
ABSTRACT
Despite advances in medical care, cancer remains a major threat to human health. Antibody-drug conjugates (ADCs) are a promising targeted therapy to overcome adverse side effects to normal tissues. In this field, the current challenge is obtaining homogeneous preparations of conjugates, where a defined number of drugs are conjugated to specific antibody sites. Site-directed cysteine-based conjugation is commonly used to obtain homogeneous ADC, but it is a time-consuming and expensive approach due to the need for extensive antibody engineering to identify the optimal conjugation sites and reduction – oxidation protocols are specific for each antibody. There is thus a need for ADC platforms that offer homogeneity and direct applicability to the already approved antibody therapeutics. Here we describe a novel approach to derive homogeneous ADCs with drug-to-antibody ratio of 2 from any human immunoglobulin 1 (IgG1), using trastuzumab as a model. The method is based on the production of heavy chains (HC) and light chains (LC) in two recombinant HEK293 independent cultures, so the original amino acid sequence is not altered. Isolated LC was effectively conjugated to a single drug-linker (vcMMAE) construct and mixed to isolated HC dimers, in order to obtain a correctly folded ADC. The relevance of the work was validated in terms of ADC homogeneity (HIC-HPLC, MS), purity (SEC-HPLC), isolated antigen recognition (ELISA) and biological activity (HER2-positive breast cancer cells cytotoxicity assays).
Introduction
Antibody-drug conjugates (ADCs) are a class of targeted therapy used in the treatment of various cancers. As of late 2019, the US Food and Drug Administration (FDA) has approved four ADCs, three ADCs are undergoing regulatory review, and over 100 ADCs are in the clinical pipeline.Citation1 Their medical potential is based on their structure, since it combines the ability of a monoclonal antibody (mAb) to specifically target a tumor-associated antigenCitation2 with the cell-killing ability of potent cytotoxic drugs via a chemical linker.
These unique targeted drugs, however, pose both technological and developmental challenges. One key characteristic of an ADC is the drug:antibody ratio (DAR), which is the number of cytotoxic drug molecules per molecule of antibody. The DAR defines the two major types of ADCs: heterogeneous (conjugated in random positions that vary between mAbs) and homogeneous (conjugation at specific sites that are defined in each mAb molecule). The four commercially available ADCs are heterogeneous due to limitations in the technology available when they were originally developed. The lack of efficient methods for producing completely homogeneous ADCs has compelled pharmaceutical companies to develop site-specific conjugation methods, which improves ADC homogeneity,Citation3 batch-to-batch consistency, and is more desirable from a regulatory perspective.Citation4
Different approaches have been developed to obtain homogeneous ADCs by site-specific conjugation, including cysteine residue(s) addition, non-natural amino acids, enzyme-mediated conjugation (including transglutaminase, sortase A, glycosyltransferase or endoglycosidase) and linker-based conjugation.Citation5-Citation12
Cysteine-engineered residues have been introduced into different antibodies by site-directed mutation to provide free thiol groups for conjugation using conventional thiol-specific maleimide linkers,Citation13 a technology developed by Seattle Genetics and used to produce the heterogeneous ADC brentuximab vedotin (Adcetris), which was approved by FDA in 2011. The process generates almost homogeneous ADCs containing approximately two drug molecules per antibody, but additional antibody reduction/oxidation steps are required for selective conjugation of the added cysteines.Citation14-Citation16
Non-recombinant methods for reducing ADC heterogeneity that enable the production of homogeneous ADCs based on existing antibodies have also been developed. For example, disubstituted maleimide linkers have been used for synthesizing homogeneous ADCs via interchain cysteine cross-linking through disulfide bonds rebridging.Citation17 Godwin and coworkers presented an alternative thiol conjugation approach where bis-sulfone-functionalized reagents are used to form a stable three-carbon bridge with disulfides.Citation18 In addition, the structure of the antibody remains intact and the ligand-to-antibody ratio can be tightly controlled, with a 78% conversion to DAR 4 ADCs.Citation19 Badescu and coworkers used a bis-sulfone linker designed to react with two antibody cysteines to conjugate an auristatin payload (monomethyl auristatin E (MMAE)) to trastuzumab and its antigen-binding fragments (Fabs).Citation20 The approaches described above are time consuming and specific for each construct, as they often require extensive antibody engineering to identify the optimal conjugation sitesCitation9,Citation21 where unique side chains can be introduced for conjugation, or they require complex linker and payload modifications that have not been clinically validated.Citation8 As a result, they are not suitable for converting existing antibodies directly into ADCs.Citation5,Citation7
In vivo, heavy chains (HCs) and light chains (LCs) are co-translationally translocated into the endoplasmic reticulum and folding begins even before the polypeptide chains are completely translated. Most IgGs assemble first as HC dimers to which LCs are added covalently via a disulfide bond between the LC constant domain and the first HC constant domain (CH1) domain.Citation22 Most FDA-approved mAbs are human(ized) immunoglobulin (Ig) G1.
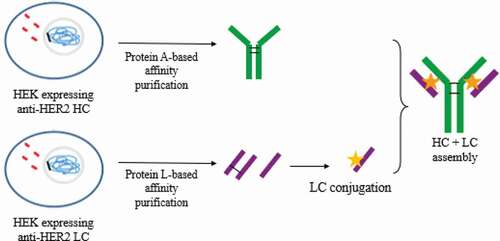
Here, we describe a novel platform to generate totally homogeneous DAR 2 ADCs, without antibody engineering, that could potentially be applied to any human IgG1 and IgG3 antibodies by using conventional maleimide linker. Trastuzumab, an IgG1 mAb that targets human epidermal growth factor receptor-2 (HER2), was used as a model. The method described is based on the production of HCs and LCs in two independent recombinant HEK293 cultures, obtained by transfection,Citation23 and the subsequent conjugation of isolated LC in the only solvent-accessible cysteine residue available, obtaining a homogeneous solution of LC-MMAE. MAb assembly was subsequently achieved by combining LC-MMAE with an isolated HC to generate a non-engineered Cys-based homogeneous ADC ().
Figure 1. Strategy to obtain DAR 2 homogeneous ADCs. anti-HER2 chains were independently produced in recombinant HEK293 cultures. Then, each chain was purified by affinity chromatography and light chain (LC) was conjugated to vcMMAE. Finally, the complete mAb was assembled
Results
Selection of the components of the study
To evaluate the technology described, trastuzumab (Herceptin) was selected as a model. This therapeutic antibody has been successfully used in the treatment of HER2-positive breast cancer, and it is the antibody component of FDA-approved ADC trastuzumab emtansine (Kadcyla). Anticancer drug MMAE and valine-citruline linker were selected for their commercial availability and their proven activity in another FDA-approved ADC, brentuximab vedotin. The method described could be applied to alternative linkers conjugated to reactive thiols with improved stability characteristics.Citation24
Structure of independently produced HC and LC
Anti-HER2 LC and HC were produced in separate HEK293 cultures and purified by affinity chromatography. Using sodium dodecyl sulfate-polyacrylamide electrophoresis gel (SDS-PAGE) under denaturing conditions (), HC is detected as a single band of approximately 100 KDa, indicating that the transfected HEK293 cell line expresses the protein forming a covalently bonded dimer. The single band together with the single peak under native conditions by SEC-HPLC () confirm that no trimers or other structures are formed Further characterization of HC by HPLC-MS () shows two peaks that correspond with 2xHC (100 KDa). This could be explained because the unpaired cysteines of the heavy chain dimers (dcHC) are capped by small cysteine-containing molecules from the culture broth, mainly glutathione, therefore avoiding unwanted reactions of the HC.
Figure 2. HC and LC conformation of proteins expressed by recombinant HEK293. (a) SDS-PAGE gel (denaturing conditions). (b) SEC-HPLC (native conditions) for HC. (c) SEC-HPLC for LC
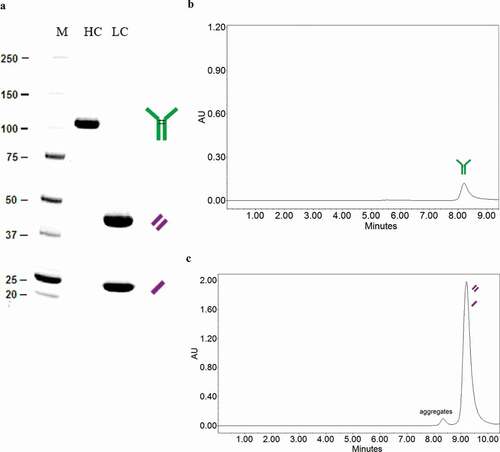
Unexpectedly, LC appears in 2 bands in the SDS-PAGE gel, representing 55% the 42 KDa band and 45% the 21 KDa band. The same sample under reducing conditions shows a single lane at 21 KDa (data not shown) and the SEC-HPLC analysis shows that there is only one peak of 42 KDa under native conditions (). Therefore, the anti-HER2 LC expressed forms dimers, but whereas 55% of them are covalently bonded, 45% of LC forms dimers without covalent unions. A small amount of aggregates (3%) are observed in the produced LC ().
mAB assembly from independently produced HC and LC
Reduction-oxidation assembly approach
Prior to conjugation, the mAb assembly was evaluated following a sequential reduction-oxidation protocol of a LC and dcHC mixture (see Materials and Methods section). SEC-HPLC results shown in demonstrate that a 73% of mAb can successfully be reassembled.
Table 1. Molecules detected under native conditions (SEC-HPLC)
Spontaneous assembly approach
Isolated HC and LC were mixed in 50 mM citrate pH 6 buffer and similar results were obtained without a reduction–oxidation procedure. In this approach, the mAb original structure was assembled because it may be thermodynamically more stable than the independent sub-units; SEC-HPLC profile confirmed the presence of 90.1% of monomer (). The resulting product recognized isolated HER2 antigen in an ELISA assay in the same proportion as in-house anti-HER2 antibody () produced by HEK293 by using a tricistronic vector, as previously described.Citation23
Table 2. Isolated antigen HER2 recognition in the ELISA test
In the reduction-oxidation approach, a lower yield was obtained, since residual LC was detected in the final product. Since both residual HC and LC are present, a more complex purification sequence must be used, as compared with the spontaneous approach, where only residual HC was detected.
The final mixture of monomer, aggregates and dcHC obtained in the spontaneous assembly approach was purified by Capto L affinity chromatography (specific for LC), since no residual LC was detected by SEC-HPLC so only the assembled mAb would be captured.
ADC assembly from independently produced HC and MMAE-conjugated LC
LC conjugation
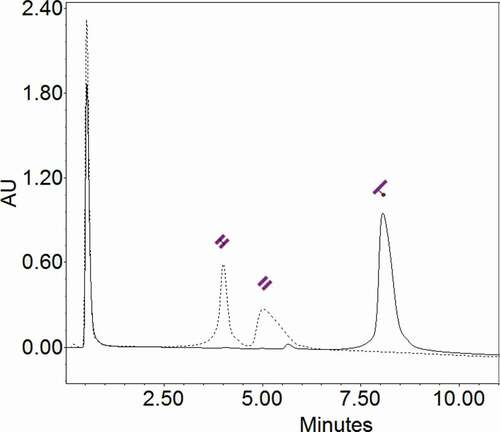
Purified LC was completely reduced with TCEP and conjugated with valine-citrulline monomethyl auristatin E (vcMMAE) at the only solvent-accessible thiol group of cysteine in the LC. Since only one thiol is available, each LC is conjugated to a single vcMMAE molecule and a homogeneous solution of LC-MMAE is obtained, as shown by HIC-HPLC (), which shows the differences in hydrophobicity between LC and conjugated LC due to the presence of the hydrophobic cytotoxic drug. A single peak was detected in the conjugated LC, showing a conjugation efficiency close to 100%.
Figure 3. HIC-HPLC chromatogram. LC (dotted line) appears in two peaks, corresponding to the (un)covalently bonded LC dimers. LC-MMAE (continuous line) appears as a single peak at 8.1 minutes
ADC assembly
The selected assembly strategy for ADC generation was the spontaneous approach due to its simplicity and efficacy. The obtained monomer recognized isolated HER2 antigen in an ELISA assay in the same proportion as in-house anti-HER2 antibody produced by HEK293 by using a tricistronic vector, as previously describedCitation23 and shown in .
DAR analysis: MS
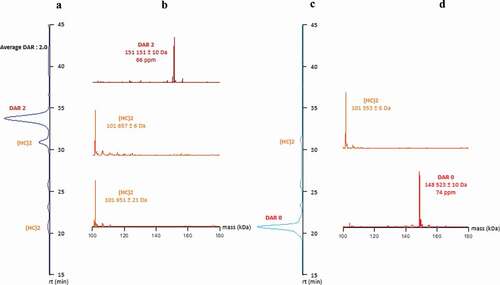
MAb and ADC were analyzed with direct hyphenation of HIC to native MS through a HICxSEC-native MS setup.Citation25 Results obtained confirm that only one main peak is present in HIC and online detected with native MS as DAR 2 for the ADC without the presence of D0 and under/over conjugate, highlighting a homogeneous ADC with an average DAR of 2.0 (). Also, dcHC were detected by confirming the excess of this sub-unit and the necessity of purification by protein L affinity chromatography.
Figure 4. Comparison of HICxSEC-native MS chromatograms of ADC and naked mAb. a) DAR 2 HIC profile. b) DAR 2 deconvoluted spectra. c) Naked mAb HIC profile. d) Naked mAb deconvoluted spectra
Identification of cysteine linked payload position
In order to confirm the homogeneous and specific conjugation of the ADC, structural information based on the identification of drug-loaded peptides was performed after classical tryptic digestion followed by a nanoLC-MS/MS analysis as previously described. To perform this manual identification, the first step was to explore the extracted ion chromatogram (XIC) of one of the two characteristic in-source fragmentation ions (m/z 718.51 or 762.51) of the MC-vc-PAB-MMAE-conjugate.Citation15 Due to the high resolution MS1, conjugated peptides were identified based on accurate mass measurement. Then, confirmation of the drug-loaded peptides was achieved through the detection of seven additional signature ions of MC-vc-PAB-MMAE-conjugate (m/z 686.49, 506.36, 411.26, 321.22, 170.11 and 134.09)Citation26 in all MS/MS spectra. Moreover, the presence of the peptide linked to the MC-vc-PAB-MMAE payload was highlighted by the detection of the conjugated peptide with the loss of the m/z 762.51 species. Through this manual interpretation, the identification of two drug-loaded peptides ([SFNRGEC] + 1 payload and [GEC] + 1 payload) containing the same cysteine at position 214 located on the LC part of the ADC was achieved without ambiguity ().
Figure 5. MS and MS/MS spectra of the two peptides containing the MMAE payload linked to the Cys214 localized on the LC part of the ADC. (a) [SFNRGEC] + 1 payload, (b) [GEC] + 1 payload
![Figure 5. MS and MS/MS spectra of the two peptides containing the MMAE payload linked to the Cys214 localized on the LC part of the ADC. (a) [SFNRGEC] + 1 payload, (b) [GEC] + 1 payload](/cms/asset/971db405-6bf8-49f6-a63b-7e4e8a5360b4/kmab_a_1702262_f0005_oc.jpg)
Biological activity – In vitro assay
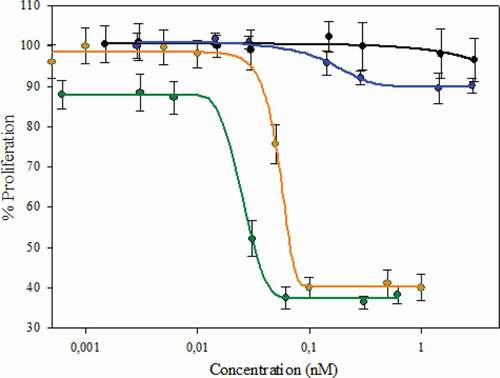
HER2-positive breast cancer cells SKBR3 were used to assess the cytotoxicity of the homogeneous DAR 2 trastuzumab conjugated to MMAE (T-MMAE) we obtained. Its biological activity was compared to an in-house heterogeneous DAR 4 T-MMAE (control) using an in vitro MTS cell-based assay. Our results () show that homogeneous DAR 2 T-MMAE has an antiproliferative effect on the target cells, and it exhibits a significantly lower cytotoxic activity than heterogeneous DAR 4 T-MMAE (IC50 DAR 2: 51.5 pM; DAR 4: 25.5 pM). These differences correspond to the differences in the drug load. Furthermore, in vitro assembled mAb behave similarly to trastuzumab folded in vivo ().
Figure 6. Biological activity of the in vitro assembled mAb and homogeneous DAR 2 ADC compared to in vivo folded mAb and heterogeneous DAR 4 ADC, respectively. Cell inhibition assay. Orange: DAR 2 homogeneous T-MMAE assembled by the strategy described. Black: assembled non-conjugated mAb by the strategy described. Green: DAR 4 heterogeneous T-MMAE (reference). Blue: in vivo folded trastuzumab. The concentration indicated is referred to the complete ADC (mainly mAb) and not to toxin:payload
Discussion
Several methods to generate homogeneous ADC have reported,Citation5,Citation7 but they require either genetic engineering of the sequence of the original mAb or complex linker technology, which have not been clinically evaluated. Here, we describe a novel method to obtain a homogeneous ADC based on cysteine conjugation without genetic engineering of the mAb sequence and by using a conventional linker-drug structure. The use of genetic engineering remains at the expression level since HCs and LCs are expressed and folded in two different cell lines, and the complete antibody with the original amino acid sequence is generated after they are isolated. The in vitro assembled mAb behaves like its correspondent in vivo assembled mAb in terms of isolated antigen binding capacity and biological activity (). The proposed method shows that no HC or LC reduction is required to achieve a functional mAb conformation despite the fact that the expressed LCs form dimers. We hypothesize that, as it is stated to occur in vivo,Citation22 it is thermodynamically more favorable for LC to be assembled with HC than to remain as dimers. The reaction is, therefore, biologically displaced to generate a monomer antibody.
Most ADCs in current clinical development were obtained by conjugation to endogenous lysine or cysteine residues of the antibody, carefully controlling the average degree of modification to yield an average DAR in the range of 3.5–4.0.Citation6 This ratio was selected in order to minimize the amount of non-conjugated antibody and to avoid very high DAR species in the mixture, which may be problematic in manufacturing and formulation because of higher hydrophobicity and lower solubility.Citation6
Furthermore, it has been stated that despite the fact that in vitro potency of ADCs increases with the DAR, ADC plasma clearance and aggregation also increase in species with a high DAR, reducing exposure and in vivo efficacy.Citation27,Citation28 An optimal DAR value and a homogeneous ADC are therefore crucial to maximize the balance of efficacy, tolerability and cytotoxic activity.Citation29 Engineered cysteine-based methods to produce homogeneous ADC targeted a DAR value of 2 or 4. ThioMabs enabled the generation of 90% DAR 2 ADCsCitation30 and dibromomaleimide linkers targeted DAR 4.Citation30 Some approaches were developed to reduce clearance of highly hydrophobic ADCs, but they require complex drug-linked design.Citation27,Citation28
Our work provides a novel strategy to obtain cysteine-based completely homogeneous ADC (no other DAR values were detected) that involves selective conjugation of the isolated mAb light chain. In this method, the antibody sequence is preserved, but HC and LC are expressed in independent cell cultures. In vivo folded and isolated chains are assembled to obtain a functional ADC. DAR 2 trastuzumab conjugated to MMAE was used as a model, but the applicability of the method might include any cysteine-based conjugation to any human IgG1 or IgG3. This method offers a generic strategy to obtain homogeneous ADCs with high efficiency and, to the best of the authors’ knowledge, such an approach has not been previously described.
The two cell lines used in this work were not industrial cell lines, so further work will include their improvement as well as optimization of the equimolar proportion of the chains in order to enhance process yield, which was outside the scope of our study. Other future steps include HC conjugation to obtain DAR 2 homogeneous ADCs with another drug-linker construct in the CH1 region cysteines and preserving the hinge disulfide bonds followed by either LC or LC-MMAE assembly (), which would allow the generation of homogeneous ADCs taking advantage of 2 different payloads. These new ADC should be assessed in vivo and compared in terms of bioavailability and pharmacokinetics
Figure 7. Future proposed strategies based on the described method to obtain homogeneous ADCs. The same conjugation could occur in either LC or HC. After assembly, the ADC could be loaded with 2 different site-directed payloads
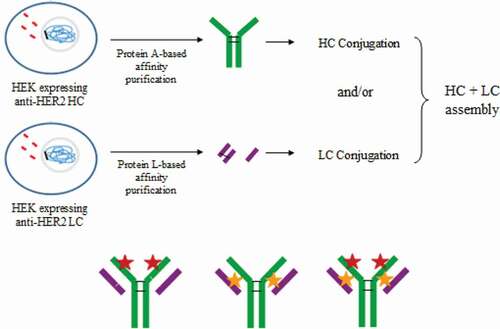
Figure 8. Reduction-oxidation assembly approach. In vivo produced and purified LC and HC are reduced to free disulfide bonds and deoxidized to obtain the assembled mAb
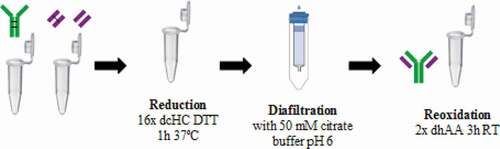
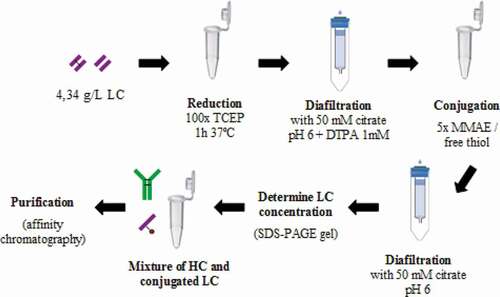
Figure 9. Spontaneous assembly approach to generate homogeneous ADC. LC are reduced to free interchain cysteines and allow the site-directed payload:linker conjugation. Then, dcHC is mixed to obtain the assembled ADC
Materials and methods
Cell lines and maintenance
Trastuzumab protein sequence for LC and HC were obtained from the Drugbank website (ref. DB00072).Citation31 The optimized signal peptides for trastuzumab were added to LC and HC.Citation32 The Trastuzumab DNA sequence for both LC and HC were obtained from the mentioned protein sequence, after being optimized to mammalian codon usage using the Codon Optimization On-line (COOL) method,Citation33 and were synthesized by Genscript (NJ, USA).
HEK293SF-3F6 producing anti-HER2 antibody were obtained by cloning the above synthetic DNA sequence in a tricistronic expression vector derived from the commercial pIRESpuro3 vector (Clontech, CA, USA) between NheI-AgeI for LC DNA sequence and BamHI-EcoRI for HC DNA sequence. HEK293SF-F6 cells were then transfected with the construct and selected by puromycin antibiotic resistance. The new anti-HER2 antibody producer cell lines were referred to as HEK293_T10.
HEK293SF-3F6 producing anti-HER2 HC or anti-HER2 LC were obtained by cloning the above-mentioned synthetic DNA sequences in the expression vector pIRESpuro3 (Clontech, CA, USA) between BamHI and NotI for HC and in the vector pIRESneo3 (Clontech) between NheI and AgeI for LC. HEK293 cells were then transfected with the different constructs and selected as described previously.Citation30 The new producer cell lines were referred to as HEK293_THC and HEK293_TLC.
Cells were subcultured at 0.3 × 106 cell/mL three times per week to keep them in exponential growing phase. Cell maintenance was performed in 125 mL polycarbonate shake flasks (Corning Inc., NY, USA), with a working volume of 12 mL, and maintained at 37°C in an incubator with a 5% CO2 humidity saturated atmosphere (Steri-cult 2000 Incubator, Forma Scientific). Flasks were continuously agitated at 110 rpm on an orbital shaking platform (Stuart SSL110).
HC and LC production
Culture media used in shake flasks and 5 L wave bag cultures for HEK293_THC and HEK293-TLC cell lines was SFM4Transfx-293 (HyClone, UT, USA) supplemented with 4 mM GlutaMAX (Gibco, Invitrogen, CA, USA), 10% (v/v) Cell Boost 5 (60 g/L solution) (HyClone), 2% (v/v) Kolliphor P188 (100 g/L solution) (Sigma, MO, USA) and 0.5% (v/v) Antifoam C (10 g/L solution) (Sigma). Selection pressure was maintained during the production step by adding 0.2% (v/v) puromycin (1 mg/mL) (Merck, NJ, USA) for HEK293_THC and 20 mL/L of neomycin (G-418 solution, Roche, Basel, Switzerland) for HEK293_TLC.
Bioreactor cell cultures were performed using Flexsafe RM 10 L bags (DFB010L, Sartorius, Göttingen, Germany) in a WAVE 20/50 EHT (GE Healthcare, IL, USA) with a working volume of 5 L. Temperature was set at 37ºC. Agitation and angle were set at 22 rpm and 8º, respectively.
Purification steps were performed when maximum cell density was reached, including solids separation by depth filtration (Clarisolve 40MS, Merck) and a bioburden reduction filter (Millipore Express SHF, Merck), broth concentration by tangential flow filtration (Hydrosart 30 KDa membrane, Sartorius), protein A affinity chromatography for HC purification (MAb Select Sure, GE) and protein L affinity chromatography for LC purification (Capto L, GE), as described by the supplier and buffer exchange by desalting columns (PD Minitrap G-25, GE). The final buffer was 50 mM sodium citrate pH 6 for both HC and LC.
mAB assembly from independently produced HC and LC
Reduction-oxidation assembly approach
The reduction-oxidation mAb assembly approach involves mixing HC and LC. Later, reduction of the disulfide bonds of the mixed chains was achieved by the addition of 16:1 molar proportion of dithiothreitol (DTT) (Sigma) with respect to dcHC, and the mixture was incubated at 37ºC for 1 h. Next, a buffer exchange column allowed glutathione/cysteine and DTT removal while the product remained in 50 mM citrate buffer. Finally, 2:1 molar proportion of dehydroascorbic acid (Sigma) with respect to DTT was added to promote disulfide bonds reoxidation for 3 hours at room temperature. This procedure is detailed in .
Spontaneous assembly approach
A simpler approach involved the incubation of a mixture of LC and HC in 50 mM citrate pH 6 buffer for 3 hours at room temperature.
ADC assembly from independently produced HC and MMAE-conjugated LC
LC conjugation
MAb assembly approach 2 was easiest, and it was applied to MMAE-conjugated LC. Since DTT reduction power is not optimal at pH 6, tris(2-carboxyethyl)phosphine (TCEP) in a 100:1 molar proportion was used to reduce purified LC. Under this condition, LC did not form covalently bonded dimers (analyzed by SDS-PAGE) and was conjugated to vcMMAE construct (MedchemExpress, NJ, USA).
ADC assembly
Working at a concentration range of 1–5 mg/mL of HC in the final mixture leads to a successful reassembly of the mAb. We applied a relation of 5.54 g dcHC/g scLC. Thereafter, affinity chromatography (GE) is used in the ADC purification so unassembled LC-MMAE will not bind to protein A ligand. This procedure is described in .
Heterogeneous T-MMAE DAR 4 (control for MTS test)
Heterogeneous T-MMAE DAR 4 was obtained by trastuzumab reduction with 4:1 molar proportion of TCEP with respect to trastuzumab followed by the addition of diethylenetriaminepentaacetic acid (Sigma) at a final concentration of 1 mM. Next, the conjugation step occurred by the reaction of free thiol groups of reduced trastuzumab to vcMMAE (MedchemExpress), used in a 10:1 molar proportion, as described before.Citation15
Analytical methods
SDS-PAGE gels
Protein mixture composition was analyzed by SDS-PAGE gels (MiniProtean TGX Stain-free gels 4-20%, Biorad, CA, USA). Band densitometry to determine the relative amount of each band in the sample was performed using the software GelDoc EZ (Biorad).
SEC-HPLC
SEC experiments were performed in Waters Alliance 2695 system using Zenix-C SEC-300 (4,6 x 300 mm, Sepax, DE, USA). The mobile phase was PBS 1x (Sigma) at a flow rate of 0.35 ml/min. The UV absorbance was measured at a wavelength of 214 nm.
HIC-HPLC
HIC experiments were performed using a Waters Alliance 2695 system using Proteomix HIC Butyl-NP5 (4,6 x 50 mm, Sepax). The mobile phase was a gradient of 25 mM sodium phosphate pH 7 (Sigma) with decreasing ammonium sulfate (2170, Merck) concentration (1.8 M to 0M) and increasing concentration of isopropanol (Scharlab, Barcelona, Spain) to improve peaks resolution, as recommended by the column manufacturer, at a flow rate of 0.8 ml/min. The UV absorbance was measured at a wavelength of 214 nm.
ELISA assay
Assembly was evaluated by comparing in vitro isolated anti-HER2 recognition. Namely, 50 µL of 50 ng/µL anti-HER2 antibody in phosphate-buffered saline (PBS, P5493, Sigma Aldrich) solution (SinoBiological, Beijing, China) was placed in the wells of 96-well Maxisorp plates for its adsorption at 4ºC overnight. After removal of the supernatants, 100 µL of 2% skim milk were added and allowed to stand at room temperature for 1 h. The plates were washed three times with PBS-Tween20 (Sigma) and 50 µL of samples at different dilutions in PBS were placed in each well. Samples were analyzed in duplicates. After standing at room temperature for 1 h, the plates were washed three times with PBS-Tween20. After the addition of 50 µL of a solution of 0.1 µg/mL of polyclonal anti-IgG1 conjugated to horseradish peroxidase (A00166, Genscript), the plates were allowed to stand at room temperature for 1 h. After three washings with PBS-Tween20, 50 µL of TMB were added. The staining reaction was carried out at room temperature for 10–15 minutes and stopped by adding 50 µL of 20% sulfuric acid addition. The absorbance at 450–630 nm was measured with a Labtech LT-4000 microplate reader (Labtech, MI, USA).
HIC-SEC native MS
The direct hyphenation of HIC to native MS consists of a combination of H-Class and I-Class liquid chromatography systems hyphenated to a Synapt G2 HDMS Q-TOF mass spectrometer. The HIC column was a MAbPac HIC-10 (100 mm x 4.6 mm, 5 µm, 1000 Å) (Thermo Scientific, Cheshire, UK). The SEC column used in the second dimension was an AdvanceBio SEC (50 mm x 4.6 mm, 2.7 µm, 300 Å) (Agilent Technologies, Wilmington, DE, USA).
For the HIC first dimension, the mobile phase A was composed of 2.5 M of ammonium acetate and 0.1 M phosphate buffer (Na2HPO4), pH 7.0 (adjusted with phosphoric acid), while the mobile phase B was composed of 0.1 M phosphate buffer (Na2HPO4 and NaH2PO4) with pH 7.0 (adjusted with sodium hydroxide solution). The following gradient was employed in HIC: 0 to 90% B in 36 min, 90 to 100% B in 21 min, followed by an isocratic step at 100% B for 8 min at a flow rate of 100 µL/min. For the second chromatographic dimension in SEC, the separation was carried out in isocratic mode with an aqueous mobile phase composed of 100 mM ammonium acetate at a flow-rate of 700 µL/min.
The Synapt G2 HDMS was operated in sensitive mode and positive polarity with a capillary voltage of 3.0 kV. The sample cone and pressure in the interface region were set to 180 V and 6 mbar, respectively. Source and desolvation temperature were set to 100 and 450°C, respectively. Desolvation and cone gas flows were set at 750 and 60 L/hr, respectively. Acquisitions were performed in the m/z range of 1000–10000 with a 1.5 s scan time. External calibration was performed using singly charged ions produced by a 2 g/L solution of cesium iodide in 2-propanol/water (50/50 v/v).
Sample preparation for peptide mapping analysis
Fifteen micrograms of conjugated antibody were solubilized in 150 mM NH4HCO3, 0.1% RapiGestTM (Waters, Milford, USA) at pH 7.4. Disulfide reduction was performed by incubating the ADC solution with 5 mM DTT for 30 min at 60°C. Alkylation was performed with 15 mM IAA for 30 min in the dark. Digestion was performed by adding trypsin (Promega, Madison, USA) to a 1:50 enzyme:substrate ratio. Sample was incubated overnight at 37°C. The reaction was quenched by adding 1% of TFA. RapiGestTM was eliminated by centrifugation at 10,000 g for 5 min.
Peptide mapping analysis
NanoLC-MS/MS analysis was performed using a nanoAcquity Ultra-Performance-LC (Waters, Milford, USA) coupled to the Q-Exactive HF-X mass spectrometer (Thermo Scientific, Bremen, Germany) with a nanoSpray source. The sample was trapped on a nanoACQUITY UPLC precolumn (C18, 180 µm x 20 mm, 5 µm particle size), and the peptides were separated on a nanoACQUITY UPLC column (C18, 75 µm x 250 mm with 1.7 µm particle size, Waters, Milford, USA) maintained at 60°C. Mobile phase A was 0.1% (v/v) formic acid in water and mobile phase B was 0.1% (v/v) formic acid in acetonitrile. A gradient (1–8% B for 2 min, 8–35% B for 58 min, 35–90% B for 1 min, 90% B for 5 min, 90–1% B for 1 min and maintained 1% B for 20 min) was used at a flow rate of 450 nL/min. The Q-Exactive HF-X source temperature was set to 250°C and spray voltage to 1.8 kV. Full scan MS spectra (300–1800 m/z) were acquired in positive mode at a nominal resolution of 60,000 with a maximum injection time of 50 ms and an AGC target value of 3 × 106 charges, with lock-mass option being enabled (polysiloxane ion from ambient air at 445.12 m/z). The 10 most intense multiply charged peptides per full scan were isolated using a 2 m/z window and fragmented using higher energy collisional dissociation (normalized collision energy of 27). MS/MS spectra were acquired with at a nominal resolution of 15,000 with a maximum injection time of 50 ms and an AGC target value of 1 x 105, and dynamic exclusion was set to 60 sec. The system was fully controlled by XCalibur software v3.0.63, 2013 (Thermo Scientific) and NanoAcquity UPLC console v1.51.3347 (Waters).
MTS assay
SK-BR-3 cells at the logarithmic growth phase were washed with sterile PBS 1x (HyClone), treated with trypsin (Sigma), re-suspended in 10% fetal bovine serum (Sigma) DMEM medium (Sigma) with cell density at 2 × 105 cells/ml and seeded in a 96-well cell culture plate (Corning) by applying 50 µL of cell suspension per well. The plates were incubated at 37ºC in a humidified 5% CO2 incubator for 24 hours. Next, samples to be analyzed were diluted to the concentration range to be tested and 50 µL were applied to the cell culture wells under sterile conditions by using decreasing sample concentrations. The last row of each plate was used as control, which consisted of cells in medium without sample to be tested. The plates were incubated at 37ºC in a humidified 5% CO2 incubator for 72 hours. After incubation, each well was treated with 20 µL MTS solution (Promega, WI, USA) followed by incubation for 3 hours. The absorbance at 405 nm was measured with a Labtech LT-4000 microplate reader. The concentration indicated is referred to the complete ADC (mainly mAb) and not to toxin:payload.
Abbreviations
| ADC | = | antibody-drug conjugate |
| dhAA | = | dehydroascorbic acid |
| IgG | = | immunoglobulin |
| mAb | = | monoclonal antibody |
| LC | = | light chain |
| HC | = | heavy chain |
| dcHC | = | heavy chains dimers |
| SDS-PAGE | = | dodecyl sodium sulfate polyacrylamide electrophoresis gel |
| vcMMAE | = | valine-citrulline monomethyl auristatin E |
| TCEP | = | tris(2-carboxyethyl)phosphine |
| SEC-HPLC | = | size exclusion chromatography - high performance liquid chromatography |
| HIC-HPLC | = | hydrophobic interaction column – high performance liquid chromatography |
| DAR | = | drug antibody ratio |
| MS | = | mass spectrometry |
| T-MMAE | = | trastuzumab conjugated to monomethyl auristatin E |
Disclosure of potential conflicts of interest
ADC are a promising therapeutic tool because they combine the selectivity of the antibody (vehicle) with the cell killing ability of cytotoxic drugs, minimizing chemo-associated side effects. The method described provides a solution to generate homogeneous compositions of ADCs without antibody sequence engineering. Results also demonstrate that ADC and mAbs can be folded in vitro from their subunit parts (LC and HC) which can be prior assembly conjugated to a cytotoxic drug. The work described herein could pave the way to easily obtaining homogeneous ADCs that could be tested in clinical trials for existing described antibodies or approved to target a disease.
Acknowledgments
This work was supported by Farmhispania SA with funding from CDTI (Spanish Government).
This work was supported by the CNRS, the University of Strasbourg, the “Agence Nationale de la Recherche” (ANR) and the French Proteomic Infrastructure (ProFI; ANR-10-INBS-08-03). The authors thank GIS IBiSA and Région Alsace for financial support in purchasing a Synapt G2 HDMS instrument. A.E. acknowledges the “Association Nationale de la Recherche et de la Technologie” (ANRT) and Syndivia for funding his PhD fellowship.
Additional information
Funding
Related Research Data
References
- Persistence Market Research. Antibody drug conjugates market: global industry analysis and forecast 2017–2025. [Accessed 04 Apr 2018]. https://www.persistencemarketresearch.com/market-research/antibody-drug-conjugates-market.asp
- Maruani A, Smith MEB, Miranda E, Chester KA, Chudasama V, Caddick S, et al. A plug-and-play approach to antibody-based therapeutics via a chemoselective dual click strategy. Nat Commun. 2015 Mar 31;6:6645. doi:https://doi.org/10.1038/ncomms7645. PMID: 25824906.
- Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MAbs. 2014 Jan-Feb;6(1):34–11. doi:https://doi.org/10.4161/mabs.27022. PMID: 24423619.
- Storz U. Antibody-drug conjugates: intellectual property considerations. mAbs. 2015 7;6:989–1009. doi:https://doi.org/10.1080/19420862.2015.1082019.
- Jackson DY. Processes for constructing homogeneous antibody drug conjugates. Org Process Res Dev. 2016:205852–866. doi:https://doi.org/10.1021/acs.oprd.6b00067.
- Grawunder U, Barth S. Next generation antibody drug conjugates (ADCs) and immunotoxins. Chapter 5 Enzyme-based strategies to generate site-specifically conjugated antibody drug conjugates. Springer; 2017.
- Kline T, Steiner AR, Penta K, Sato AK, Hallam TJ, Yin G. Methods to make homogenous antibody drug conjugates. Pharm Res. 2015;32(11):3480–93. doi:https://doi.org/10.1007/s11095-014-1596-8.
- Jain N, Smith SW, Ghone S, Tomczuk B. Current ADC linker chemistry. Pharm Res. 2015;32(11):3526–40. doi:https://doi.org/10.1007/s11095-015-1657-7.
- Sochaj AM, Świderska KW, Otlewski J. Current methods for the synthesis of homogeneous antibody–drug conjugates. Biotechnol Adv. 2015;33(6):775–784. doi:https://doi.org/10.1016/j.biotechadv.2015.05.001.
- Agarwal P, Bertozzi CR. Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug Chem. 2015;26:176–192. doi:https://doi.org/10.1021/bc5004982.
- Behrens CR, Liu B. Methods for site-specific drug conjugation to antibodies. mAbs. 2014;6:46–53. doi:https://doi.org/10.4161/mabs.26632.
- Dan N, Setua S, Kashyap V, Khan S, Jaggi M, Yallapu M and Chauhan S. Antibody-drug conjugates for cancer therapy: chemistry to clinical implications. Pharmaceuticals (Basel). 2018;11(2):32. doi:https://doi.org/10.3390/ph11020032.
- Bhakta S, Raab H, Junutula JR. Engineering THIOMABs for site-specific conjugation of thiol-reactive linkers. Methods Mol Biol. 2013;1045:189–203.
- Junutula J, Hans Erickson Rachana V, Ohri S, Bhakta H, Raab R, Vandlen JJ, Erickson H, A high-throughput conjugation strategy for the selection of THIOMABTM antibodies with desired properties for antibody-drug conjugation; 2015; AACR 106th Annu. Meet. in Philadelphia
- Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–932. doi:https://doi.org/10.1038/nbt.1480.
- Kim EG, Kim KM. Strategies and advancement in antibody-drug conjugate optimization for targeted cancer therapeutics. Biomolecules Ther. 2015;23(6):493–509. doi:https://doi.org/10.4062/biomolther.2015.116.
- Behrens CR, Ha EH, Chinn LL, Bowers S, Probst G, Fitch-Bruhns M, Monteon J, Valdiosera A, Bermudez A, Liao-Chan S, et al. Antibody–drug conjugates (ADCs) derived from interchain cysteine cross-linking demonstrate improved homogeneity and other pharmacological properties over conventional heterogeneous ADCs. Mol Pharm. 2015;12(11):3986–3998. doi:https://doi.org/10.1021/acs.molpharmaceut.5b00432.
- Dennler P, Fischer E, Schibli R. Antibody conjugates: from heterogeneous populations to defined reagents. Antibodies. 2015;4:197–224. doi:https://doi.org/10.3390/antib4030197.
- Badescu G, Bryant P, Bird M, Henseleit K, Swierkosz J, Parekh V, Tommasi R, Pawlisz E, Jurlewicz K, Farys M, et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug Chem. 2014 18;25(6):1124–36. doi:https://doi.org/10.1021/bc500148x.
- Badescu G, Bryant P, Swierkosz J, Khayrzad F, Pawlisz E, Farys M, Cong Y, Muroni M, Rumpf N, Brocchini S, et al. A new reagent for stable thiol-specific conjugation. Bioconjug Chem. 2014;25(3):460–469. doi:https://doi.org/10.1021/bc400245v.
- Shinmi D, Taguchi E, Iwano J, Yamaguchi T, Masuda K, Enokizono J, Shiraishi Y. One-step conjugation method for site-specific antibody–drug conjugates through reactive cysteine-engineered antibodies. Bioconjug Chem. 2016;27(5):1324–1331. doi:https://doi.org/10.1021/acs.bioconjchem.6b00133.
- Feige MJ, Hendershot LM, Buchner J. How antibodies fold. Trends Biochem Sci. 2010;35(4):189–98. doi:https://doi.org/10.1016/j.tibs.2009.11.005.
- Miret RRJ, Roura A, Moreno C, Arboix G, Farràs M, Cancelliere D, Di Gesù C, Casablancas A, Lecina M, Cairó JJ, “PO276 comparison of bicistronic and tricistonic expression strategies for trastuzumab and trastuzumab-interferon-a2b production in CHO and HEK293 cells; 2017; 25th ESACT meeting in Lausanne.
- Lyon R, Setter J, Bovee T, Doronina SO, Hunter JH, Anderson ME, Balasubramanian CL, Duniho SM, Leiske CI, Li F, et al. Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-dug-conjugates. Nat Biotechnol. 2014;32:1059–1062. doi:https://doi.org/10.1038/nbt.2968.
- Ehkirch A, D’Atri V, Rouviere F, Hernandez-Alba O, Goyon A, Colas O, Sarrut M, Beck A, Guillarme D, Heinisch S, et al. An online four-dimensional HIC×SEC-IM×MS methodology for proof-of-concept characterization of antibody drug conjugates. Anal Chem. 2018;90(3):1578–1586. doi:https://doi.org/10.1021/acs.analchem.7b02110.
- Said N, Gahoual R, Kuhn L, Beck A, Yannis-Nicolas F. Structural characterization of antibody drug conjugate by a combination of intact, middle-up and bottom-up techniques using sheathless capillary electrophoresis - Tandem mass spectrometry as nanoESI infusion platform and separation method. Anal Chim Acta. 2016;918:50–9. doi:https://doi.org/10.1016/j.aca.2016.03.006.
- Lyon RP, Bovee TD, Doronina SO, Burke PJ, Hunter JH, Neff-LaFord HD, Jonas M, Anderson ME, Setter JR, Senter PD, et al. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol. 2015;33:733–735. doi:https://doi.org/10.1038/nbt.3212.
- Burke PJ, Hamilton JZ, Jeffrey SC, Hunter JH, Doronina SO, Okeley NM, Miyamoto JB, Anderson ME, Stone IJ, Ulrich ML, et al. Optimization of a PEGylated glucuronide-monomethylauristatin E linker for antibody–drug conjugates. Mol Cancer Ther. 2017;16:116–123. doi:https://doi.org/10.1158/1535-7163.
- Tsuchikama K, An Z. Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell. 2018;9:33–46. doi:https://doi.org/10.1007/s13238-016-0323-0.
- Román R, Miret J, Scalia F, Casablancas A, Lecina M, Cairó JJ. Enhancing heterologous protein expression and secretion in HEK293 cells by means of combination of CMV promoter and IFNα2 signal peptide. J Biotechnol. 2016;239:57–60. doi:https://doi.org/10.1016/j.jbiotec.2016.10.005.
- Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–D1082. doi:https://doi.org/10.1093/nar/gkx1037.
- Haryadi R, Ho S, Kok YJ, Pu HX, Zheng L, Pereira NA, Li B, Bi X, Goh L-T, Yang Y, et al. Optimization of heavy chain and light chain signal peptides for high level expression of therapeutic antibodies in CHO cells. PLoS One. 2015 Feb 23;10(2):e0116878. doi:https://doi.org/10.1371/journal.pone.0116878.
- Chin JX, Chung BKS, Lee DY. Codon Optimization OnLine (COOL): a web-based multi-objective optimization platform for synthetic gene design. Bioinformatics. 2014;30(15):2210–2212. doi:https://doi.org/10.1093/bioinformatics/btu192.