
ABSTRACT
The maintenance of the homeostasis of immunoglobulin G (IgG) represents a fundamental aspect of humoral immunity that has direct relevance to the successful delivery of antibody-based therapeutics. The ubiquitously expressed neonatal Fc receptor (FcRn) salvages IgG from cellular degradation following pinocytic uptake into cells, conferring prolonged in vivo persistence on IgG. However, the cellular sites of FcRn function are poorly defined. Pinocytic uptake is a prerequisite for FcRn-mediated IgG salvage, prompting us to investigate the consequences of IgG uptake and catabolism by macrophages, which represent both abundant and highly pinocytic cells in the body. Site-specific deletion of FcRn to generate mice harboring FcRn-deficient macrophages results in IgG hypercatabolism and ~threefold reductions in serum IgG levels, whereas these effects were not observed in mice that lack functional FcRn in B cells and dendritic cells. Consistent with the degradative activity of FcRn-deficient macrophages, depletion of these cells in FcRn-deficient mice leads to increased persistence and serum levels of IgG. These studies demonstrate a pivotal role for FcRn-mediated salvage in compensating for the high pinocytic and degradative activities of macrophages to maintain IgG homeostasis.
Introduction
Macrophages are tissue-resident cells of the innate immune system that are essential for the maintenance of tissue homeostasis and host defense.Citation1,Citation2 For example, macrophages engulf apoptotic cells throughout the body in an immunologically silent fashion.Citation3 These innate cells also protect the host from pathogens by playing a crucial role in the clearance and initiation of an adaptive immune response against foreign molecules or organisms.Citation4 For both tissue homeostasis and inflammatory responses, macrophages use receptor-dependent and -independent mechanisms to internalize particulates, soluble proteins and other macromolecules for subsequent degradation. Further, both pinocytosis and macropinocytosis, which involve vesicles/vacuoles of ~100 nm and up to 5 μm diameter, respectively, by these cells is very efficient,Citation5–Citation7 resulting in uptake of about 26% of the cellular volume per hour.Citation6 In combination with the high relative abundance of macrophages in organs or tissues in the body,Citation8,Citation9 this raises the question as to how the homeostasis of prevalent soluble proteins such as immunoglobulin G (IgG), which are essential for normal health, is maintained despite the high pinocytic and degradative capacity of these cells.
At the level of subcellular trafficking, the neonatal Fc receptor, FcRn, which is broadly expressed in hematopoietic and parenchymal cells, maintains IgG homeostasis by salvaging its IgG ligand from lysosomal degradation.Citation10,Citation11 This process involves the endosomal sorting of FcRn-bound IgG, following internalization into cells, into recycling or transcytotic pathways followed by exocytic release.Citation12–Citation14 FcRn binds relatively tightly to IgG at acidic, endosomal pH, but with negligible to very low affinity at pH ~7 for almost all IgG isotypes/allotypes.Citation15,Citation16 Consequently, in the majority of cells that are bathed at near neutral pH, FcRn-mediated salvage of IgG is preceded by receptor-independent, pinocytic uptake of IgG into cells. The expression of FcRn in highly pinocytic macrophagesCitation17–Citation19 raises questions concerning the capacity of this salvage pathway to rescue internalized IgG from degradation in these cells. In this context, although earlier studies using bone marrow transfers of FcRn-sufficient or deficient cells have shown that FcRn-mediated recycling in hematopoietic cells contributes to IgG homeostasis,Citation18,Citation20–Citation22 it was not possible to elucidate the role of this receptor in individual cell subsets within this class. Further, bone marrow transfers can result in differing extents of reconstitution, particularly of long-lived tissue-resident macrophages that are derived from yolk sac erythro-myeloid progenitors (EMPs) or fetal hematopoietic stem cells (HSCs).Citation23–Citation25
Here, we have used complementary approaches to establish that FcRn activity in macrophages plays an indispensable role in regulating IgG homeostasis. Site-specific deletion of FcRn reveals that, in the absence of FcRn expression, macrophages become a degradative sink for IgG. Consistent with this, depletion of macrophages in FcRn-deficient mice results in increased persistence and serum levels of IgG, further supporting the degradative activity of FcRn-deficient macrophages. However, macrophage depletion in FcRn-sufficient (wild type) mice does not affect IgG persistence, demonstrating that in the presence of FcRn, the pinocytic uptake of IgG by macrophages is compensated for by FcRn-mediated recycling of IgG by these cells. Collectively, our studies demonstrate the importance of FcRn-mediated salvage in macrophages to avoid hypercatabolism of IgG.
Results
The effect of macrophage depletion on the dynamic behavior of IgG
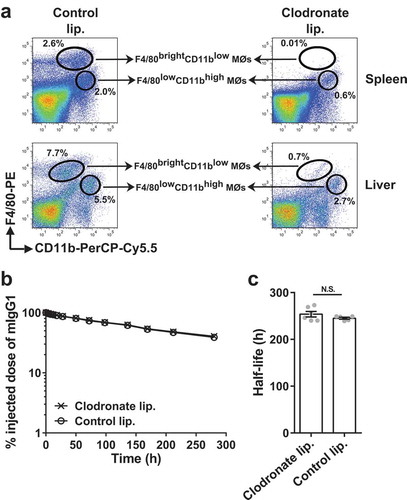
Depletion of macrophages using clodronate encapsulated in liposomes can be instrumental for the determination of the role of macrophages in a physiological process.Citation26 We, therefore, used this approach to investigate the effect of macrophage depletion on the persistence of injected, radiolabeled mIgG1 in C57BL/6J mice. Due to their size, intravenously delivered liposomes are primarily taken up by macrophages in the liver and spleen.Citation26 Following intravenous (i.v.) delivery of clodronate liposomes, 95% depletion of F4/80brightCD11blow [yolk sac EMP- or fetal HSC-derived] and ~50–65% depletion of F4/80lowCD11bhigh [bone marrow HSC-derived] macrophagesCitation23-Citation25 was observed in liver and spleen (), indicating that clodronate liposomes are efficient in depleting both subtypes of macrophages. Notably, clodronate liposome-mediated reduction in macrophage numbers did not affect the in vivo half-life of mIgG1 [~254 ± 6 (SEM) h with depletion and ~245 ± 2 h without depletion; ,].
Figure 1. Depletion of splenic and liver macrophages in wild type mice does not affect the half-life of IgG. (a) C57BL/6J mice (2–3 mice/group) were intravenously treated with clodronate (1.5 mg/dose) or PBS (control) liposomes at 0 h and 48 h, and single cell suspensions from spleens and livers were isolated at 120 h. Macrophage populations were analyzed using flow cytometry and data for one representative mouse from each group is shown. The gating strategies used for the identification of macrophage (MØ) populations are shown in Supplementary Figure 9C and D. (b) C57BL/6J mice (5–6 mice/group) were intravenously treated with clodronate (1.5 mg/dose) or PBS (control) liposomes at 0 h and 48 h. 125I-labeled mIgG1 was injected (i.v.) at 18 h following the first injection of clodronate liposomes, and whole body radioactivity levels were determined at the indicated times. (c) β-phase half-lives of mIgG1 in different mice obtained by fitting the pharmacokinetic data to a decaying bi-exponential model. Error bars indicate SEM. N.S., no significant difference (p > .05; two-tailed Student‘s t-test). Data shown in panels b and c is representative of two independent experiments.
Immunohistochemistry was used to further investigate the fluid phase pinocytic uptake of macromolecules by macrophages in vivo. An engineered human IgG1 (D265A; to ablate binding to FcγRs)Citation27,Citation28 was delivered intravenously into mice that lack FcRn expression [FcRn knockout (KO)Citation;29 global KO, G-KO]. FcRn-deficient mice were used to avoid a contribution of FcRn to recycling, and, combined with the loss of FcγR binding, hIgG1D265A, therefore, represents a fluid phase marker in these analyses. hIgG1D265A was predominantly detected within macrophages in the liver, skin, muscle and intestine and liver sinusoidal cells (Supplementary Figures 1–4). The high pinocytic activity of macrophages, together with our observation that clodronate liposome-mediated reduction of macrophage numbers does not affect the in vivo half-life of mIgG1, suggest that the pinocytic activity of these cells is compensated for by IgG salvage.
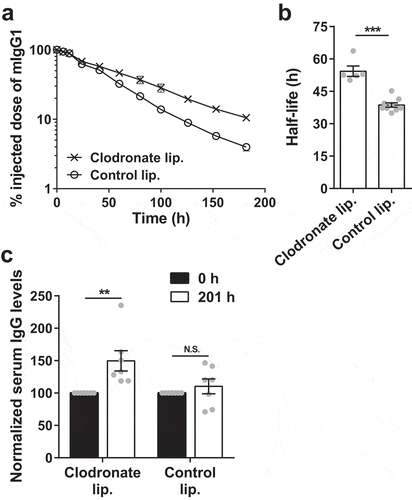
Compensatory salvage of IgG following pinocytic uptake by macrophages is most likely mediated by FcRn, which is known to be expressed in all hematopoietic cells.Citation17-Citation19 This led us to hypothesize that the depletion of macrophages in mice that do not express FcRn (G-KO mice) would remove a degradative compartment and result in increased IgG levels and persistence. The clearance rate of mIgG1 and steady state IgG levels were therefore determined in G-KO mice following the depletion of liver and splenic macrophages. Significantly, the β-phase half-life of mIgG1 was higher in G-KO mice treated with clodronate liposomes (~54 ± 2 h) compared to mice treated with control liposomes (~39 ± 1 h; ,). The steady state levels of serum IgG in the clodronate liposome-treated mice were also increased by ~50 ± 16% (). Thus, the depletion of macrophages in the liver and spleen, which are believed to constitute less than 35% of the total macrophage number in the body,Citation8 results in higher in vivo persistence and steady state IgG levels in FcRn-deficient mice.
Figure 2. Depletion of splenic and liver macrophages in G-KO mice results in an increase in half-life and serum levels of IgG. G-KO mice were treated with clodronate (1.5 mg/dose) or PBS (control) liposomes at 0 h and 48 h. (a, b) 125I-labeled mIgG1 was injected (i.v.) at 18 h, and whole body radioactivity levels determined at the indicated times (a; 3–4 mice/group) post-antibody injection. Data shown is representative of two independent experiments. (b) β-phase half-lives of mIgG1 were obtained by fitting the pharmacokinetic data to a decaying mono-exponential model. (c) Normalized IgG levels in serum samples collected 0 h prior to and 201 h post-first injection of clodronate liposomes. Error bars indicate SEM. Significant differences (**, p < .01; ***, p < .001; two-tailed Student‘s t-test) between the groups are indicated. N.S., no significant difference (p > .05; two-tailed Student‘s t-test). Data shown for panels b and c is combined from two independent experiments (5–9 mice/group).
Generation of mice with selective loss of FcRn in macrophages
To analyze the ability of FcRn to counteract the high pinocytic activity of macrophages, we next studied the effects of specific deletion of FcRn in macrophages on IgG homeostasis. We first generated site-specific, FcRn KO mice using Cre-loxP technology. This approach involves the crossing of mice expressing Cre recombinase under the control of cell type-specific promoters with floxed mice that are engineered to harbor exons 5–7 of the FcRn gene flanked by loxP sequences.Citation30 We intercrossed FcRn-floxed miceCitation19 with mice transgenic for LysM-CreCitation31 with the goal of generating mice that specifically lack FcRn in macrophages. Our earlier study indicated that hematopoietic cells and/or endothelial cells are the primary contributors to FcRn-mediated IgG homeostasis in vivo.Citation19 Therefore, for comparative purposes, we also used CD11c-Cre,Citation32 CD19-CreCitation33 and Tie2e-CreCitation34 mice in intercrosses with FcRn-floxed mice, with the goal of generating mice with FcRn deletion in dendritic cells (DCs), B cells and endothelial cells, respectively. To determine the specificity of FcRn deletion in these conditional FcRn KO mice, flow cytometry was used to assess the accumulation of a fluorescently labeled mutated human IgG1 (MST-HN, M252Y/S254T/T256E/H433K/N434F,Citation35 specific for hen egg lysozyme) that binds to mouse FcRn with increased affinity at acidic and near-neutral pH (Kd = 1.2 nM at pH 6.0; Kd = 7.4 nM at pH 7.2).Citation35 In contrast to wild type human IgG1, this mutated human IgG1 is efficiently endocytosed into FcRn-expressing cells by receptor-mediated uptake, and can, therefore, be used as an indicator of the levels of functional FcRn.Citation35 The high affinity of this mutated IgG for mouse FcRn at near neutral pH also results in inefficient release during exocytic events and retention within cells. As a control for fluid phase accumulation, a fluorescently labeled mutated human IgG1 variant (H435A) of the same antigen binding specificity, but with substantially reduced affinity for FcRn relative to its wild type parent antibody,Citation36 was used.
To demonstrate FcRn-dependent accumulation of MST-HN within cells, and show a correlation between levels of FcRn expression and cell-associated MST-HN, we used a human endothelial cell line (HMEC-1)Citation37 that expresses very low or negligible levels of endogenous FcRn. HMEC-1 cells were co-transfected with expression plasmids encoding mouse FcRn tagged with an enhanced green fluorescent protein (mFcRn-GFP) and mouse β2-microglobulin (mβ2m). In addition, HMEC-1 cells were co-transfected with expression plasmids encoding a human FcRn variant tagged with an enhanced green fluorescent protein (hFcRn-L136-GFP) and human β2-microglobulin (hβ2m). Since HMEC-1 cells are of human origin, a mutated variant of hFcRn, hFcRn-L136, which resembles mFcRn in binding specificity,Citation38 was used in addition to mFcRn to exclude the possibility that cross-species differences in cytosolic tail sequences might affect subcellular trafficking. Following incubation with fluorescently labeled MST-HN or H435A, HMEC-1 cells transfected with mFcRn or hFcRn-L136 had high levels of cell-associated MST-HN, whereas the fluorescence signal of H435A within these cells, or accumulation of either proteins within untransfected cells, was similar to background autofluorescence levels (Supplementary Figure 5). Importantly, the accumulation of MST-HN was proportional to the level of FcRn-GFP expression. These analyses, therefore, validated the use of MST-HN and H435A to assess FcRn expression levels in different cell types.
Consistent with the analyses shown in Supplementary Figure 5, the levels of fluorescently labeled MST-HN and H435A were similar (i.e., very low) in all splenic immune cells derived from G-KO mice ( and ). In contrast, the accumulation of fluorescently labeled MST-HN was greater than that observed for H435A in all wild type (C57BL/6J) splenic immune cells analyzed, except in neutrophils, which do not express functional FcRn.Citation19 Importantly, based on the difference in the uptake of MST-HN and H435A, the functional FcRn expression levels in C57BL/6J mice can be ranked in the following order: macrophages > monocytes > DCs ≥ B cells.
Table 1. Summary of FcRn expression, half-lives of mouse IgG1 and serum IgG levels in mice expressing FcRn in different cell types.
Figure 3. Specificity of loss of FcRn function in FcRnflox/flox mice with site-specific Cre expression. (a) Splenocytes were isolated, pooled (from 2 to 3 mice/genotype) and incubated with anti-FcγRIIB/III (2.4G2) antibody at 4°C followed by Alexa 647-labeled MST-HN or H435A mutant at 37°C to assess FcRn-mediated uptake. Fluorescence levels associated with each of the indicated cell types were determined using flow cytometry. Cell populations were identified as follows: macrophages, F4/80brightCD11blow; monocytes, Ly6ChighCD11bint; DCs, CD11c+CD11b+; B cells, CD23highCD21low and neutrophils, Ly6CintCD11bhigh. The gating strategies used for the identification of these cell types are shown in Supplementary Figure 8A-C. Data shown is representative of at least two independent experiments. (b) Mice were perfused and kidneys, lungs and livers were harvested. Single-cell suspensions from these organs were isolated, pooled (from 2 mice/genotype) and treated as in panel A except that CD45+F4/80brightCD11blow and CD45+F4/80lowCD11bhigh macrophages were analyzed. These macrophage populations were identified using the gating strategy shown in Supplementary Figure 8D. M-KO, LysM-Cre-FcRnflox/flox (macrophage-specific FcRn KO); M-Het, LysM-Cre-FcRnflox/+ (control); B-DC-KO, CD19-Cre-FcRnflox/flox (B cell- and DC-specific FcRn KO); B-DC-Het, CD19-Cre-FcRnflox/+ (control); Tie2e-KO, Tie2e-Cre-FcRnflox/flox (multiple cell type-specific FcRn KO); Tie2e-Het, Tie2e-Cre-FcRnflox/+ (control); G-KO, FcRn-/- (global FcRn KO); MST-HN, mutated human IgG1 with increased affinity for FcRn;Citation35 H435A, mutated (control) human IgG1 with negligible binding for FcRn.Citation36 Data shown is representative of at least two independent experiments.
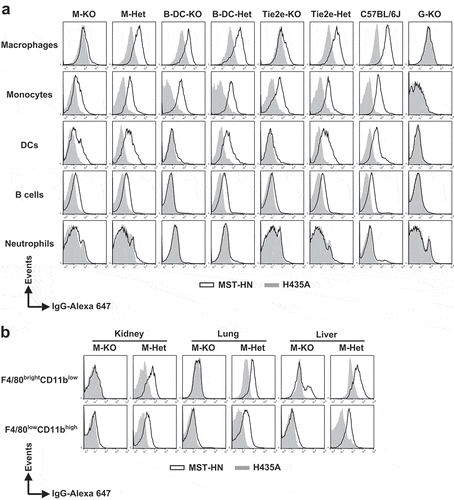
In LysM-Cre-FcRnflox/flox mice (M-KO mice), complete loss of FcRn was observed in splenic macrophages, whereas partial loss of FcRn activity was observed in splenic monocytes and DCs ( and ). We also analyzed the levels of functional FcRn in F4/80brightCD11blow and F4/80lowCD11bhigh macrophagesCitation23 in lung, kidney, and liver in M-KO mice (). In control mice, both of these cell subsets had functional FcRn, whereas loss of FcRn activity was observed in M-KO mice, with the exception of a fraction of hepatic F4/80brightCD11blow macrophages. In addition, FcRn function in both heart and lung endothelial cells in M-KO mice was similar to that in control mice (). Consistent with the observations of others using the LysM-Cre strain to target other genes,Citation31,Citation39 our analyses show that FcRn deletion in M-KO mice occurs predominantly in macrophages.
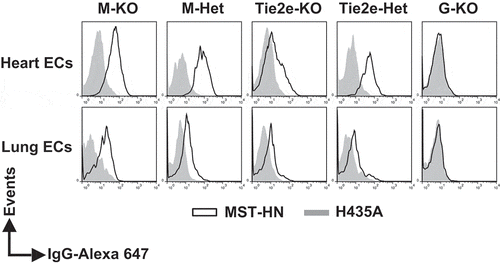
Figure 4. The levels of functional FcRn are reduced in endothelial cells in Tie2e-KO mice but not in M-KO mice. Single cell suspensions from heart and lung were isolated, pooled (from 3–5 mice/genotype) and incubated with anti-FcγRIIB/III (2.4G2) antibody at 4°C followed by Alexa 647-labeled MST-HN or H435A mutant at 37°C to assess FcRn-mediated uptake. Fluorescence levels associated with heart- or lung-derived CD31+CD105+isolectin B4+ endothelial cells (ECs) were determined using flow cytometry. ECs were identified according to the gating strategies shown in Supplementary Figure 9A and B. M-KO, LysM-Cre-FcRnflox/flox (macrophage-specific FcRn KO); M-Het, LysM-Cre-FcRnflox/+ (control); Tie2e-KO, Tie2e-Cre-FcRnflox/flox (FcRn KO in multiple different cell types); Tie2e-Het, Tie2e-Cre-FcRnflox/+ (control); G-KO, FcRn-/- (global FcRn KO); MST-HN, mutated human IgG1 with increased affinity towards FcRn;Citation35 H435A, mutated (control) human IgG1 with negligible binding towards FcRn.Citation36 Data shown are representative of at least two independent experiments.
In contrast to the macrophage specificity of FcRn deletion in M-KO mice, for CD11c-Cre-FcRnflox/flox mice, complete loss of FcRn activity was observed in macrophages and DCs (Supplementary Figure 6), indicating that CD11c-Cre-mediated deletion is of broad specificity. Consequently, CD11c-Cre-FcRnflox/flox mice were not used further in these studies. For CD19-Cre-FcRnflox/flox mice (B-DC-KO mice), loss of FcRn function was observed in both DCs and B cells ( and ). Further analyses revealed that the homozygous FcRnflox/flox (Cre−) strain, but not the heterozygous strain, has substantially reduced levels of functional FcRn in DCs compared with wild type mice. In addition, the generation of several non-DC-targeting Cre-expressing strains that are homozygous for floxed FcRn alleles resulted in strains that have different levels of functional FcRn in DCs. For example, we observed higher levels of functional FcRn in DCs in M-KO mice compared with B-DC-KO mice ( and ). B-DC-KO mice, therefore, provided a model for selective loss of FcRn in both B cells and DCs.
Cre expression in Tie2e-Cre mice has been reported to be specific to endothelial cells.Citation34 However, Tie2e-Cre-FcRnflox/flox (Tie2e-KO) mice exhibited partial or complete loss of FcRn function in macrophages, B cells, DCs and heart endothelial cells, although this activity was retained in lung endothelial cells (, and ), excluding their possible use as endothelial-specific knockouts. However, these mice exhibited the normal activity of FcRn in monocytes compared with the corresponding control mice, Tie2e-FcRnflox/+ (Tie2e-Het; and ), and therefore provided a model to differentiate the contribution of monocytes and macrophages to IgG homeostasis.
FcRn expression in macrophages compensates for their high degradative capacity
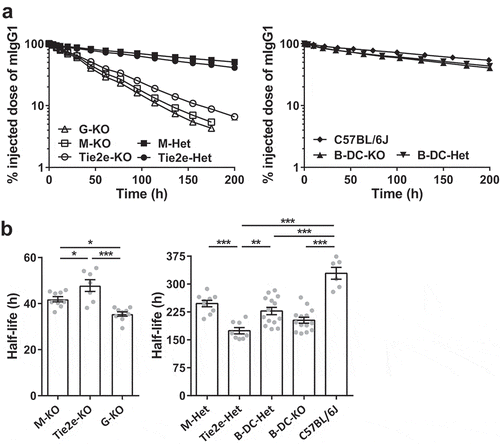
In order to assess the effect of site-specific deletion of FcRn in macrophages on IgG homeostasis in vivo, we next analyzed the pharmacokinetics of mIgG1 and steady state serum IgG levels in M-KO, Tie2e-KO, B-DC-KO, their corresponding controls and, as comparators, G-KO, and wild type C57BL/6J mice. The β-phase half-life of mIgG1 was the longest in C57BL/6J mice (~329 ± 16 h; and ). Notably, the β-phase half-life of mIgG1 in M-KO mice (~42 ± 1 h) was substantially reduced compared with that in the corresponding control mice (M-Het, LysM-Cre-FcRnflox/+; ~248 ± 9 h), and was slightly longer than that observed in G-KO mice (~36 ± 1 h) ( and ). In addition, the half-life of mIgG1 in Tie2e-KO mice (~48 ± 3 h), which have normal levels of FcRn activity in monocytes but greatly reduced levels in macrophages, was substantially lower compared with that in the respective control mice (Tie2e-Het; ~175 ± 8 h) and marginally higher than that in M-KO mice ( and ). In contrast, the half-life of mIgG1 in B-DC-KO mice (~203 ± 8 h) was not significantly different from that in the corresponding control mice (B-DC-Het, CD19-Cre-FcRnflox/+; ~228 ± 10 h) ( and ), indicating that FcRn expression in DCs and B cells does not contribute to IgG homeostasis.
Figure 5. Loss of FcRn function in macrophages results in substantial reductions in the half-life of IgG. (a) Different FcRn KO and control mice (3–6 mice/genotype) were injected (i.v.) with 125I-labeled mIgG1 and whole body radioactivity levels determined at the indicated times. Data shown are representative of at least two independent experiments. (b) β-phase half-lives of mIgG1 in different FcRn KO and control mice were determined by fitting the pharmacokinetic data to a mono-exponential (M-KO, Tie2e-KO, and G-KO) or bi-exponential model (M-Het, Tie2e-Het, B-DC-Het, B-DC-KO and C57BL/6J). Error bars indicate SEM. Significant differences (*, p < .05; **, p < .01; ***, p < .001; one-way ANOVA followed by Tukey‘s multiple comparisons test) between the groups are indicated. M-KO, LysM-Cre-FcRnflox/flox (macrophage-specific FcRn KO); M-Het, LysM-Cre-FcRnflox/+ (control); B-DC-KO, CD19-Cre-FcRnflox/flox (B cell- and DC-specific FcRn KO); B-DC-Het, CD19-Cre-FcRnflox/+ (control); Tie2e-KO, Tie2e-Cre-FcRnflox/flox (FcRn KO in multiple different cell types); Tie2e-Het, Tie2e-Cre-FcRnflox/+ (control); G-KO, FcRn-/- (global FcRn KO). Data shown in panel b is combined from at least two independent experiments (n = 6–14 mice/group).
The steady state levels of IgG in the sera of M-KO, G-KO, Tie2e-KO, and B-DC-KO mice were also determined. The IgG levels in M-KO and G-KO mice were ~0.48 mg/ml and ~0.13 mg/ml, respectively, compared with ~1.43 mg/ml in the control mice (M-Het) ( and ). The IgG levels in Tie2e-KO mice were ~0.54 mg/ml, and 1.34 mg/ml in the control mice (Tie2e-Het) ( and ). This represents ~threefold and ~2.5-fold reductions in IgG levels in M-KO and Tie2e-KO mice, respectively, compared with corresponding controls. Consistent with the lack of effect of FcRn deletion in B cells and DCs on IgG pharmacokinetics ( and ), the steady state serum IgG levels were similar between B-DC-KO and B-DC-Het mice (Supplementary Figure 7 and ). In addition to the regulation of IgG homeostasis, FcRn maintains albumin levels.Citation40 The serum albumin levels were also reduced in M-KO and Tie2e-KO mice, although the decreases relative to corresponding controls were lower compared with those observed for IgG (). This difference is possibly due to feedback regulation of albumin synthetic rates.Citation41 The levels of IgG and albumin are ~3.8-fold and ~1.7-fold higher, respectively, in M-KO mice relative to G-KO mice, indicating FcRn-mediated salvage of these ligands by FcRn-sufficient cells in M-KO mice. Collectively, our observations using conditional knockout mice indicate that FcRn-mediated salvage of IgG in macrophages plays a major role in maintaining IgG levels.
Figure 6. Loss of FcRn function in macrophages results in ~2–3 fold reductions in steady state levels of serum IgG and albumin and reduces the effects of FcRn inhibition. Serum IgG (a) and albumin (b) levels in the different mouse strains. Data shown are derived from 5 to 13 mice/genotype. (c) M-KO, M-Het, and G-KO mice were treated (i.v.) with 1 mg MST-HN mutant and serum IgG levels determined at the indicated times post-injection. Error bars indicate SEM. Significant differences [*, p < .05; **, p < .01; ***, p < .001; one-way ANOVA followed by Tukey‘s (a, b) or Dunnett‘s (c) multiple comparisons test] between the groups are indicated. M-KO, LysM-Cre-FcRnflox/flox (macrophage-specific FcRn KO); M-Het, LysM-Cre-FcRnflox/+ (control); Tie2e-KO, Tie2e-Cre-FcRnflox/flox (multiple cell type-specific FcRn KO); Tie2e-Het, Tie2e-Cre-FcRnflox/+ (control); G-KO, FcRn-/- (global FcRn KO); MST-HN, FcRn inhibitor. Data shown in panel c is combined from two independent experiments (n = 8–9 mice/group).
![Figure 6. Loss of FcRn function in macrophages results in ~2–3 fold reductions in steady state levels of serum IgG and albumin and reduces the effects of FcRn inhibition. Serum IgG (a) and albumin (b) levels in the different mouse strains. Data shown are derived from 5 to 13 mice/genotype. (c) M-KO, M-Het, and G-KO mice were treated (i.v.) with 1 mg MST-HN mutant and serum IgG levels determined at the indicated times post-injection. Error bars indicate SEM. Significant differences [*, p < .05; **, p < .01; ***, p < .001; one-way ANOVA followed by Tukey‘s (a, b) or Dunnett‘s (c) multiple comparisons test] between the groups are indicated. M-KO, LysM-Cre-FcRnflox/flox (macrophage-specific FcRn KO); M-Het, LysM-Cre-FcRnflox/+ (control); Tie2e-KO, Tie2e-Cre-FcRnflox/flox (multiple cell type-specific FcRn KO); Tie2e-Het, Tie2e-Cre-FcRnflox/+ (control); G-KO, FcRn-/- (global FcRn KO); MST-HN, FcRn inhibitor. Data shown in panel c is combined from two independent experiments (n = 8–9 mice/group).](/cms/asset/d06bd5cb-28c6-4c54-b3c6-5be86a249505/kmab_a_1602459_f0006_b.gif)
The effect of FcRn inhibition is reduced in mice bearing FcRn-deficient macrophages
To further investigate the activity of FcRn in M-KO mice, serum IgG levels in M-KO, control M-Het, and G-KO mice were determined following the treatment of these mice with the MST-HN mutantCitation19 (‘Abdeg’) that is engineered to bind with increased affinity to mouse FcRn. This mutant is a specific inhibitor of FcRn-mediated recycling of IgG, and therefore enhances the degradation of endogenous antibodies when delivered into mice.Citation35 As expected, MST-HN treatment did not modulate the already low serum IgG levels in G-KO mice (). Serum IgG levels in control, M-Het mice were reduced by ~33%, 50%, 68% and 62% at 6, 12, 24 and 48 h post-treatment, respectively, and the IgG levels recovered to only ~51% of the levels in untreated mice at 120 h post-treatment (). In M-KO mice, a ~38% decrease in the relatively low serum IgG levels was observed at 24 h post-treatment (). These observations indicate that in the presence of FcRn inhibitors, macrophages represent a major degradative sink for IgG.
Discussion
Antibodies of the IgG class are essential for humoral immunity. In addition, the use of IgG-based therapeutics to treat cancer, autoimmune and infectious diseases, and prevent transplant rejection has expanded enormously over the past several decades.Citation42 FcRn-mediated salvage of IgG from degradation provides a homeostatic mechanism to regulate IgG levels and transport in the body. The majority of IgG isotypes are internalized into cells by fluid phase, pinocytic uptake due to their very low binding affinity for FcRn at extracellular, near neutral pH.Citation15,Citation16 This raises the question as to whether pinocytically active cells such as macrophages, which are also of relatively high abundance, have compensatory FcRn-mediated salvage pathways to avoid dysregulation of IgG homeostasis and consequent immunodeficiency. Here, we demonstrate that such compensatory pathways exist, and this is supported by our observations that, although macrophage depletion in FcRn-sufficient mice does not affect IgG clearance rate, specific loss of FcRn expression in macrophages results in IgG hypercatabolism.
Consistent with the activity of FcRn-deficient macrophages as a degradative sink, the steady state serum levels and half-life of IgG are increased in FcRn-/- mice following clodronate liposome-mediated depletion of splenic and liver macrophages. The lack of such an effect in wild type mice indicates that FcRn function counteracts the high pinocytic activity in macrophages to maintain IgG homeostasis. Such a compensatory mechanism may inhibit major changes to IgG homeostasis under pathological conditions such as infection, obesity, and lymphedema, which are characterized by significant increases in macrophage numbers.Citation43–Citation45
In this study, we used Cre-loxp technology to generate cell type-specific FcRn KO mice. To produce macrophage-specific FcRn KO mice, LysM-Cre miceCitation31 were crossed with FcRnflox/flox mice.Citation19 Although it is established that Cre expression in LysM-Cre mice is limited to macrophages and neutrophils,Citation31 neutrophils in C57BL/6J mice do not express FcRn.Citation19 Analysis of the efficiency and specificity of FcRn deletion in LysM-Cre-FcRnflox/flox mice revealed almost complete loss of FcRn function in splenic macrophages and partial loss of FcRn function in monocytes and DCs. In contrast to the specificity of Cre-mediated deletion in LysM-Cre-FcRnflox/flox mice, the use of CD11c-Cre miceCitation32 resulted in complete loss of FcRn activity in macrophages in addition to DCs. These observations are consistent with several recent reports highlighting the lack of specificity of Cre expression in multiple mouse lines, including CD11c-Cre.Citation39,Citation46 Although ‘off-target’ deletion has not been reported for CD19-Cre mice,Citation33 crossing these mice with FcRn-floxed mice generated a mouse strain with loss of FcRn activity in both B cells and DCs. The loss of FcRn function in DCs in these mice can be explained by our observation that Cre+-FcRnflox/flox mice have different levels of functional FcRn in DCs, dependent on the particular Cre strain (non-DC targeting) used. The mechanistic basis for this differential reduction in FcRn levels, which is not seen in the corresponding mice heterozygous for the floxed FcRn allele, is currently not understood. Nevertheless, CD19-Cre-FcRnflox/flox mice provided a model to investigate the effect of reduced FcRn function in both DCs and B cells. The generation of a mouse line in which deletion of FcRn is specific to B cells was not pursued further, since the pharmacokinetic behavior and steady state serum levels of IgG were not affected in the double B cell/DC FcRn knockout strain, indicating that FcRn in these cells does not significantly contribute to the maintenance of IgG levels. Further, the use of Tie2e-Cre-FcRnflox/flox mice, which exhibit partial loss of FcRn function in heart endothelial cells and in all hematopoietic cells except monocytes, which are less pinocytically active than macrophagesCitation47 and are present in relatively low numbers,Citation48 allowed us to exclude a major contribution of FcRn in monocytes to IgG homeostasis.
Earlier studies using bone marrow chimeras of FcRn-/- and wild type mice demonstrated an important contribution of FcRn activity in the hematopoietic compartment to IgG homeostasis, although variability in this contribution was observed.Citation18,Citation20–Citation22 Further, it was not possible to elucidate the role of individual hematopoietic cell subsets in these analyses. In contrast to our observations using mice that specifically lack FcRn activity in macrophages, loss of FcRn function in B cells and DCs does not have a significant effect on IgG homeostasis. The low pinocytic rates of B cells and mature DCs,Citation47 combined with the relatively small numbers of highly pinocytic (immature) DCs in the body, could provide an explanation for the substantial difference in the contribution of FcRn in B cells and DCs to maintaining IgG levels compared with macrophages. In addition to steady state IgG levels, we analyzed serum albumin levels in mouse strains that lack FcRn activity in macrophages. Although substantial reductions of albumin levels occur in these mice, the decreases are not as marked as for IgG. Earlier studies demonstrated that albumin biosynthesis by hepatocytes increases by 20% in FcRn-/- mice,Citation41 suggesting that the reduced effect on albumin levels in our study is most likely due to compensatory upregulation of albumin biosynthesis.
The inhibition of FcRn using (engineered) antibodies that interact with high affinity with FcRn through their Fc region or variable domains is effective in the treatment of autoantibody-mediated pathologies in various animal models of disease.Citation49–Citation51 Further, recent clinical trials have indicated that these FcRn inhibitors have promise for use as therapeutics.Citation52–Citation54 The results of this study suggest that FcRn blockade in macrophages is a major contributor to the decrease in IgG levels that are observed.Citation52–Citation54 In addition to a role in IgG homeostasis, studies have shown that FcRn plays diverse roles in other processes, such as antigen presentation and IgG transport across epithelial barriers.Citation10,Citation55 In particular, the role of FcRn in both cross-presentation and delivery of immune complex-associated antigen to MHC Class II processing/loading compartments in DCs is well documented,Citation22,Citation56,Citation57 and leads to the suggestion that in DCs, the primary function of FcRn may be to serve this role rather than to regulate IgG homeostasis. In contrast, although the involvement of FcRn in antigen presentation in macrophages has to date not been explored, it is likely that this receptor serves dual functions, namely in both recycling (‘monomeric’) IgG and, for immune complexes, antigen presentation in this cell type.
In summary, this study underscores a major contribution of FcRn-mediated recycling in macrophages to compensate for their extraordinarily high fluid phase pinocytic activity. Importantly, loss of FcRn function in these cells leads to IgG hypercatabolism, whereas depletion of FcRn-deficient macrophages removes a degradative sink, resulting in an increase in IgG levels. These findings not only have relevance to understanding the dynamic behavior of therapeutic and diagnostic antibodies, but also to the homeostatic regulation of humoral immunity.
Materials and methods
Mice
FcRn-/-,Citation29 LysM-Cre,Citation31 CD11c-Cre,Citation32 CD19-CreCitation33, and C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice with a floxed FcRn allele have been described previously;Citation19 these mice will be available at the Jackson Laboratory (Bar Harbor, ME) with the stock number 032630. Tie2e-Cre miceCitation34 were a kind gift from Dr. Xiaoxia Li (Cleveland Clinic, OH). Mice were bred in a specific pathogen-free facility at the University of Texas Southwestern Medical Center or Texas A&M University, and were handled in compliance with institutional policies and protocols approved by the Institutional Animal Care and Use Committees.
Antibodies and labeling
A mutated variant, hIgG1D265A, which does not bind to FcγRs,Citation27,Citation28 was purchased from Crown Bioscience (cat# C0020-3; San Diego, CA). NS0 transfectants expressing mutated derivatives of humanized anti-hen egg lysozyme IgG1 (HuLys10),Citation58 MST-HN (M252Y/S254T/T256E/H433K/N434F) and H435A, which bind to FcRn with increased affinity or negligible affinity, respectively, were generated and cultured as described previously.Citation35,Citation36 The recombinant antibodies were purified from culture supernatants using lysozyme-Sepharose.Citation58 mIgG1 (anti-hen egg lysozyme, D1.3Citation59) was purified from hybridoma culture supernatants using lysozyme-Sepharose.Citation58
Iodination (125I) of mIgG1 was carried out using Iodogen as previously described.Citation16 MST-HN and H435A were labeled with Alexa Fluor 647 (Alexa 647) carboxylic acid (succinimidyl ester; Life Technologies, Grand Island, NY) using previously described methods.Citation13 The binding behavior of Alexa 647-labeled antibodies was analyzed using surface plasmon resonance (BIAcore).
Pharmacokinetic experiments
6–10-week old male and female mice were fed 0.1% Lugol (Sigma-Aldrich, St. Louis, MO) in drinking water starting from 72 h prior to i.v. injection in the tail vein with 125I-labeled mIgG1 (10–15 μg per mouse). Levels of whole body radioactivity were measured at the indicated times using an Atom Lab 100 dose calibrator (Biodex Medical Systems, Shirley, NY).
To investigate the effects of splenic and liver macrophage depletion on mIgG1 pharmacokinetics, C57BL/6J and GKO mice were intravenously injected with clodronate (1.5 mg/dose) or control (phosphate-buffered saline (PBS)) liposomes (Encapsula NanoSciences, Brentwood, TN) at 0 h and 48 h. 125I-labeled mIgG1 was injected (i.v.) at 18 h and whole body clearance analyzed as described above.
To determine the half-lives of injected mIgG1 in mice that exhibit relatively slow clearance of injected antibody (M-Het, Tie2e-Het, B-DC-Het, B-DC-KO, and C57BL/6J), the whole body counts over time were fitted to bi-exponential decay curves. In mice that exhibit rapid clearance of injected mIgG1 (M-KO, Tie2e-KO, and G-KO), the α- and β-phases cannot be clearly defined. Consequently, the whole body counts obtained from these mice over time were fitted to mono-exponential decay curves. The α- and β-phase half-lives were determined using a non-linear least squares minimization method implemented using custom scripts written in MATLAB (Mathworks, Natick, MA).
Flow cytometry analyses
CDC/EU.HMEC-1 (HMEC-1),Citation37 a human dermal microvascular-derived endothelial cell line, was generously provided by F. Candal (Centers for Disease Control, Atlanta, GA). The cells were transiently co-transfected with expression plasmids encoding mouse FcRn tagged with enhanced green fluorescent protein (mFcRn-GFP)Citation35 and mouse β2-microglobulin (mβ2m) or mutated human FcRn variant tagged with enhanced green fluorescent protein (hFcRn-L136-GFP)Citation60 and human β2-microglobulin (hβ2m) using previously described methods.Citation13 Following culture (~18 h) in phenol red-free, IgG-depleted Ham’s F-12K medium, transfected cells were pulsed with 5 µg/ml Alexa 647-labeled MST-HN or H435A for 45 min at 37°C in medium adjusted to pH 7.2–7.4. The cells were washed with Dulbecco’s phosphate-buffered saline (DPBS), harvested by trypsinization and analyzed using flow cytometry.
For the analysis of functional FcRn levels in hematopoietic cell types in the spleen, 6–12 week old male and female mice were euthanized, and spleens were harvested and used to make single cell suspensions by mechanical disruption and forcing cells through 70 µm cell strainers (Becton-Dickinson, San Jose, CA). The cell suspensions were depleted of erythrocytes using red blood cell lysis buffer and washed with DPBS. Subsequently, the cells were incubated with anti-FcγRIIB/III antibody (purified from the 2.4G2 hybridoma; HB-197 available from the ATCC) for 15 min at 4°C, followed by incubation in IgG-depletedCitation13 phenol red-free cDMEM (pH 7.2–7.4) containing 5 μg/ml Alexa 647-labeled MST-HN or H435A mutant for 45 min at 37°C. Following one wash with DPBS, the cells were incubated on ice with fluorescently labeled antibodies to identify the following cell types: macrophages (F4/80brightCD11blow), monocytes (Ly6ChighCD11bint), classical DCs (CD11c+CD11b+), follicular B cells (CD23highCD21low) and neutrophils (Ly6CintCD11bhigh). The gating strategies employed for the identification of these cell types are shown in Supplementary Figure 8A-C.
To evaluate functional FcRn levels in heart and lung endothelial cells, a modified version of a previously described protocolCitation61 was followed to prepare single-cell suspensions from these tissues. Briefly, the animals (6–12-week old male and female mice) were euthanized, and the heart and lungs were harvested and washed with calcium- and magnesium-free Hank‘s Balanced Salt Solution (HBSS; Life Technologies, Grand Island, NY) to remove excess blood. The washed tissues were then mechanically dissociated into small pieces. To obtain single cell suspensions, the diced tissues were incubated in calcium- and magnesium-free HBSS containing 1 mg/ml of collagenase type 1 (Worthington Biochemical Corp., Lakewood, NJ) and 2.4 mg/ml of dispase (Life Technologies, Grand Island, NY) for 45 min at 37°C. Following incubation, the tissue homogenates were filtered through 100 μm cell strainers (Becton-Dickinson, San Jose, CA) and the resulting cell suspensions were depleted of erythrocytes using red blood cell lysis buffer. Cells were washed with HBSS and incubated with anti-FcγRIIB/III (2.4G2) antibody for 15 min at 4°C, followed by incubation in IgG-depletedCitation13 phenol red-free cDMEM (pH 7.2–7.4) containing 5 μg/ml Alexa 647-labeled MST-HN or H435A mutants for 45 min at 37°C. The cells were subsequently washed, and, while on ice, stained with fluorescently labeled isolectin B4 (cat# FL-1201; Vector Laboratories, Burlingame, CA) and antibodies to identify endothelial cells (CD31+CD105+isolectin B4+) according to the gating strategies shown in Supplementary Figure 9A and B.
Analysis of functional FcRn expression in tissue-resident macrophages was carried out using the protocol described above for endothelial cells, except that the animals (6–12 week old male and female mice) were anesthetized and intracardially perfused with 10–20 ml 10 U/ml heparin in DPBS before the collection of organs (kidneys, lungs and livers) and washes were performed with calcium- and magnesium-free DPBS. Yolk sac EMP- (or fetal HSC-) and bone marrow HSC-derived macrophages were identified as CD45+F4/80brightCD11blow and CD45+F4/80lowCD11bhigh, respectively (Supplementary Figure 8D).Citation23 To evaluate the percentage of macrophages in the spleen and liver following treatment with clodronate or control liposomes, the animals were euthanized, and the spleens and livers were harvested and then used to make single cell suspensions using the protocol described above. Since the mice were not perfused, macrophage subsets were identified following the exclusion of monocytes and neutrophils (Ly6ChighCD11bint and Ly6CintCD11bhigh, respectively) as follows: spleen yolk sac EMP- or fetal HSC-derived macrophages, F4/80brightCD11blow; spleen bone marrow HSC-derived macrophages, F4/80lowCD11bhigh; liver yolk sac EMP- or fetal HSC-derived macrophages, CD45+F4/80brightCD11blow; liver bone marrow HSC-derived macrophages, CD45+F4/80lowCD11bhigh.Citation23 The gating strategies used for the identification of these cells are shown in Supplementary Figure 9C and D.
Flow cytometry analyses were performed using a FACSCalibur (Becton-Dickinson, San Jose, CA), LSRFortessa (Becton-Dickinson, San Jose, CA) or Accuri C6 (Becton-Dickinson, San Jose, CA), and data were analyzed using FlowJo (Tree Star, Ashland, OR). The following antibodies for flow cytometry analyses were purchased from Becton-Dickinson (BD, San Jose, CA), eBioscience (San Diego, CA) or Biolegend (San Diego, CA): anti-Ly-6C-FITC (BD cat# 553104), anti-F4/80-PE (eBioscience cat# 12–4801-82), anti-CD11b-PerCP-Cy5.5 (BD cat# 550993), anti-CD11b–FITC (BD cat# 553310), anti-CD11c–PE (BD cat# 557401), anti-B220 – PerCP (BD cat# 553093), anti-CD23-FITC (BD cat# 553138), anti-CD21-PE (BD cat# 552957), anti-CD45-FITC (BD cat# 553079), anti-CD45-APC (BD cat# 559864), anti-CD105-PE (eBioscience cat# 12–1051-82) and anti-CD31-PerCP-efluor710 (eBioscience cat# 46–0311-82).
Quantitation of serum IgG and albumin levels
Serum IgG and albumin levels were assessed using sandwich ELISAs and previously described methods.Citation19 Polyclonal rabbit anti-mouse IgG (cat# 616000; Life Technologies, Grand Island, NY) and polyclonal goat anti-mouse albumin (cat# ab19194; Abcam, Cambridge, MA) were used as coating antibodies. Horseradish peroxidase (HRP)-conjugated polyclonal rabbit anti-mouse IgG (cat# 616520; Life Technologies, Grand Island, NY) and HRP-conjugated polyclonal goat anti-mouse albumin (cat# ab19195; Abcam, Cambridge, MA) were used as secondary antibodies. Mouse IgG (cat# 015–000-002) and albumin (cat# A3139) standards were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA) and Sigma-Aldrich (St. Louis, MO), respectively.
To study the effects of splenic and liver macrophage depletion on serum IgG levels, 8–10 week old male and female G-KO mice were intravenously injected with clodronate (1.5 mg/dose) or control (PBS) liposomes (Encapsula NanoSciences, Brentwood, TN) at 0 h and 48 h, and IgG levels determined in serum samples at 0 h (before administration of liposomes) and 201 h.
For analyzing the effects of FcRn inhibition on serum IgG levels, 10 week old male and female M-KO, M-Het and G-KO mice were intravenously treated with 1 mg MST-HN and serum samples were collected at 0 (immediately prior to administration of MST-HN), 6, 12, 24, 48 and 120 h to determine IgG levels.
Immunofluorescence analyses
To analyze the accumulation of intravenously administered IgG in vivo, 8 week old male and female mice were injected (i.v.) with 1.5 mg hIgG1D265A (cat# C0020-3; Crown Bioscience, San Diego, CA). Ten hours later, mice were anesthetized and intracardially perfused with 10–20 ml 10 U/ml heparin in DPBS, followed by excision of organs or tissues. The organs/tissues were immediately embedded in Tissue-Tek® OCT compound (Sakura Finetek USA, Torrance, CA), frozen and stored at −80°C. Eight-micrometer thick sections were prepared, and either used immediately for fixation/staining or stored at −80°C for subsequent fixation/staining. Tissue sections were dried for ~16–20 h, fixed in acetone (−20°C) for 2.5 min, and dried again for ~16–20 h. After washing with DPBS, sections were blocked using 3% bovine serum albumin (Fisher Scientific, Fair Lawn, NJ), followed by incubation with rabbit anti-human IgG (cat# SA5-10223; Thermo Fisher Scientific, Rockford, IL) and rat anti-mouse CD31 (clone 390 (cat# 102412) and/or MEC13.3 (cat# 102501); Biolegend, San Diego, CA) or rat anti-mouse F4/80 (cat# ab6640; Abcam, Cambridge, MA) diluted in 3% bovine serum albumin. Following washes with DPBS containing 0.05% Tween 20 (Fisher Scientific, Fair Lawn, NJ), the sections were incubated with 1% goat serum (Sigma-Aldrich, St. Louis, MO). Bound primary antibodies were detected using cross-adsorbed Alexa 555-labeled polyclonal goat anti-rat IgG (cat# 405420; Biolegend, San Diego, CA) and Alexa 647-labeled polyclonal goat anti-rabbit IgG (cat# A21245; Life Technologies, Grand Island, NY) diluted in 5% goat serum. Subsequent to washes with DPBS containing 0.05% Tween 20, coverslips were mounted using Vectashield mounting medium containing DAPI (Vector Laboratories, Burlingame, CA).
Sections were imaged using a Zeiss Axio Observer Z1 inverted epifluorescence microscope (Zeiss, Oberkochen, Germany) equipped with a Zeiss 20X, 0.8 NA Plan-Apochromat objective, a Hamamatsu Orca-ER camera (Hamamatsu Photonics, Hamamatsu city, Japan) as detector and a broadband LED lamp (X-Cite 110LED, Excelitas Technologies, Waltham, MA) as excitation source. Images were acquired with standard filtersets for Alexa 555 (Cy3-4040C-ZHE M327122; Semrock, Rochester, NY), Alexa 647 (Cy5-4040C-ZHE M327126; Semrock, Rochester, NY) and DAPI (DAPI-5060C-ZHE M327119; Semrock, Rochester, NY). The data were processed and displayed using in-house written software, MIATool.Citation62 To allow comparison between different organs, the same lamp intensity and exposure times were used for acquiring data for the Alexa 647 channel (hIgG). No intensity adjustments were carried out for the Alexa 647 channel, and the images are presented as acquired. For adjustment of background levels for the Alexa 555 channel (CD31 or F4/80), the mean of the 50th percentile pixel values from all images obtained using secondary antibody only for each individual organ was used as the threshold, except for muscle. For muscle, due to the relatively high background signal, independent linear adjustments were made for the Alexa 555 channel for display purposes. Images were exported into Inkscape for final composition of the figures.
Statistical analyses
Statistical analyses of data were carried out using two-tailed Student’s t-test or one-way ANOVA (GraphPad Software, La Jolla, CA). p values of less than 0.05 were taken to be significant.
Data availability
The data that supports the findings in this study are available upon request from the corresponding author.
Code availability
Software is available upon request at www.wardoberlab.com/software/miatool.
Author contributions
D.K.C., R.J.O. and E.S.W. designed the experiments. D.K.C. and H.P.M. performed the ex vivo and in vivo experiments. D.K.C., X.W. and R.V. performed the microscopy analyses. D.K.C., X.W., R.V., R.J.O. and E.S.W. analyzed the data. D.K.C., R.J.O. and E.S.W. wrote the manuscript that was edited by all other authors.
Abbreviations
Supplemental Material
Download MS Word (7.9 MB)Acknowledgments
We are indebted to Dr. Werner Müller (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) and Martin Hafner (University of Cologne, Cologne, Germany) for generating the FcRn-floxed mice.19 We are grateful to Dr. Xiaoxia Li (Cleveland Clinic, OH) for generously providing Tie2e-Cre mice. We thank Dr. Priyanka Khare for assistance with mouse husbandry and serum IgG quantitation. This work was supported in part by grants from the National Institutes of Health (RO1 AI 39167 and RO1 AR 56478).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
Related Research Data
References
- Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41(1):21–35. doi:10.1016/j.immuni.2014.06.013.
- Lavin Y, Mortha A, Rahman A, Merad M. Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol. 2015;15(12):731–44. doi:10.1038/nri3920.
- Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, Barton GM. Tissue-resident macrophages are locally programmed for silent clearance of apoptotic cells. Immunity. 2017;47(5):913–27. doi:10.1016/j.immuni.2017.10.006.
- Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16(4):343–53. doi:10.1038/ni.3123.
- Lim JP, Gleeson PA. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol. 2011;89(8):836–43. doi:10.1038/icb.2011.20.
- Steinman RM, Brodie SE, Cohn ZA. Membrane flow during pinocytosis. A stereologic analysis. J Cell Biol. 1976;68:665–87.
- Abrass CK. Measurement of the rates of basal pinocytosis of horseradish peroxidase and internalization of heat-aggregated IgG by macrophages from normal and streptozotocin-induced diabetic rats. Immunology. 1988;65:411–15.
- Lee SH, Starkey PM, Gordon S. Quantitative analysis of total macrophage content in adult mouse tissues. Immunochemical studies with monoclonal antibody F4/80. J Exp Med. 1985;161:475–89.
- Yu YR, O‘Koren EG, Hotten DF, Kan MJ, Kopin D, Nelson ER, Que L, Gunn MD. A protocol for the comprehensive flow cytometric analysis of immune cells in normal and inflamed murine non-lymphoid tissues. PLoS One. 2016;11(3):e0150606. doi:10.1371/journal.pone.0150606.
- Challa DK, Velmurugan R, Ober RJ, Ward ES. FcRn: from molecular interactions to regulation of IgG pharmacokinetics and functions. Curr Top Microbiol Immunol. 2014;382:249–72. doi:10.1007/978-3-319-07911-0_12.
- Pyzik M, Rath T, Lencer WI, Baker K, Blumberg RS. FcRn: the architect behind the immune and nonimmune functions of IgG and albumin. J Immunol. 2015;194(10):4595–603. doi:10.4049/jimmunol.1403014.
- Ober RJ, Martinez C, Lai X, Zhou J, Ward ES. Exocytosis of IgG as mediated by the receptor, FcRn: an analysis at the single-molecule level. Proc Natl Acad Sci USA. 2004;101:11076–81. doi:10.1073/pnas.0402970101.
- Ober RJ, Martinez C, Vaccaro C, Zhou J, Ward ES. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J Immunol. 2004;172:2021–29.
- Claypool SM, Dickinson BL, Wagner JS, Johansen FE, Venu N, Borawski JA, Lencer WI, Blumberg RS. Bidirectional transepithelial IgG transport by a strongly polarized basolateral membrane Fcγ-receptor. Mol Biol Cell. 2004;15:1746–59. doi:10.1091/mbc.e03-11-0832.
- Raghavan M, Gastinel LN, Bjorkman PJ. The class I major histocompatibility complex related Fc receptor shows pH-dependent stability differences correlating with immunoglobulin binding and release. Biochemistry. 1993;32:8654–60.
- Kim JK, Tsen MF, Ghetie V, Ward ES. Localization of the site of the murine IgG1 molecule that is involved in binding to the murine intestinal Fc receptor. Eur J Immunol. 1994;24(10):2429–34. doi:10.1002/eji.1830241025.
- Zhu X, Meng G, Dickinson BL, Li X, Mizoguchi E, Miao L, Wang Y, Robert C, Wu B, Smith PD, et al. MHC class I-related neonatal Fc receptor for IgG is functionally expressed in monocytes, intestinal macrophages, and dendritic cells. J Immunol. 2001;166:3266–76.
- Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol. 2007;179:4580–88.
- Perez-Montoyo H, Vaccaro C, Hafner M, Ober RJ, Mueller W, Ward ES. Conditional deletion of the MHC Class I-related receptor, FcRn, reveals the sites of IgG homeostasis in mice. Proc Natl Acad Sci USA. 2009;106(8):2788–93. doi:10.1073/pnas.0810796106.
- Kobayashi K, Qiao SW, Yoshida M, Baker K, Lencer WI, Blumberg RS. An FcRn-dependent role for anti-flagellin immunoglobulin G in pathogenesis of colitis in mice. Gastroenterology. 2009;137(5):1746–56. doi:10.1053/j.gastro.2009.07.059.
- Rath T, Baker K, Dumont JA, Peters RT, Jiang H, Qiao SW, Lencer WI, Pierce GF, Blumberg RS. Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit Rev Biotechnol. 2013;35(2):235–54. doi:10.3109/07388551.2013.834293.
- Qiao SW, Kobayashi K, Johansen FE, Sollid LM, Andersen JT, Milford E, Roopenian DC, Lencer WI, Blumberg RS. Dependence of antibody-mediated presentation of antigen on FcRn. Proc Natl Acad Sci USA. 2008;105(27):9337–42. doi:10.1073/pnas.0801717105.
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336(6077):86–90. doi:10.1126/science.1219179.
- Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–51. doi:10.1038/nature13989.
- Sheng J, Ruedl C, Karjalainen K. Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity. 2015;43(2):382–93. doi:10.1016/j.immuni.2015.07.016.
- Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93.
- Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6(4):443–46. doi:10.1038/74704.
- Baudino L, Shinohara Y, Nimmerjahn F, Furukawa J, Nakata M, Martinez-Soria E, Petry F, Ravetch JV, Nishimura S, Izui S. Crucial role of aspartic acid at position 265 in the CH2 domain for murine IgG2a and IgG2b Fc-associated effector functions. J Immunol. 2008;181:6664–69.
- Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, Petkova S, Avanessian L, Choi EY, Shaffer DJ, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J Immunol. 2003;170(7):3528–33.
- Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–06.
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–77.
- Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8− dendritic cells in the spleen. J Exp Med. 2007;204(7):1653–64. doi:10.1084/jem.20062648.
- Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–18.
- Kano A, Wolfgang MJ, Gao Q, Jacoby J, Chai GX, Hansen W, Iwamoto Y, Pober JS, Flavell RA, Fu XY. Endothelial cells require STAT3 for protection against endotoxin-induced inflammation. J Exp Med. 2003;198(10):1517–25. doi:10.1084/jem.20030077.
- Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol. 2005;23(10):1283–88. doi:10.1038/nbt1143.
- Firan M, Bawdon R, Radu C, Ober RJ, Eaken D, Antohe F, Ghetie V, Ward ES. The MHC class I related receptor, FcRn, plays an essential role in the maternofetal transfer of gammaglobulin in humans. Int Immunol. 2001;13:993–1002.
- Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, Lawley TJ. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–90.
- Zhou J, Mateos F, Ober RJ, Ward ES. Conferring the binding properties of the mouse MHC class I-related receptor, FcRn, onto the human ortholog by sequential rounds of site-directed mutagenesis. J Mol Biol. 2005;345(5):1071–81. doi:10.1016/j.jmb.2004.11.014.
- Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods. 2014;408:89–100. doi:10.1016/j.jim.2014.05.009.
- Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC, Anderson CL. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J Exp Med. 2003;197:315–22.
- Kim J, Bronson CL, Hayton WL, Radmacher MD, Roopenian DC, Robinson JM, Anderson CL. Albumin turnover: FcRn-mediated recycling saves as much albumin from degradation as the liver produces. Am J Physiol Gastrointest Liver Physiol. 2006;290(2):G352–G360. doi:10.1152/ajpgi.00286.2005.
- Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit‘. Nat Rev Drug Discov. 2018;17(3):197–223. doi:10.1038/nrd.2017.227.
- Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS, Allen JE. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332(6035):1284–88. doi:10.1126/science.1204351.
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808. doi:10.1172/JCI19246.
- Ghanta S, Cuzzone DA, Torrisi JS, Albano NJ, Joseph WJ, Savetsky IL, Gardenier JC, Chang D, Zampell JC, Mehrara BJ. Regulation of inflammation and fibrosis by macrophages in lymphedema. Am J Physiol Heart Circ Physiol. 2015;308(9):H1065–H1077. doi:10.1152/ajpheart.00598.2014.
- Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nat Immunol. 2007;8(7):665–68. doi:10.1038/ni0707-665.
- Weissleder R, Nahrendorf M, Pittet MJ. Imaging macrophages with nanoparticles. Nat Mater. 2014;13(2):125–38. doi:10.1038/nmat3780.
- Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325(5940):612–16. doi:10.1126/science.1175202.
- Liu L, Garcia AM, Santoro H, Zhang Y, McDonnell K, Dumont J, Bitonti A. Amelioration of experimental autoimmune myasthenia gravis in rats by neonatal FcR blockade. J Immunol. 2007;178:5390–98.
- Patel DA, Puig-Canto A, Challa DK, Perez Montoyo H, Ober RJ, Ward ES. Neonatal Fc receptor blockade by Fc engineering ameliorates arthritis in a murine model. J Immunol. 2011;187(2):1015–22. doi:10.4049/jimmunol.1003780.
- Challa DK, Bussmeyer U, Khan T, Montoyo HP, Bansal P, Ober RJ, Ward ES. Autoantibody depletion ameliorates disease in murine experimental autoimmune encephalomyelitis. MAbs. 2013;5(5):655–59. doi:10.4161/mabs.25439.
- Kiessling P, Lledo-Garcia R, Watanabe S, Langdon G, Tran D, Bari M, Christodoulou L, Jones E, Price G, Smith B, et al. The FcRn inhibitor rozanolixizumab reduces human serum IgG concentration: A randomized phase 1 study. Sci Transl Med. 2017;9(414):1208. doi:10.1126/scitranslmed.aan1208.
- Ulrichts P, Guglietta A, Dreier T, van Bragt T, Hanssens V, Hofman E, Vankerckhoven B, Verheesen P, Ongenae N, Lykhopiy V, et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J Clin Invest. 2018;128(10):4372–86. doi:10.1172/JCI97911.
- Ling L, Hillson JL, Tiessen RG, Bosje T, van Iersel MP, Nix DJ, Markowitz L, Cilfone NA, Duffner J, Streisand J, et al. M281, an anti-FcRn antibody: pharmacodynamics, pharmacokinetics, and safety across the full range of IgG reduction in a first-in-human study. Clin Pharmacol Ther. 2018. doi:10.1002/cpt.1276.
- Baker K, Rath T, Pyzik M, Blumberg RS. The role of FcRn in antigen presentation. Front Immunol. 2014;5:408. doi:10.3389/fimmu.2014.00408.
- Baker K, Qiao SW, Kuo TT, Aveson VG, Platzer B, Andersen JT, Sandlie I, Chen Z, de Haar C, Lencer WI, et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8−CD11b+ dendritic cells. Proc Natl Acad Sci USA. 2011;108(24):9927–32. doi:10.1073/pnas.1019037108.
- Baker K, Rath T, Flak MB, Arthur JC, Chen Z, Glickman JN, Zlobec I, Karamitopoulou E, Stachler MD, Odze RD, et al. Neonatal Fc receptor expression in dendritic cells mediates protective immunity against colorectal cancer. Immunity. 2013;39(6):1095–107. doi:10.1016/j.immuni.2013.11.003.
- Foote J, Winter G. Antibody framework residues affecting the conformation of the hypervariable loops. J Mol Biol. 1992;224:487–99.
- Amit AG, Mariuzza RA, Phillips SE, Poljak RJ. Three-dimensional structure of an antigen-antibody complex at 2.8 Å resolution. Science. 1986;233:747–53.
- Gan Z, Ram S, Vaccaro C, Ober RJ, Ward ES. Analyses of the recycling receptor, FcRn, in live cells reveal novel pathways for lysosomal delivery. Traffic. 2009;10(5):600–14. doi:10.1111/j.1600-0854.2009.00887.x.
- Marelli-Berg FM, Peek E, Lidington EA, Stauss HJ, Lechler RI. Isolation of endothelial cells from murine tissue. J Immunol Methods. 2000;244:205–15.
- Chao J, Ward ES, Ober RJ. A software framework for the analysis of complex microscopy image data. IEEE Trans Inf Technol Biomed. 2010;14(4):1075–87. doi:10.1109/TITB.2010.2049024.