
ABSTRACT
Chinese hamster ovary (CHO) cells are the biopharmaceutical industry’s primary means of manufacturing therapeutic proteins, including monoclonal antibodies. The major challenge in cell line development for the production of recombinant biopharmaceuticals lies in generating and isolating rare high-producing stable clones, amongst thousands of low-producing or unstable clones, in a short period of time. One approach to accomplish this is to use the glutamine synthetase (GS) selection system, together with the GS inhibitor, methionine sulfoximine (MSX). However, MSX can only increase protein productivity to a limited extent. Often productivity will drop when MSX is removed from the system. We evaluated a congenital GS mutation, R324C, which causes glutamine deficiency in human as an attenuated selection marker for CHO cell line generation. We also created a panel of GS mutants with diminished GS activity. Our results demonstrated that using attenuated GS mutants as selection markers significantly increased antibody production of stably transfected pools. Furthermore, these stably transfected pools sustained high productivity levels for an extended period of time, whereas cells transfected with wild-type GS lost considerable protein productivity over time, particularly after MSX was removed. In summary, the use of attenuated GS as a selection marker in CHO cell line development bypasses the need for MSX, and generates stable clones with significantly higher antibody productivity.Abbreviations: CHO: Chinese hamster ovary; CMV: Cytomegalovirus; DHFR: Dihydrofolate reductase; GFP: Green fluorescent protein; GOI: gene-of-interest; GS: Glutamine synthetase; IRES: internal ribosomal entry site; MSX: Methionine sulfoximine; MTX: Methotrexate; psGS: pseudoGS; RVDs: Repeated variable di-residues; TALENs: transcription activator-like effector nucleases; VCD: Viable cell density; ZFNs: zinc finger nucleases.
Introduction
The Chinese hamster ovary (CHO) cell line is one of the most commonly employed mammalian cell lines used for the production of therapeutic recombinant biologics.Citation1 This is due to its adaptability to suspension, protein-free media culture systems, rapid proliferation in large-scale suspension, ease of genetic manipulation, and glycosylation profile, which is similar to human.Citation2–Citation5 The stable cell line generation process is a critical part of biotherapeutics production. To obtain a stable and high-producing cell line, several rounds of selection are performed to generate stable pools. Subsequently, hundreds to thousands of single clones from such bulk transfected cultures are isolated and screened for stable high producers. This process is time-consuming and laborious.
Of CHO cell expression systems, the dihydrofolate reductase (DHFR) and the glutamine synthetase (GS) selection systems are the most commonly used.Citation3 In cell lines deficient in the DHFR gene, e.g., CHO-DG44 cells, selection is performed in the medium without hypoxanthine and thymidine.Citation6,Citation9 Amplification steps through rounds of increasing concentration of methotrexate (MTX) are often included to improve productivity.Citation6–Citation8 However, this step increases the timeline for cell line generation, and introduces potential instability upon removal of MTX selection pressure.Citation10–Citation13 The GS selection system offers a time advantage because it requires fewer gene copies for the survival of the stable cells, and thus allows faster selection of high producers.Citation3,Citation14 The GS selection system uses the essential activity of GS in catalyzing the ATP-dependent condensation of glutamate and ammonia to generate glutamine. The use of ammonia in the glutamine production also aids in the reduction of toxic ammonia waste accumulation. In cell lines that do not express sufficient level of endogenous GS, such as mouse myeloma lines, removal of glutamine supplementation serves as sufficient selection pressure to isolate productive stable cells.Citation14,Citation15 In cell lines with sufficient level of GS, such as CHO cells, addition of the GS inhibitor, MSX allows the selection of productive stable cells.Citation16,Citation17 Initially, a 2-step selection approach was employed with the first round of 25–50 µM MSX for enhanced stringency and a second round of 100–1000 µM for amplification.Citation16,Citation18–Citation20 However, selection in such CHO cell lines may result in the survival of a large number of poor producers, as some CHO cells are able to survive up to 5 mM MSX.Citation21 To improve selection stringency for the GS system, a CHO GS knockout host cell line was generated,Citation22,Citation23 and enhanced productivity has been observed. To further improve selection stringency and eliminate the use of MSX, the SV40E promoter driving the GS gene expression was weakened to reduce the GS level.Citation24 This enhanced the selection pressure, generating even higher producers in the absence of MSX.
Apart from productivity, the stability of production of a high-producing clone is a critical attribute. In the cell line generation process, after rounds of selections for high-producing clones, the stability of production has to be monitored over at least 60 generations. Production stability is important because the cell line undergoes a substantial number of generations when going from a vial of cells from the working cell bank to a large commercially relevant scale of production. It has been observed that a significant percentage of GS-selected CHO production cell lines lost more than 20–30% of their original productivity.Citation23,Citation25 Thus, the process of trying to obtain a high-producing, and yet stable, clone can be time-consuming and laborious.
In a bid to improve stability and achieve high productivity without the use of MSX, we evaluated the idea of using an attenuated GS selection marker. Two attenuated GS mutants containing R324C and R341C mutations were identified in two unrelated infants with congenital GS deficiency.Citation26 To further enhance the efficiency of the cell line generation process to generate high-producing and stable clones, we evaluated the impact of using these attenuated GS mutants as selection markers in a vector configuration where the expression of the gene of interest was linked directly to the selection marker via an internal ribosomal entry site (IRES). We tested the congenital GS patient mutation R324C as the selection marker. We generated our in-house CHO-GS knockout cell line using transcription activator-like effector nucleases (TALENs) and zinc finger nucleases (ZFNs) technologies. In this setup, a single step of glutamine removal from the media is sufficient to generate high-producing stable pools. In order to identify additional novel attenuating mutations of GS, we generated a panel of GS mutants using alanine scanning mutagenesis at the substrate- and ATP-binding sites based on the mammalian GS crystal structure.Citation27 We identified several additional single point mutations at either of these sites that significantly diminished GS activity. Using these attenuated GS mutants as selection markers, we demonstrated enhanced productivity of green fluorescent protein (GFP) and a monoclonal anti-CD20 antibody GA101 (obinutuzumab) compared to using the wild-type GS (GSwt) as a selection marker. Single clones derived from GS mutant-selected stable pools produced higher titers of the antibody, and more importantly, a larger percentage of these single clones remained stable over time compared to those obtained using GSwt with MSX-selected stable pools.
Results
Targeted knockout of the GS gene in CHO-K1 cells
To knock out the GS gene, adherent CHO-K1 cells were transfected with plasmids encoding the GS left and right TALENs. The GS TALENs are specific to exon 6 of the GS gene (). The activity of the GS TALENs was confirmed by the T7E1 mismatch assay (). To enhance the process of isolating successful GS knockout mutants among transfected cells, we used a selection procedure that included the use of the glutamine-free medium.
Figure 1. Generation of CHO-GS−/- cells using TALENs DNA-editing technology. (a) Generation and characterization of TALENs targeting CHO GS. A suitable TALEN target site was found in exon 6 of the GS gene with the TALEN binding sites separated by a spacer of 15-bp. Graphic was generated with the aid of CRISPy software.Citation28 TALENs each with 18.5 RVDs and 20-bp specificity were assembled by Golden Gate Cloning. The first 5ʹ T of each binding site is recognized by the N-terminus of TALEN protein. The NH monomer was used to recognize G for increased specificity. (b) T7E1 mismatch assay indicates that the designed GS TALENs has gene modification activity of 25.8%. (c) DNA sequence analysis of the GS gene from CHO WT cells and CHO-GS−/- cells for clones 1, 2 and 3. The TALEN binding sites are in red, the 15-bp spacer is in black and underlined. The resulting deletions identified in different CHO mutants are shown on the right. Clone 3 showed two different deletions in the two alleles. (d) Western blot analysis confirms the successful knockout of GS in the CHO cells. Endogenous GS was detected in cell lysates. Whole cell proteins were resolved through SDS-PAGE under reducing conditions, and, after transfer onto PVDF membrane, detected with mouse monoclonal anti-GS antibody and probed with secondary HRP-conjugated goat anti-mouse IgG + IgM (H + L) antibody. Actin was used as a loading control. (e) TALENs-inactivated GS mutants show dependency for glutamine. Cells were seeded overnight in six-well plates at 200 000 cells per well in normal media and replaced with the respective media as indicated. Five days later, cell growth and morphology were observed. Both parental CHO-K1 and GS knockout cells showed normal growth and morphology in normal media. However, in glutamine-deprived media, GS-mutant demonstrated growth-arrest characteristics with rounded and shrunk morphology. On the other hand, CHO-K1 WT cells showed slower growth in glutamine-deprived media, but maintained its characteristic fibroblastic morphology.
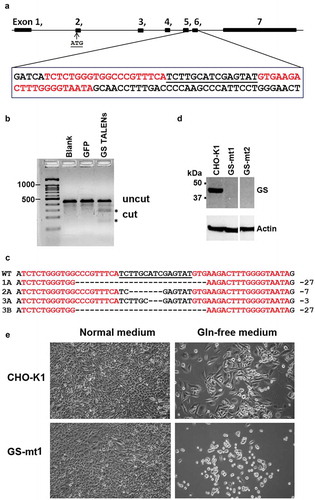
GS TALEN-transfected cells were single-cell sorted three days after transfection and cultured in 96-well plates in the presence of L-glutamine. The single cell clones were duplicated to separate 96-well plates upon confluency. In a normal medium, both CHO-K1 and GS knockout mutants (GS−/-) exhibited comparable growth (data not shown). In a glutamine-free medium, mutant cells with successfully inactivated GS activity showed dependency on glutamine and perish upon glutamine deprivation. In contrast, wild-type CHO cells continued to grow, although slower, in the glutamine-free medium due to its ability to synthesize endogenous glutamine (). Further genetic analysis revealed that CHO cells with only one allele of the GS gene knocked out also showed a glutamine-dependent phenotype. Nonetheless, this method helped us to quickly rule out the wild type cells and focus on potential mutants.
Molecular characterization of more than 20 of these putative GS knockout clones was carried out by amplifying the exon 6 containing the targeted locus using PCR. Sequencing results from three such clones (clones 1, 2, and 3) are shown in . They have insertion or deletion mutations characteristic of TALEN-mediated DNA cleavage and non-homologous end joining (NHEJ) repair (). In some clones, only one mutation was detected, which may be due to identical mutations. Other possibilities include chromosomal translocation or deletion of a larger fragment of the chromosome that includes one of the PCR primer sites. Other clones are bi-allelic with two different mutations. Western blot analysis verified that these clones were indeed deficient in GS protein (). We confirmed the GS knockout in clones 1 and 2.
Evaluation of attenuated GS mutant R324C as a selection marker for stable cell line generation
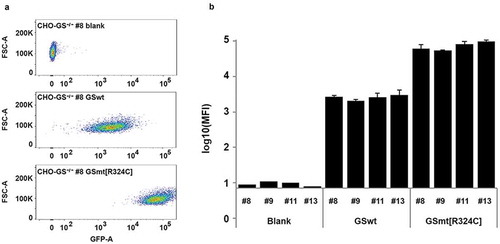
With the CHO-GS−/- lines isolated, we could use the GS gene as the selection marker in expression vectors for the production of biologics. As the congenital attenuated GS mutant R324C reported in some patients showed a significantly reduced GS activity, we wondered whether GS mutant R324C could enhance the selection stringency, and therefore increase productivity. We compared GSwt and R324C as selection markers for the generation of stable lines that expressed GFP as the reporter protein in CHO-GS−/- lines. Either the GFP-IRES-GSwt or GFP-IRES-GS R324C construct was transfected into various CHO-GS−/- single clones – clone number 8, 9, 11 and 13. There was a clear enhancement of GFP intensity levels in the stable pools generated with R324C compared to those generated with GSwt as the selection marker (). The results were consistent across three transfection pools and across the different CHO-GS−/- clones ().
Figure 2. Evaluation of GS R324C as a selection marker for the generation of GFP-expressing stable cells. (a) Comparison of GSwt with congenital mutant R324C on the generation of stable GFP expressing CHO cells. GFP-IRES-GS and GFP-IRES-GS R324C constructs were transfected into GS−/- CHO cells – clones #8, #9, #11, and #13. The GS gene in these clones was previously knocked out with targeted zinc-finger nucleases (data not shown). The resulting stable pools were analyzed for GFP expression using FACSAriaIII. Representative area dot blots for GFP fluorescence vs FSC are shown. (b) GFP mean fluorescence intensity (MFI) is expressed as a mean of biological replicates (n = 2–3 + SD).
In order to translate the application of GS mutant R324C into the production of antibody, we compared the GSwt and R324C mutant as selection markers for the generation of antibody GA101 in CHO-GS−/- clone 1 cells. In the 2-promoter bicistronic vector configuration (Figure S1), the expression of the light and heavy chains of antibody GA101 is driven by a cytomegalovirus (CMV) promoter in an IRES-mediated bicistronic expression cassette while the GS selection marker is driven by a separate SV40 promoter. The antibody titers of each stable pool generated by using GSwt, GSwt treated with 25 µM MSX (GSwt + MSX), or R324C as the respective selection markers is shown in . Addition of 25 µM MSX on GSwt stable pools that survived in L-glutamine free media resulted in an average of threefold increment of antibody titer. The R324C stable pools had an average of 10-fold titer enhancement over GSwt pools (). Overall, these results show that it is possible to use an attenuated GS selection marker to increase the stringency of the selection process, generating stable cells with higher productivity.
Figure 3. Evaluation of GS R324C as a selection marker for the generation of anti-CD20 antibody GA101 expressing stable cells. (a) Antibody GA101 titers of CHO-GS−/- stable pools have been generated using GSwt or R324C as the selection marker in a bicistronic vector. At least three independent stable pools were generated under each condition in CHO-GS−/- clone 1. The GSwt pools that survived the L-glutamine deprivation selection were further treated with 25 μM MSX (GSwt + MSX) to improve the antibody production. Batch culture titers were analyzed using nephelometer IgG reagent measurement. Stability assessment of single clones isolated from one of the bicistronic GSwt + MSX (b) or R324C (c) stable pools over at least 60 generations for antibody GA101 production. IgG titers were normalized to week 0 (MSX removal) for each clone in glutamine-free media.
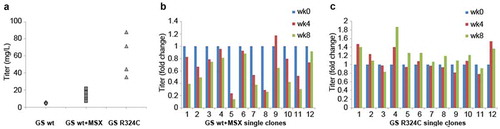
In addition to productivity, another major concern for cell line development is the production stability of the cells. Twelve single clones randomly isolated from the best-producing stable pools generated by GSwt + MSX and R324C selections were subjected to continuous culture for at least 60 generations (approximately eight weeks). Antibody titers were measured every four weeks. A cell line is typically considered stable if its productivity after 60 generations is at least 70% of its original productivity. The productivity of all of the R324C single clones remained stable after 60 generations, but only 5 of the 12 GSwt + MSX single clones maintained their stability after this period (), demonstrating the improved efficiency of generating stable single clones using the R324C mutant as a selection marker.
Identification of novel amino acid residues critical for GS activity
Structure-based sequence alignment of mammalian, plant and bacteria GS established that most of the substrate binding sites for ATP, glutamate, and ammonia are conserved.Citation27 Alignment of human and Chinese hamster GS protein sequences revealed that these residues are conserved, including the congenital mutation residues R324 and R341 (). To further support our hypothesis that GS activity regulates selection stringency, and in a bid to evaluate other GS mutants with diminished activity and possibly enhanced selection marker properties, we proceeded to identify novel residues critical for GS activity.
Figure 4. Identification of novel GS residues critical to GS activity. (a) Alignment of human and Chinese hamster GS protein sequences. Conserved sites involved in ammonium, glutamate and ATP binding are highlighted in green, red and blue as depicted together with their residue number.Citation27 The congenital disease mutations R324C and R341C are pointed out with the black arrowheads. R341C residue is not involved in neither the ammonium, glutamate nor ATP binding, and is highlighted in yellow. (b) Comparison of GSwt activity with the various alanine mutants of the conserved residues highlighted in (a). The GS activities were normalized to luciferase activity to minimize expression level variations. Each GS construct was linked to a luciferase gene via IRES in a bicistronic manner. CHO-GS−/- clone 1 cells were transiently transfected with the constructs and total cell lysates were harvested. The GS activities were evaluated using the standard GS activity assay whereby GS–catalyzed formation of γ -glutamylhydroxamate from glutamine and hydroxylamine was measured photometrically at 500 nm. The activities of the mutants were represented as fold-change to GSwt.
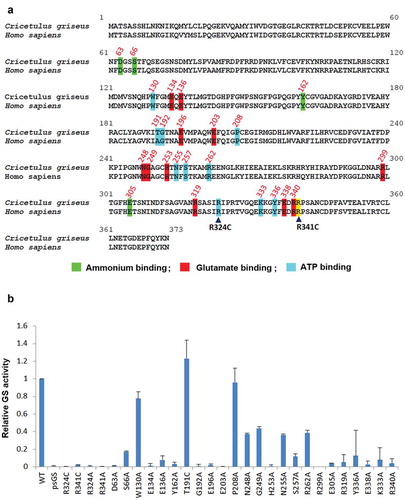
We performed alanine scanning site-directed mutagenesis of these conserved substrate-binding residues and measured their GS activity levels. Residue T191 was mutated to cysteine, as human GS carries alanine in this position. Analysis of the in vitro GS activity using a transient transfection cell-based assay showed that many of these substrate-binding sites are critical for GS activity with the exception of W130, T191 and P208 ()). The congenital mutations – R324C and R341C – were included as controls with attenuated activities, and had less than 5% of GSwt activity. Mutating R324 and R341 to alanine instead of cysteine resulted in similar levels of attenuated activities. From this assay, several other mutations were identified to be critical for GS activity. GS mutations of D63A, E134A, Y162A, G192A, E196A, E203A, H253A, R299A, E305A, E338A, and R340A resulted in a drop of GS activity level to less than 5%. The second tier of attenuated mutations at E136, S257, R319, and K333 had 5–15% of GSwt activity. The third tier of mutants that had GS activity levels between 15%-50% of GSwt is S66A, N248A, G249A, N255A, R262A, and Y336A. All three substrate-binding sites seem to be important for GS activity.
In the Chinese hamster NCBI database, a continuous stretch of genomic DNA is highly similar to the open reading frame of the functional GS gene. We cloned and sequenced this region from CHO-K1 genomic DNA. We termed this sequence pseudoGS (psGS) and aligned its translated product with GSwt (Figure S2). The sequences are mostly similar, except for a number of mutations including the R341C mutation in the psGS. We confirmed that the psGS is not expressed in CHO-K1 cells (data not shown). As R341 is critical for GS activity, the psGS indeed displayed attenuated activity compared to GSwt ()). The psGS gene is interesting because it is akin to the cDNA version of GS mRNA except that it contains numerous mutations. The mutations arise probably because it is normally not expressed, and therefore lacks selection pressure.
Evaluation of novel attenuated GS mutants on stable cell line generation
Previously, we tested and compared the antibody titer generated by GSwt and R324C selection markers in a 2-promoter bicistronic vector configuration. To further improve the selection stringency, we used a tricistronic IRES-mediated vector with a single CMV promoter driving the expression of antibody GA101 followed by the GS selection marker in the last cistron (Figure S1).Citation29 Novel GS mutants of varying activity levels were tested to demonstrate the effect of GS activity on selection pressure and titer level. Randomly, six GS mutants, D63A, E134A, E136A, G192A, E203A, and E305A, belonging to the first tier of <5% activity and involved in either ATP, glutamate or ammonia binding were selected. The GS mutants with higher activity, S257A (~12%) and N248A (~37%) were selected from the second and third tiers, respectively, as well.
Among the six GS mutants in tier 1, only pools generated with either D63A or E305A survived the selection (). Their average titers were 162 mg/L and 128 mg/L, respectively. The other four pools did not survive, perhaps due to the GS enzyme being completely inactivated. The GS activity assay might not have been sensitive enough to differentiate between highly attenuated and inactivated GS. Using the GS mutants with higher activity, S257A, and N248A, as selection markers resulted in an average titer of 91 mg/L and 28 mg/L, respectively. These results show a clear inverse correlation between GS activity level and bulk titer.
Figure 5. Comparison of the novel GS mutants to GSwt as a selection marker in generating stable antibody-producing cell lines. (a) Comparison of the antibody GA101 titer production in CHO-GS−/- clone 1 stable pools generated using GSwt, GSwt + 25 µM MSX, and the various GS mutants (as labeled) as the selection condition. The antibody GA101 was expressed in a tricistronic vector configuration. (b) Stability assessment of antibody titer from single clones isolated from one of the GSwt + MSX stable pools over at least 60 generations. Titers were normalized to week 0 (MSX removal) for each clone in L-glutamine-free media. (c) Stability assessment of antibody titer from single clones isolated from one of the GS (D63A) stable pools over at least 60 generations. Titers were normalized to week 0 for each clone in L-glutamine-free media. (d) Stability assessment of antibody titer from single clones isolated from one of the GS (S257A) stable pools over at least 60 generations. Titers were normalized to week 0 for each clone in L-glutamine-free media.
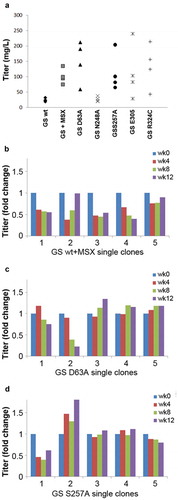
Next, we randomly isolated five single clones from each of the best-performing stable pools of GSwt + MSX, GS D63A and GS S257A and assessed their antibody GA101 productivity over at least 60 generations. Only 2 of 5 single clones from GSwt + MSX selected stable pool were able to maintain stable production of antibody GA101 (). In contrast, 4 of 5 clones from D63A and S257A stable pools were stable (). This demonstrates that using attenuated GS as selection markers can enhance the efficiency of identifying stable clones.
Growth and productivity analysis of single clones derived from GSwt and GS mutant R324C generated stable pools
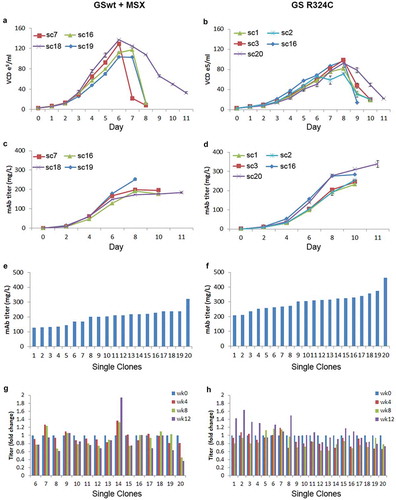
In stable cell line development, besides productivity and stability, growth rate and maximum cell density of the cell line must also be evaluated. Two best-performing stable pools using GSwt + MSX and GS R324C as selection markers in the tricistronic vector configuration were selected. We evaluated the growth curve and productivity of a few randomly selected clones from these two pools under batch culture shake flask conditions. The GSwt + MSX single clones reached a maximum viable cell density of about 10.3–12.5 x106/ml on day 6–7 (). The R324C single clones reached a maximum viable cell density of 7.1–9.3 x106/ml on day 8 (). The bulk productivity of the GSwt + MSX single clones reached a maximum of 184–252 mg/L whereas GS R324C single clones produced 234–340 mg/L of antibody GA101 (). Although the growth rate of the GS R324C single clones was slower, their productivity was higher. Batch culture in shake flasks of the top 20 single clones isolated from GSwt + MSX and GS R324C stable pools showed that the GS R324C single clones on average had a higher titer of antibody GA101 (). The stability of antibody GA101 production in these single clones was also evaluated. Titer assessment showed that all of the GS R324C-generated single clones were stable whereas only 67% of the GSwt + MSX single clones was stable after at least 60 generations ().
Figure 6. Growth and productivity assessment of single clones derived from GSwt + MSX and GS (R324C) selection in batch culture analysis. The stable pools were generated from CHO-GS−/- clone 2. Growth curves of 4 and 5 randomly selected single clones isolated from GSwt + MSX (a) and R324C (b) stable pools, respectively, are shown. Viable cell density (VCD) was measured every day. The antibody GA101 titer of clones derived from GSwt + MSX (c) and R324C (d) stable pools were measured using nephelometer every other day. Growth and productivity were assessed until the viability dropped below 50%. Antibody GA101 titer measurement of top 20 producing clones from GSwt + MSX (e) and R324C (f) stable pools were assessed in a 14-day batch culture. Stability assessment was done for the single clones with productivity more than 150 mg/L of the antibody isolated from GSwt + MSX (g) and R324C (h) stable pools. The single clones were seeded at 3x103/ml in glutamine-free 50/50 medium for a 14-day batch culture analysis of antibody GA101 production stability at week 0, 4, 8, and 12. Titers were analyzed via nephelometer.
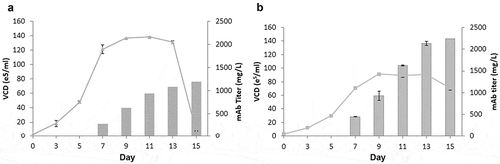
Next, we evaluated the growth and productivity of GSwt + MSX stable clone 18 and GS R324C stable clone 20 obtained from the best-producing pools () in a simple fed-batch shake flask experiment. The maximum cell density of each clone was similar to that under batch culture conditions, but with prolonged survival (). Antibody GA101 production of GSwt + MSX clone 18 only reached 1.2g/L, whereas R324C clone 20 reached 2.2g/L. These data demonstrate that the use of attenuated GS as a selection marker can enhance antibody titer production to levels comparable to industry standards.Citation30
Figure 7. Fed-batch analysis of top producing stable clone derived from GSwt + MSX and GS (R324C) selection. The top producing and stable single clones isolated from GSwt + MSX (a) and R324C (b) stable pools derived from CHO-GS−/- clone 2 were seeded in glutamine-free 50/50 medium. Cells were fed with EX-CELL Advanced Feed 1 at 7.5% (v/v) at Day 3, 5, 7, 9 and 11. Glucose levels were maintained at above 4 g/L. Viable cell numbers, viability, and antibody titers were assessed at the time points indicated in the graph, and fed-batch cultures were terminated when the viability dropped below 50%.
Discussion
One major challenge in the cell line generation process for biologics production is to efficiently identify high producing and stable clones. Several methods such as Cell Xpress technology,Citation29 transcriptome-based mRNA screening, and fluorescence-activated cell sorting (FACS)-based selection systems have been used to increase the identification of high-producing clones in a less labor-intensive way.Citation31–Citation34 Another approach is to increase the selection stringency to improve the percentage of high-producing clones.
As reported previously, the selection stringency for the GS selection system can be enhanced via the use of a CHO-GS−/- host cell line.Citation22,Citation23 With the use of a low amount of 25 μM MSX on CHO-K1SV GS−/- cells, the bulk culture productivity was doubled when compared to parental CHO-K1SV cells. The efficiency of finding high-producing clones (>2g/L) was increased sixfold. To further eliminate low- to medium-producing clones, the SV40 promoter driving the expression of the GS selection marker was weakened.Citation24 Their data suggested that the selection could be performed in the absence of MSX to improve the productivity to levels similar to those treated with MSX.
However, the use of 2-promoter vector configuration has the possibility of generating clones whereby only the GS selection marker cassette is integrated, while the gene-of-interest (GOI) could be lost or integrated into less active sites. This might result in the survival of non- to low-producing clones. In addition, in the case where the expression of more than one GOI is required, as in the case of bispecific antibody, the second promoter could not be attenuated. We sought an alternative method to improve the selection stringency by attenuating the GS activity. With an attenuated selection marker, our method can be applied directly in IRES- or F2A-mediated expression cassettes whereby the expression of the GOI is linked directly to the selection marker. With a linked configuration, the chances of selecting GS positive, but GOI-negative, clones are lower. In addition, the attenuated GS marker can be used in a 2-promoter vector configuration for the expression of more than one GOI.
Our preliminary experiments using the congenital mutation R324CCitation26 in the generation of stable GFP expression in CHO-GS−/- cells showed a significant enhancement in the mean GFP fluorescence intensity compared to GSwt selected pools (). In a 2-promoter antibody, GA101 construct, use of GS R324C also enhanced the bulk productivity over GSwt with 25 µM MSX (), supporting the use of an attenuated GS selection marker to improve selection stringency. In a single promoter tricistronic GA101 vector configuration, the titers of GS R324C selected stable pools were also enhanced (). This supported the use of attenuated GS selection marker in various vector configurations and showed it was capable of enhancing productivity.
To further prove the significance of GS activity on selection stringency, we wanted to identify new GS attenuated mutants. In addition, such novel GS mutants may potentially show further improvement in stable productivity levels, and therefore be highly valuable for cell line development. Through a comprehensive evaluation of the residues involved in ATP, glutamate and ammonia binding,Citation27 we identified a series of residues that are critical for GS activity (). In a tricistronic IRES-mediated vector configuration, some GS mutant stable pools had similar or even higher productivity than GSwt with 25 µM MSX. Among the four novel GS mutants tested, N248A retained the most GS activity and correlated with the lowest bulk productivity. We have identified two additional GS mutants, D63A, and E305A that generated similar average bulk productivity compared to that of congenital mutant R324C.
Apart from productivity, the stability of production is a critical attribute. The stability of the GS selection system in CHO cells has previously been evaluated.Citation23,Citation25,Citation35,Citation36 Evaluation of high-producing GS-selected clones demonstrated that instability stems from promoter silencing, antibody gene copy loss, and generation of secondary poor-producing populations.Citation25,Citation36 Gene copy loss could be due to unstable chromosomal structures, as shown previously in GS-selected CHO cells.Citation19 Unstable clones seemed to be more prone to apoptosis, and early prediction of unstable clones via measurement of Annexin V, and caspase 3 activities may serve as indicators for instability.Citation25 Typically, stability assessment in DHFR or GS selection systems is performed via drug removal and titer assessment for at least 60 generations. It is often observed that high-producing clones lose their productivity upon drug removal.Citation23,Citation35,Citation36 To enhance the selection stringency of GS selection system in CHO-GS−/- host cells such that MSX is dispensable, the SV40 promoter driving the expression of the GS selection marker was weakened.Citation24 The authors suggested that the absence of MSX would help to improve the production stability. However, in a comprehensive study on GS-mediated selection system in CHO cells, it has been reported that, regardless of the host cell lines of either CHO-K1 or CHO-GS−/- cells and the use of MSX concentration of up to 50 µM, the production stability of most of the stable clones decreased over 30 passages even for clones with no signs of GS-mediated gene amplification.Citation23 The authors suggested that the transcriptional activity of the transgenes is more important than copy numbers.
A key advantage of our attenuated GS system is the improvement in the efficiency of identifying a stable clone. In the 2-promoter bicistronic vector configuration, the percentage of stable clones from GSwt selection was 42% compared to 100% from the R324C selection (). In the single promoter tricistronic configuration, the percentage of stable clones from GSwt selection was 67% compared to 100% from the R324C selection (). Because GS activity is attenuated, which is akin to the use of a GS activity inhibitor, MSX is not needed in the selection process. In glutamine-free media, the clones are under constant selection pressure to maintain enough production of GS for survival, which significantly enhances the stability of production. Furthermore, elimination of MSX from the cell line generation process and manufacturing process is desirable because it negates potential regulatory concerns about the presence of trace amount of MSX in the product.
Growth analysis of GS R324C clones suggested that the attenuated GS system reached a lower maximum cell density compared to the GSwt clones (), but it was offset by higher productivity. Single clones obtained from the GS R324C selection were able to produce >2 g/L under a shake flask fed-batch condition. Even though the maximum cell density is lower in the GS mutant clones (80x105/ml to 100x105/ml), it is within a normal range and media optimization can be used to enhance the cell density. The lower growth rate we observed could be due to the stress induced by the higher productivity or altered metabolites production, which warrants further study.
In summary, we evaluated the use of attenuated GS as a selection marker for the generation of a stable cell line. We also identified novel GS mutations that resulted in varying degrees of attenuated GS activity. The use of attenuated GS enhanced the bulk productivity of stable pools generated in the absence of MSX to levels similar or higher than the typical 25 µM MSX selection. More importantly, the efficiency of identifying stable clones was significantly enhanced. Therefore, it is less time consuming to identify high-producing stable clones. The productivity and growth assessment demonstrated that using attenuated GS allows the development of a cell line that can produce at least at industry standards.
Materials and methods
TALEN design
TALENs with 18.5 repeats and 20-bp specificity were generated based on a modified Golden-Gate cloning methodology.Citation37,Citation38 To inactivate GS gene in CHO cells, a potential TALEN target site was found within GS exon 6 using the online tool TAL Effector Nucleotide Targeter 2.0 (https://talent.cac.cornell.edu/node/add/talen).Citation39,Citation40 The two 20-bp TALE binding sites are separated by a 15-bp spacer. Repeated variable di-residues (RVDs) used for the TALENs are as follows: Left TALEN: HD NG HD NG NH NH NH NG NH NH HD HD HD NH NG NG NG HD NI;
Right TALEN: NI NG NG NI HD HD HD HD NI NI NI NH NG HD NG NG HD NI HD.
T7E1 mismatch assay
Genomic DNA of CHO cells transfected with GS TALEN was extracted using DNeasy Blood & Tissue Kit (Cat: 69506, Qiagen) 72-h post-transfection. PCR amplification of GS exon 6 region encompassing the TALEN target site was carried out using AccuPrime Taq DNA Polymerase High Fidelity kit (Cat: 12346086, Thermo Fisher Scientific) and the primer pair 5ʹ- GAGGTAAGTAGAACAAGCTAGGAGC −3ʹ and 5ʹ- ACTGTCACTTCTCCAACGGGTAC −3ʹ. Purified PCR products were then heated and re-annealed slowly for heteroduplex formation. The reannealed DNA was then treated with 5 U of T7E1 (Cat: M03025, New England Biolabs) for 15 min at 37°C and resolved on 2.5% TBE agarose gel. Gene modification activities were calculated using the formula: % gene modification = 100 × (1 − (1 − fraction cleaved)1/2) as described previously.Citation41
Isolation and screening for GS knockout CHO clones
GS TALEN transfected CHO-K1 cells were single-cell sorted three days post-transfection using FACSAria III cell sorter (Becton Dickinson Biosciences) and grown in 96-well plates. Upon confluency, duplicate set of the clones were made in separate 96-well plates. Cell growth was then compared in Dulbecco’s modified Eagle’s medium (DMEM) (Cat: 10313–021, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) (Cat: 10093–177, Thermo Fisher Scientific) with or without L-glutamine supplementation (Cat: 25030–081, Thermo Fisher Scientific). In the presence of glutamine, both wild-type and mutant cells grow well. Without glutamine, unlike wild-type cells, mutant cells shrink and die. Clones showing glutamine dependency were then selected and scaled up for further characterization. CHO-GS knockout clones used in this study are as follows. Clone number 1 and 2 were isolated from the parental CHO-K1 (ATCC), and clone number 8, 9, 11 and 13 were obtained from another CHO-K1 cell line that was originally obtained from Dr. Donald K. MacCallum (University of Michigan Medical School, Ann Arbor, MI) and has been with one of us (ZS) since 1992.
Molecular characterization of mutant clones
Genomic DNA from selected clones was extracted using DNeasy Blood & Tissue Kit as per the manufacturer’s protocol. PCR amplification of GS exon 6 was carried out using AccuPrime Pfx DNA Polymerase (Cat: 12344024, Thermo Fisher Scientific) with the forward primer 5ʹ- GAGGTAAGTAGAACAAGCTAGGAGC −3ʹ and the reverse primer 5ʹ- ACTGTCACTTCTCCAACGGGTAC −3ʹ. PCR products were gel-purified and cloned into pCR-Blunt II TOPO® (Cat: K280040, Thermo Fisher Scientific). Ten bacterial clones from each TOPO reaction were sequenced to characterize the mutation.
Western blot analysis of GS knockout clones
Untransfected CHO cells and GS knockout clones were lysed with Ripa buffer to obtain total cell lysates. An equivalent amount of total cell lysates was resolved in SDS-PAGE under reducing conditions and electroblotted to PVDF membrane. The membrane was blocked with 5% nonfat dry milk in 1x phosphate-buffered saline (PBS)/0.05% Tween-20. GS protein was detected with the primary antibody, mouse anti-GS (Cat: 610517, BD Biosciences) and secondary antibody, peroxidase-conjugated goat anti-mouse IgG + IgM (H + L) (Cat: 115–035-044, Jackson ImmunoResearch Laboratories). β-actin level was detected as a loading control by using mouse anti-actin (Cat: MAB1501, Merck Millipore) and the same secondary antibody. The blots were developed with chemiluminescent substrates (Cat: 34578, Thermo Fisher Scientific) and visualized in ChemiDoc™ Touch Imaging System (Bio-Rad Laboratories, Hercules, CA).
Generation of bicistronic and tricistronic antibody GA101 expression vectors
To generate the bicistronic mAb GS vector, the mammalian expression vector pcDNA3.1 (+) (Invitrogen) was used as the vector backbone. The full-length light and heavy chains of antibody GA101 were cloned into the MCS of the vector as a bicistronic construct: light chain-IRES-heavy chain. The neomycin resistance gene after the SV40 promoter was replaced with the GSwt construct. The forward and reverse primers used for amplifying the GS gene from the tricistronic mAb GS vector were 5ʹ – GTGCTCCCGGGAGCTTGTATATCCATTTTCGGATCTGATCAAGAGACAGGATGAGGATCGGCCACCATGGCCACCTCAGCAAGTTCCCACTTGAAC −3ʹ and 5ʹ – GGTCATTTCGAACCCCAGAGTCCCGCTTAGTTTTTGTATTGGAAGGGCTCGTCG-3ʹ, respectively.
To generate the tricistronic mAb GS vector, the GS gene was first amplified from the mRNA of CHOK1 using the RNAeasy kit (Cat: 74104, Qiagen) and Superscript III One-step RT-PCR System with Platinum Taq™ DNA polymerase (Cat: 12574018, Thermo Fisher Scientific). The DHFR gene in the tricistronic DHFR vector (kindly provided by Dr. Yuangsheng Yang from Bioprocessing Technology Institute, Agency for Science Technology and Research (A*STAR), Singapore) was replaced with the GS gene via standard cloning methods. The forward and reverse primers used were 5ʹ – GTGTGACCCGGGAGATGAGGATCGTTTCGCATGGCCACCTCAGCAAGTTCCCACTTG-3ʹ and 5ʹ – GAATTCTTCGAATTAGTTTTTGTATTCGAAGGGCTCGTCGCC- 3ʹ, respectively. The antibody GA101 heavy and light chains were synthesized commercially (Integrated DNA Technologies) and cloned into the region downstream of the CMV promoter of the tricistronic GS vector in the following orientation: Light chain-IRES-heavy chain-IRESatt GS. The IRES and IRESatt sequences have been described previously.Citation27
Generation of the GS-IRES-luciferase construct
The GS gene was first cloned into the multiple cloning sites (MCS) region of the pcDNA3.1 (+) vector. The IRES construct was cloned in downstream of the GS gene and followed by the firefly luciferase gene. This expression cassette of GS-IRES-Luciferase was used to evaluate the GS activity by using luciferase activity as a surrogate reporter to normalize the GS expression levels.
Generation of the pseudo GS-IRES-luciferase construct
The coding sequence of the pseudo GS gene (psGS gene) identified in CHO cells was amplified from the genomic DNA of CHO-K1 cells using DNeasy Blood & Tissue Kit. The psGS gene was then cloned into the GS-IRES-Luciferase construct (as discussed above) to replace the wild-type GS gene, to form the expression cassette of psGS-IRES-Luciferase.
Generation of the GFP-IRES-GS construct
The coding sequence of eGFP was cloned into a minimal pcDNA3.1 vector (lacking BGHpA, f1 origin, SV40 promoter and NeoR) with NheI and NgoMIV restriction enzymes (Cat: R31315 and R05645, respectively, New England Biolabs), together with the PCR amplified and digested IRES-GSwt insert in frame with the CMV promoter and the SV40 polA sequences.
Mutagenesis of the GS gene
Mutagenesis of the specific sites on the wild-type GS sequence was achieved by designing primers with the specific mutations (Supplementary Table 1). Sites targeted were conserved between human and Chinese hamster GS. Site-directed mutagenesis reactions were performed using the QuikChange II XL site-directed mutagenesis kit (Cat: 200522, Agilent Technologies). Sequencing was performed to ensure that the mutations were achieved.
GS activity assay
Adherent CHO-K1 GS−/- cells were cultured in DMEM (Cat: 10569044, Thermo Fisher Scientific) with 10% FBS in 37°C, 5% CO2 incubator. The cells were transfected with the wild-type GS-IRES-Luc or various GS mutant-IRES-Luc constructs using LTX transfection reagent (Cat: 15338100, Thermo Fisher Scientific). The cells were harvested 24-h post-transfection with the Reporter Lysis buffer (Cat: E3971, Promega), and luciferase activity was measured according to the protocol of the Luciferase Assay kit (Cat: E1500, Promega). GS activity was measured using the standard GS activity assay whereby GS–catalyzed formation of γ-glutamylhydroxamate from glutamine and hydroxylamine was measured at 500 nm. Briefly, the cell lysate supernatant was incubated while shaking at 37°C for 45 min with 100 mM (pH 7) imidazole-HCl (Cat: I5513, Sigma-Aldrich), 50 mM L-glutamine (Cat: 25030–081, Thermo Fisher Scientific), 0.4 mM MnCl2 (Cat: 244589, Sigma-Aldrich), 62.5 mM (pH 7) hydroxylamine (Cat: 159417, Sigma-Aldrich) and 10 mM sodium arsenate (Cat: 35000, Fluka Analytical) to a final volume of 250 µl. The reaction was terminated by the addition of 250 µl FeCl3 reagent consisting of 0.37 M FeCl3 (Cat: 157740, Sigma-Aldrich), 0.2 M trichloroacetic acid (Cat: T6399, Sigma-Aldrich) and 0.67 M HCl. Precipitate was removed by centrifugation at 10,000 rpm for 5 min and the absorbance of the supernatant was measured at 500 nm using a spectrophotometer. The measured luciferase activity was used as a surrogate reporter for GS expression level for normalization.
FACS analysis of GFP-IRES-GS transfectants
When cells showed >90% viability over two to three passages after transfection with different GFP-IRES-GS constructs, they were analyzed for GFP expression using a FACSAriaIII cell sorter (BD Biosciences) equipped with FACSDiva software. Ten million cells per stable pool were washed in PBS without calcium and magnesium (Cat: 14190250, Thermo Fisher Scientific) and re-suspended in 2% FBS in PBS. Cell pools were gated for live cells in a forward versus side scatter (FSC vs SSC) area dot plot, and doublets excluded in an FSC/SSC height vs width dot plot. The analysis gate for GFP expression was a combination of the 3 dot plots.
Cell culture, transfection and stable pool generation
Suspension CHO-GS−/- cells were cultured in the 50/50 medium consisting of 1:1 ratio mix of HyClone PF-CHO Multi-powder system (Cat: SH30333.01, GE Healthcare) and CD CHO medium (Cat: 10743029, Thermo Fisher Scientific) and incubated in 37°C, 8% CO2 shaking incubator. The 50/50 medium was supplemented with 0.05% Pluronic F-68 acid (Cat: 24040032, Thermo Fisher Scientific) and 6 mM L-glutamine (Cat: 25030–081, Thermo Fisher Scientific). Ten million cells were transfected with 5 μg of different DNA constructs via electroporation (SG Cell line 4D-Nucleofector® X Kit) (Cat: V4XC-3024, LONZA). Two days after transfection, the transfected cells were placed under L-glutamine removal selection until their viability recovered to more than 90%. GSwt stable pools were further subjected to 25 µM MSX (Cat: M5379, Sigma-Aldrich) selection. Batch culture was then performed on the recovered pools to measure the levels of IgG (antibody GA101) production using the nephelometer (Siemens Healthcare). The nephelometry method is a liquid-based immunoassay based on measuring the amount of scattered light generated by the immunocomplexed particles in the sample. It is a sensitive and reliable method for IgG measurement, often used in clinical settings, and allows the screening of a large number of samples in a short period of time. The IgG assay reagent (Cat: OSAS15, Siemens Healthcare), which is the IgG-specific reagent, was routinely used calibrated to ensure accuracy.
Single clone isolation and stability assessment
Cells were seeded at a density of 600 cells/ml in soft agar, CloneMedia™ CHO Growth A with L-GLN (Cat: K8840, Molecular Devices) and incubated in a 37°C, 5% CO2 static incubator for more than a week to form colonies. Single colonies were picked manually under the microscope into 96-well plates containing the 50/50 medium without L-glutamine supplementation. Single clones in the 96-well plates were scaled up into shake flasks. For the generation of high titer single clones, an overgrowth assay in 24-well format was performed to select for the top 30 producing clones to be scaled up into shake flasks and subsequent batch culture analysis to choose the top 15–20 producing clones for stability assessment. The overgrowth assay was performed by duplicating the single clones in 24-well formats and incubated for 14 days in 37°C, 5% CO2 incubator. Batch culture analysis of the titer production was assessed by seeding the cells at 3x105/ml in the 50/50 medium without L-glutamine and incubated for 14 days in 37°C, 8% CO2 shaking incubator. Supernatants were harvested by centrifugation at 8000 rpm for 10 min and measured using the IgG reagent in the nephelometer. Stability assessment was performed by subculturing the cells in the 50/50 medium without L-glutamine for 8–12 weeks. Seeding for batch culture was performed at week 0 and subsequently every 4 weeks. Titer analysis was performed at Day 14 using nephelometer.
Batch culture growth curve and fed-batch analysis
Single clones were seeded at 3x105/ml in the 50/50 medium without L-glutamine and incubated in 37°C, 8% CO2 shaking incubator. Cell count was measured at the indicated time points.
For fed-batch analysis, single clones were seeded at 3x105/ml in 50/50 medium without L-glutamine and incubated in 37°C, 8% CO2 shaking incubator. Cell count was measured at the indicated time points. Cells were fed with EX-CELL Advanced CHO Feed 1 without glucose (Cat: 24368C, Sigma-Aldrich) at 7.5% v/v on day 3, 5, 7, 9, and 11. Glucose analysis of the supernatants was measured using the BioProfile 100 Plus System (Nova Biomedical) and glucose (Cat: G8769, Sigma-Aldrich) was added up to 4 g/L. Titer measurement was performed using the nephelometer.
Supplemental Material
Download Zip (308.8 KB)Acknowledgments
This work was supported by A*STAR BMRC Strategic Positioning Fund. The authors would like to thank Dr. Yun Lei Tan and Mr. Ryan Haryadi for critical review of the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Related Research Data
References
- Kim JY, Kim YG, Lee GM. CHO cells in biotechnology for production of recombinant proteins: current state and further potential. Appl Microbiol Biotechnol 2012; 93:917–30.
- Hacker DL, De Jesus M, Wurm FM. 25 years of recombinant proteins from reactor-grown cells - where do we go from here? Biotechnol Adv 2009; 27:1023–7.
- Rita Costa A, Elisa Rodrigues M, Henriques M, Azeredo J, Oliveira R. Guidelines to cell engineering for monoclonal antibody production. Eur J Pharm Biopharm 2010; 74:127–38.
- Lai T, Yang Y, Ng SK. Advances in Mammalian cell line development technologies for recombinant protein production. Pharmaceuticals (Basel) 2013; 6:579–603.
- Ghaderi D, Zhang M, Hurtado-Ziola N, Varki A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev 2012; 28:147–75.
- Ringold G, Dieckmann B, Lee F. Co-expression and amplification of dihydrofolate reductase cDNA and the Escherichia coli XGPRT gene in Chinese hamster ovary cells. J Mol Appl Genet 1981; 1:165–75.
- Kaufman RJ, Sharp PA. Amplification and expression of sequences cotransfected with a modular dihydrofolate reductase complementary dna gene. J Mol Biol 1982; 159:601–21.
- Scahill SJ, Devos R, Van der Heyden J, Fiers W. Expression and characterization of the product of a human immune interferon cDNA gene in Chinese hamster ovary cells. Proc Natl Acad Sci U S A 1983; 80:4654–8.
- Chusainow J, Yang YS, Yeo JH, Toh PC, Asvadi P, Wong NS, et al. A study of monoclonal antibody-producing CHO cell lines: what makes a stable high producer? Biotechnol Bioeng 2009; 102:1182–96.
- Weidle UH, Buckel P, Wienberg J. Amplified expression constructs for human tissue-type plasminogen activator in Chinese hamster ovary cells: instability in the absence of selective pressure. Gene 1988; 66:193–203.
- Pallavicini MG, DeTeresa PS, Rosette C, Gray JW, Wurm FM. Effects of methotrexate on transfected DNA stability in mammalian cells. Mol Cell Biol 1990; 10:401–4.
- Cossons NH HP, Tuite MF, Jenkins N. Stability of amplified DNA in Chinese hamster ovary cells. In: Spider JB, Meigner B, ed. Production of biologicals from animal cells in culture. United Kingdom: Butterworth-Heinemann, 1991.
- Fann CH, Guirgis F, Chen G, Lao MS, Piret JM. Limitations to the amplification and stability of human tissue-type plasminogen activator expression by Chinese hamster ovary cells. Biotechnol Bioeng 2000; 69:204–12.
- Brown ME, Renner G, Field RP, Hassell T. Process development for the production of recombinant antibodies using the glutamine synthetase (GS) system. Cytotechnology 1992; 9:231–6.
- Bebbington CR, Renner G, Thomson S, King D, Abrams D, Yarranton GT. High-level expression of a recombinant antibody from myeloma cells using a glutamine synthetase gene as an amplifiable selectable marker. Biotechnology (N Y) 1992; 10:169–75.
- Cockett MI, Bebbington CR, Yarranton GT. High level expression of tissue inhibitor of metalloproteinases in Chinese hamster ovary cells using glutamine synthetase gene amplification. Biotechnology (N Y) 1990; 8:662–7.
- Wurm FM. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat Biotechnol 2004; 22:1393–8.
- Kingston RE, Kaufman RJ, Bebbington CR, Rolfe MR. Amplification using CHO cell expression vectors. Curr Protoc Mol Biol 2002; Chapter 16.23: 1–13.
- Jun SC, Kim MS, Hong HJ, Lee GM. Limitations to the development of humanized antibody producing Chinese hamster ovary cells using glutamine synthetase-mediated gene amplification. Biotechnol Prog 2006; 22:770–80.
- Liu W, Wei H, Liang S, Zhang J, Sun R, Tian Z. A balanced expression of two chains of heterodimer protein, the human interleukin-12, improves high-level expression of the protein in CHO cells. Biochem Biophys Res Commun 2004; 313:287–93.
- Sanders PG, Wilson RH. Amplification and cloning of the Chinese hamster glutamine synthetase gene. Embo J 1984; 3:65–71.
- Fan L, Kadura I, Krebs LE, Hatfield CC, Shaw MM, Frye CC. Improving the efficiency of CHO cell line generation using glutamine synthetase gene knockout cells. Biotechnol Bioeng 2012; 109:1007–15.
- Noh SM, Shin S, Lee GM. Comprehensive characterization of glutamine synthetase-mediated selection for the establishment of recombinant CHO cells producing monoclonal antibodies. Sci Rep 2018; 8:5361.
- Fan L, Kadura I, Krebs LE, Larson JL, Bowden DM, Frye CC. Development of a highly-efficient CHO cell line generation system with engineered SV40E promoter. J Biotechnol 2013; 168:652–8.
- Dorai H, Corisdeo S, Ellis D, Kinney C, Chomo M, Hawley-Nelson P, et al. Early prediction of instability of Chinese hamster ovary cell lines expressing recombinant antibodies and antibody-fusion proteins. Biotechnol Bioeng 2012; 109:1016–30.
- Haberle J, Gorg B, Rutsch F, Schmidt E, Toutain A, Benoist JF, et al. Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med 2005; 353:1926–33.
- Krajewski WW, Collins R, Holmberg-Schiavone L, Jones TA, Karlberg T, Mowbray SL. Crystal structures of mammalian glutamine synthetases illustrate substrate-induced conformational changes and provide opportunities for drug and herbicide design. J Mol Biol 2008; 375:217–28.
- Ronda C, Pedersen LE, Hansen HG, Kallehauge TB, Betenbaugh MJ, Nielsen AT, et al. Accelerating genome editing in CHO cells using CRISPR Cas9 and CRISPy, a web-based target finding tool. Biotechnol Bioeng 2014; 111:1604–16.
- Ho SC, Bardor M, Feng H, Mariati, Tong YW, Song Z, et al. IRES-mediated Tricistronic vectors for enhancing generation of high monoclonal antibody expressing CHO cell lines. J Biotechnol 2012; 157:130–9.
- Kelley B. Industrialization of mAb production technology: the bioprocessing industry at a crossroads. MAbs 2009; 1:443–52.
- Richardson GA, Lin, N., Lacy, K.A.,Davis, L.A.,Gray, M.I., Cresswell, J., Gerber, M.A., Caple, M.V., Kayser, K.J.. Cell Xpress™ Technology Facilitates High-Producing Chinese Hamster Ovary Cell Line Generation Using Glutamine Synthetase Gene Expression System. In: Thomas. N, ed. Cells and Culture ESACT Proceedings Springer, Netherlands: Dordrecht, 2010; 45–48.
- Clarke C, Doolan P, Barron N, Meleady P, O'Sullivan F, Gammell P, et al. Predicting cell-specific productivity from CHO gene expression. J Biotechnol 2011; 151:159–65.
- Pichler J, Galosy S, Mott J, Borth N. Selection of CHO host cell subclones with increased specific antibody production rates by repeated cycles of transient transfection and cell sorting. Biotechnol Bioeng 2011; 108:386–94.
- Nakamura T, Omasa T. Optimization of cell line development in the GS-CHO expression system using a high-throughput, single cell-based clone selection system. J Biosci Bioeng 2015; 120:323–9.
- Barnes LM, Bentley CM, Dickson AJ. Stability of protein production from recombinant mammalian cells. Biotechnol Bioeng 2003; 81:631–9.
- Kim M, O'Callaghan PM, Droms KA, James DC. A mechanistic understanding of production instability in CHO cell lines expressing recombinant monoclonal antibodies. Biotechnol Bioeng 2011; 108:2434–46.
- Chan KF, Shahreel W, Wan C, Teo G, Hayati N, Tay SJ, et al. Inactivation of GDP-fucose transporter gene (Slc35c1) in CHO cells by ZFNs, TALENs and CRISPR-Cas9 for production of fucose-free antibodies. Biotechnol J 2016; 11:399–414.
- Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc 2012; 7:171–92.
- Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic acids research 2011; 39:e82.
- Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, et al. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic acids research 2012; 40:W117–22.
- Guschin DY, Waite AJ, Katibah GE, Miller JC, Holmes MC, Rebar EJ. A rapid and general assay for monitoring endogenous gene modification. Methods in molecular biology 2010; 649:247–56.