrifugation at 500 x g, diluted in culture media (stimulation experiments 1:2, competition 1:25, 33 nM TNF control 1:75) and analyzed for IL-6/IL-8 concentration by ELISA as recommended by the manufacturer (IL-6, 31670069, IL-8, 31670089, ImmunoTools, Friesoythe, Germany).
Cytotoxicity assay
Kym-1 cells were incubated overnight at 37°C and 5% CO2 in 96-well microtiter plates (1 x 104 per well) and subsequently treated with serial dilutions of the protein samples in culture medium (5% RPMI 1640 + 10% FCS) alone or in combination with a constant concentration of TNF (0.01 nM) for another 24 h at 37°C and 5% CO2. The supernatants were discarded and 50 µl crystal violet solution (0.5% crystal violet, 20% methanol in H2O) was added to each well and incubated for 20 min at RT. Plates were washed 20 times with ddH2O, dried at RT, and residual crystal violet was dissolved in 50 µl methanol per well. The absorption at 595 nm was measured after shaking the plates for 10 min at RT.
Pharmacokinetics
Animal care and experiments were performed in accordance with federal guidelines and have been approved by university and state authorities. PK properties of Atrosimab were determined after injection of 400 µg protein into the tail vein of transgenic C57BL/6J mice expressing a chimeric TNFR1, composed of the extracellular domain of human TNFR1 and the murine intracellular region from the locus of the particular mouse gene (C57BL/6J-huTNFRSF1Aecdtm1UEG/izi).Citation39 Blood samples were collected at the indicated time points from the tail and incubated on ice for 20 min. Serum was separated by centrifugation (13,000 x g, 4°C, 10 min) and frozen to −20°C prior to analysis. Binding of remaining protein in the serum samples to human TNFR1was analyzed by ELISA and the resulting data were analyzed using the PKSolver Excel add-in.Citation70
Disclosure of Potential Conflicts of Interest
F.R., O.S., A.H., K.P., and R.E.K. are named inventors on patent applications covering Fc heterodimerization modules and monovalent inhibitors of TNFR1 interaction.
Abbreviations
| ADA | = | anti-drug antibodies |
| ATROSAB | = | antagonistic TNF receptor one specific antibody |
| AUC | = | area under the curve |
| CH1 | = | first constant domain of the IgG1 heavy chain |
| CH3 | = | third constant domain of the IgG1 heavy chain |
| CH3κ | = | CH3kappa |
| CHO | = | Chinese hamster ovary |
| CIA | = | collagen-induced arthritis |
| CLκ | = | constant domain of the kappa light chain |
| CRS | = | cytokine release syndrome |
| dAb | = | domain antibody |
| EAE | = | experimental autoimmune encephalomyelitis |
| ELISA | = | enzyme-linked immunosorbent assay |
| ESI | = | electrospray ionization |
| FA | = | formic acid |
| Fab | = | antigen-binding fragment |
| Fc1κ | = | Fc-one/kappa |
| FcRn | = | neonatal Fc receptor |
| Fv | = | fragment variable |
| HRP | = | horseradish peroxidase |
| Ig | = | immunoglobulin |
| IL-6 | = | interleukin-6 |
| IL-8 | = | interleukin-8 |
| LPS | = | lipopolysaccharides |
| LT | = | Lymphotoxin |
| M | = | Marker |
| NR | = | non-reducing conditions |
| PBS | = | phosphate-buffered saline |
| PBST | = | PBS containing Tween20 |
| PEG | = | polyethylene glycol |
| PK | = | pharmacokinetic |
| QCM | = | quartz crystal microbalance |
| R | = | reducing conditions |
| RA | = | rheumatoid arthritis |
| RT | = | room temperature |
| scFv | = | single-chain variable fragment |
| SEC | = | size-exclusion chromatography |
| SEED | = | strand-exchange engineered domain |
| Tm | = | melting/aggregation temperature |
| TNF | = | tumor necrosis factor |
| TNFR1 | = | TNF-receptor one |
| VH | = | variable domain of the heavy chain |
| VH1C | = | VH fused to the CH2-CH31chain |
| VHκC | = | VH fused to the CH-2CH3κ chain |
| VL | = | variable domain of the light chain |
| VL1C | = | VL fused to the CH2-CH31 chain |
| VLkC | = | VL fused to the CH2-CH3κ chain. |
Supplemental Material
Download MS Word (321.9 KB)Acknowledgments
We would like to thank Nadine Heidel and Doris Göttsch for excellent technical assistance. We further express our appreciation to the team at Catalent Pharma Solutions for the expression of Atrosimab and especially to Xin Chen for performing the Mass Spectrometry analysis.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Havell EA. Evidence that tumor necrosis factor has an important role in antibacterial resistance. J Immunol. 1989;143:2894–99.
- Pfeffer K, Matsuyama T, Kündig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Krönke M, Mak TW. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73(3):457–67. doi:10.1016/0092-8674(93)90134-C.
- Rothe J, Lesslauer W, Lötscher H, Lang Y, Koebel P, Köntgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to IMF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364(6440):798–802. doi:10.1038/364798a0.
- Dumitru CD, Ceci JD, Tsatsanis C, Kontoyiannis D, Stamatakis K, Lin JH, Patriotis C, Jenkins NA, Copeland NG, Kollias G, et al. TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell. 2000;103(7):1071–83. doi:10.1016/S0092-8674(00)00210-5.
- Segueni N, Benmerzoug S, Rose S, Gauthier A, Bourigault ML, Reverchon F, Philippeau A, Erard F, Le Bert M, Bouscayrol H, et al. Innate myeloid cell TNFR1 mediates first line defence against primary Mycobacterium tuberculosis infection. Sci Rep. 2016;6:22454. doi:10.1038/srep22454.
- Cope AP, Liblau RS, Yang XD, Congia M, Laudanna C, Schreiber RD, Probert L, Kollias G, McDevitt HO. Chronic tumor necrosis factor alters T cell responses by attenuating T cell receptor signaling. J Exp Med. 1997;185(9):1573–84. doi:10.1084/jem.185.9.1573.
- Aspalter RM, Wolf HM, Eibl MM. Chronic TNF-α exposure impairs TCR-signaling via TNF-RII but not TNF-RI. Cell Immunol. 2005;237(1):55–67. doi:10.1016/j.cellimm.2005.10.001.
- Chen X, Baumel M, Mannel DN, Howard OMZ, Oppenheim JJ. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol. 2007;179(1):154–61. doi:10.4049/jimmunol.179.1.154.
- Jacob CO, Lewis GD, McDevitt HO. MHC class II-associated variation in the production of tumor necrosis factor in mice and humans: relevance to the pathogenesis of autoimmune diseases. Immunol Res. 1991;10(2):156–68. doi:10.1007/BF02918162.
- Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A. 1994;91(11):4854–58. doi:10.1073/pnas.91.11.4854.
- Emanuelli B, Peraldi P, Filloux C, Chavey C, Freidinger K, Hilton DJ, Hotamisligil GS, Van Obberghen E. SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-α in the adipose tissue of obese mice. J Biol Chem. 2001;276(51):47944–49. doi:10.1074/jbc.M104602200.
- Adegbola SO, Sahnan K, Warusavitarne J, Hart A, Tozer P. Anti-TNF Therapy in Crohn’s Disease. Int J Mol Sci. 2018;19:8. doi:10.3390/ijms19082244.
- Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. 2002;2(5):364–71. doi:10.1038/nri802.
- Romas E, Gillespie MT, Martin TJ. Involvement of receptor activator of NFκB ligand and tumor necrosis factor-α in bone destruction in rheumatoid arthritis. Bone. 2002;30(2):340–46. doi:10.1016/S8756-3282(01)00682-2.
- Müssener Å, Litton MJ, Lindroos E, Klareskog L. Cytokine production in synovial tissue of mice with collagen-induced arthritis (CIA). Clin Exp Immunol. 1997;107(3):485–93. doi:10.1046/j.1365-2249.1997.3181214.x.
- Kontermann RE, Scheurich P, Pfizenmaier K. Antagonists of TNF action: clinical experience and new developments. Expert Opin Drug Discov. 2009;4(3):279–92. doi:10.1517/17460440902785167.
- Sherman M, Tsynman DN, Kim A, Arora J, Pietras T, Messing S, St Hilaire L, Yoon S, Decross A, Shah A, et al. Sustained improvement in health-related quality of life measures in patients with inflammatory bowel disease receiving prolonged anti-tumor necrosis factor therapy. J Dig Dis. 2014;15(4):174–79. doi:10.1111/1751-2980.12125.
- Ding NS, Hart A, De Cruz P. Systematic review: predicting and optimising response to anti-TNF therapy in Crohn’s disease - Algorithm for practical management. Aliment Pharmacol Ther. 2016;43(1):30–51. doi:10.1111/apt.13445.
- Desai SB, Furst DE. Problems encountered during anti-tumour necrosis factor therapy. Best Pract Res Clin Rheumatol. 2006;20(4):757–90. doi:10.1016/j.berh.2006.06.002.
- Steeland S, Libert C, Vandenbroucke RE. A new venue of TNF targeting. Int J Mol Sci. 2018;19(5):1–55. doi:10.3390/ijms19051442.
- Fontaine V, Mohand-Said S, Hanoteau N, Pfizenmaier K. Neurodegenerative and neuroprotective effects of Tumor Necrosis Factor (TNF) in retinal ischemia : opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. 2002;22(7):RC216.
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10(1):45–65. doi:10.1038/sj.cdd.4401189.
- Qu Y, Zhao G, Forward LH. Reverse signaling mediated by transmembrane tumor necrosis factor-alpha and TNF receptor 2: potential roles in an immunosuppressive tumor microenvironment. Front Immunol. 2017;8:1675. doi:10.3389/fimmu.2017.01675.
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JPY. TNFα promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4(11):1116–22. doi:10.1038/nn738.
- Kassiotis BG, Kollias G. Uncoupling the proinflammatory from the immunosuppressive properties of Tumor Necrosis Factor (TNF) at the p55 TNF receptor level : implications for pathogenesis and therapy of autoimmune demyelination. J Exp Med. 2001;193(4):427–34.
- Van Hauwermeiren F, Vandenbroucke RE, Libert C. Treatment of TNF mediated diseases by selective inhibition of soluble TNF or TNFR1. Cytokine Growth Factor Rev. 2011;22(5–6):311–19. doi:10.1016/j.cytogfr.2011.09.004.
- Williams SK, Maier O, Fischer R, Fairless R, Hochmeister S, Stojic A, Pick L, Haar D, Musiol S, Storch MK, et al. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS One. 2014;9(2):e90117. doi:10.1371/journal.pone.0090117.
- Shibata H, Yoshioka Y, Ohkawa A, Abe Y, Nomura T, Mukai Y, Nakagawa S, Taniai M, Ohta T, Mayumi T, et al. The therapeutic effect of TNFR1-selective antagonistic mutant TNF-α in murine hepatitis models. Cytokine. 2008;44(2):229–33. doi:10.1016/j.cyto.2008.07.003.
- Shibata H, Yoshioka Y, Abe Y, Ohkawa A, Nomura T, Minowa K, Mukai Y, Nakagawa S, Taniai M, Ohta T, et al. The treatment of established murine collagen-induced arthritis with a TNFR1-selective antagonistic mutant TNF. Biomaterials. 2009;30(34):6638–47. doi:10.1016/j.biomaterials.2009.08.041.
- Kitagaki M, Isoda K, Kamada H, Kobayashi T, Tsunoda S, Tsutsumi Y, Niida T, Kujiraoka T, Ishigami N, Ishihara M, et al. Novel TNF-α receptor 1 antagonist treatment attenuates arterial inflammation and intimal hyperplasia in mice. J Atheroscler Thromb. 2012;19(1):36–46. doi:10.5551/jat.9746.
- Bertok S, Wilson MR, Morley PJ, de Wildt R, Bayliffe A, Takata M. Selective inhibition of intra-alveolar p55 TNF receptor attenuates ventilator-induced lung injury. Thorax. 2012;67(3):244–51. doi:10.1136/thoraxjnl-2011-200590.
- Steeland S, Van Ryckeghem S, Van Imschoot G, De Rycke R, Toussaint W, Vanhoutte L, Vanhove C, De Vos F, Vandenbroucke RE, Libert C. TNFR1 inhibition with a Nanobody protects against EAE development in mice. Sci Rep. 2017;7(1):1–17. doi:10.1038/s41598-017-13984-y.
- Richter F, Zettlitz KA, Seifert O, Herrmann A, Scheurich P, Pfizenmaier K, Kontermann RE. Monovalent TNF receptor 1-selective antibody with improved affinity and neutralizing activity. MAbs. 2018;25:1–12. doi:10.1080/19420862.2018.1524664.
- Martin WL, West AP, Gan L, Bjorkman PJ. Crystal structure at 2.8 Å of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell. 2001;7(4):867–77. doi:10.1016/S1097-2765(01)00230-1.
- Atwell S, Ridgway JB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol. 1997;270(1):26–35. doi:10.1006/jmbi.1997.1116.
- Armour KL, Clark MR, Hadley AG, Williamson LM. Recombinant human IgG molecules lacking Fcγ receptor I binding and monocyte triggering activities. Eur J Immunol. 1999;29(8):2613–24. doi:10.1002/(ISSN)1521-4141.
- Zettlitz KA, Lorenz V, Landauer K, Münkel S, Herrmann A, Scheurich P, Pfizenmaier K, Kontermann RE. ATROSAB, a humanized antagonistic anti-tumor necrosis factor receptor one-specific antibody. MAbs. 2010;2(6):639–47. doi:10.4161/mabs.2.6.13583.
- Richter F, Liebig T, Guenzi E, Herrmann A, Scheurich P, Pfizenmaier K, Kontermann RE. Antagonistic TNF receptor one-specific antibody (ATROSAB): receptor binding and in vitro bioactivity. PLoS One. 2013;19(8):e72156. doi:10.1371/journal.pone.0072156.
- Dong Y, Fischer R, Naudé PJW, Maier O, Nyakas C, Duffey M, Van der Zee EA, Dekens D, Douwenga W, Herrmann A, et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc Natl Acad Sci. 2016;113(43):12304–09. doi:10.1073/pnas.1605195113.
- Ha JH, Kim JE, Kim YS. Immunoglobulin Fc heterodimer platform technology: from design to applications in therapeutic antibodies and proteins. Front Immunol. 2016;7:394. doi:10.3389/fimmu.2016.00394.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9(2):182–212. doi:10.1080/19420862.2016.1268307.
- Skegro D, Stutz C, Ollier R, Svensson E, Wassmann P, Bourquin F, Monney T, Gn S, Blein S. Immunoglobulin domain interface exchange as a platform technology for the generation of Fc heterodimers and bispecific antibodies. J Biol Chem. 2017;292(23):9745–59. doi:10.1074/jbc.M117.782433.
- Feige MJ, Groscurth S, Marcinowski M, Shimizu Y, Kessler H, Hendershot LM, Buchner J. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol Cell. 2009;34(5):569–79. doi:10.1016/j.molcel.2009.04.028.
- Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo KM, Huston JS. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel. 2010;23(4):195–202. doi:10.1093/protein/gzp094.
- Elkabetz Y, Argon Y, Bar-Nun S. Cysteines in CH1 underlie retention of unassembled Ig heavy chains. J Biol Chem. 2005;280(15):14402–12. doi:10.1074/jbc.M500161200.
- De Nardis C, Hendriks LJA, Poirier E, Arvinte T, Gros P, Bakker ABH, De Kruif J. A new approach for generating bispecific antibodies based on a common light chain format and the stable architecture of human immunoglobulin G1. J Biol Chem. 2017;292(35):14706–17. doi:10.1074/jbc.M117.793497.
- Rathore AS, Kumar D, Kateja N. Recent developments in chromatographic purification of biopharmaceuticals. Biotechnol Lett. 2018;40(6):895–905. doi:10.1007/s10529-018-2552-1.
- Li Y. A brief introduction of IgG-like bispecific antibody purification: methods for removing product-related impurities. Protein Expr Purif. 2019;155:112–19. doi:10.1016/j.pep.2018.11.011.
- Martin N, Ma D, Herbet A, Boquet D, Winnik FM, Tribet C. Prevention of thermally induced aggregation of igg antibodies by noncovalent interaction with poly(acrylate) derivatives. Biomacromolecules. 2014;15(8):2952–62. doi:10.1021/bm5005756.
- Brader ML, Estey T, Bai S, Alston RW, Lucas KK, Lantz S, Landsman P, Maloney KM. Examination of thermal unfolding and aggregation profiles of a series of developable therapeutic monoclonal antibodies. Mol Pharm. 2015;12(4):1005–17. doi:10.1021/mp400666b.
- Röthlisberger D, Honegger A, Plückthun A. Domain interactions in the Fab fragment: A comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J Mol Biol. 2005;347(4):773–89. doi:10.1016/j.jmb.2005.01.053.
- Wozniak-Knopp G, Stadlmayr G, Perthold JW, Stadlbauer K, Gotsmy M, Becker S, Rüker F. An antibody with fab-constant domains exchanged for a pair of CH3 domains. PLoS One. 2018;13(4):e0195442. doi:10.1371/journal.pone.0195442.
- Quintero-Hernández V, Juárez-González VR, Ortíz-León M, Sánchez R, Possani LD, Becerril B. The change of the scFv into the Fab format improves the stability and in vivo toxin neutralization capacity of recombinant antibodies. Mol Immunol. 2007;44(6):1307–15. doi:10.1016/j.molimm.2006.05.009.
- Lord DM, Bird JJ, Honey DM, Best A, Park A, Wei RR, Qiu H. Structure-based engineering to restore high affinity binding of an isoform-selective anti-TGFβ1 antibody. MAbs. 2018;10(3):444–52. doi:10.1080/19420862.2018.1426421.
- Geng SS, Feng J, Li Y, Sun Y, Gu X, Huang Y, Wang Y, Kang X, Chang H, Shen B. Binding activity difference of anti-CD20 scFv-Fc fusion protein derived from variable domain exchange. Cell Mol Immunol. 2006;3:439–43.
- Wajant H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 2015;22(11):1727–41. doi:10.1038/cdd.2015.109.
- Turaj AH, Cox KL, Penfold CA, French RR, Mockridge CI, Willoughby JE, Tutt AL, Griffiths J, Johnson PWM, Glennie MJ, et al. Augmentation of CD134 (OX40)-dependent NK anti-tumour activity is dependent on antibody cross-linking. Sci Rep. 2018;8(1):2278. doi:10.1038/s41598-018-20656-y.
- Lundkvist Ryner M, Farrell RA, Fogdell-Hahn A. The case for measuring anti-drug antibodies in people with multiple sclerosis. Expert Rev Clin Immunol. 2014;10(6):697–99. doi:10.1586/1744666X.2014.914852.
- Mok CC, van der Kleij D, Wolbink GJ. Drug levels, anti-drug antibodies, and clinical efficacy of the anti-TNFα biologics in rheumatic diseases. Clin Rheumatol. 2013;32(10):1429–35. doi:10.1007/s10067-013-2336-x.
- Holland MC, Wurthner JU, Morley PJ, Birchler MA, Lambert J, Albayaty M, Serone AP, Wilson R, Chen Y, Forrest RM, et al. Autoantibodies to Variable Heavy (VH) chain Ig sequences in humans impact the safety and clinical pharmacology of a VH domain antibody antagonist of TNF-α receptor 1. J Clin Immunol. 2013;33(7):1192–203. doi:10.1007/s10875-013-9915-0.
- Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507–32. doi:10.1023/A:1014414520282.
- Hansen L, Petersen LC, Lauritzen B, Clausen JT, Grell SN, Agersø H, Sørensen BB, Hilden I, Almholt K. Target-mediated clearance and bio-distribution of a monoclonal antibody against the Kunitz-type protease inhibitor 2 domain of tissue factor pathway inhibitor. Thromb Res. 2014;133(3):464–71. doi:10.1016/j.thromres.2013.12.015.
- Kamath AV, Yip V, Gupta P, Boswell CA, Bumbaca D, Haughney P, Castro J, Tsai SP, Pacheco G, Ross S, et al. Dose dependent pharmacokinetics, tissue distribution, and anti-tumor efficacy of a humanized monoclonal antibody against DLL4 in mice. MAbs. 2014;6(6):1631–37. doi:10.4161/mabs.36107.
- Suen WE, Bergman CM, Hjelmström P, Ruddle NH. A critical role for lymphotoxin in experimental allergic encephalomyelitis. J Exp Med. 1997;186(8):1233–40. doi:10.1084/jem.186.8.1233.
- O’Rourke KP, O’Donoghue G, Adams C, Mulcahy H, Molloy C, Silke C, Molloy M, Shanahan F, O’Gara F. High levels of Lymphotoxin-Beta (LT-Beta) gene expression in rheumatoid arthritis synovium: clinical and cytokine correlations. Rheumatol Int. 2008;28(10):979–86. doi:10.1007/s00296-008-0574-z.
- Buch MH, Conaghan PG, Quinn MA, Bingham SJ, Veale D, Emery P. True infliximab resistance in rheumatoid arthritis: A role for lymphotoxin alpha? Ann Rheum Dis. 2004;63(10):1344–46. doi:10.1136/ard.2003.014878.
- Ji H, Cao R, Yang Y, Zhang Y, Iwamoto H, Lim S, Nakamura M, Andersson P, Wang J, Sun Y, et al. TNFR1 mediates TNF-α-induced tumour lymphangiogenesis and metastasis by modulating VEGF-C-VEGFR3 signalling. Nat Commun. 2014;5(1):4944. doi:10.1038/ncomms5944.
- Oshima H, Ishikawa T, Yoshida GJ, Naoi K, Maeda Y, Naka K, Ju X, Yamada Y, Minamoto T, Mukaida N, et al. TNF-/TNFR1 signaling promotes gastric tumorigenesis through induction of Noxo1 and Gna14 in tumor cells. Oncogene. 2014;33(29):3820–29. doi:10.1038/onc.2013.356.
- Müller D, Trunk G, Sichelstiel A, Zettlitz KA, Quintanilla M, Kontermann RE. Murine endoglin-specific single-chain Fv fragments for the analysis of vascular targeting strategies in mice. J Immunol Methods. 2008;339(1):90–98. doi:10.1016/j.jim.2008.08.008.
- Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99(3):306–14. doi:10.1016/j.cmpb.2010.01.007.