
ABSTRACT
The immunoglobulin (Ig) CH2 domain is a promising scaffold for the development of candidate therapeutics. We have previously shown that the stability of isolated CH2 could be increased by the introduction of an additional disulfide bond and removal of seven N-terminal residues (m01s). However, both isolated CH2 and m01s aggregate, likely due to the existence of aggregation-prone regions (APRs) that we identified by using computational methods. This knowledge was used to generate a phage display library of mutants. The library was incubated at high temperature to remove aggregating CH2 domains, and then panned against a mouse anti-human CH2 monoclonal antibody targeting a conformational epitope to remove misfolded CH2s. After two rounds of panning, one clone, m01s5, with smaller APRs, was identified. After additional mutagenesis one clone, m01s5.4, which aggregated much less than m01s as measured by a turbidity assay and dynamic light scattering, was identified. m01s5.4 also exhibited much lower nonspecific binding than m01s. Engineering of a previously identified m01s-based tumor antigen-specific binder led to a dramatic reduction of its aggregation without affecting its binding. In summary, we describe a new approach for reducing aggregation based on a combination of computational and phage display methodologies, and show that aggregation of CH2-based scaffolds can be significantly reduced by the newly identified mutants, which can improve the developability of potential CH2-based therapeutics.
Introduction
Monoclonal antibodies (mAbs) are currently widely used for the treatment of diseases such as cancers, immune disorders, and infections.Citation1 However, a major problem encountered during the manufacture of these antibodies is aggregation, which can occur during production, purification and long-term storage, and in the high concentration formulations required for disease treatment.Citation2–Citation5 Aggregated antibodies have decreased activity and can elicit an immunological response, which may hamper clinical development.Citation6–Citation8 Although aggregation may be decreased by the improvement of extrinsic factors, such as careful manipulation, and optimization of the storage and formulation,Citation9–Citation12 decreasing the aggregation tendency of the antibodies themselves is a better solution. For an immunoglobulin (Ig) molecule, the intrinsic factors for aggregation propensity include framework regions,Citation13 complementarity-determining regions,Citation14 constant domains,Citation15–Citation21 glycosylation patterns,Citation22,Citation23 as well as many others.Citation24–Citation29 Therefore, solving the aggregation challenge requires an understanding of the fundamental mechanisms involved in aggregation, which could be helpful for stabilizing the therapeutic antibodies to prevent aggregation in vitro and in vivo.
The antibody CH2 domain has been proposed as a promising scaffold for the development of novel antigen recognition units termed C-based single domain antibodies (C-sdAbs) or nanoantibodies (nAbs) as next-generation candidate therapeutics. Compared to other antibody fragments, such as antigen-binding fragments (Fabs), single-chain variable fragments (scFVs) and heavy chain variable domains (VHs), the CH2 domain contains binding sites or portions of binding sites conferring neonatal Fc receptor and effector binding.Citation17,Citation30–Citation32 The autonomous CH2 domain mainly consists of two β-sheets that contain seven β-strands connected with three loops and two helices. There is a native disulfide bond between strand B and F. We stabilized a human IgG1 CH2 domain by the introduction of an additional disulfide bond and removal of the seven N-terminal amino acids in CH2 (m01s).Citation32–Citation34 However, we found that most clones tend to aggregate and become sticky after the introduction of foreign sequences in CH2 or m01s for library construction, which constrained the selection of high-affinity binders based on CH2 (m01s).Citation35,Citation36
To decrease the aggregation propensity, we used a combination of computational, structural and phage display methodologies. Since several computational programs have been developed to predict aggregation-prone regions (APRs), we used one of them, TANGO,Citation37–Citation39 for the prediction of APRs in CH2 (m01s), which resulted in the identification of three major APRs. We also analyzed the crystal structure of an isolated CH2 domain and found a potential hotspot involved in aggregation besides the three APRs. These aggregation-related regions were selected for optimization by phage display technique and site directed mutagenesis. Finally, we identified a mutant, m01s5.4, with dramatically increased aggregation resistance compared to m01s. Interestingly, m01s5.4 showed no or very low nonspecific binding to other proteins. We introduced the corresponding mutated residues to a binder (D3-m01s) derived from an m01s-based phage display libraryCitation40 to make a D3 mutant (D3-m01s5.4). As expected, D3-m01s5.4 aggregated much less than D3-m01s. Importantly, the binding was still maintained. Therefore, m01s5.4 could be a good candidate as a scaffold for the selection of C-sdAbs. These binders may have increased drugability as candidate CH2-based therapeutics.
Results
Design and construction of an m01s mutant library
TANGO is an online program (http://tango.crg.es/) based on simple physicochemical principles of secondary structure formation extended by the assumption that the core regions of an aggregate are fully buried. Here, it was used to predict the ARPs in the CH2 domain. Three major APRs (1–3) in CH2 were identified at strand B, C, and E, respectively (). All the three clusters contain a relatively high percentage of hydrophobic amino acids, indicating that these residues could contribute to misfolding and aggregation. APR3 is the largest region, and it is located on the same side (one β-sheet) with APR1, while APR2 is on the other side (the other β-sheet). m01s, as mentioned above, has the same APRs predicted by TANGO as CH2 (). According to the crystal structure of an isolated CH2 domain (PDB entry: 3DJ9Citation41), the residues T260 (EU numberingCitation42), V262 and V264 in APR1, and R301, V303 and V305 in APR3 are exposed on the surface of one β-sheet, while the residues N276 and Y278 in APR2 protrude on the surface of the other β-sheet (). The directions of these residues were also confirmed by the analysis of the crystal structure of the CH2 domain in the context of an intact human IgG (PDB entry: 1HZHCitation43) (Figure S1). We also noticed that two hydrophobic residues F241 and F243 in strand A (), which are exposed to the solvent, could also be involved in aggregation formation.
Figure 1. Analysis of residues involved in aggregation in CH2 for optimization. (a). Prediction of APRs in CH2 and m01s by the online program TANGO. Three major APRs (APR1-3, highlighted by purple, green and red, respectively) that located at three different β-strands in two different sides are identified. (b). The analysis of the crystal structure of CH2 domain (PDB entry: 3DJ9). The exposed residues F241 and F243 in strand A (blue), T260, V262, V264 in APR1 (purple), N276, Y278 in APR2 (green) and R301, V303, V305 in APR3 (red) are shown. (c). The conceptual sketch of the CH2 optimization approach. Six residues in APR1 (T260, V262, and V264) and APR3 (R301, V303 and V305) and two exposed “phenylalanine” on strand A (F241 and F243) in one side were randomly mutated for library construction. Those resides in APR2 (from F275 to V279) on the other side were scanned by site mutagenesis of lysine
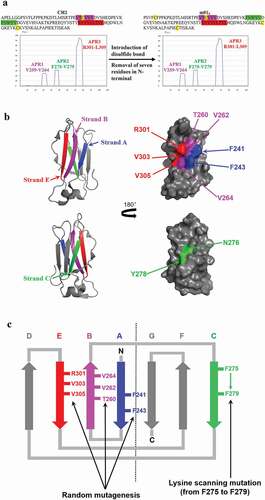
We selected the “sticking out” residues in one β-sheet, including F241 and F243; T260, V262 and V264; and R301, V303 and V305 for random mutagenesis (). It should be noted that, although T260 and R301 were not hydrophobic, they were still selected for phage display construction because we wanted to increase the diversity of the library to allow selection of the best combination of amino acids. To reduce the aggregation propensity of APR2, we simply did full substitution from F275 to V279 by lysine scanning to try in spite of exposure or burying ().
Identification of one soluble clone m01s5 with reduced APRs
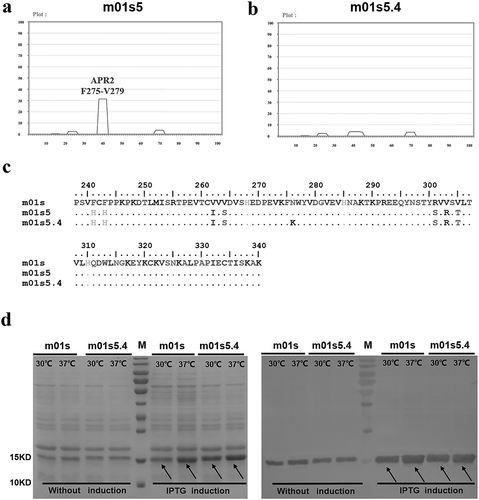
To obtain mutants with reduced APRs, we used a heat-denaturation panning approach for isolating non-aggregating mutants from the m01s library. After two rounds of panning against a mouse anti-human CH2 IgG, several clones with positive binding activity were obtained by monoclonal phage enzyme-linked immunosorbent assay (ELISA). The candidate clones were selected for expression and sequencing. Among these clones, the soluble expression level in E. coli of one clone, m01s5 was higher than or equal to that of m01s. Hence, the mutant m01s5 was selected for further characterization. TANGO was applied for the prediction of the APRs in m01s5. Compared to APRs in m01s, APR1 and APR3 were significantly reduced in m01s5 (). However, APR2 was not changed because this region was not mutated.
Figure 2. Selection of clone m01s5.4. (a). Prediction of APRs in m01s5. Only one major APR (APR2 in m01s in ) could be identified after panning. (b). Prediction of APRs in m01s5.4. No obvious cluster is identified. (c). Sequence alignment of m01s, m01s5 and m01s5.4. (d). Soluble expression of m01s and m01s5.4. Left: SDS-PAGE, right: Western blot. m01s5.4 shows good soluble expression as m01s at 37°C
Elimination of APR2 in m01s5
As a basic amino acid, lysine has been widely used in protein engineering to reduce aggregation. To decrease APR2 in m01s5, lysine scanning was performed to mutate the residues one by one in this region (Figure S2a). After scanning, only one mutant (m01s5.4) with the replacement of N276 by lysine still maintained a good soluble expression level (Figure S2b). TANGO analysis showed that the APR2 in m01s5.4 was significantly reduced compared to that in m01s5 ( vs. ). Sequence alignment of m01s, m01s5, and m01s5.4 was also performed (). It could be observed that many hydrophobic residues in APRs in m01s were replaced by hydrophilic or charged amino acids, whereas the original hydrophilic residue T260 in APR1 and the basic residue R301 in APR3 were either unchanged or changed to hydrophilic serine, respectively (). The soluble expression levels of m01s and m01s5.4 were compared side by side at 30°C and 37°C. In general, both of them could be expressed at comparable levels (). m01s5.4 was subjected to further characterization.
Well-folded β-structural monomer of m01s5.4
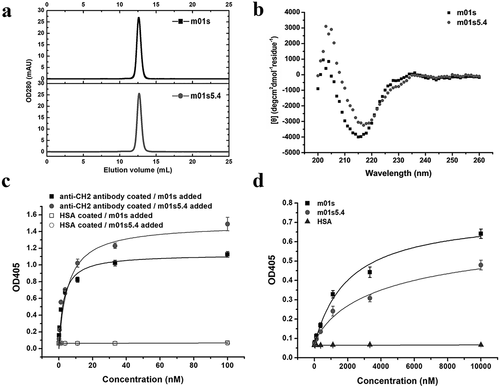
Size exclusion chromatography (SEC) was performed to determine the molecular weight of m01s5.4 in solution. Only a unique peak was eluted in the position that corresponded to a monomer according to a standard curve ( and Figure S3). The secondary structure of m01s5.4 determined by circular dichroism (CD) showed a maximum negative peak at 216 nm, which represents typical β-sheet structure (). In addition, m01s5.4 could still be recognized by a mouse anti-human CH2 mAb targeting the conformation epitope described above () and a human Fab, m01m1, specific for CH2 used previouslyCitation34 (). Taken together, m01s5.4 is a well-folded β-strand rich monomer similar to m01s.
Figure 3. Characterization of m01s5.4. (a). Oligomer formation of m01s5.4 and m01s measured by SEC. m01s5.4 exists as a monomer. (b). CD for secondary structure measurement. Typical maximum negative peak appears at 216 nm indicating a β-sheet structure. (c). Binding of m01s5.4 and m01s to mouse anti-human CH2 monoclonal antibody. HSA was used as a negative control. (d). Binding of m01s5.4 and m01s to anti-human CH2 Fab (m01m1). HSA was also used as a negative control. In general, m01s and m01s5.4 show similar binding to these two tested antibodies
m01s5.4 is less stable against heating and urea
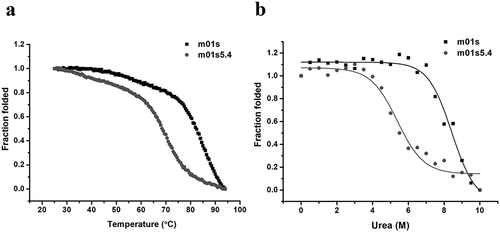
The thermal stability of m01s and m01s5.4 were also monitored by CD. Notably, the melting temperature (Tm) of m01s was 82.0 ± 0.1°C as reported previously,Citation34 which was higher than that of m01s5.4 (Tm = 68.5 ± 0.3°C) (). The stability against the chemical reagent-induced unfolding of m01s and m01s5.4 was also compared in the presence of urea. The urea concentration at the middle point of urea-induced unfolding of m01s was 8.4 ± 0.1 M, which was higher than that of m01s5.4 (5.6 ± 0.2 M) (). Therefore, m01s5.4 is less stable than m01s.
Figure 4. The stability of m01s and m01s5.4. (a). Thermo-induced unfolding curve measured by CD. The CD signal was recorded at 216 nm in the temperature range from 25–94°C with a heating rate of 0.5°C/min. Tm of m01s (82.0 ± 0.1°C) is higher than that of m01s5.4 (68.5 ± 0.3°C). (b). Urea-induced unfolding of m01s and m01s5.4. m01s (8.4 ± 0.1 M) is more stable than m01s5.4 (5.6 ± 0.2 M) in urea-induced unfolding
Aggregation resistance of m01s5.4 as measured by a turbidity assay
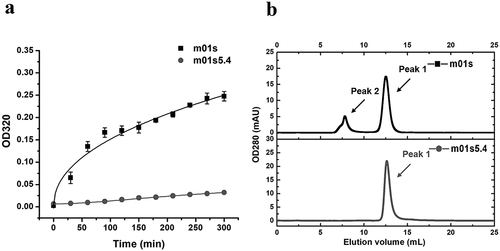
Despite the change of stability, it was still interesting to compare the aggregation propensity of m01s and m01s5.4. A turbidity assay was first used for comparison of the aggregation between m01s and m01s5.4. The optical density (OD) at 320 nm was measured after 60°C incubation of prepared samples at different time points (). Turbidity of m01s increased faster than that of m01s5.4, which showed m01s aggregated more than m01s5.4. This result was confirmed by SEC at the end of incubation, which showed that no obviously soluble large oligomers formed in the case of m01s5.4 (). In contrast, a remarkable second peak that eluted earlier in m01s was observed, indicating the formation of aggregates. In addition, we incubated m01s and m01s5.4 at 15 mg/mL in two tubes at 37°C. After a 3-day incubation, the tube containing m01s was visibly cloudy, while that containing m01s5.4 was still clear. Obvious precipitate was observed in the m01s-containing tube after centrifugation, while no precipitate formed in the tube containing m01s5.4.
Figure 5. Comparison of aggregation tendency between m01s and m01s5.4. (a). Turbidity assay of m01s and m01s5.4 after 60°C incubation at different time points. OD320 of m01s increases obviously while that of m01s5.4 only changes slightly. (b). Evaluation of soluble aggregation formation by SEC. Besides Peak 1, there is an obvious aggregation peak (Peak 2) in the case of m01s at the end of 60°C incubation. In contrast, only one peak (Peak 1) is observed in the case of m01s5.4
Aggregation resistance of m01s5.4 as measured by dynamic light scattering
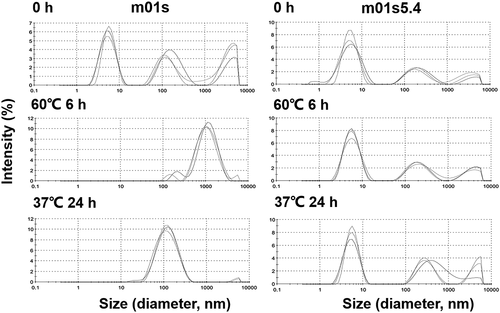
We also used dynamic light scattering (DLS) to monitor the existence of soluble oligomers at different temperatures and incubation times (). After 6 h incubation at 60°C, the peaks in the case of m01s shifted to a larger size, indicating the formation of large soluble oligomers. In contrast, no obvious change of the peaks was observed in the case of m01s5.4, showing that m01s5.4 was stable and resistant to aggregation. Similar results were observed at 37°C (). As expected, these results indicate that m01s5.4 is more aggregation-resistant than m01s.
Figure 6. Measurement of aggregation formation in m01s and m01s5.4 after incubation at 60°C and 37°C by DLS. In general, the peak in the case of m01s becomes larger after incubation, indicating the formation of large soluble oligomers, while the peak in the case of m01s5.4 does not obviously change
Low nonspecific binding of m01s5.4
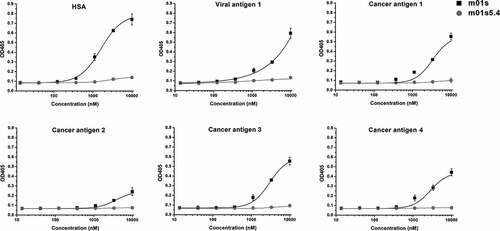
In order to explore the relationship between aggregation and nonspecific binding, we also compared the binding of m01s and m01s5.4 to human serum albumin (HSA), and a panel of viral and cancer-related antigens (Viral antigen 1, Cancer antigen 1, Cancer antigen 2, Cancer antigen 3 and Cancer antigen 4). Interestingly, m01s displayed relatively strong nonspecific binding to all tested proteins, especially at the highest concentration tested, while no obvious binding of m01s5.4 to these antigens was found (). The nonspecific binding could be caused by the hydrophobic clusters in APRs. Therefore, reduction of hydrophobicity could decrease the nonspecific binding.
Figure 7. Binding of m01s5.4 and m01s to HSA and a panel of viral or cancer-related antigens. m01s displays relatively strong nonspecific binding to all tested proteins especially at the high concentration, while m01s5.4 has no obvious binding to these antigens at the same concentration, indicating the low nonspecific binding capacity of m01s5.4
Improving aggregation of a previously selected m01s-based binder
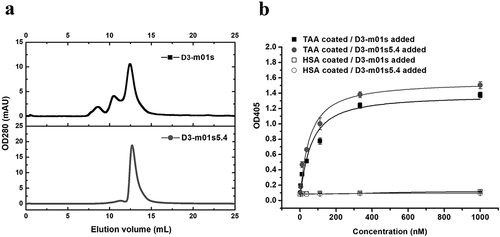
To find whether the aggregation propensity of antigen-specific binders based on m01s could be reduced using the same mutations as those in m01s5.4, we mutated a tumor-associated antigen (TAA) specific binder (D3-m01s) that was selected from an m01s-based phage display library.Citation40 As expected, D3-m01s mutant (D3-m01s5.4) existed as a monomer after direct affinity purification, whereas large oligomer formation was observed in the case of D3-m01s (). Importantly, the binding ability of D3-m01s5.4 to the corresponding tumor antigen was still maintained (). These results indicate that the m01s5.4-specific mutations could be used for the development of better CH2-based therapeutics.
Figure 8. Application of m01s5.4 in a specific m01s-based binder (D3-m01s) against a tumor-associated antigen (TAA). (a). Oligomer formation of the parental clone D3-m01s and its mutant D3-m01s5.4 measured by SEC. D3-m01s5.4 exists as a monomer, while the mixture is observed in D3-m01s. (b). Comparison of binding of D3-m01s and D3-m01s5.4 to TAA measured by ELISA. The EC50 of binding of D3-m01s and D3-m01s5.4 to TAA is about 63 and 45 nM, respectively, indicating the promising application value of m01s5.4
Discussion
Aggregation is a fundamental problem that can impede the development of therapeutic mAbs. The CH2 domain is prone to aggregation. In one example, the location and size of APRs in an intact IgG were identified using spatial aggregation propensity technology.Citation44 After mutation of L235 and L309 in the CH2 domain to two lysines, the aggregation resistance and thermostability of the IgG1 significantly increased.Citation15 The role of CH2 glycans in modulating Fc aggregation has also been revealed.Citation16 Interestingly, the CH2 domain could also be stabilized by engineering an enhanced aromatic sequon (Q295F/Y296A) into the N-glycosylated DE loop, which also confers enhanced stability against thermal- and low pH-induced aggregation in the context of a full-length monoclonal IgG1.Citation19
In an intact IgG or Fc fragment, several APRs in CH2 are buried inside the molecule due to the dimerization. In contrast, when expressed in isolation, more aggregation-related residues, such as hydrophobic amino acids in the CH2 β-strands, are exposed, which increases the risk of aggregation. We found that the N-terminal and C-terminal residues in the CH2 domain are important for stabilization of the molecule, and could be truncated or substituted, respectively, to increase the aggregation resistance.Citation17,Citation21 However, little is known about the effects on aggregation propensity after mutation of the residues in the β-strand regions. In this study, we first performed computational predication of APRs in CH2 by TANGO, which resulted in the identification of three APRs in the CH2 domain. Although two hydrophobic residues F241 and F243 were not recognized as APRs by TANGO, we still selected them for random mutagenesis due to their exposure in the crystal structure of an isolated CH2 domain. In the identified mutant m01s5.4, most of the selected residues are mutated to hydrophilic or charged amino acids. Hence, the hydrophobicity of the exposed residues is significantly decreased, which leads to the improvement of aggregation resistance.
We also noticed that the stability against thermo- and urea-induced unfolding was weakened after mutation (m01s5.4 vs. m01s). In m01s5.4, many hydrophilic and charged amino acids have been introduced to substitute the hydrophobic residues, which might result in an increase in the flexibility of the molecule, as well as a decrease of its rigidity and the total β-sheet-forming propensity. Therefore, it is reasonable that the stability of m01s5.4 is lower than that of m01s. Interestingly, while it is believed that in general stability correlates with aggregation resistance, here we found an example that suggests an inverse correlation. This phenomenon was observed previously.Citation45 In addition, m01s5.4 does not show nonspecific binding to tested proteins, in contrast to m01s, possibly because of the replacement of hydrophobic residues and subsequent nonspecific interactions that could confer advantages in selection of specific binders (e.g., reduction of “sticky” clones). Recently, we reported that several residues at the C-terminus of CH2 could be optimized by the introduction of triple mutations (K338I, A339K, and K340S), which resulted in the identification of a mutant CH2-IKS with increased stability and aggregation resistance.Citation21 Additional work is needed to determine whether m01s5.4 could be further improved by C-terminal optimization.
In our previous study, using an analysis of potential APRs and solvent accessible surface areas, we found two hydrophobic residues, V264 and L309, involved in the aggregation propensity of different m01s-based binders against nucleolin.Citation36 After the mutation of both of them to two lysines, the formation of aggregation was significantly reduced in one of the binders, NCL2H2. However, the percent of the monomer was about 40%. There was no obvious change in aggregation formation of the other binder, NCL2H9. Therefore, the elimination of CH2 aggregates is complex and our current study will hopefully help reduce aggregation for some, if not all, binding molecules.
Since the overall secondary structure of m01s and m01s5.4 is quite similar, use of the m01s5.4-specific mutations for an m01s-based specific binder could successfully eliminate the aggregation formation without loss of the binding activity. This, however, may not be true in the general case because slight conformational changes may lead to a significant decrease of binding. When we performed panning, we used an antibody-guided strategy to enhance the preservation of the CH2 conformation. Hence, m01s5.4-specific mutations could be used for the optimization of m01s-derived binders or for the design of new scaffolds for the selection of binders with better drugability. Our strategy for reducing aggregation of an isolated CH2 domain could also be helpful for the optimization of other scaffolds and binders.
Materials and methods
Design and construction of m01s mutant library
The CH2 domain used in this study was from the human IgG1 as reported previously.Citation33 The online program TANGOCitation37–Citation39 was used to identify the APRs in CH2 and m01s. The crystal structures from an isolated CH2 (PDB entry: 3DJ9) and CH2 in the intact human IgG (PDB entry: 1HZH) were used to determine the directions of related residues. Structural analysis was processed by PyMOL software. For m01s mutant phage display library construction, F241 and F243 in strand A, residues from V259 to V264 in APR1, including T260, V262 and V264, and residues from R301 to L309 in APR3, including R301, V303 and V305 were randomly mutated by PCR according to our published protocols.Citation21 The phagemid vector pComb3XSS (Addgene, USA) with the replacement of HA tag by FLAG tag was used for library construction.
Identification of m01s mutant with reduced APRs
The panning steps were performed as previously described.Citation21 The m01s mutant phage display library was heated at 80°C for 10 min to denature phage-displayed mutants, cooled at 4°C for 20 min for refolding, and subsequently added to wells pre-coated with an anti-human CH2 mAb.Citation34 After two rounds of panning, the candidate clones were screened by monoclonal phage ELISA. The clones with positive binding signal were sequenced and the plasmids were extracted for expression in E. coli strain HB2151. The clone with the best soluble expression level was further modified by “lysine” scanning in its APR2.
Analysis of expression by western blot
The cell lysate of E. coli strain HB2151 used for protein expression was clarified by centrifugation at 14,000 rpm for 30 min at 4ºC. Then, 20 μL of each clarified supernatant was loaded in SDS-PAGE (15% gel), and then transferred onto a polyvinylidene fluoride membrane (Millipore, ISEQ00010). After blocking with TBST buffer containing 5% w/v nonfat milk for 1.5 h at room temperature, the membrane was washed three times for 5 min each time in TBST and incubated with mouse anti-His tag mAb (Proteintech, 66005-1-Ig) at a 1:3000 dilution at room temperature for another 1.5 h. The membrane was washed and incubated with alkaline phosphatase (AP)-conjugated goat anti-mouse antibody (Proteintech, SA00002-1) at a 1:3000 dilution for 1 h at room temperature. Finally, the membrane was washed again, and the signal detection was performed by BCIP/NBT Alkaline Phosphatase Color Development Kit (Beyotime, C3206) according to the manufacturer’s instructions.
Size exclusion chromatography
One clone (m01s5.4) that showed high-level soluble expression was expressed and purified as previously describedCitation33 for characterization. The purified m01s5.4 was loaded into the Superdex 75 10/300 GL column (GE Healthcare, USA) running on ÄKTA pure system (GE Healthcare, USA) to assess possible oligomer formation. Phosphate-buffered saline (PBS), pH 7.4 was selected as the mobile phase. The molecular mass standards, including bovine serum albumin (67 kDa), β-lactoglobulin (35 kDa), cytochrome C (13.6 kDa), aprotinin (6.512 kDa) and vitamin B12 (1.355 kDa), were used to calculate the molecular weight of the proteins. m01s was used for comparison.
Circular dichroism
The secondary structure and thermal stability of all proteins were determined by CD spectroscopy. The purified m01s and m01s5.4 were diluted in PBS at the final concentration of 0.4 mg/mL, and the CD spectra were recorded on Applied Photophysics Chirascan-SF.3 spectrophotometer (Applied Photophysics Ltd, UK). Wavelength spectra were recorded at 25°C using a 1 mm path-length cuvette in the range of 200–260 nm. The scan rate was 1 nm/s, and the final spectrum was averaged over three scans. Thermal stability was measured by recording the CD signal at 216 nm in the temperature range of 25–94°C with a heating rate of 0.5°C/min. The experiments were repeated twice.
Conformation of m01s5.4 detected by the corresponding antibody
ELISA was used for further comparison of the structures of m01s and m01s5.4. Briefly, a commercial mouse anti-human CH2 mAb (Bio-Rad, MCA647) was coated on 96-well plate with a concentration of 2 µg/mL. After blocking by PBS + 3% milk, then three-fold serially diluted proteins were added with concentrations from 0 to 1 µM. 1:2000 diluted horseradish peroxidase (HRP)-conjugated anti-FLAG antibody (Sigma-Aldrich, A8592) was used as secondary antibody. To confirm the result, an anti-human CH2 Fab (m01m1)Citation34 targeting the conformational epitope in CH2 selected from a human naïve phage display library was also used for ELISA. m01s, m01s5.4 and HSA at concentrations of 2 µg/mL were coated on 96-well plates. The serially diluted anti-human CH2 Fab (m01m1) from 0 to 10 µM in PBS + 1% milk were added. HRP-conjugated anti-human Fab antibody (Sigma-Aldrich, A0293) was used as the secondary antibody.
Urea-induced denaturation
The purified m01s and m01s5.4 were diluted into PBS (pH 7.4) with urea from 0 to 10 M. Intrinsic fluorescence measurements were performed using a protein concentration of 100 µg/mL with excitation wavelength at 280 nm, and emission spectra at 340 nm at 25°C. After incubation at 4°C overnight, the intrinsic fluorescence intensity was recorded on EnVision™ (PerkinElmer, USA) to compare their stability against chemical denaturant. With all samples, fluorescence spectra were corrected for the background fluorescence of the solution (buffer + denaturant). The fraction folded (ff) of the protein is calculated as ff = ([F] − [Fu])/([Ff] − [Fu]). [Ff] and [Fu] are the intrinsic fluorescence with excitation wavelength at 280 nm and emission wavelength at 340 nm at 25°C of the folded state in the absence of urea and the unfolded state in the presence of urea concentration of 10 M. The experiments were repeated twice. Four parameter logistic curve analysis was used to fit the data.
Turbidity assay
A turbidity assay was carried out on purified and filtered m01s and m01s5.4 with a concentration of 1 mg/mL in PBS (pH 7.4) after different time points of incubation at 60°C. Turbidity was quantified by recording the absorbance values at 320 nm with ultraviolet spectrophotometer (Beijing Liuyi Biotechnology Co., Ltd, China). After incubation at 60°C for 300 min as the final point, the protein samples were centrifuged at 4°C, 13,000 × g for 15 min, then the supernatant was injected into the Superdex 75 10/300 GL column running on ÄKTA pure system to assess oligomer formation.
Dynamic light scattering
In order to compare the tendency of soluble aggregation formation, both m01s and m01s5.4 protein samples were incubated at 60°C (6 h) and 37°C (1 d). Then, the samples were taken out and centrifuged at 13,000 × g for 10 min. The supernatants of samples were used for DLS measurement to determine the size of protein particles. Measurements were carried out on Zetasizer Nano ZS90 (Malvern Panalytical Ltd, UK). Each sample was measured three times.
Comparison of the nonspecific binding of m01s and m01s5.4
ELISA was used to test the binding of m01s and m01s5.4 to HSA and viral and cancer-related antigens to compare their nonspecific binding activities. Antigens were coated on 96-well ELISA plates overnight with a concentration of 2 µg/mL in PBS at 4°C, and blocked with 100 µl per well of protein-free blocking buffer (Pierce, 37584). The serially diluted m01s and m01s5.4 in protein-free blocking buffer only from 0 to 10 µM were added. 1:2000 diluted HRP-conjugated anti-FLAG antibody (Sigma-Aldrich, A8592) in protein-free blocking buffer only was used as secondary antibody.
Use of m01s5.4-specific mutations to improve an m01s-based binder
An m01s-based binder D3-m01s selected in our lab against a TAA was used here. Then, the corresponding mutated residues in m01s5.4 were introduced into the D3-m01s binder by multi-step PCR to get D3-m01s5.4. The oligomeric state of these two binders was assessed by SEC as described above. ELISA was used to determine the binding capability of the selected binders to the corresponding TAA. Briefly, the TAA (2 μg/mL) was coated on 96-well ELISA plate, while HSA (2 μg/mL) was used for the detection of nonspecific binding as a negative control. After blocking by PBS + 3% milk, the three-fold serially diluted D3-m01s and D3-m01s5.4 were added with concentrations from 0 to 1 µM in 1% milk. HRP-conjugated anti-FLAG antibody (Sigma-Aldrich, A8592) was used as the secondary antibody. ELISA data analysis with four parameter logistic curve fit.
Abbreviations
| mAbs | = | monoclonal antibodies |
| CH2 | = | human immunoglobulin constant domain 2 |
| C-sdAb | = | C-based single domain antibody |
| Ig | = | immunoglobulin |
| APRs | = | aggregation prone regions |
| ELISA | = | enzyme linked immunosorbent assay |
| AP | = | alkaline phosphatase |
| HRP | = | horseradish peroxidase |
| SEC | = | size exclusion chromatography |
| CD | = | circular dichroism |
| Tm | = | melting temperature |
| OD | = | optical density |
| DLS | = | dynamic light scattering |
| HSA | = | human serum albumin |
| TAA | = | tumor-associated antigen |
Author Contributions
The manuscript was written by all authors. All authors have given approval for the final version of the manuscript.
Supplemental Material
Download Zip (3.1 MB)Acknowledgments
We thank the Core Facility and Technical Support, Wuhan Institute of Virology, Chinese Academy of Sciences; Wuhan Institute of Biotechnology; Wuhan Key Laboratory on Emerging Infectious Diseases and Biosafety; Wuhan National Bio-Safety Level 4 Lab of the Chinese Academy of Sciences; as well as UPMC Enterprises and the University of Pittsburgh for their support.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Dimitrov DS. Therapeutic antibodies, vaccines and antibodyomes. MAbs. 2010;2:347–11. doi:https://doi.org/10.4161/mabs.2.3.11779.
- Shire SJ, Shahrokh Z, Liu J. Challenges in the development of high protein concentration formulations. J Pharm Sci. 2004;93:1390–402. doi:https://doi.org/10.1002/jps.20079.
- Agrawal NJ, Kumar S, Wang X, Helk B, Singh SK, Trout BL. Aggregation in protein-based biotherapeutics: computational studies and tools to identify aggregation-prone regions. J Pharm Sci. 2011;100:5081–95. doi:https://doi.org/10.1002/jps.22705.
- Vazquez E, Corchero JL, Villaverde A. Post-production protein stability: trouble beyond the cell factory. Microb Cell Fact. 2011;10:60. doi:https://doi.org/10.1186/1475-2859-10-60.
- Skamris T, Tian X, Thorolfsson M, Karkov HS, Rasmussen HB, Langkilde AE, Vestergaard B. Monoclonal antibodies follow distinct aggregation pathways during production-relevant acidic incubation and neutralization. Pharm Res. 2016;33:716–28. doi:https://doi.org/10.1007/s11095-015-1821-0.
- Richard J, Prang N. The formulation and immunogenicity of therapeutic proteins: product quality as a key factor. IDrugs. 2010;13:550–58.
- Singh SK. Impact of product-related factors on immunogenicity of biotherapeutics. J Pharm Sci. 2011;100:354–87. doi:https://doi.org/10.1002/jps.22276.
- Majumdar R, Esfandiary R, Bishop SM, Samra HS, Middaugh CR, Volkin DB, Weis DD. Correlations between changes in conformational dynamics and physical stability in a mutant IgG1 mAb engineered for extended serum half-life. mAbs. 2015;7:84–95. doi:https://doi.org/10.4161/19420862.2014.985494.
- Cromwell ME, Hilario E, Jacobson F. Protein aggregation and bioprocessing. Aaps J. 2006;8:E572–9. doi:https://doi.org/10.1208/aapsj080366.
- Obrezanova O, Arnell A, de la Cuesta RG, Berthelot ME, Gallagher TR, Zurdo J, Stallwood Y. Aggregation risk prediction for antibodies and its application to biotherapeutic development. mAbs. 2015;7:352–63. doi:https://doi.org/10.1080/19420862.2015.1007828.
- Ho SC, Wang T, Song Z, Yang Y. IgG aggregation mechanism for CHO cell lines expressing excess heavy chains. Mol Biotechnol. 2015;57:625–34. doi:https://doi.org/10.1007/s12033-015-9852-7.
- Respaud R, Marchand D, Parent C, Pelat T, Thullier P, Tournamille J-F, Viaud-Massuard M-C, Diot P, Si-Tahar M, Vecellio L, et al. Effect of formulation on the stability and aerosol performance of a nebulized antibody. mAbs. 2014;6:1347–55. doi:https://doi.org/10.4161/mabs.29938.
- Jung S, Honegger A, Pluckthun A. Selection for improved protein stability by phage display . J Mol Biol. 1999;294:163–80. doi:https://doi.org/10.1006/jmbi.1999.3196.
- Jespers L, Schon O, Famm K, Winter G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat Biotechnol. 2004;22:1161–65. doi:https://doi.org/10.1038/nbt1000.
- Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci U S A. 2009;106:11937–42. doi:https://doi.org/10.1073/pnas.0904191106.
- Latypov RF, Hogan S, Lau H, Gadgil H, Liu D. Elucidation of acid-induced unfolding and aggregation of human immunoglobulin IgG1 and IgG2 Fc. J Biol Chem. 2012;287:1381–96. doi:https://doi.org/10.1074/jbc.M111.297697.
- Gong R, Wang Y, Ying T, Feng Y, Streaker E, Prabakaran P, Dimitrov DS. N-terminal truncation of an isolated human IgG1 CH2 domain significantly increases its stability and aggregation resistance. Mol Pharm. 2013;10:2642–52. doi:https://doi.org/10.1021/mp400075f.
- Yageta S, Lauer TM, Trout BL, Honda S. Conformational and colloidal stabilities of isolated constant domains of human immunoglobulin G and their impact on antibody aggregation under acidic conditions. Mol Pharm. 2015;12:1443–55. doi:https://doi.org/10.1021/mp500759p.
- Chen W, Kong L, Connelly S, Dendle JM, Liu Y, Wilson IA, Powers ET, Kelly JW, et al. Stabilizing the CH2 domain of an antibody by engineering in an enhanced aromatic sequon. ACS Chem Biol. 2016;11:1852–61. doi:https://doi.org/10.1021/acschembio.5b01035.
- Zeng F, Yang C, Gao X, Li X, Zhang Z, Gong R. Comprehensive elucidation of the structural and functional roles of engineered disulfide bonds in antibody Fc fragment. J Biol Chem. 2018;293:19127–35. doi:https://doi.org/10.1074/jbc.RA118.005367.
- Gao X, Conard A, Yang C, Zhan Y, Zeng F, Shi J, Li W, Dimitrov DS, Gong R. Optimization of the C-terminus of an autonomous human IgG1 CH2 domain for stability and aggregation resistance. Mol Pharm. 2019;16:3647–56. doi:https://doi.org/10.1021/acs.molpharmaceut.9b00544.
- Schaefer JV, Pluckthun A. Engineering aggregation resistance in IgG by two independent mechanisms: lessons from comparison of Pichia pastoris and mammalian cell expression. J Mol Biol. 2012;417:309–35. doi:https://doi.org/10.1016/j.jmb.2012.01.027.
- Courtois F, Agrawal NJ, Lauer TM, Trout BL. Rational design of therapeutic mAbs against aggregation through protein engineering and incorporation of glycosylation motifs applied to bevacizumab. mAbs. 2016;8:99–112. doi:https://doi.org/10.1080/19420862.2015.1112477.
- Perchiacca JM, Tessier PM. Engineering aggregation-resistant antibodies. Annu Rev Chem Biomol Eng. 2012;3:263–86. doi:https://doi.org/10.1146/annurev-chembioeng-062011-081052.
- Lee CC, Perchiacca JM, Tessier PM. Toward aggregation-resistant antibodies by design. Trends Biotechnol. 2013;31:612–20. doi:https://doi.org/10.1016/j.tibtech.2013.07.002.
- Buchanan A, Clementel V, Woods R, Harn N, Bowen MA, Mo W, Popovic B, Bishop SM, Dall’Acqua W, Minter R, et al. Engineering a therapeutic IgG molecule to address cysteinylation, aggregation and enhance thermal stability and expression. mAbs. 2013;5:255–62. doi:https://doi.org/10.4161/mabs.23392.
- Chakroun N, Hilton D, Ahmad SS, Platt GW, Dalby PA. Mapping the aggregation kinetics of a therapeutic antibody fragment. Mol Pharm. 2016;13:307–19. doi:https://doi.org/10.1021/acs.molpharmaceut.5b00387.
- Chen X, Zeng F, Huang T, Cheng L, Liu H, Gong R. Optimization on Fc for improvement of stability and aggregation resistance. Curr Pharm Biotechnol. 2016;17:1353–59. doi:https://doi.org/10.2174/1389201017666161117145312.
- Yang C, Gao X, Gong R. Engineering of Fc fragments with optimized physicochemical properties implying improvement of clinical potentials for Fc-Based therapeutics. Front Immunol. 2017;8:1860. doi:https://doi.org/10.3389/fimmu.2017.01860.
- Dimitrov DS. Engineered CH2 domains (nanoantibodies). mAbs. 2009;1:26–28. doi:https://doi.org/10.4161/mabs.1.1.7480.
- Ying T, Gong R, Ju TW, Prabakaran P, Dimitrov DS. Engineered Fc based antibody domains and fragments as novel scaffolds. Biochim Biophys Acta. 2014;1844:1977–82. doi:https://doi.org/10.1016/j.bbapap.2014.04.018.
- Chen W, Gong R, Ying T, Prabakaran P, Zhu Z, Feng Y, Dimitrov D. Discovery of novel candidate therapeutics and diagnostics based on engineered human antibody domains. Curr Drug Discov Technol. 2014;11:28–40. doi:https://doi.org/10.2174/15701638113109990032.
- Gong R, Vu BK, Feng Y, Prieto DA, Dyba MA, Walsh JD, Prabakaran P, Veenstra TD, Tarasov SG, Ishima R, et al. Engineered human antibody constant domains with increased stability. J Biol Chem. 2009;284:14203–10. doi:https://doi.org/10.1074/jbc.M900769200.
- Gong R, Wang Y, Feng Y, Zhao Q, Dimitrov DS. Shortened engineered human antibody CH2 domains: increased stability and binding to the human neonatal Fc receptor. J Biol Chem. 2011;286:27288–93. doi:https://doi.org/10.1074/jbc.M111.254219.
- Xiao X, Feng Y, Vu BK, Ishima R, Dimitrov DS. A large library based on a novel (CH2) scaffold: identification of HIV-1 inhibitors. Biochem Biophys Res Commun. 2009;387:387–92. doi:https://doi.org/10.1016/j.bbrc.2009.07.044.
- Li D, Gong R, Zheng J, Chen X, Dimitrov DS, Zhao Q. Engineered antibody CH2 domains binding to nucleolin: isolation, characterization and improvement of aggregation. Biochem Biophys Res Commun. 2017;485:446–53. doi:https://doi.org/10.1016/j.bbrc.2017.02.058.
- Linding R, Schymkowitz J, Rousseau F, Diella F, Serrano L. A comparative study of the relationship between protein structure and beta-aggregation in globular and intrinsically disordered proteins. J Mol Biol. 2004;342:345–53. doi:https://doi.org/10.1016/j.jmb.2004.06.088.
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22:1302–06. doi:https://doi.org/10.1038/nbt1012.
- Rousseau F, Schymkowitz J, Serrano L. Protein aggregation and amyloidosis: confusion of the kinds? Curr Opin Struct Biol. 2006;16:118–26. doi:https://doi.org/10.1016/j.sbi.2006.01.011.
- Gong R, Wang Y, Ying T, Dimitrov DS. Bispecific engineered antibody domains (nanoantibodies) that interact noncompetitively with an HIV-1 neutralizing epitope and FcRn. PLoS One. 2012;7:e42288. doi:https://doi.org/10.1371/journal.pone.0042288.
- Prabakaran P, Vu BK, Gan J, Feng Y, Dimitrov DS, Ji X. Structure of an isolated unglycosylated antibody C(H)2 domain. Acta Crystallogr Sect D Biol Crystallogr. 2008;64:1062–67. doi:https://doi.org/10.1107/S0907444908025274.
- Edelman GM, Cunningham BA, Gall WE, Gottlieb PD, Rutishauser U, Waxdal MJ. The covalent structure of an entire gammaG immunoglobulin molecule. Proc Natl Acad Sci U S A. 1969;63:78–85. doi:https://doi.org/10.1073/pnas.63.1.78.
- Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science. 2001;293:1155–59. doi:https://doi.org/10.1126/science.1061692.
- Chennamsetty N, Helk B, Voynov V, Kayser V, Trout BL. Aggregation-prone motifs in human immunoglobulin G. J Mol Biol. 2009;391:404–13. doi:https://doi.org/10.1016/j.jmb.2009.06.028.
- Sahin E, Jordan JL, Spatara ML, Naranjo A, Costanzo JA, Weiss WF, Robinson AS, Fernandez EJ, Roberts CJ, et al. Computational design and biophysical characterization of aggregation-resistant point mutations for gammaD crystallin illustrate a balance of conformational stability and intrinsic aggregation propensity. Biochemistry. 2011;50:628–39. doi:https://doi.org/10.1021/bi100978r.