
ABSTRACT
Antibody-drug conjugates (ADCs) that exploit the active metabolite SN-38, which is derived from the popular anticancer drug, irinotecan (a camptothecin that inhibits the nuclear topoisomerase I enzyme, inducing double-stranded DNA breaks during the mitotic S-phase of affected cells), represent a substantial advance in the ADC field. SN-38 has been conjugated to a humanized antibody against trophoblast cell surface antigen 2 (TROP-2), which is involved in cancer signaling pathways and has increased expression by many cancer cell types, yielding the ADC sacituzumab govitecan. By conjugating a higher number of SN-38 molecules to the immunoglobulin (drug-to-antibody ratio = 7–8:1), and giving higher (10 mg/kg) and repeated therapy cycles (Days 1 and 8 of 21-day cycles), enhanced drug uptake by the targeted cancer cells is achieved. Based on a unique conjugation method, the lactone ring of the SN-38 molecule is stabilized and the molecule is protected from glucuronidation, a process that contributes to the untoward late diarrhea experienced with irinotecan. Finally, while the ADC is internalized, the use of a moderately stable linker permits release of SN-38 in an acidic environment of the tumor cell and its microenvironment, contributing to a bystander effect on neighboring cancer cells. Here, we discuss the development of sacituzumab govitecan and clinical results obtained using it for the management of patients with advanced, refractive breast, lung, and urinary bladder cancers. Sacituzumab govitecan, which is undergoing accelerated approval review by the US Food and Drug Administration while also being studied in Phase 3 clinical studies, was granted Breakthrough Therapy status from the FDA for advanced, refractory, metastatic triple-negative breast cancer patients.
Introduction
Although nearly a half-century in development, antibody-drug conjugates (ADCs) in the past decade have become an area of intensive research as a more selective cancer therapy, with four approved ADCsCitation1−Citation5 and over 80 in clinical trials (Janice Reichert, personal communication, April 8, 2019). Early conjugates were thought to have failed because they incorporated some of the more contemporary anticancer drugs of their era, such as doxorubicin and methotrexate, and began with immunogenic murine antibodies. Success was achieved ultimately with the introduction of human and humanized antibodies, as well as so-called ultratoxic agents, drugs that were several logs more potent than those used first, with activity in the picomolar range. The combination of drug, selective linker and tumor-targeting antibody was designed to reduce off-target toxicities. Issues of antibody-target specificity, antigen-antibody internalization, and conjugate pharmacokinetics and dosing also required attention in ADC development.Citation5
The ADCs currently approved, and most agents under development, use one of the three basic compounds: (1) calicheamicin (an enediyne antibiotic that causes double-stranded DNA breaks, e.g., gemtuzumab ozogamicin),Citation6 (2) auristatin (most commonly monomethyl auristatin E (MMAE), but also the F derivative (MMAF), with anti-mitotic activity by inhibiting polymerization of tubulin, e.g., brentuximab vedotin),Citation7 and (3) maytansine (maytansinoid analogues DM1 or DM4, which also are microtubule inhibitors, e.g., trastuzumab emtansine).Citation8,Citation9 These drugs are so potent that they cannot be used as stand-alone therapeutics, causing more toxicity than therapeutic gain. In order to use these ultratoxic drugs in ADCs, new chemistries were required, including specialized linkers, with the primary goal to sustain the bond between the antibody and the drug while the conjugate was in the circulation, but enabling release of the active drug when the conjugate was internalized by, or released on or adjacent to, the target cells (e.g., when trafficked to the lysosome). Thus, while bound to the antibody-linker, the drug should be inactive, but once released within or in the immediate environment of the tumor cell, it should effectively kill at relatively low concentrations due to its extreme potency. This resulted in ultratoxic ADCs that had relatively low ratios of drug to antibody (<4) and low dosing in order to control off-target toxicity.Citation5,Citation10,Citation11
ADCs that incorporate ultratoxic drugs require a high level of stability between the antibody-drug bond when in the blood, with activity only possible if the conjugate is internalized by the tumor cell. This places a specific requirement on the target, i.e., the antigen must be on the surface of the tumor cells, in high concentration, and accessible, and a high level of tumor specificity is needed in order to preclude off-target toxicities. The antigen-antibody must be internalized, and in some cases, the conjugate needs to be trafficked selectively to sites within the cells that will enable the cleavage of the linker-drug bond. While the advent of stably linked ultratoxic agents conjugated to relatively cancer-specific human or humanized monoclonal antibodies led to the approval of several ADCs,Citation12 the testing of new ADCs has shown that success is not guaranteed based solely on the use of any one conjugate platform or drug. Other factors, such as the selection and level of tumor expression of the target, target antigen biosynthesis and heterogeneity during the course of therapy, tumor progression (all influencing therapeutic resistance), and specific properties of the cancer type and stage also play important roles.
This perspective presents our experience in the development of an alternative ADC platform utilizing a drug of lower potency than the ultratoxic agents (i.e., with nanomolar toxicity) that is coupled to antibodies with a moderately stable linker, but one that essentially releases most of the drug while the conjugate is in the circulation over a period of about 3 days. The linker is particularly susceptible to pH changes, thereby accelerating drug release when the conjugate is internalized and delivered to the acidic lysosomes, or even if the conjugate is bound to the antigen within the tumor microenvironment, where a lower pH would facilitate drug release. We focus on sacituzumab govitecan (IMMU-132), a conjugate targeting the tumor-associated antigen, TROP-2 (trophoblast cell surface antigen 2, or TROP-2), conjugated with a topoisomerase-I inhibitor (or TOPO-I),Citation13,Citation14 developed by Immunomedics, Inc.
TROP-2
TROP-2 was first described about 40 years ago as a surface marker of trophoblast cellsCitation15 and was rediscovered years later as tumor-associated calcium signal transducer 2 (TACSTD2), membrane component chromosome 1 surface marker 1 (M1S1), gastrointestinal antigen 733–1 (GA733-1), and epithelial glycoprotein-1 (EGP-1).Citation16,Citation17 We first identified the RS7 murine monoclonal antibody, developed against human non-small-cell lung cancer (NSCLC), finding it also bound to human breast, colon, renal, and prostate cancers, with staining also in many normal tissues.Citation18 Subsequently, we determined the molecular properties of the antigen bound by RS7 as a 46-kDa glycoprotein (35 kDa when deglycosylated), showing it was phosphorylated by protein kinase C on serine-303 in the cytoplasmic domain, suggesting a role in signal transduction across the cell membrane.Citation19,Citation20 RS7 was subsequently classified as binding to an antigen that at the time was given the name epithelial glycoprotein-1.Citation20,Citation21 The RS7 antibody also showed rapid cell internalization.Citation18,Citation22 TROP-2 and its biological functions have been reviewed recently.Citation14
Development and properties of sacituzumab govitecan (IMMU-132)
Early ADCs used payloads composed of common chemotherapeutic agents, but these had potencies in the micromolar range, whereas the successful ultratoxic agents were all active at picomolar levels (). A potential concern for the stably linked ultratoxic conjugates was that there was limited information on the free drug’s pharmacological activity. Therefore, we examined more conventional drugs whose pharmacological activity was known, hypothesizing that agents with cytotoxic activity in the low nanomolar range would be preferred.
Figure 1. Relative range of potency for some of the more commonly used ADC payloads against human cancer cell line in vitro (adapted from Nakada et al.Citation60).
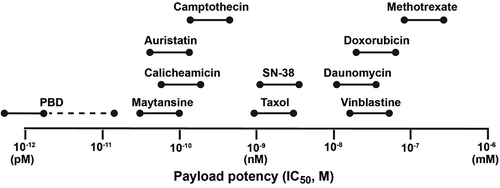
SN-38, a semi-synthetic camptothecin that is the active component of irinotecan, was chosen because irinotecan’s clinical properties were well known. Camptothecins kill cells by interacting with TOPO-I, which in turn introduces double-stranded DNA breaks while cells are in the S-phase. In our experience, SN-38 had an IC50 of approximately 1.0–6.0 nM against several human cancer cell lines of different origins.Citation13,Citation23 While SN-38 had the desired potency, the preparation of an active conjugate was challenging, since SN-38 is highly hydrophobic, with a limited number of coupling sites that could be used without jeopardizing its activity. In addition, while SN-38’s pharmacology was known, it is complex, with modifications to its structure that naturally occur in the body significantly reducing SN-38’s potency, such as glucuronidation and the opening of the lactone ring.Citation24,Citation25 In the case of SN-38, a dipiperidino side chain was added at the hydroxyl on the C10 position to improve water solubility, yielding the well-known chemotherapeutic agent, irinotecan.Citation24 The carbamate bond of this side chain was cleavable by carboxylesterases found primarily in the liver, releasing SN-38, which is as much as 1000-fold more potent than irinotecan.Citation24 However, once cleaved, this site also could be quickly glucuronidated, primarily by UGT-1A, a uridine diphosphate glucuronosyltransferase enzyme.Citation24
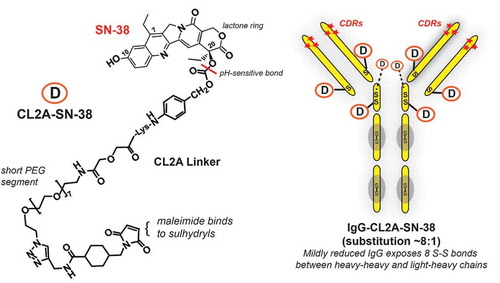
Our group investigated several types of linkers coupled to C10, as well as the C20 position, which is off the lactone ring ().Citation13,Citation26–Citation28 Short polyethylene glycol (PEG) groups that enhanced solubility and minimized aggregation were a key component. A maleimide group was added so that the linker-drug complex reacts with the sulfhydryl groups generated on the immunoglobulin (IgG), thereby forming a stable thioether bond between the linker and the IgG. Several derivatives contained a cathepsin B cleavage site (Phe-Lys) to ensure SN-38 would be released from the linker, with SN-38 bound to the linker by a pH-sensitive benzyl carbonate bond.Citation26
Figure 2. Sacituzumab govitecan (IMMU-132) composition. Sacituzumab govitecan is composed of the humanized monoclonal IgG (designated hRS7) that binds to TROP-2. The IgG is mildly reduced to expose 8 sulfhydryl-binding sites that are subsequently coupled to the CL2A-SN-38 linker-drug through the maleimide moiety of the CL2 linker. As indicated, the CL2A linker has a short PEG (polyethylene glycol) residue to aid in solubility and is coupled to SN-38 at the 20th position of the lactone ring, which stabilizes the ring from opening to the less active carboxylate form. The bond between CL2A and SN-38 is pH sensitive, being more susceptible to release in a low pH environment (e.g., found in lysosome or even in the microenvironment of tumors). The 10th position of SN-38 is protected from glucuronidation while SN-38 is bound to the IgG. Thus, SN-38, while bound to the antibody, remains in its most potent form until released.
Critical assessments included determination of conjugate yield, aggregate formation, and retention of immunoreactivity. Conjugates with reasonable production properties were subjected to additional testing. Interestingly, studies examining in vitro stability in serum revealed the conjugate of choice, with a linker designated CL2, was neither the most nor least stably linked (e.g., SN-38 release half-life >7 days to only 0.5 days, respectively), but rather was one with an intermediate stability of 1–2 days.Citation27 This initial linker was modified further to remove phenylalanine at the cathepsin B cleavage site (CL2A derivative), since this did not affect the rate of SN-38 release and improved product quality and yields.Citation13,Citation29 The resulting conjugate of CL2A-SN-38 coupled to a humanized anti-TROP-2 antibody was designated IMMU-132, and later named sacituzumab govitecan by the United States Adopted Names Council (USAN).
Coupling the linker-SN-38 complex to the IgG was performed by mild reduction, revealing eight thiol sites on the reduced IgG. Liquid chromatography-mass spectroscopy determined that the coupling procedure substituted all eight sites, four generated between cysteine residues within the hinge region (2 per heavy chain) and another four generated on cysteines that bridged the heavy chains to each of the light chains in the CH1-CL region, thus rendering a linkage that was site-specific and some distance from the antigen-binding complementarity-determining regions of the antibody ().Citation13 Since we found the native IgG and the IgG portion of the conjugate cleared at similar rates, comparable to the reduced and blocked IgG,Citation23 this suggested that the heavy-heavy and heavy-light chains remain associated even after disruption of the interchain disulfide bonds.
Analysis of several conjugate lots indicated an average SN-38:IgG ratio of 7.6, with a slightly lower ratio likely a result of a small amount of SN-38 released during the manufacturing process.Citation13 The conjugate retained immunoreactivity and, in mice, the IgG portion of the conjugate cleared similar to the native IgG,Citation23 with the exception that SN-38 was released at a rate predicted from in vitro serum stability studies; no SN-38 was detected after 72 h.Citation30 This suggested that its rapid clearance would preclude off-target toxicity.
Specificity was revealed using an assay to detect double-stranded DNA breaks.Citation13 Studies examining the conjugate’s mechanism of action identified upregulation of an early pro-apoptotic signaling effect with subsequent poly ADP ribose polymerase (PARP) cleavage, expected events occurring in cells with significant DNA damage.Citation23,Citation29
While the native hRS7 IgG showed antibody-dependent cell-mediated cytotoxicity (ADCC),Citation23,Citation31–Citation33 the conjugate’s ADCC activity was decreased by as much as 60%.Citation23 This loss was due to the reduction of the IgG and not the insertion of the linker-SN-38, since the reduced and blocked IgG had levels of ADCC that were similar to the conjugate. No complement-dependent cytotoxicity (CDC) was detectable using the native IgG or conjugate by in vitro assays.Citation23 Others have reported the direct anti-tumor activity with a phage-display derived anti-Trop-2 Fab.Citation34,Citation35 This anti-Trop-2 Fab was also conjugated with doxorubicin, which was shown to have anti-tumor activity in vitro against human pancreatic cancer cell lines.Citation36
Human tumor xenograft models in mice were used to assess activity in a variety of solid tumor cell lines, including breast, gastrointestinal, and lung.Citation13,Citation23,Citation29,Citation37 Again, enhanced activity was found in most cases when compared to irinotecan or free SN-38, as well as when compared to a non-targeting SN-38 ADC, but activity did not necessarily correlate with antigen expression measured in the cell line. This outcome most likely is based on differences in their sensitivity to a given drug (in this case, SN-38). In order to evaluate the role of TROP-2 expression on efficacy, the MDA-MB-231 breast cell line, which we found to be resistant to sacituzumab govitecan treatment, in part due to its increased expression of Rad51, was transfected with the TROP-2 gene.Citation37 Two transfectants with 4- and 25-fold higher expression of TROP-2 than the parental cell line were isolated. Both had similar Rad51 levels as the parental cell line, but they were more susceptible to sacituzumab govitecan treatment, presumably because higher TROP-2 levels increased SN-38 exposure. While these studies suggest TROP-2 expression can influence the therapeutic response, immunohistology studies found examples of heterogeneous antigen expression that also could have contributed to the effect.Citation23 This is consistent with our clinical observations that determination of TROP-2 expression levels in clinical tumor specimens may not be predictive of therapeutic outcome, but this needs further study.Citation38–Citation41
While the anti-TROP-2 antibody used in sacituzumab govitecan internalizes,Citation18,Citation22 given the tendency for the conjugate to release SN-38 with a half-life measured in serum of ~1 day, we speculate that some portion of the therapeutic activity of sacituzumab govitecan could result from bystander effects, manifested by the local release of SN-38 from the conjugate bound to antigen in the tumor microenvironment, being released before the intact conjugate is internalized. However, because sacituzumab govitecan had improved therapeutic responses over an irrelevant conjugate, specific targeting and internalization are likely the major contributing factors. This is an important distinction between this SN-38 ADC and the ultratoxic ADC platforms, where the stable linkage prevents drug release unless internalized, presumably by antigen-expressing cells, albeit IgG can be absorbed non-specifically by cells. Therefore, while bystander effects might be possible with stably linked conjugates (e.g., when active drug is released after targeted cell death),Citation42 the slow, local release of SN-38 represents a distinctly different mechanism than with most ultratoxic ADCs. Indeed, since SN-38 is active in the cell-cycle S-phase, maintaining low concentrations of SN-38 in tumors over time could improve potency, which is consistent with the clinical protocol of administering repeated high doses of sacituzumab govitecan, as discussed below.
Studies quantifying SN-38 concentrations in tumor xenografts indicated that the drug was detected over 3 days with sacituzumab govitecan compared to no more than 8 h for irinotecan-treated animals. Thus, despite administering 28-fold less mole-equivalents of SN-38 with sacituzumab govitecan compared to irinotecan, analysis of area-under-the-curve (AUC) indicated sacituzumab govitecan enhanced SN-38 concentrations in tumor xenografts by 20-fold to as much as 136-fold, compared to the AUC for SN-38 delivered by irinotecan.Citation30 This finding is perhaps even more significant because mice are able to process irinotecan to SN-38 much more efficiently than in humans,Citation43 and therefore SN-38 levels released from irinotecan in mice would be inflated. Thus, these findings support the view that the ADC’s selective targeting enhances the delivery of SN-38.
Another important finding was that levels of SN-38G, the inactivated, glucuronidated form of SN-38, were significantly lower in the serum of sacituzumab govitecan-treated mice.Citation30 SN-38G (glucuronidated SN-38)/SN-38 AUC ratios in humans given irinotecan are most often >4:1,Citation44 while in mice, this ratio for irinotecan was about 1:1,Citation30 with the estimated irinotecan-to-SN-38 conversion rate in mice was about 25%, whereas it is ~5% in humans.Citation24 However, in sacituzumab govitecan-treated animals, the AUC for SN-38G was about 5- to 10-times lower than the AUC for SN-38;Citation30 subsequently, human data showed an SN-38G/SN-38 AUC ratio of 1:5.Citation45 In patients given irinotecan therapy, the formation of SN-38G is a well-known process for the elimination of SN-38 and has been implicated in the development of so-called “late” diarrhea in patients, a significant complication with irinotecan. With irinotecan, a large portion of the injected dose of irinotecan and SN-38 is eliminated very quickly by the liver into the intestinal tract, inducing “early” diarrhea, whereas SN-38G recirculates by the hepatoenteric pathway, delaying its elimination; but when it enters the intestine, bacteria convert SN-38G to SN-38.Citation46
In sacituzumab govitecan, the C10 position is available for enzymatic glucuronidation after the conjugate is injected; however, in-vitro studies confirmed that the C10 is protected from glucuronidation while SN-38 remains bound on the IgG.Citation30 Another important detoxification process for SN-38 involves the opening of the lactone ring, leading to the carboxylate form.Citation24 This is a dynamic process that occurs naturally for both irinotecan and SN-38, with levels of the carboxylate form averaging ~35% of the total product.Citation24 Coupling the linker to the C20 position of SN-38 stabilizes the lactone ring,Citation47 which we confirmed with another TOPO-I-based ADC targeting carcinoembryonic antigen cell-adhesion molecule-5 (CEACAM5).Citation48
Thus, the SN-38 that remains bound to the IgG/linker is protected until released, which means that SN-38 released locally at the tumor will be in its fully active form, while SN-38 released from the conjugate in the serum or other tissues will be detoxified in a similar manner as found with irinotecan.
Clinical experience
Clinical studies with sacituzumab govitecan began in 2012, first as a dose-finding basket trial (NCT01631552) with patients having diverse epithelial cancers who failed conventional treatments, initially including colorectal, gastric, hepatocellular, NSCLC, small-cell lung cancer (SCLC), ovarian, pancreatic, prostate, triple-negative breast (TNBC), and urothelial cancers (UC).Citation41 Since over 80% of the tumor tissue specimens evaluated by immunohistochemistry showed positive staining for TROP-2, its expression was not a criterion for inclusion.
This trial was designed for patients to receive multiple cycles of sacituzumab govitecan, at a starting dose of 8 mg/kg given on Days 1 and 8 of a 21-day cycle. Treatment continued until disease progression or drug intolerance. Responses were determined every 8 weeks, requiring confirmation every 4–6 weeks, in accordance with RECIST 1.1 criteria for objective responses.Citation41
Although the maximum-tolerated dose from the Phase 1 study was 12 mg/kg, for repeated therapy with multiple cycles, starting doses of 8 and 10 mg/kg were preferred.Citation41 However, the final starting dose of 10 mg/kg, given twice (days 1 and 8) in each 21-day cycle of a multiple-cycle therapy was determined after further studies at these two dose levels.Citation45 Safety and pharmacokinetics (PK) were reported in 81 patients first given a starting dose of 8 mg/kg, followed by 97 patients enrolled with a starting dose of 10 mg/kg.Citation45 There was no difference in PK for these two dose levels, but assays monitoring the clearance of the intact IgG and the ADC showed the ADC cleared more quickly (e.g., half-life of 12 to 14 h), confirming that SN-38 was released from the conjugate, while the IgG remained in the circulation (e.g., half-life of 103 to 114 h). Further, as discussed above, levels of glucuronidated SN-38 were much less than reported for therapy with irinotecan,Citation44,Citation49 which may explain the lower rate of diarrhea (grade ≥3 in only 10% of patients given 10 mg/kg), compared with irinotecan therapy.Citation46,Citation49 Dose delays or reduction were due almost exclusively to neutropenia, which was similar to that experienced with irinotecan therapy. There was no evidence at this time for a higher risk of diarrhea or neutropenia based on uridine diphosphate glucuronosyltransferase haplotype status,Citation45 but additional data are necessary. Given the relative similarity between the dosing tolerability and toxicity, but with a suggestion of improved therapeutic response in one indication (triple-negative breast cancer), the 10 mg/kg dose level given weekly for the first 2 weeks of a 3-week treatment cycle was selected for future clinical trials.
The expanded Phase 2 clinical trial results at the 10 mg/kg starting dose have been reported for patients with TNBC, hormone-positive, HER2-negative breast cancer (HR+/HER2− BC), NSCLC, SCLC, and UC,Citation38–Citation40,Citation50–Citation54 summarized in . Given that these patients failed or relapsed after multiple prior therapies, the confirmed objective response rates, as well as median duration of responses, median progression-free survival, and the median overall survival rates are very encouraging, especially in the context of a very acceptable safety profile consisting of neutropenia, fatigue, diarrhea, and anemia.
Table 1. Summary of published results on phase 2 trials with sacituzumab govitecan.
An example of the efficacy of sacituzumab govitecan in patients with TNBC, an indication that was awarded Breakthrough Therapy Designation by the US Food and Drug Administration and is the potential first therapeutic target being sought for commercial approval, is presented in . Sacituzumab govitecan is being evaluated in a Phase 3 study of patients with refractory/relapsed TNBC (NCT02574455) while it is undergoing FDA review for accelerated approval, in addition to being studied in patients with HR+/HER2− BC who have failed at least two prior chemotherapy regimens (NCT03901339), in metastatic urothelial cancers after failure of platinum-based regimen or anti-PD-1/PD-L1 immunotherapy (NCT03547973), and in metastatic NSCLC, SCLC, head, and neck squamous carcinomas, endometrial, and hepatocellular carcinoma (NCT03964727).
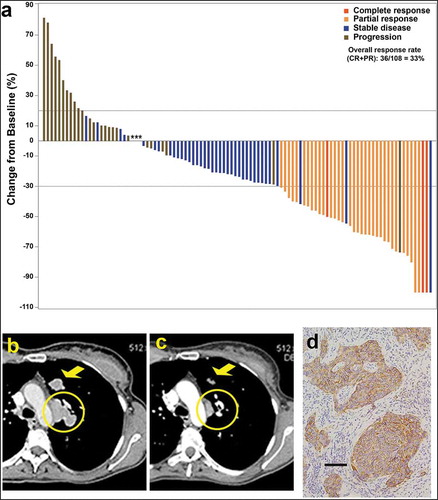
Figure 3. (a) Waterfall plot illustrating the best response data in 108 triple-negative breast cancer patients treated with sacituzumab govitecan (adapted from Bardia et al.,Citation50 with permission). (b–d) Tumor shrinkage by CT in triple-negative breast cancer patient given sacituzumab govitecan (from Bardia et al.Citation38 with permission). Case study: A 48-year-old woman with an initial diagnosis of triple-negative breast cancer in received four prior lines of treatment (including an anti-PD-L1 immune checkpoint inhibitor) and presented with lung and lymph node metastases at enrollment. She achieved a partial response that started 1.7 months after initiation of treatment with sacituzumab govitecan, with the best response of 54% reduction at 9.0 months and progression occurring at 14.4 months (partial response duration, 12.7 months). (b) Baseline image of two of the three target lesions: a 24 x 19-mm left-upper-lung mass (arrow) and a mediastinal lymph node (17 x 29-mm; circle). (c) After 16 doses, these two target lesions decreased to 13 × 7 and 9 × 19 mm, respectively. (d) TROP-2 expression in an archived tumor specimen by immunohistochemistry that shows 1+ to 2+ staining (overall, 2+).
In summary, based on the study results currently available, the TROP-2-targeted ADC sacituzumab govitecan appears to show good efficacy and manageable toxicity (mostly neutropenia and diarrhea) as a monotherapy in several cancer indications. We believe it represents a paradigm change in ADCs through its: (1) use of a drug with moderate (nM) toxicity, SN-38; (2) use of a moderately stable linker; (3) high ratio (~8:1) of drug to antibody; and (4) short conjugate half-life. The administration of repeated high doses of the ADC over prolonged periods is another differentiating factor. All of these attributes contribute, we believe, to enhanced drug targeting and concentration in the tumor, resulting in less off-target toxicity and possible bystander effects to cancer cells with reduced target antigen expression.
Other TROP-2 ADCs
It should be noted that TROP-2 as a target for ADCs has attracted the interest of additional companies, and several other anti-TROP-2 ADCs have been evaluated in clinical studies, although none has progressed further than Phase 1.
RN927C (PF-06664178, pfizer)
This ADC is composed of a different anti-TROP-2 antibody conjugated with a potent auristatin microtubule inhibitor, with a substitution ratio of 2.0.Citation55 The Phase 1 trial of 31 patients with diverse metastatic cancers, where the ADC was given every 21 days with doses escalating from 0.15 mg/kg to 4.8 mg/kg, showed unacceptable toxicities of fatigue, constipation, nausea, chills, infusion reactions, neutropenia, rash, weight loss, arthralgia, decreased appetite, diarrhea, dyspnea, mucosal inflammation, and pruritis, with neutropenia and rash being the most common grade ≥3 adverse events.Citation56 At tolerable doses, stable disease was the best response. There was also an anti-drug antibody response in almost all patients. Apparently, this ADC has been abandoned.
BAT8003 (bio-thera solutions, ltd.)
Preclinical studies were reported recently with another microtubule inhibitor (maytansine) conjugated using a novel uncleavable linker to an anti-TROP-2 antibody that is glycoengineered to have enhanced ADCC activity.Citation57 It is currently listed as starting Phase 1 clinical testing in patients with epithelial cancers that are positive for TROP-2 expression (NCT03884517).
DS-1062a (Daiichi-sankyo)
The anti-TROP-2 ADC DS-1062a is composed of an enzymatically cleavable peptide linker conjugated to a topoisomerase-I inhibitor, DXd,Citation58 that is a derivative of the camptothecin Exatecan, which is said to be 10-fold more potent than SN-38.Citation59,Citation60 This ADC showed dose-dependent tumor-growth inhibition in human cancer xenografts, as well as favorable PK profiles and safety in Cynomolgus monkeys.Citation58 Preliminary data from a Phase 1 clinical trial in patients with relapsed NSCLC (NCT03401385) indicate that the agent has been safely tolerated at doses of up to 2 mg/kg, with 1 partial response in 18 tumor evaluable patients.Citation61 An update of the data presented at the 2019 ASCO meeting included 39 patients given doses as high as 8.0 mg/kg.Citation62 At this time, 10 partial responses were reported, with 41% of the patients having at least one treatment-emergent grade ≥3 adverse event; 1 dose-liming maculopapular rash at 6 mg/kg.Citation62 Unlike its companion ADC, [fam-] trastuzumab deruxtecan (DS-8201a), which targets HER2 and uses a substitution ratio of 8 DXd/antibody,Citation59,Citation63–Citation66 DS-1062a has a substitution ratio of 4 DXd/antibody.Citation62 Thus, DS-1062a represents a second ADC platform utilizing a TOPO 1 inhibitor, but unlike sacituzumab govitecan, it employs a drug considered to have “ultratoxic” potency (i.e., 10-times higher than SN-38Citation59,Citation60), requiring a linker with high stability in serum and a reduced substitution ratio.
Conclusion
In addition to the observation that TROP-2 is a prognosticator for a number of cancers and has a role in controlling cancer growth and spread by various signaling pathways,Citation14 its enhanced expression in a large number of cancers, with minimal expression in normal tissues, suggests that it is a unique target for precision cancer medicine approaches, including ADCs.Citation14 It is anticipated that sacituzumab govitecan will achieve its first clinical use in the treatment of advanced TNBC, followed by advanced HR+, ER− breast and urothelial cancers.Citation38,Citation50–Citation53 This ADC also can be incorporated into combination therapy with poly(ADP-ribose)polymeraseCitation67 and immune checkpoint inhibitors, which may improve clinical outcomes.
Competing Interests
The authors own Immunomedics stock or stock options. Dr. Goldenberg has royalty-bearing patented inventions. Dr. Goldenberg is the founder and retired Chief Scientific Officer of Immunomedics, Inc. Dr. Sharkey is a consultant to Immunomedics.
Grant Support
The authors’ studies were supported in part by an Outstanding Investigator Grant to DMG from the National Cancer Institute, NIH (CA39841), NIH grant S07-RR05903, American Cancer Society Grant EDT-16, New Jersey Commission on Cancer Research, Claire Sullivan Memorial Fund at the Garden State Cancer Center, Garden State Cancer Center Foundation, Escalon Foundation, and Immunomedics, Inc.
Acknowledgments
We thank Rhona Stein, Ph.D., for contributing to the development and characterization of anti-TROP-2 antibodies, and Serengulam V. Govindan, Ph.D., for his contributions to SN-38 chemistry. Cynthia L. Sullivan kindly reviewed and critiqued the manuscript.
Additional information
Funding
Related Research Data
References
- Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. doi:10.1146/annurev-med-050311-201823.
- Leal M, Sapra P, Hurvitz SA, Senter P, Wahl A, Schutten M, Shah, D.K., Haddish‐Berhane, N. and Kabbarah, O. Antibody-drug conjugates: an emerging modality for the treatment of cancer. Ann N Y Acad Sci. 2014;1321:41–54. doi:10.1111/nyas.12499.
- Govindan SV, Sharkey RM, Goldenberg DM. Prospects and progress of antibody-drug conjugates in solid tumor therapies. Expert Opin Biol Ther. 2016;16:883–93. doi:10.1517/14712598.2016.1173203.
- Abdollahpour-Alitappeh M, Lotfinia M, Gharibi T, Mardaneh J, Farhadihosseinabadi B, Larki P, Faghfourian B, Sepehr KS, Abbaszadeh-Goudarzi K, Abbaszadeh-Goudarzi G, et al. Antibody-drug conjugates (ADCs) for cancer therapy: strategies, challenges, and successes. J Cell Physiol. 2019;234:5628–42. doi:10.1002/jcp.27419.
- Birrer MJ, Moore KN, Betella I, Bates RC. in press. Antibody-drug conjugate-based therapeutics: state of the Science. J Natl Cancer Inst. 2019 Mar 11. pii: djz035. doi:10.1093/jnci/djz035. [Epub ahead of print]
- Ricart AD. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin Cancer Res. 2011;17:6417–27.
- Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Curr Opin Chem Biol. 2010;14:529–37.
- Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram, M, Oh, DY, Diéras, V and Guardino, E, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–91.
- Moore KN, Vergote I, Oaknin A, Colombo N, Banerjee S, Oza A, Pautier, P, Malek, K and Birrer, MJ. FORWARD I: A Phase III study of mirvetuximab soravtansine versus chemotherapy in platinum-resistant ovarian cancer. Future Oncol. 2018;14:1669–78.
- Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler, KM, Bernhardt, SX, Kopcha, AK, Zabinski, RF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10:7063–70. doi:10.1158/1078-0432.CCR-04-0789.
- Strop P, Delaria K, Foletti D, Witt JM, Hasa-Moreno A, Poulsen K, Casas, MG, Dorywalska, M, Farias, S, Pios, A, et al. Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat Biotechnol. 2015;33:694–96.
- Carter PJ, Senter PD. Antibody-drug conjugates for cancer therapy. Cancer J. 2008;14:154–69. doi:10.1097/PPO.0b013e318172d704.
- Goldenberg DM, Cardillo TM, Govindan SV, Rossi EA, Sharkey RM. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget. 2015;6:22496–512. doi:10.18632/oncotarget.4318.
- Goldenberg DM, Stein R, Sharkey RM. The emergence of trophoblast cell-surface antigen 2 (TROP-2) as a novel cancer target. Oncotarget. 2018;9:28989–9006. doi:10.18632/oncotarget.25615.
- Lipinski M, Parks DR, Rouse RV, Herzenberg LA. Human trophoblast cell-surface antigens defined by monoclonal antibodies. Proc Natl Acad Sci USA. 1981;78:5147–50. doi:10.1073/pnas.78.8.5147.
- Cubas R, Li M, Chen C, Yao Q. Trop2: A possible therapeutic target for late stage epithelial carcinomas. Biochim Biophys Acta. 2009;1796:309–14. doi:10.1016/j.bbcan.2009.08.001.
- Shvartsur A, Bonavida B. Trop2 and its overexpression in cancers: regulation and clinical/therapeutic implications. Genes Cancer. 2015;6:84–105. doi:10.18632/genesandcancer.40.
- Stein R, Basu A, Chen S, Shih LB, Goldenberg DM. Specificity and properties of MAb RS7-3G11 and the antigen defined by this pancarcinoma monoclonal antibody. Int J Cancer. 1993;55:938–46.
- Basu A, Goldenberg DM, Stein R. The epithelial/carcinoma antigen EGP-1, recognized by monoclonal antibody RS7-3G11, is phosphorylated on serine 303. Int J Cancer. 1995;62:472–79.
- Stein R, Basu A, Goldenberg DM, Lloyd KO, Mattes MJ. Characterization of cluster 13: the epithelial/carcinoma antigen recognized by MAb RS7. Int J Cancer Suppl. 1994;8:98–102.
- De Leij L, Helrich W, Stein R, Mattes MJ. SCLC-cluster-2 antibodies detect the pancarcinoma/epithelial glycoprotein EGP-2. Int J Cancer Suppl. 1994;8:60–63.
- Shih LB, Xuan H, Aninipot R, Stein R, Goldenberg DM. In vitro and in vivo reactivity of an internalizing antibody, RS7, with human breast cancer. Cancer Res. 1995;55:5857s–63s.
- Cardillo TM, Govindan SV, Sharkey RM, Trisal P, Arrojo R, Liu D, Rossi EA, Chang C-H, Goldenberg DM. Sacituzumab govitecan (IMMU-132), an anti-Trop-2/SN-38 antibody-drug conjugate: characterization and efficacy in pancreatic, gastric, and other cancers. Bioconjug Chem. 2015;26:919–31. doi:10.1021/acs.bioconjchem.5b00223.
- Mathijssen RH, van Alphen RJ, Verweij J, Loos WJ, Nooter K, Stoter G, Sparreboom A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin Cancer Res. 2001;7:2182–94.
- Garcia-Carbonero R, Supko JG. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin Cancer Res. 2002;8:641–61.
- Moon SJ, Govindan SV, Cardillo TM, D’Souza CA, Hansen HJ, Goldenberg DM. Antibody conjugates of 7-ethyl-10-hydroxycamptothecin (SN-38) for targeted cancer chemotherapy. J Med Chem. 2008;51:6916–26. doi:10.1021/jm800719t.
- Govindan SV, Cardillo TM, Moon SJ, Hansen HJ, Goldenberg DM. CEACAM5-targeted therapy of human colonic and pancreatic cancer xenografts with potent labetuzumab-SN-38 immunoconjugates. Clin Cancer Res. 2009;15:6052–61. doi:10.1158/1078-0432.CCR-09-0586.
- Govindan SV, Cardillo TM, Goldenberg DM. Chapter 8: topoisomerase inhibitors as antibody-drug conjugate payloads. In: Thurston DE, Jackson PJM, editors. Cytotoxic payloads for antibody-drug conjugates. Cambridge (UK): Royal Society of Chemistry; 2019. p. 164–84.
- Cardillo TM, Govindan SV, Sharkey RM, Trisal P, Goldenberg DM. Humanized anti-Trop-2 IgG-SN-38 conjugate for effective treatment of diverse epithelial cancers: preclinical studies in human cancer xenograft models and monkeys. Clin Cancer Res. 2011;17:3157–69. doi:10.1158/1078-0432.CCR-10-2939.
- Sharkey RM, McBride WJ, Cardillo TM, Govindan SV, Wang Y, Rossi EA, Chang C-H, Goldenberg DM. Enhanced delivery of SN-38 to human tumor xenografts with an anti-Trop-2-SN-38 antibody conjugate (sacituzumab govitecan). Clin Cancer Res. 2015;21:5131–38. doi:10.1158/1078-0432.CCR-15-0670.
- Bignotti E, Ravaggi A, Romani C, Falchetti M, Lonardi S, Facchetti F, Pecorelli S, Varughese J, Cocco E, Bellone S, et al. Trop-2 overexpression in poorly differentiated endometrial endometrioid carcinoma: implications for immunotherapy with hRS7, a humanized anti-trop-2 monoclonal antibody. Int J Gynecol Cancer. 2011;21:1613–21. doi:10.1097/IGC.0b013e318228f6da.
- Raji R, Guzzo F, Carrara L, Varughese J, Cocco E, Bellone S, Betti, M, Todeschini, P, Gasparrini, S, Ratner, E, et al. Uterine and ovarian carcinosarcomas overexpressing Trop-2 are sensitive to hRS7, a humanized anti-Trop-2 antibody. J Exp Clin Cancer Res. 2011;30:106. doi:10.1186/1756-9966-30-24.
- Varughese J, Cocco E, Bellone S, Ratner E, Silasi DA, Azodi M, Schwartz PE, Rutherford TJ, Buza N, Pecorelli S, et al. Cervical carcinomas overexpress human trophoblast cell-surface marker (Trop-2) and are highly sensitive to immunotherapy with hRS7, a humanized monoclonal anti-Trop-2 antibody. Am J Obstet Gynecol. 2011;205:567.e1-7. doi:10.1016/j.ajog.2011.06.093.
- Lin H, Zhang H, Wang J, Lu M, Zheng F, Wang C, Tang X, Xu N, Chen R, Zhang D, et al. A novel human Fab antibody for Trop2 inhibits breast cancer growth in vitro and in vivo. Int J Cancer. 2014;134:1239–49. doi:10.1002/ijc.28451.
- Liu J, Yang D, Yin Z, Gao M, Tong H, Su Y, Zhu, J, Ye, C and Zhang, H, et al. A novel human monoclonal Trop2-IgG antibody inhibits ovarian cancer growth in vitro and in vivo. Biochem Biophys Res Commun. 2019 Apr 30;512(2):276–282. doi:10.1016/j.bbrc.2019.03.028. Epub 2019 Mar 14.
- Mao Y, Wang X, Zheng F, Wang C, Tang Q, Tang X, Xu N, Zhang H, Zhang D, Xiong L, et al. The tumor-inhibitory effectiveness of a novel anti-Trop2 Fab conjugate in pancreatic cancer. Oncotarget. 2016;7:24810–23. doi:10.18632/oncotarget.8529.
- Cardillo TM, Mostafa AA, Rossi DL, Liu D, Chang CH, Sharkey RM, and Goldenberg, DM. Treatment of high Trop-2-expressing triple-negative breast cancer (TNBC) with sacituzumab govitecan (IMMU-132) overcomes homologous recombination repair (HRR) rescue mediated by Rad51. Cancer Res. 2017;77(abstract):3193.
- Bardia A, Mayer IA, Diamond JR, Moroose RL, Isakoff SJ, Starodub AN, Shah NC, O’Shaughnessy J, Kalinsky K, Guarino M, et al. Efficacy and safety of anti-Trop-2 antibody drug conjugate sacituzumab govitecan (IMMU-132) in heavily pretreated patients with metastatic triple-negative breast cancer. J Clin Oncol. 2017;35:2141–48. doi:10.1200/JCO.2016.70.8297.
- Gray JE, Heist RS, Starodub AN, Camidge DR, Kio EA, Masters GA, Purcell WT, Guarino MJ, Misleh J, Schneider CJ, et al. Therapy of small cell lung cancer (SCLC) with a topoisomerase-I-inhibiting antibody-drug conjugate (ADC) targeting Trop-2, sacituzumab govitecan. Clin Cancer Res. 2017;23:5711–19. doi:10.1158/1078-0432.CCR-17-0933.
- Heist RS, Guarino MJ, Masters G, Purcell WT, Starodub AN, Horn L, Scheff RJ, Bardia A, Messersmith WA, Berlin J, et al. Therapy of advanced non-small-cell lung cancer with an SN-38-anti-Trop-2 drug conjugate, sacituzumab govitecan. J Clin Oncol. 2017;35:2790–97. doi:10.1200/JCO.2016.72.1894.
- Starodub AN, Ocean AJ, Shah MA, Guarino MJ, Picozzi VJ Jr., Vahdat LT, Thomas SS, Govindan SV, Maliakal PP, Wegener WA, et al. First-in-human trial of a novel anti-Trop-2 antibody-SN-38 conjugate, sacituzumab govitecan, for the treatment of diverse metastatic solid tumors. Clin Cancer Res. 2015;21:3870–78. doi:10.1158/1078-0432.CCR-14-3321.
- Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer. 2017(117):.1736–42
- Morton CL, Wierdl M, Oliver L, Ma MK, Danks MK, Stewart CF, Eiseman JL, Potter PM. Activation of CPT-11 in mice: identification and analysis of a highly effective plasma esterase. Cancer Res. 2000;60:4206–10.
- Xie R, Mathijssen RH, Sparreboom A, Verweij J, Karlsson MO. Clinical pharmacokinetics of irinotecan and its metabolites: a population analysis. J Clin Oncol. 2002;20:3293–301. doi:10.1200/JCO.2002.11.073.
- Ocean AJ, Starodub AN, Bardia A, Vahdat LT, Isakoff SJ, Guarino M, Messersmith WA, Picozzi VJ, Mayer IA, Wegener WA, et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: safety and pharmacokinetics. Cancer. 2017;123:3843–54. doi:10.1002/cncr.30789.
- Stein A, Voigt W, Jordan K. Chemotherapy-induced diarrhea: pathophysiology, frequency and guideline-based management. Ther Adv Med Oncol. 2010;2:51–63. doi:10.1177/1758834009355164.
- Zhao H, Lee C, Sai P, Choe YH, Boro M, Pendri A, Guan S, Greenwald RB. 20-O-acylcamptothecin derivatives: evidence for lactone stabilization 1. J Org Chem. 2000;65:4601–06.
- Sharkey RM, Govindan SV, Cardillo TM, Donnell J, Xia J, Rossi EA, Chang C-H, Goldenberg DM. Selective and concentrated accretion of SN-38 with a CEACAM5-targeting antibody-drug conjugate (ADC), labetuzumab govitecan (IMMU-130). Mol Cancer Ther. 2018;17:196–203. doi:10.1158/1535-7163.MCT-17-0442.
- Xie R, Mathijssen RH, Sparreboom A, Verweij J, Karlsson MO. Clinical pharmacokinetics of irinotecan and its metabolites in relation with diarrhea. Clin Pharmacol Ther. 2002;72:265–75.
- Bardia A, Mayer IA, Vahdat LT, Tolaney SM, Isakoff SJ, Diamond JR, O’Shaughnessy, J, Moroose, RL, Santin, AD, Abramson, VG, et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N Engl J Med. 2019;380:741–51.
- Bardia A, Tolaney SM, Juric D, Mayer IA, Vahdat LT, Diamond JR, Kalinsky K, O'Shaughnessy J, Moroose RL, Wahskowitz S, et al. Efficacy of sacituzumab govitecan (anti-Trop-2-SN-38 antibody-drug conjugate) for endocrine-refractory hormone-receptor positive (HR+) metastatic breast cancer (mBC). J Clin Oncol. 2018;36(Abstr):1004.
- Faltas B, Goldenberg DM, Ocean AJ, Govindan SV, Wilhelm F, Sharkey RM, Hajdenberg, J, Hodes, G, Nanus, DM, Tagawa, ST. Sacituzumab govitecan, a novel antibody-drug conjugate, in patients with metastatic platinum-resistant urothelial carcinoma. Clin Genitourin Cancer. 2016;14:e75–9.
- Tagawa ST, Faltas B, Lam ET, Saylor P, Bardia A, JHajdenberg J, Morgans, AK, Lim, EA, Kalinsky, K, Simpson, PS, et al. Sacituzumab govitecan (IMMU-132) in patients with previously treated metastatic urothelial cancers (mUC): results from a Phase I/II study. J Clin Oncol. 2019;37(abst):354.
- Tagawa ST, Petrylak DP, Grivas P, Agarwal N, Sternberg CN, Hernandez C, Siemon-Hryczyk P, Goswami T, Loriot Y. TROPHY-U-01: A phase II open-label study of sacituzumab govitecan (IMMU-132) in patients with advanced urothelial cancer after progression on platinum-based chemotherapy and/or anti-PD-1/PD-L1 checkpoint inhibitor therapy. (suppl). J Clin Oncol. 2019;37:abstrTPS3153.
- Strop P, Tran TT, Dorywalska M, Delaria K, Dushin R, Wong OK, Ho, WH, Zhou, D, Wu, A, Kraynov, E, et al. RN927C, a site-specific Trop-2 antibody-drug conjugate (ADC) with enhanced stability, is highly efficacious in preclinical solid tumor models. Mol Cancer Ther. 2016;15:2698–708.
- King GT, Eaton KD, Beagle BR, Zopf CJ, Wong GY, Krupka HI, Hua, SY, Messersmith, WA and El-Khoueiry, AB. A phase 1, dose-escalation study of PF-06664178, an anti-Trop-2/Aur0101 antibody-drug conjugate in patients with advanced or metastatic solid tumors. Invest New Drugs. 2018;36:836–47.
- Tang W, Huang X, Ou Z, Yan H, Gan J, Dong Q, Tan, B, Yang, Y, Guo, Y, Li, S, et al. 2019. BAT8003, a potent anti-Trop-2 antibody-drug conjugate, for the treatment of triple negative breast cancer [abstract]. Cancer Res. 79(Issue 4). (suppl; In: The Proceedings of the 2018 San Antonio Breast Cancer Symposium, 2018): Abstractnr P6-20-16
- Okajima D, Yasuda S, Yokouchi Y, Fujitani T, Sakurai K, Yamaguchi J, Kitamura, M, Terauchi, T, Shibutani, T, Aida, T, et al. Preclinical efficacy studies of DS-1062a, a novel TROP2-targeting antibody-drug conjugate with a novel DNA topoisomerase I inhibitor DXd. J Clin Oncol (Suppl). 2018;36(abstract):e24206.
- Ogitani Y, Aida T, Hagihara K, Yamaguchi J, Ishii C, Harada N, Soma M, Okamoto H, Oitate M, Arakawa S, et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin Cancer Res. 2016;22:5097–108.
- Nakada T, Sugihara K, Jikoh T, Abe Y, Agatsuma T. The latest research and development into the antibody-drug conjugate, [fam-] trastuzumab deruxtecan (DS-8201a), for HER2 cancer therapy. Chem Pharm Bull (Tokyo). 2019;67:173–85.
- Sands JM, Shimizu T, Garon EB, Greenberg J, Guevara FM, Heist RS, Kobayashi, F, Noguchi, Y, Okajima, D, Tajima, N, et al. First-in-human phase 1 study of DR-1062a in patients with advanced solid tumors. J Clin Oncol (Suppl). 2019;37(abstr):9051.
- Brennan J Daiichi Sankyo presents preliminary phase 1 data for TROP2 targeting ADC DS-1062 in patients with non-small cell lung cancer at 2019. ASCO annual meeting 2019; Chicago, IL.
- Doi T, Shitara K, Naito Y, Shimomura A, Fujiwara Y, Yonemori K, Shimizu, C, Shimoi, T, Kuboki, Y, Matsubara, N, et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody-drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: a phase 1 dose-escalation study. Lancet Oncol. 2017;18:1512–22.
- Iwata TN, Ishii C, Ishida S, Ogitani Y, Wada T, Agatsuma T. A HER2-targeting antibody-drug conjugate, trastuzumab deruxtecan (DS-8201a), enhances antitumor immunity in a mouse model. Mol Cancer Ther. 2018;17:1494–503.
- Ogitani Y, Hagihara K, Oitate M, Naito H, Agatsuma T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016;107:1039–46.
- Takegawa N, Nonagase Y, Yonesaka K, Sakai K, Maenishi O, Ogitani Y, Tamura, T, Nishio, K, Nakagawa, K and Tsurutani, J. DS-8201a, a new HER2-targeting antibody-drug conjugate incorporating a novel DNA topoisomerase I inhibitor, overcomes HER2-positive gastric cancer T-DM1 resistance. Int J Cancer. 2017;141:1682–89.
- Cardillo TM, Sharkey RM, Rossi DL, Arrojo R, Mostafa AA, Goldenberg DM. Synthetic lethality exploitation by an anti-Trop-2-SN-38 antibody-drug conjugate, IMMU-132, plus PARP inhibitors in BRCA1/2-wild-type triple-negative breast cancer. Clin Cancer Res. 2017;23:3405–15. doi:10.1158/1078-0432.CCR-16-2401.