
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Despite recent advances in the development of tools to predict immunogenicity risk of biotherapeutic molecules, the ability of a protein to elicit the formation of anti-drug antibodies (ADA) remains one of the most common causes for termination of clinical development programs. In this study, we use ADA assays to detect and measure pre-existing reactivity or the ability of a molecule to produce an ADA-like response in serum from treatment-naïve, healthy donors. We report herein that the magnitude of pre-existing reactivity evaluated pre-clinically and expressed as the 90th percentile of Tier 2 inhibition correlates with the subsequent rate of ADA emergence in the clinic. Furthermore, a multi-domain biotherapeutic (IgG-scFv bispecific antibody) showed the highest pre-existing reactivity and incidence of treatment-emergent ADA (TE-ADA) (57% and 93%, respectively). Using the components of the multidomain molecule in the Tier 2 step of the ADA assay, we were able to identify the scFv as the target of the serum pre-existing reactivity. Most importantly, the domain specificity of pre-existing ADA was the same as that of the TE-ADA from patients treated with the molecule. Based on these data, we propose the evaluation of the magnitude and of the domain specificity of pre-existing reactivity as a powerful tool to understand the immunogenic potential of novel biotherapeutics.
Introduction
The assessment of pre-existing reactivity to a biotherapeutic is performed routinely by laboratories across the industry as an essential part of immunogenicity assay development to establish screening and confirmatory cut points to be used later in the clinic. As part of the above-mentioned assessment, normal human serum samples from treatment-naïve donors are tested in the immunogenicity assay, and the presence of pre-existing anti-drug reactivity (pre-ADA) is represented by a signal above the background of the assay. At present, little is known about how pre-ADA originate; one possibility is that they may be part of the normal antibody population originally directed towards common antigens or proteins that happened to be homologous to a certain biotherapeutic. Nonetheless, the ability of a biotherapeutic to elicit an ADA-like response in treatment-naïve individuals is poorly understood. A better understanding of pre-existing reactivity could prove helpful in estimating the immunogenicity risk of a biotherapeutic, aid in the development of an appropriate clinical immunogenicity strategy and, ultimately, improve the chances that therapeutic proteins will be successfully developed.
Numerous groups have tried to review, compile and understand the available data around pre-existing reactivity.Citation1-Citation4 A variety of matrix components can contribute to reactivity, including high concentrations of circulating endogenous target, heterophilic antibodies, anti-host cell protein antibodies, and rheumatoid factor. Most commonly, reactivity is due to pre-ADA, which are immunoglobulins possessed by a percentage of normal individuals that cross-react to a certain biotherapeutic in the absence of any previous exposure. The detection of a high pre-ADA signal is not a rare event. A survey conducted in 2013 in association with the American Association of Pharmaceutical Scientists reported that at least 70% of the interviewed scientists have observed it at least once, either pre-clinically or clinically.Citation1 In addition, the high specificity of pre-ADA binding to the biotherapeutic has been shown through confirmatory assays by addition of the excess unlabeled drug. Furthermore, although pre-ADA have been detected both in pre-clinical and clinical studies, it appears that, once in the clinic, the observed incidence of pre-ADA is higher in specific disease states such as rheumatoid arthritis and other autoimmune disorders.Citation1,Citation5 The structure of the biotherapeutic protein product could also increase the prevalence of pre-existing antibodies. Thus, novel antibody-based constructs have been reported to result in up to 41% of the study subjects showing some degree of pre-ADA positivity.Citation5 Most importantly, it remains to be understood whether the presence of pre-ADA increases the risk of treatment-emergent ADA (TE-ADA) development, which occurs as if the patient was “primed” to respond to the drug by memory B-cells.
Frequently, pre-ADA are considered to be biological outliers that need to be eliminated from the data set by the statistical approach used to calculate the cut points, since they may artificially raise the risk of false negatives.Citation6,Citation7 Here, however, we demonstrate that pre-ADA are more than just outliers. We retrospectively investigated the magnitude and the epitope specificity of the pre-existing reactivity observed pre-clinically for a bispecific antibody (bsAb). We demonstrate that the magnitude and the epitope specificity of the pre-ADA correlated with the incidence of TE-ADA and their epitope preference, respectively. Ultimately, these data suggest that increased pre-ADA observed for novel biotherapeutics may increase the risk of TE-ADA in the clinic.
Results
Case studies of low and high pre-existing reactivity
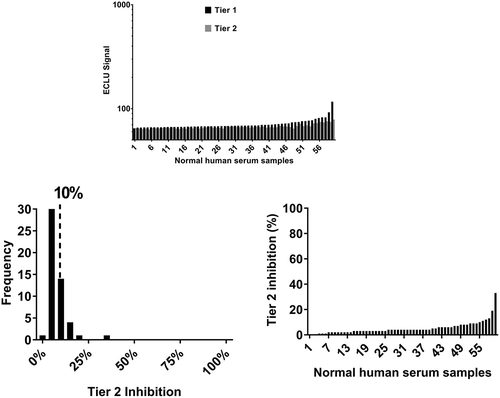
An electrochemiluminescence (ECL)-based ADA Affinity Capture Elution (ACE)-bridging assayCitation8 was developed to evaluate the ADA response against a representative humanized IgG1-EN (carrying effector-null mutations in the Fc constant region) monoclonal, monospecific antibody (mAb1) that binds to a target expressed as a transmembrane protein, but that also circulates. To determine the screening and confirmatory assay cut points, the assay signal in ECL units (ECLU) from 60 serum samples was evaluated. The majority of the ADA assay signals were low, suggesting limited pre-existing reactivity. Human monoclonal, monospecific antibodies are commonly associated with low ADA signals as shown in , in which >90% of the samples were within 25% of the buffer background. In addition, the screening ADA signal was consistently minimally inhibited by excess unlabeled drug; this resulted in overall low average Tier 2 inhibition (5.2% ± 5.0%) () and a narrow frequency distribution where 59 of 60 samples yielded Tier 2 inhibitions between 0% and 30% (). To quantify the pre-existing reactivity, the 90th percentile of Tier 2 inhibition was calculated on the entire data set of 60 samples (with outliers included). As shown in , the 90th percentile of Tier 2 inhibition for mAb1 was 10%.
Figure 1. Overall pre-existing reactivity to monoclonal human antibodies is often minimal. Pre-existing reactivity is expressed as Tier 2 (confirmatory) inhibition calculated from Tier 1 and Tier 2 signal from 60 normal human sera from treatment-naïve individuals obtained in the ACE-Bridge for a monoclonal, monospecific, fully human antibody directed against a soluble protein (mAb1). (a) The pattern of Tier 1 and Tier 2 signal (ECLU) for each normal human serum sample tested. (b) Tier 2 inhibition and frequency distribution show overall low mean Tier 2 inhibition, low biological variability and low 90th percentile of Tier 2 percent inhibition.
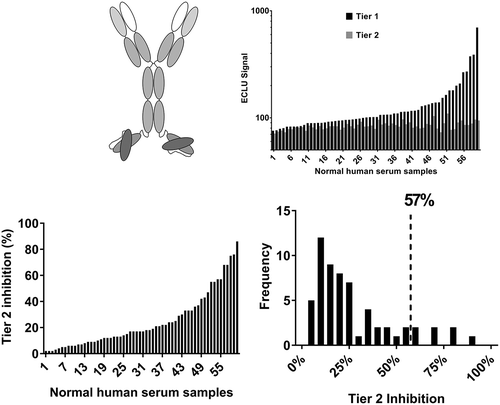
A bispecific antibody with IgG-single-chain variable fragment (scFv) architecture (bsAb1) was tested next. BsAb1 consists of two components: a human monoclonal, monospecific IgG4-PAA antibody that binds a target that is present in the circulation and on cell membranes (not the same target as mAb1), and an scFv made from variable regions (VH and VL) targeting a molecule that circulates as a dimer. The VH and VL regions are produced connected to each other by a (G4S)4 linker; the scFv is then linked to the heavy chain of the monoclonal antibody by a second linker (G4S)3 (). In contrast to mAb1, when the same experiments were performed with bsAb1, high Tier 1 assay signals were noted in the majority of the tested samples, and the signals were greatly inhibited by the excess unlabeled drug, indicating the presence of pre-existing ADA to bsAb1 (). Consistently with the high screening signal, the average Tier 2 inhibition was also elevated (25% ± 21%) () and the frequency distribution was wide, resulting in the 90th percentile of Tier 2 inhibition of 57% ().
Figure 2. Pre-existing reactivity to a bispecific antibody with the IgG-scFv architecture shows high Tier 2 inhibition and a wide distribution of the reactivity across donors. (a) Schematic representation of the IgG-scFv architecture, shared by bsAb1 and bsAb2. (b) The pattern of Tier 1 and Tier 2 signal (ECLU) for each normal human serum sample tested in the ACE-Bridge ADA assay. (c) Tier 2 inhibition and frequency distribution show overall high mean Tier 2 inhibition, high biological variability and high 90th percentile of Tier 2 percent inhibition.
Characterization of the TE-ADA response to bsAb1
Serum samples for immunogenicity testing were collected from patients in a Phase 1, single ascending dose (SAD) trial in healthy adult subjects. As expected, pre-existing reactivity was observed in the pre-dose samples from the enrolled patients with the magnitude slightly higher compared to assay validation. Whereas the incidence of TE-ADA was minimal in the case of mAb1 (12%, data not shown), bsAb1 treatment triggered an ADA response in 94% of the dosed patients (). Furthermore, close to 50% of all patients developed titers higher than 1:1,000 by day 15 post-dose, further confirming that bsAb1 was a highly immunogenic molecule. In addition, the lowest dose resulted in the highest titer (2 mg, 1:20,480) and in the 30 mg cohort, administration of the biotherapeutic subcutaneously resulted in a higher incidence of TE-ADA compared to the same dose administered by infusion (100% and 80%, respectively) and a higher median titer (1:960 compared to 1:320).
Figure 3. Administration of bsAb1 resulted in a high incidence of immunogenicity rate. (a) The overall incidence of TE-ADA was 94% in the phase 1 single-dose study, with the majority of the patients developing high titers by day 15 post-dose. B-D, the numbers 1 through 5 in the legends refer to the same patient in all 4 panels. (b) ADA titers measured on 1, 8, 15, 29 and 85 days post-injection, in Cohort 2 (8 mg dose). (c) Mean serum bsAb1 concentration versus time as measured by scFv-target capture assay in cohort 2. For ease of comparison with adjacent panels, in the interval 0–1 day, only the data collected at 20-h post-injection are shown, labeled as Day 1 measurement. All values below the LLOQ of 75 ng/mL are shown as 75 ng/mL; each symbol represents the same patient in all four panels. (d), Target engagement of the scFv target (left) and mAb Target (right); all values below the LLOQ of 18.4 pg/mL (scFv target) and 0.6 pg/mL (mAb Target) are shown as the respective assays’ LLOQ. (e), Characterization of the domain specificity was performed using the parent molecules of bsAb1 in Tier 2 in TE-ADA samples from Cohort 2. An excess of scFv alone or scFv linked to human Fc (hFc-scFv) could suppress the Tier 1 signal as efficiently as bsAb1 itself; each bar is the average of all five patients in cohort 2. (f), Target interference was assessed in the bsAb1 ADA assay using the scFv target (left) and the mAb target (right). Both proteins do not interfere in the assay when tested at physiological concentrations and in the presence of bsAb1.
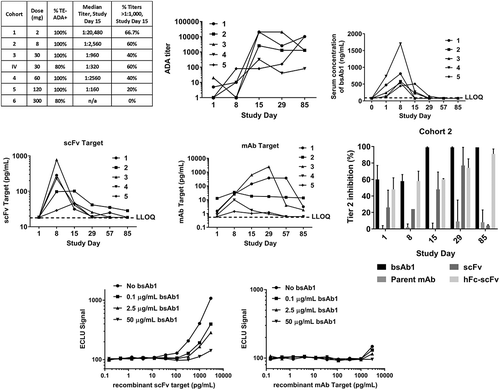
In the clinical trial, ADA titers () correlated with a reduction in circulating bsAb1 levels () and in a diminished engagement of the soluble protein targeted by the scFv portion of the bsAb1 (), suggesting that the majority of the ADA were directed against the scFv component of the molecule. Data collected from cohort 2 (8 mg dose) are shown as representative of all the cohorts. To investigate the specificity of the ADA, patient samples were analyzed in the presence of a structurally related hFc-scFv molecule that contained the same scFv as in bsAb1 but fused to an irrelevant human Fc IgG (). In addition, the parent mAb and free scFv alone were also tested in Tier 2. In the presence of hFc-scFv, the ADA signals were inhibited in a similar manner as with bsAb1 (). When the parent mAb was added, the ADA signals were not inhibited (). In the presence of the scFv alone, the Tier 2 inhibition was consistently above 60%, indicating that the ADA in the clinical trial patients were mainly anti-scFv antibodies. In order to assess the potential contribution of target interference to the detection of both pre-existing reactivity and TE-ADA, a target interference experiment was performed using recombinant forms of the scFv target and the mAb target. The results showed that the scFv target, a dimeric protein, was not able to generate a signal in the ADA assay when tested at concentrations below the physiological levels that are measurable in serum (<18.4 pg/mL, the lower limit of quantification (LLOQ) in the target engagement assay). Similarly, the mAb target, a monomer, could interfere in the assay only at supra-physiological concentrations (). Furthermore, as expected, target interference was significantly reduced when the target was tested in a complex with bsAb1 ().
We next sought to investigate whether high pre-existing reactivity was a recurring feature of scFv bispecific antibodies. In order to do this, we tested bsAb2, another biotherapeutic bearing the IgG-scFv architecture and sharing the same monoclonal antibody as bsAb1, but in the IgG1 isotype, linked to a completely different scFv via the same type of linkers. In the bsAb2-specific ACE-Bridge ADA assay, the average screening signal calculated from 60 serum samples was 250 ECLU with values ranging from 67.5 ECLU to 2374 ECLU. Consequently, the average Tier 2 inhibition was high (44%, ) and the calculated inhibitions broadly distributed with the 90th percentile of Tier 2 inhibition of 83% ().
Figure 4. High pre-existing reactivity to a second IgG-scFv bispecific antibody, but not to its heteromab counterpart. (a) Pre-existing reactivity to another IgG-scFv bsAb (bsAb2) was elevated. Percent Tier 2 inhibition in normal human serum samples and frequency of distribution reveal a high mean Tier 2 inhibition, high biological variability, and high 90th percentile of Tier 2 percent inhibition. (b), Characterization of the domain specificity on bsAb2 was performed on four normal serum samples; the hFc-scFv alone could inhibit the reactivity. (c), schematic representation of Heteromab1, composed of the same parent antibodies as bsAb2. (d), low pre-existing reactivity of Heteromab1. (e), Mass spectrometry analysis of serum proteins enriched by the IgG-scFv (bsAb2) or the heteromab showed preferential binding to immunoglobulins for bsAb2 in all four of the normal human serum samples tested.
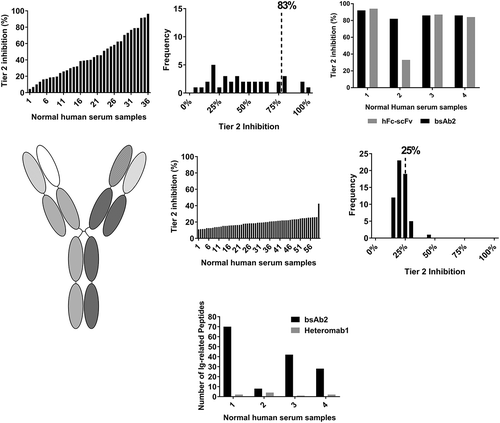
To investigate whether treatment-naïve samples contained pre-existing anti-scFv antibodies, the signal from a subset of four donor samples was tested in the presence of bsAb2 and a hFc-scFv. These Tier 2 studies revealed that, similar to bsAb1, pre-existing reactivity was directed mainly against the scFv portion of the molecule (). We then asked the question whether the incorporation of the variable regions present in bsAb2 into an antibody architecture structurally more similar to a natural IgG (Heteromab1, ) would reduce the observed reactivity to bsAb2. Pre-existing reactivity to Heteromab1 was much lower compared to bsAb2, and the Tier 2 inhibitions were more tightly distributed, with the 90th percentile of Tier 2 inhibition of 25% ().
To characterize further the serum reactivity to bsAb2, mass spectrometry (MS) was used as an unbiased approach to identify proteins bound to bsAb2 and responsible for the high signal. Serum samples collected from four of the donors with varying degrees of reactivity to bsAb2 were subjected to immunoprecipitation with bsAb2 and Heteromab1. The proteins enriched from human serum were identified using liquid chromatography-MS/MS after proteolytic digestion. Data showed the only peptides with significant enrichment in the serum immunoprecipitated with bsAb2 compared to Heteromab1 () were unrelated to bsAb2 and derived from donor-specific IgG and IgM immunoglobulins, further confirming the presence of pre-existing ADA specific to the drug in treatment-naïve samples.
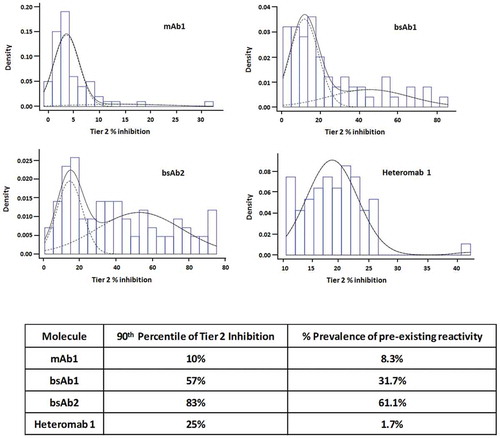
We then wondered whether describing the pre-existing reactivity using prevalence, as it is more commonly done, would result in a different ranking of the four tested molecules based on reactivity. In order to define the individuals as having pre-existing reactivity and thus calculate prevalence, we fitted a two-component Normal mixture to the Tier 2 percent Inhibition data sets, under the assumption that the percent inhibition values come from two populations: 1) a true negative (lower mean), and 2) a true positive (higher mean, individuals with pre-existing reactivity) (). Using this data distribution, the prevalence of pre-existing reactivity was calculated and compared to the proposed method of the 90th percentile of Tier 2 inhibition (). The comparison indicated that the prevalence is indeed a suitable method to describe the reactivity, and it adequately discriminated among the various molecules.
Figure 5. Pre-existing reactivity data reported as “prevalence”. (a), Histograms of the distribution of Tier 2 inhibition for each molecule; the dashed curves show the two mixture components; the black curve shows the sum of the two dashed curves. (b), Table summarizing the pre-existing reactivity as the 90th percentile of Tier 2 inhibition and as prevalence.
Discussion
In this study, we describe and characterize pre-clinical pre-existing reactivity as a novel parameter to aid in the prediction of clinical immunogenicity risk of biotherapeutics. To do this, we utilized Tier 1 and Tier 2 data from ADA screening assays to determine the 90th percentile of Tier 2 inhibition, which our data show is a reliable measure of pre-existing reactivity, and thus a predictor of the overall treatment-emergent immunogenicity risk of a molecule. Our results indicate that a correlation may exist between the magnitude of the reactivity of a given molecule in naïve serum and its overall risk to trigger TE-ADA when administered to humans. In addition, characterizing the reactive epitopes through Tier 2 inhibition studies can reveal potentially immunogenic portions of novel multi-domain biotherapeutics that are more likely to be targeted by TE-ADA. The importance of characterizing the ADA specificity to biotherapeutics with multiple domains has been reviewed in detail by Gorovits et al.Citation9 However, although evaluation of ADA specificity is routinely performed on clinical samples, there are no reports to date that propose a similar analysis on pre-existing ADA.
The development of biologic therapies, such as monoclonal antibodies and recombinant proteins, has seen remarkable growth in the past two decades,Citation10 due to the high specificity and long half-life of these molecules. However, despite the progress in the development of new tools to predict and evaluate the immunogenicity risk of biotherapeutics, the ability of a biologic drug to elicit the production of ADA by the recipient’s immune system remains one of the most common causes for the halting of clinical development programs. The mechanisms through which biotherapeutics elicit the formation of ADA are only partly understood. Thus, identification of new assays able to foresee and accurately quantify the risk will substantially improve the chances of a molecule’s clinical success.
The molecule that served as our case study underwent an internal process of immunogenicity risk evaluation that included established tools such as in silico EpiVax screening to predict the presence of T-cell epitopes. Of note, the approach we proposed here provides a novel means to potentially predict the presence of B-cell epitopes, and thus complements risk assessment tools based on T-cell epitopes.
Although the existence of pre-ADA to selected therapeutic proteins has been known for some time, the biological relevance and potential predictive value have remained unclear. The most current theory on the origin of pre-ADA draws similarities with natural antibodies, low affinity, primarily IgM, produced as part of an early defense against a wide repertoire of antigens, but without prior exposure to them.Citation11
A cross-industry survey conducted in 2013 reported that 32% of the interviewed scientists reported an increase of TE-ADA associated with the presence of pre-existing reactivity.Citation1 According to another publication, the same incidence of TE-ADA could be found regardless of the presence or the absence of pre-existing reactivity.Citation5 Interestingly, if the biotherapeutics were tested in rheumatoid arthritis, pre-existing antibodies were associated with an increased incidence of TE-ADA in 100% of the examined cases. Of note, the data reported in our experiments were collected on normal human serum samples, less susceptible to potentially confounding factors such as rheumatoid factor or other autoantibodies.
In addition, the research described here departed from the published literature on the way pre-existing reactivity was quantified and described. Rather than using prevalence of incidence of pre-ADA, the magnitude of the reactivity, expressed as Tier 2 inhibition, was used, and a correlation was found between the extent of reactivity to the drug and its immunogenicity in the clinic. Since the extent of the pre-existing reactivity is not discussed in detail in the previous literature, it is not possible to determine whether others have seen a correlation similar to the one reported in this study. We also report that the 90th percentile of Tier 2 inhibition correlated well with the more traditional prevalence. However, prevalence could fail to adequately describe datasets in which the true negative distribution was located at high percent inhibition values, and thus it could result in a misleading low prevalence of pre-existing reactivity. In addition, the use of prevalence was based on the assumption that the data were distributed as a mixture of Normal distributions, whereas the 90th percentile of Tier 2 inhibition was not based on any assumption. In addition, we leveraged the information obtainable from Tier 2 inhibition studies and demonstrated that it could predict immunogenic domains where other, more established tools could not. The relative ease of execution of ligand binding ADA assays and the wide availability of normal human serum samples suggest that this approach could complement the in silico tools and established in vitro assays to assess immunogenicity risk preclinically.
In our hands, factors such as IgG subclass or the nature of the drug target (whether transmembrane or soluble) did not appear to predispose a therapeutic antibody to pre-existing reactivity. However, the use of novel antibody architectures (i.e., IgG-scFv) and the expression of wild-type proteins linked to unnatural amino acid sequences appeared to be a clear risk factor that predisposed the molecule to react to naïve serum. In the former case, unconventional architectures that deviate from the natural structure of antibodies can expose neo-epitopes that are recognized by specific cross-reactive pre-existing antibodies. Although there are no examples of neoepitopes generated specifically by the scFv structure in the literature, there are reports of pre-existing human anti-hinge autoantibodies reactive to therapeutic monoclonal Fab or F(ab’)2.Citation4,Citation12,Citation13 Information on safety and immunogenicity of bifunctional antibodies is limited and, specifically, no clinical data are available for the IgG-scFv architecture. One IgG-based bsAb approved for human dosing, the anti-EpCAM×anti-CD3 antibody catumaxomab, is a mouse/rat chimera that was highly immunogenic.Citation14 Another IgG-based bsAb, emicizumab, a humanized anti-activated Factor IX-X antibody, was reported to have relatively low immunogenicity.Citation15 Both of these molecules, however, were variants of the Heteromab1 type architecture mentioned in this report and, therefore were more structurally similar to a natural IgG.
In our observations, the reactive epitope in the scFv-containing bispecific antibodies was the scFv portion. It remains to be determined whether the high immunogenic potential of the scFv was inherently due to its sequence or structure, or whether the IgG may have played a role by causing the internalization of the scFv by antigen-presenting cells,Citation16 or whether it was a synergistic combination of the two factors. The latter hypothesis is supported by the discovery that a potentially large number of healthy treatment-naïve individuals produce pre-existing human anti-VH antibodies (HAVH).Citation17 Furthermore, high levels of HAVH were associated with agonistic effects of a biotherapeutic molecule to the receptor.Citation17
A retrospective evaluation of our historical data allowed us to see the emerging pattern of a correlation between the extent of pre-existing reactivity and the incidence of TE-ADA in the clinic, in the absence of any other known mechanisms of immunogenicity or high baseline ADA signals. Based on these observations, we conclude that the detection of pre-ADA is a significant risk factor for clinical TE-ADA, and that pre-existing ADA should be evaluated as early as possible during the course of molecule development. Pre-existing reactivity to a biotherapeutic molecule is thus a novel, previously untapped source of information on its potential immunogenic risk and should, therefore, be part of a more holistic approach to inform protein engineering efforts in the design of less immunogenic molecules.
Materials and methods
Human serum samples
Samples tested for pre-existing reactivity were from treatment-naïve, normal human individuals, equally distributed between genders and 20 to 50 years of age (purchased commercially from Bioreclamation IVT) or obtained via the Eli Lilly Research Blood Donor program. Clinical trial samples were obtained from patients after obtaining protocol approval from an Institutional Review Board and proper informed consent, with all samples de-identified to protect patient privacy. Serum samples were stored at −80°C prior to analysis.
BsAb1 SAD study design and bioanalytical methods
Samples from 41 subjects with inflammatory arthritis enrolled in a Phase 1 single dose study for bsAb1 were obtained after patients gave their permission for serum samples to be stored for analysis. Samples were collected for immunogenicity evaluation at baseline, day 8, 15, 29 and 85. Samples were assessed for pre-existing reactivity using an ACE-Bridge assay format. BsAb1 was administered by subcutaneous injection in all the cohorts except the 30 mg intravenous cohort, using 1 to 4 1.5 mL injections.
Human serum samples obtained during this study were analyzed for bsAb1 using a validated antigen capture enzyme-linked immunosorbent assay (ELISA) method using the target of the scFv. The LLOQ was 75.00 ng/mL, and the upper limit of quantification was 2000.00 ng/mL. The samples above the limit of quantification were diluted to yield results within the calibrated range. In addition, bsAb1 concentrations were measured by a second antigen capture ELISA method using the target of the mAb portion; however, the scFv-target-based assay had a lower LLOQ, and thus it allowed a more complete description of the concentration-time profile of bsAb1.
The effect of bsAb1 on the accumulation of the two target proteins was assessed in serum samples by the respective drug-tolerant assays that measured levels of the two analytes in the presence of bsAb1. Briefly, the two analytes were captured in the presence of added excess bsAb1 by specific antibodies immobilized on a plate and recognizing non-overlapping epitopes to bsAb1. After the plate was washed, the analytes are detected through the bound bsAb1 using a ruthenium-labeled anti-human IgG4 monoclonal antibody.
Labeling of biotherapeutic molecules
Two mg of antibody was biotinylated using EZ-Link Sulfo-NHS-Biotin (ThermoScientific, 21217) and an additional 2 mg labeled with ruthenium using MSD SULFO-TAG NHS-Ester (Mesoscale, R91AO-1). Labeling of antibodies was confirmed by Matrix-assisted laser desorption/ionization time-of-flight MS; on average, antibodies were labeled with 4–6 biotins or rutheniums per molecule. Labeled antibodies were diluted in 50% glycerol and stored at −20°C prior to use.
Pre-existing reactivity assessment using the affinity capture elution bridging assay (ACE-bridge)
To generate the Tier 1 (also known as screening) signal, an ACE-Bridge approach was used.Citation8 Briefly, streptavidin-coated 96-well plates (Pierce, 15500) were washed with 1X TBST (Boston BioProducts, IBB-181X) and subsequently coated using 100 μL per well of biotinylated antibody at a concentration of 30 nM diluted in TBST with 0.1% bovine serum albumin (BSA; Sigma, A7888) for 1 h at room temperature. Coated plates were washed three times with TBST, samples were diluted 1:10 with TBS (Fisher, BP2471-1), and 100 μL of the samples were added to the coated plates and allowed to incubate overnight at 4°C. The following day, plates were washed three times with TBST and captured ADA were acid eluted using 65 μL per well of 300 mM acetic acid (fisher scientific, A38-500) for 5 min at room temperature. Polypropylene 96-well plates (Corning, 3359) were then loaded with 50 μL of 1 μg/mL each of biotinylated antibody and ruthenium-labeled antibody in neutralizing buffer (0.375 M Tris, 300 mM NaCl, pH 9). Next, 50 μL of the acid eluted samples were added to the polypropylene plate containing the mixture in neutralizing buffer and ADA and were allowed to bridge the labeled antibodies for 1 h at room temperature. At the same time, MSD Gold 96-well streptavidin plates (Mesoscale, L15SA-1) were washed and blocked with TBS+1% BSA for 1 h at room temperature. Afterward, blocked MSD plates were washed, and 80 μL of bridged samples were added to the plate for 1 h. Afterward, the wells were washed three times with TBST, and 150 μL per well of 2 × MSD Read Buffer (Mesoscale, R92TC-2) was added to the wells. Plates were then read on an MSD SQ120 reader to provide the Tier 1 signal expressed as electrochemiluminescent units (ECLU).
Confirmatory (tier 2) assays
To generate the Tier 2 (also known as confirmatory) signal, used for competition assays, unlabeled biotherapeutic molecule was added during the detection step in the ACE-Bridge at a 10-fold excess molar concentration compared to the labeled biotherapeutic. Tier 2 percent inhibition was then calculated using Tier 1 and Tier 2 signal with the following formula:
Target interference assessment
The ability of the two target proteins to generate a signal above background was evaluated in the bsAb1 ADA assay. Briefly, the two target proteins were serially diluted in buffer (PBS 1X, 1% BSA) starting from a 3 ng/mL concentration, and then combined with 0 µg/mL, 0.1 µg/mL, 2.5 µg/mL and 50 µg/mL of bsAb1 and let to incubate for 30 min at room temperature. The curves were then tested in the bsAb1 ADA assay according to the Method. The concentrations of targets and bsAb1 were selected based on the target engagement and pharmacokinetic profiles observed in the bsAb1 SAD study and covered a wide range of conditions found in patient samples.
Identification of serum-binding partners by mass spectrometry
Streptavidin-coated agarose beads (ThermoFisher Scientific 20357) were incubated with biotinylated biotherapeutic molecules, washed and blocked with 1% BSA. Serum was diluted 10-fold and incubated with biotherapeutic-coated beads overnight at 4°C with agitation. Beads were washed 5 times with 5 mL of PBS, and enriched proteins were eluted 2 times with 50 μL of 300 mM acetic acid incubated at room temperature for 5 min. Eluates were combined for buffer exchange and concentrated using 10K microcon filters (Millipore Sigma MRCPRT010). Samples were reduced and alkylated for 1 h at 37°C and dried using a SpeedVac overnight. Dried samples were digested with trypsin (10 μL from 100 μg/mL solution) overnight at 37°C. Digested peptides were passed on C18 microcolumn (Millipore Sigma ZTC18S096) and reconstituted in 10 μL of 1% acetonitrile/0.1% trifluoroacetic acid solution for LC-MS/MS analysis using Easy-nLC1200 (ThermoScientific) coupled with Velos Pro orbitrap mass spectrometer (ThermoScientific). Log2AUC values were calculated for all identified peptides across all the biotherapeutic/human sera donor combinations using a suite of internal analysis platforms as previously described.Citation18
Data analysis
Peptides enriched with specific biotherapeutic/donor serum combinations were identified using Dunnett’s test with the following criteria: (1) Benjamini-Hochberg (across peptides) multiplicity adjusted minimum Dunnett’s p-value (within peptide) <0.05; (2) maximum Dunnett’s p-value (within peptide) <0.05; and (3) Dunnett’s reference group peptide AUC > 10-folder higher than all other compared groups. The protein annotations for the enriched peptides were then examined to determine the identity of the serum proteins identified. All Graphs were generated using GraphPad Prism. To establish the prevalence of donors with pre-existing reactivity, the Tier 2 percent inhibition data were modeled under the assumption that the percent inhibition values originated from two populations: true negative donors and donors with pre-existing reactivity. For each molecule, a two-component Normal mixture was fit to the Tier 2 percent inhibition data; one component, described by a lower mean, was interpreted as the true negative population of donors, while the other component, described by higher mean, was interpreted as the “biological outliers” or the donors with pre-existing reactivity.
Abbreviations
ADA Anti-drug antibodies
ACE Bridge Affinity Capture Elution-Bridge
scFv single-chain variable fragment.
Disclosure of Potential Conflicts of Interest
All authors are employees of Eli Lilly and Company. This work was supported and funded entirely by Eli Lilly and Company.
Acknowledgments
The authors thank Donmienne Leung for the critical review of the manuscript, and Stuart Friedrich for providing the pharmacokinetics data.
Related Research Data
References
- Xue L, Fiscella M, Rajadhyaksha M, Goyal J, Holland C, Gorovits B, Morimoto A. Pre-existing biotherapeutic-reactive antibodies: survey results within the American Association of pharmaceutical scientists. Aaps J. 2013;15:852–55. doi:10.1208/s12248-013-9492-4.
- Gorovits B, Clements-Egan A, Birchler M, Liang M, Myler H, Peng K, Purushothama S, Rajadhyaksha M, Salazar-Fontana L, Sung C, et al. Pre-existing antibody: biotherapeutic modality-based review. Aaps J. 2016;18:311–20. doi:10.1208/s12248-016-9878-1.
- van Schie KA, Wolbink GJ, Rispens T. Cross-reactive and pre-existing antibodies to therapeutic antibodies–effects on treatment and immunogenicity. MAbs. 2015;7:662–71. doi:10.1080/19420862.2015.1048411.
- Kim HS, Kim I, Zheng L, Vernes JM, Meng YG, Spiess C. Evading pre-existing anti-hinge antibody binding by hinge engineering. MAbs. 2016;8:1536–47. doi:10.1080/19420862.2016.1196521.
- Xue L, Rup B. Evaluation of pre-existing antibody presence as a risk factor for posttreatment anti-drug antibody induction: analysis of human clinical study data for multiple biotherapeutics. Aaps J. 2013;15:893–96. doi:10.1208/s12248-013-9497-z.
- Kumar SC, DelCarpini JA, Qu Q, Kane M, Gorovits B. Mitigation of pre-existing antibodies to a biotherapeutic in non-clinical species when establishing anti-drug antibody assay cutpoint. Aaps J. 2017;19:313–19. doi:10.1208/s12248-016-0011-2.
- Schneider AK, Vainshtein I, Roskos LK, Chavez C, Sun B, Liang M. An immunoinhibition approach to overcome the impact of pre-existing antibodies on cut point establishment for immunogenicity assessment of moxetumomab pasudotox. J Immunol Methods. 2016;435:68–76. doi:10.1016/j.jim.2016.05.007.
- Chen YQ, Pottanat TG, Carter QL, Troutt JS, Konrad RJ, Sloan JH. Affinity capture elution bridging assay: A novel immunoassay format for detection of anti-therapeutic protein antibodies. J Immunol Methods. 2016;431:45–51. doi:10.1016/j.jim.2016.02.008.
- Gorovits B, Wakshull E, Pillutla R, Xu Y, Manning MS, Goyal J. Recommendations for the characterization of immunogenicity response to multiple domain biotherapeutics. J Immunol Methods. 2014;408:1–12. doi:10.1016/j.jim.2014.05.010.
- Kaplon H, Reichert JM. Antibodies to watch in 2019. mAbs. 2019;11(2):219–38. doi:10.1080/19420862.2018.1556465.
- Lutz HU, Binder CJ, Kaveri S. Naturally occurring auto-antibodies in homeostasis and disease. Trends Immunol 2009;30:43–51. doi:10.1016/j.it.2008.10.002.
- Knight DM, Wagner C, Jordan R, McAleer MF, DeRita R, Fass DN, Coller BS, Weisman HF, Ghrayeb J. The immunogenicity of the 7E3 murine monoclonal Fab antibody fragment variable region is dramatically reduced in humans by substitution of human for murine constant regions. Mol Immunol. 1995;32:1271–81. doi:10.1016/0161-5890(95)00085-2.
- Nasu H, Chia DS, Knutson DW, Barnett EV. Naturally occurring human antibodies to the F(ab‘)2 portion of IgG. Clin Exp Immunol. 1980;42:378–86.
- Ruf P, Kluge M, Jager M, Burges A, Volovat C, Heiss MM, Hess J, Wimberger P, Brandt B, Lindhofer H. Pharmacokinetics, immunogenicity and bioactivity of the therapeutic antibody catumaxomab intraperitoneally administered to cancer patients. Br J Clin Pharmacol. 2010;69:617–25. doi:10.1111/j.1365-2125.2010.03635.x.
- Shima M, Hanabusa H, Taki M, Matsushita T, Sato T, Fukutake K, Kasai R, Yoneyama K, Yoshida H, Nogami K. Long-term safety and efficacy of emicizumab in a phase 1/2 study in patients with hemophilia A with or without inhibitors. Blood Adv. 2017;1:1891–99. doi:10.1182/bloodadvances.2017006684.
- Deora A, Hegde S, Lee J, Choi CH, Chang Q, Lee C, Eaton L, Tang H, Wang D, Lee D, et al. Transmembrane TNF-dependent uptake of anti-TNF antibodies. MAbs. 2017;9:680–95. doi:10.1080/19420862.2017.1304869.
- Holland MC, Wurthner JU, Morley PJ, Birchler MA, Lambert J, Albayaty M, Serone AP, Wilson R, Chen Y, Forrest RM, et al. Autoantibodies to variable heavy (VH) chain Ig sequences in humans impact the safety and clinical pharmacology of a VH domain antibody antagonist of TNF-alpha receptor 1. J Clin Immunol. 2013;33:1192–203. doi:10.1007/s10875-013-9915-0.
- Higgs RE, Knierman MD, Gelfanova V, Butler JP, Hale JE. Comprehensive label-free method for the relative quantification of proteins from biological samples. J Proteome Res. 2005;4:1442–50. doi:10.1021/pr050109b.