
ABSTRACT
Through reactivating tumor-infiltrating lymphocytes, therapeutics targeting programmed cell death protein 1 (PD-1) demonstrate impressive clinical efficacy in the treatment of multiple cancers. In this report, we characterize HX008, a humanized IgG4S228P anti-PD-1 monoclonal antibody with an engineered Fc domain, in a series of in vitro assays and in vivo studies. In vitro, HX008 binds to human PD-1 with high affinity and potently suppresses the interaction of PD-1 with PD-L1 and PD-L2. The lack of detectable binding to complement C1q and Fc gamma receptor III-a (FcγRIIIa) suggested that HX008 maintained reduced antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity. A comparable enhancement of cytokine production and NFAT-driven luciferase expression in cell-based assays confirmed that HX008 could promote T-cell function as effectively as Nivolumab. In vivo antitumor activity studies were carried out within two special tumor models: 1) the MiXeno model with an adoptive transfer of human peripheral blood mononuclear cells into HCC827 xenograft mice; and 2) HuGEMM with human PD-1 gene knock-in syngeneic MC38-bearing mice. In both models, HX008 significantly inhibits tumor growth and shows an effective antitumor response comparable to approved anti-PD-1 drugs. Furthermore, in a pharmacokinetics study performed in cynomolgus monkeys, HX008 induced no immune-related adverse events when administered at 10 mg/kg. Although some anti-drug antibody effects were observed in the primate PK study, the safety and favorable pharmacokinetics demonstrated in human clinical trials validate HX008 as a suitable candidate for cancer immunotherapy. Taken together, our studies provide a fairly thorough characterization of HX008 and strong support for its further clinical research and application.
Introduction
The immune checkpoint inhibitors have been intensively explored in the last decade, and some of those targeting programmed cell death-1 (PD-1) or programmed death-ligand 1 (PD-L1) have demonstrated encouraging antitumor response with acceptable toxicity in the treatment of a diverse array of cancers.1–Citation3 PD-1 is an inhibitory receptor of the Ig superfamily related to CD28, the expression of which has been found on activated T and B lymphocytes, dendritic cells (DCs), monocytes, and macrophages and is often up-regulated in tumor-infiltrating lymphocytes (TILs).Citation4–Citation7 PD-1 has two ligands, PD-L1 and PD-L2. Typically, PD-1-mediated regulation is essential for maintaining T-cell tolerance and preventing excessive immune response.Citation8 PD-1 knockout mice develop a variety of autoimmune pathologies, indicating an important role of PD-1 in down modulating immune response and lymphocyte homeostasis.Citation9,Citation10 However, in the tumor microenvironment, this mechanism can be exploited by tumor cells to evade immune surveillance. The interaction between PD-1 and PD-L1 or PD-L2 tends to inhibit T-cell proliferation and cytokine secretion, leading to dysfunction and exhaustion of T cells.Citation5,Citation11–Citation13 From this perspective, blockade of the PD-L1/PD-1 signaling would be a sound strategy to restore T cell function and enhance immune response to tumor cells.
Nivolumab (Opdivo®, MDX-1106/BMS-936558, Bristol-Meyers Squibb) and pembrolizumab (Keytruda, MK-3475, Merck) are the first two PD-1 inhibitors approved by the US Food and Drug Administration, and both have achieved great success in the treatment of a diverse array of cancers.Citation14–Citation16 One of the characteristics they have in common is that they are both IgG4 monoclonal antibodies (mAbs). In comparison with IgG1 and IgG3 subtypes, IgG4 antibodies cannot activate the classical complement pathway and have a generally lower affinity for Fc receptors and complement C1q, so they exhibit considerably decreased antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC).Citation17–Citation20 Therefore the combination of an antigen-binding fragment (Fab) targeting PD-1 and an IgG4 Fc backbone can prevent depletion of PD-1-expressing lymphocytes while retaining their antitumor activity. In addition to a S228P mutation in the hinge region of HX008 created to prevent the IgG4 Fab arm exchange, we also introduced a triple mutation, S254T/V308P/N434A (TPA, with substitutions of Ser254 to Thr254, Val308 to Pro308 and Asn434 to Ala434), in its Fc domain based on our analysis of reported IgG-FcRn interacting complex structure.Citation21–Citation23 A substantial number of studies have provided evidence that FcRn plays a pivotal role in maintaining the homeostasis of serum IgG levels through its pH-dependent binding to IgG Fc, which protects IgGs from lysosomal degradation and results in prolonged half-life.Citation24,Citation25 The mutated residues we introduced have profound implications for FcRn interaction to enhance in vivo pharmacokinetics (PK) of antibody therapeutics.Citation23,Citation26
While syngeneic mouse tumor models have been validated as an experimental model for testing surrogate immune-oncology therapy,Citation27 the lack of adequate in vivo animal models for testing human-specific therapeutics technically hampers the preclinical evaluation of immunotherapeutics. Herein, we introduced two elaborate tumor model to overcome this challenge, MiXeno and HuGEMM, which are human genetically engineered mouse models.Citation28 MiXeno models allow the study of immunotherapeutics within a human tumor microenvironment. The reconstitution of human immunity is accomplished by engraftment of adult peripheral blood mononuclear cells (PBMC) into NSG® (NOD, Prkdcscid, IL2rg null) mice, which then are capable of displaying human immune cells, including T cells (CD4+, CD8+), B cells (CD19+), as well as lower levels of human natural killer cells (CD56+) and macrophages (CD14+).Citation29–Citation32 HuGEMM TM are immune-competent chimeric mouse models, in which the mice are engineered to express humanized drug targets such as immune checkpoint proteins. The major hurdle in evaluating anti-PD-1 therapeutics within syngeneic mouse tumor models is the low homology between the extracellular domains of human and mouse PD-1 (61% identity). However, the chimeric h/mPD-1 protein expressed in HuGEMM mice, developed by knocking-in human exon 2 and 3 to replace its mouse counterpart, is able to bind to PD-L1 of both mouse and human origins, as well as anti-human PD-1 antibodies.Citation33 Thus, we can evaluate the in vivo activity of HX008 within mice with a partial functional human immune system. Knock-in mice with human immune checkpoint genes and human immunity reconstituted mice both have been widely used to support in vivo efficacy evaluation of immunotherapeutics such as cytotoxic T-lymphocyte-associated protein-4, PD-1, and PD-L1 inhibitors.Citation34–Citation36
Here, we report the characterization of HX008, which was fully developed in-house, from immunization to structure design. HX008, a humanized IgG4 anti-PD-1 mAb with an S228P hinge mutation and an engineered Fc domain, includes different complementarity-determining region sequences compared to the approved PD-1 inhibitors. It has high affinity for human PD-1 and efficiently blocks its engagement of PD-L1 and PD-L2. Using nivolumab as a reference, comparable improvements in the level of effector molecules were both observed in mixed lymphocyte reaction (MLR) and luciferase reporter assays. To evaluate the ADCC and CDC effect of the antibody, we also determined the binding kinetics of HX008 to C1q and FcγRIIIa using Octet systems. In the antitumor activity studies within tumor graft HCC827 and MC38 (used in the MiXeno model and HuGEMM, respectively), HX008 showed significant inhibition of tumor growth and remarkable complete response rates. Our preclinical results presented here suggest that HX008 is a promising candidate for cancer immunotherapy.
Results
HX008 binds specifically to PD-1 and competitively blocks the PD-L1 and PD-L2 engagement
HX008 was derived from a process that involved immunizing mice with recombinant human PD-1, and then screening a panel of hybridoma cells secreting mAbs capable of binding to human PD-1 protein with high affinity and specificity. The humanized antibody was generated by grafting hypervariable regions of murine antibody onto a human kappa and IgG4 format, which contains an S228P hinge mutation and a TPA substitution in the Fc domain.
Using nivolumab as a reference, a set of enzyme-linked immunosorbent assays (ELISAs) were performed to evaluate whether HX008 was a potential therapeutic agent. Affinity results (Fig. S1) showed that HX008 has a similar PD-1 binding affinity to nivolumab, with an EC50 value of 0.067 nmol/L for HX008 and 0.053 nmol/L for nivolumab, which is consistent with equilibrium dissociation constants (KD, 0.075 nmol/L) of HX008 for PD-1 determined by surface plasmon resonance (Fig S2). The affinity for cynomolgus PD-1 protein was also similar, with an EC50 value of 0.219 nmol/L for HX008 and 0.399 nmol/L for nivolumab (Fig S3).
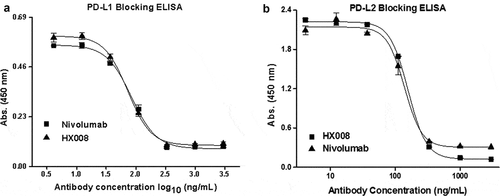
In the PD-L1 and PD-L2 blocking assays (), HX008 and nivolumab could both effectively block the interaction of PD-1 with PD-L1 at a low concentration, with an IC50 value of 0.49 nmol/L and 0.56 nmol/L, respectively. HX008- or nivolumab-mediated inhibition of PD-1 binding to PD-L2 was determined in the same way, with an IC50 value of 1.05 nmol/L and 0.92 nmol/L, respectively. These binding results are in line with published data of nivolumab.Citation37
Figure 1. HX008 efficiently blocks the interaction of PD-1 with PD-L1 and PD-L2. (a) HX008 and nivolumab both inhibit PD-L1 from binding to PD-1 in a competition ELISA. Binding of 0.3 μg/mL PD-L1-mFc to plate-coated PD-1-hFc in the presence of increasing concentrations (4.12 ~ 3000 ng/mL) of HX008 or nivolumab. (b) Binding of 1 μg/mL human PD-L2 to plate-coated human PD-1 was inhibited by serially diluted HX008 or nivolumab (4.12 ~ 3000 ng/mL). Results were shown as mean ± SD of one representative experiment out of three
FcγRIIIa and C1q binding activity relative to ADCC and CDC
Unlike other subclasses of IgG (e.g., IgG1 or IgG3), the IgG4 subclass has negligible binding to the C1q protein complex as well as reduced binding to the low-affinity Fc-gamma receptors (FcγRIIIa).Citation38,Citation39 Therefore, limited ADCC and CDC activity are observed with IgG4 antibodies.Citation35,Citation37 Since FcγRIIIa (aka CD16a) and C1q are the major receptors associated with ADCC and CDC, and with the consideration of three site mutations (TPA) being introduced to the Fc region, binding activities of HX008 for the two molecules were tested to evaluate its ADCC and CDC activity.
Binding kinetics were determined using an Octet system (Fig. S4 A-F). When antibodies were loaded with relatively high concentration (125 ~ 4000 nM), no detectable binding to FcγRIIIa or C1q were observed with HX008 and nivolumab, while the IgG1 subclass exhibited significant binding to both of the effector molecules. The results suggest that HX008 bearing an IgG4 backbone shared similar FcγRIIIa and C1q binding characteristics with nivolumab, which has been reported to have limited ADCC and CDC activity.Citation37
HX008 promotes cytokine production in MLR by PD-1 blockade
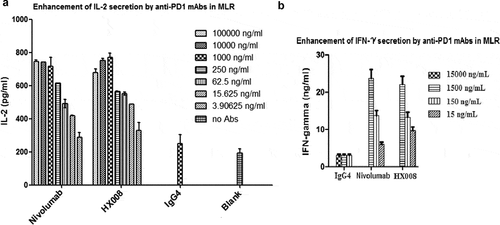
To evaluate whether HX008 exhibited any functional effect on T cells, we first confirmed that HX008 was able to bind to PD-1 receptors on engineered 293T cells expressing human PD-1 protein, with an EC50 value of 0.313 nmol/L (Fig S5). The MLR was determined by mixing matured dendritic cells with T cells from different donors to induce allogenic T-cell activation. Compared with the isotype control, HX008 and nivolumab both significantly improved the release of interleukin (IL)-2 and interferon (IFN)γ about 2 ~ 5 fold at concentrations of 150 ~ 1500 ng/mL (1 ~ 10 nM/L), and there was no obvious difference between the capacity of HX008 and nivolumab to stimulate cytokine secretion of the activated T cells (). These data suggested HX008 could promote the T cell function as efficiently as nivolumab. Moreover, it was noteworthy that there might be an upper limit of the enhancement of IL-2 production at concentrations higher than 1 μg/mL.
Figure 2. Concentration-dependent stimulation of IL-2 and IFNγ secretion induced by HX008 in mixed lymphocyte reaction (MLR) assays, using nivolumab and human IgG4 as positive and negative controls, respectively. T cells and DCs from different donors were co-cultured in the presence of a titration of anti-PD-1 mAbs or isotype control antibodies, then the level of IL-2 and IFNγ in supernatant was determined after 5 days. (a) enhancement of IL-2 secretion by anti-PD-1 mAbs. (b) enhancement of IFNγ secretion by anti-PD-1 mAbs. Representative data from multiple DC/T cell pairs of different donors were shown
HX008 improves T-cell response as effectively as nivolumab in a luciferase reporter assay
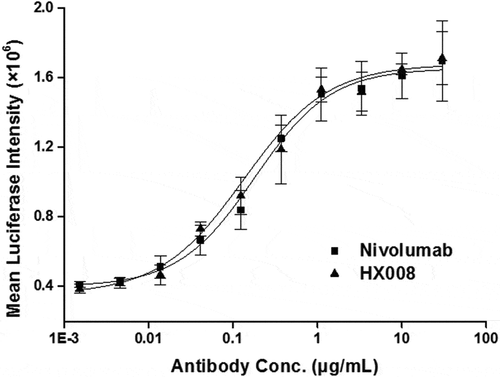
The biological function of HX008 was further investigated in a cell-based luciferase reporter assay, where engineered CHO-PD-L1-CD3L cells and the Jurkat-PD-1-NFAT cells were used as target cells and effector cells, respectively. In the presence of HX008 or nivolumab, the NFAT-driven expression of luciferase showed a dose-dependent increase with a similar EC50 of 1.15 nmol/L and 1.30 nmol/L, respectively (). The data show that HX008 can, at relatively low concentrations (0.1 ~ 1 μg/mL), enhance T-cell reactivity via PD-1 blockade as effectively as nivolumab, which confirms the ELISA and MLR results.
Figure 3. Biological activity evaluation of HX008 in a cell-based luciferase reporter assay. CHO-PD-L1-CD3L cells and the Jurkat-PD-1-NFAT cells were seeded into plates at a ratio of 1:2. After 6 h of incubation, levels of luciferase activity were determined in the presence of serially diluted anti-PD-1 antibodies. All the points in the figures (from one representative data out of three experiments) are average of triplicate with standard deviation
HX008 inhibits tumor growth in vivo
A substantial amount of preclinical and clinical data suggest that inhibition of PD-1/PD-L1 signaling enhances antitumor immune responses in various cancer treatments.Citation37,Citation40 In this study, we examined the in vivo antitumor effect of HX008 in two different tumor models.
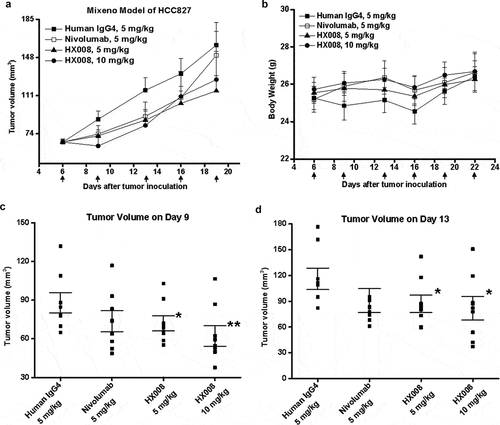
In the first in vivo study, a Mixeno model was created by mixing human PBMC into the tumor cell line-derived xenograft model. Although the tumor growth rate was relatively slow due to the particular tumor model (), we could still observe a statistically significant difference of average tumor volume change in a dose-dependent manner between the HX008-treated group and the IgG4 analogue group, with a TGIRTV value of 18% (, p = .049, 5 mg/kg) and 30% (, p = .007, 10 mg/kg) on Day 9, and 25% (, p = .041, 5 mg/kg) and 30% (, p = .039, 10 mg/kg) on Day 13, respectively. On the other hand, we found less or no statistically significant difference between the nivolumab-treated group and isotype control group, with a TGIRTV value of 17% on Day 9 (, p = .084) and 23% on Day 13 (, p = .073), respectively, suggesting nivolumab might be less effective in this Mixeno model. Moreover, no treatment-related toxicities (such as severe weight loss or death, ) occurred in all four treatment groups within 16 days post dose (i.e., within 22 days after tumor inoculation), indicating that HX008 was well tolerated. These results confirmed the potent antitumor response of HX008 to HCC827-bearing mice within the Mixeno model.
Figure 4. Antitumor response of HX008 in Mixeno model. (a) tumor growth curves of HCC827 tumors in NSG mice. Human PBMC were intravenously (i.v.) injected into mice prior to a subcutaneous HCC827 inoculation. The mice were treated with HX008 (10 mg/kg, or 5 mg/kg; n = 8/group) or nivolumab (5 mg/kg, n = 8) or isotype control antibody (5 mg/kg, n = 8) on Day 6, 9, 13, 16, 19 and 22 after inoculation, respectively. Each group had eight mice, and tumor volume was measured twice weekly. (b) the results of the body weight changes in the tumor-bearing mice. (c) group mean tumor volume measured on Day 9. (d) group mean tumor volume measured on Day 13. For comparisons between groups in the study, we used one-way ANOVA with the Dunn-Sidak post hoc test. A P value of <0.05 was considered to be statistically significant (compared with isotype control). Data were shown as SEM of 8 mice per group; *, P < .05; **, P < .01
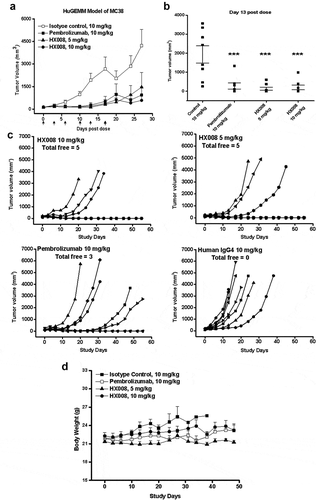
To further investigate the antitumor activity of HX008, another in vivo study was performed with an MC38 (murine colon cancer cells derived from C57BL/6 mice) cell-implanted HuGEMM model. The HuGEMM PD-1 knock-in mouse was developed by replacing the region of murine PD-1 protein that interacts with PD-L1 protein with its human counterpart, which enabled us to test the therapeutic efficacy of HX008 directly in this mouse model. It has been demonstrated that human PD-1 functionally binds to human and mouse PD-L1 with similar affinity,Citation5,Citation35 so HuGEMM would be an ideal model for in vivo evaluation of HX008. Single dose of HX008 (5 mg/kg or 10 mg/kg), positive control pembrolizumab (10 mg/kg) and isotype control (10 mg/kg) were intravenously (i.v.) administered twice weekly after the inoculation. Tumor volume of each group was measured until the mice were humanely euthanized at maximally allowed tumor burden.
The data () showed that anti-PD-1 mAbs dramatically reduced MC38 tumor growth when compared with IgG4 analogue. On Day 13 after grouping, the mean tumor volume of control group reached 1933.67 mm3. At this time point, the tumor growth inhibition value (%) of the 10 mg/kg pembrolizumab-treated group and the 5 mg/kg and 10 mg/kg HX008-treated groups were 85%, 93%, and 90%, respectively. Compared with isotype-treated controls, all three treated groups showed a statistically significant inhibition of tumor growth (P < .001) (). Strikingly, we observed complete regression with 5 of 8 mice both in the 5 mg/kg and 10 mg/kg HX008-treated groups, while none of the isotype control-treated mice was tumor-free. Pembrolizumab was slightly less efficacious, with three tumor-free mice at the same dose (). At the endpoint of the study (Day 55), all the tumor-free mice showed impressively durable response lasting for one month or more after treatment, indicating a robust antitumor effect induced by anti-PD-1 therapeutics. Similarly, the tumor-bearing mice in each treatment group had no significant weight loss () or death. In the late stage of this study, the body weight curves fluctuated because some mice with large tumors were euthanized.
Figure 5. Antitumor response of HX008 in HuGEMM Model. Human PD-1 knock-in mice were subcutaneously implanted with MC38 cells (1 × 106 per mouse) and were randomized into treatment groups after mean tumor volume reached 134 mm3. Single i.v. administration of HX008 (5 mg/kg or 10 mg/kg, n = 8/group) or positive control pembrolizumab (10 mg/kg, n = 8) and isotype control (10 mg/kg, n = 8) were performed twice weekly as indicated, six times in total. Tumor volume was measured twice a week and mice were euthanized at maximum allowed tumor burden. (a) tumor growth curves. (b) group mean tumor volume measured on Day 13 post dose. (c) individual tumor growth curves in each treatment group. The number of total free mice per group was indicated. (d) Body weight of HuGEMM MC38 bearing mice during the treatment. For comparisons between groups in the study, we used one-way ANOVA with the Dunn-Sidak post hoc test. Data were shown as SEM of 8 mice per group; ***, P < .001 (compared with control)
PK study in primates
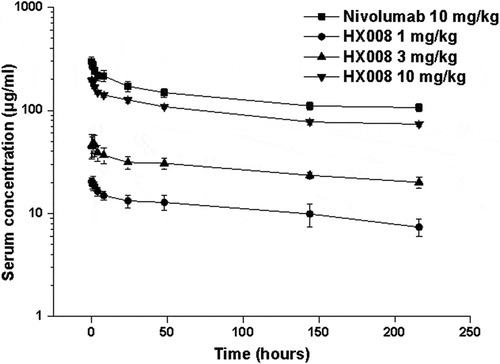
In a single-dose preclinical PK study, four randomized groups of cynomolgus monkeys (3 males and 3 females per group) received i.v. administration of HX008 at 1, 3, 10 mg/kg and nivolumab at 10 mg/kg dosing, respectively. No obvious body weight loss or unexpected deaths were observed with the four groups during the treatment. Serum drug concentration levels of all individuals prior to administration were below the lower limit of quantitation (LLOQ). The concentration-time profiles of HX008 was similar to nivolumab at 10 mg/kg and showed linear kinetic characteristics in the dose range of 1 ~ 10 mg/kg. The drug serum PK parameter estimates are shown in . Drug concentration in the cynomolgus monkey’s serum increased almost dose-proportionally at dose 1, 3, 10 mg/kg, of which the mean C5min was 20.35, 46.16, and 195.25 μg/mL, respectively, with a ratio of 1: 2.27: 9.59, and the mean AUClast was 3180.89, 9313.68, and 33777 h*μg/mL, respectively, with a ratio of 1: 2.93: 10.52. Additionally, the exposure ratio of females to males in the three dose groups of HX008 ranged from 0.92 to 1.28, indicating that there was almost no gender difference in drug exposure of cynomolgus monkeys. As demonstrated by the Vss value (48.37 ~ 66.34 mL/kg), HX008 was mainly in blood with limited extravascular distribution, which was consistent with the plasma volume of macaques (about 44.3 ~ 66.6 mL/kg).
Table 1. Serum PK parameter estimates for HX008 following single i.v. administration to cynomolgus monkey
However, HX008-treated groups showed some anti-drug antibodies (ADA) responses during the administration, which resulted in accelerated elimination of drugs in the terminal phase and had a considerable impact on the PK assessment. In the ADA test, the signal of blank serum matrixes was less than the optical density of the LLOQ, indicating that no significant endogenous interfering substances or nonspecific binding affected the analyte detection. The serum ADA is inversely proportional to dose level in this case, with a higher positive rate for low dose group (1 mg/kg, 83.3%) and lower positive rate for 3 mg/kg and 10 mg/kg (66.7% and 50.0%, respectively). Due to the ADA effect, the value of mean retention time (MRT), mean clearance (CL) and terminal elimination half-life (#t1/2) of HX008 were all obviously lower than that of nivolumab. But if concentration points affected by ADA were excluded from the concentration-time profiles () and only the mean apparent half-life (*t1/2) were taken into account, we found that HX008-treated groups had a comparable *t1/2 with a value of 215.72, 288.78, 268.92 h at doses of 1, 3, 10 mg/kg, respectively, which approximated to the *t1/2 of nivolumab (224.00 h) at the same dose (10 mg/kg). On the other hand, the PK parameter assessment of nivolumab was not affected by ADA, consistent with the reported data.Citation37
Figure 6. Mean (± SD) serum concentration-time profiles following a single i.v. administration of 1, 3, 10 mg/kg HX008 and 10 mg/kg nivolumab to cynomolgus monkeys. N = 8 monkeys per time point per group. Serum concentrations were determined using a validated antigen capture ELISA for the two mAb. PK data were shown without effect of anti-drug antibodies (ADA)
Human pharmacokinetics in phase 1 clinical trial
In an open-label, single-arm Phase 1a clinical trial (CTR20180125) conducted to investigate safety and PK in Chinese patients with advanced solid tumors, HX008 exhibited good safety profile and impressive PK properties. Thirty-nine patients received a single i.v. infusion of HX008 every 3 weeks in dose-escalating 6, 8, 6, 10-patient cohorts at 1 mg/kg, 3 mg/kg, and 10 mg/kg or a fixed dose of 200 mg/patient, respectively. HX008 was well tolerated at 10 mg/kg with no reported grade 4 or 5 drug-related adverse events. Moreover, only one case of ADA was reported within all patient cohorts after multiple administrations (1 ~ 19 treatments) up to 1 ~ 13 months in Phase 1a study, suggesting that HX008 has low immunogenicity in human. Notably, HX008 showed a long half-life of 17.0 to 23.5 days and extended retention time of 621 to 793 h for all dose groups (), indicating HX008’s favorable PK.
Table 2. Summary statistics for PK parameters of HX008 in patients with advanced solid tumors
Discussion
Blocking PD-1/PD-L1 regulatory signal has been validated as an effective strategy to reinvigorate antitumor immune responses in advanced cancers. In this study, we ab initio screened a humanized anti-PD-1 mAb (HX008) that binds to human PD-1 with high affinity and potently blocks the PD-1 engagement with PD-L1 and PD-L2. We further characterized the functional properties and antitumor activity of HX008 in a series of in vitro assays and in vivo animal models.
HX008 is a humanized IgG4 S228P mAb with an engineered Fc conserving a relative low affinity for Fcγ receptors and complement component 1Q. The S228P substitution in the hinge region was introduced to prohibit Fab arm exchanges within endogenous IgG4 antibodies.Citation41 Our primary goal is to evaluate the bioactivity of the therapeutics, which determines whether HX008 is capable of promoting T-cell function or activating antitumor responses. Two approved anti-PD-1 antibody drugs, nivolumab and pembrolizumab, were used as positive controls in our experiments. Either in the mixed lymphocyte reaction or luciferase reporter assays, HX008 and nivolumab both significantly improved T-cell response with similar effectiveness. In terms of cytokine production or luciferase activity, HX008 can enhance T-cell function as efficiently as nivolumab by disrupting the inhibitory signaling of PD-1. All the results of ELISAs and cell-based assays are consistent and display great response values, suggesting a moderate dose of HX008 might be effective in cancer patients.
Activation of C1q by antigen/IgG immune complexes triggers the classical complement cascade,Citation18 and substantial evidence indicates that the affinity of mAbs for FcγRIIIa plays a central role in mediating ADCC.Citation42 The FcγRIIIa and Clq binding properties of HX008 were therefore also tested to evaluate the ADCC and CDC effect mediated by its mutated IgG4 Fc. No detectable binding to FcγRIIIa or C1q were observed in bio-layer interferometry (BLI) assays, suggesting that HX008 is very likely to retain attenuating CDC and ADCC activity in the same way as nivolumab and wildtype IgG4 mAbs.Citation37,Citation43
For in vivo evaluation of HX008, two established tumor models, Mixeno and HuGEMM, were used, and both of them had been validated as effective tools for preclinical assessment of anti-PD-1 therapeutics.Citation33,Citation44 The first model partially reconstituted human immunity in NSG mice, developing by engrafting xenograft tumors and human immune cells sequentially. To remove the obstacle that HX008 does not cross-react with mouse PD-1, we also used the human PD-1 knock-in HuGEMM model, in which the chimeric PD-1 receptor can be recognized by human antibodies of interest and bind efficiently to both mouse and human PD-L1. However, the two models do have limitations. For example, the reconstituted immunity in the Mixeno model is still far different from the real human tumor microenvironment, and the transferred PBMCs relied solely on tumor xenografts for the cytokine and chemokine supply critical for their activation and proliferation. Although we replaced the ectodomain of mouse PD-1 with human homologue in HuGEMM, the endogenous PD-L1 and PD-L1 in the MC38 graft were murine. Therefore, we set positive controls using nivolumab and pembrolizumab, which had been validated for their in vivo anti-tumor activity. The responses observed in tested animals confirmed that the two models worked well and could provide valid evaluation for HX008.
In the Mixeno model, the mean tumor volume of HCC827 did not display a stark contrast between treatment groups and control group, which might be due to the influence from graft-versus-host disease and graft versus tumor. The tumors grew slower or even shrank over time (19 days in this case), and the maximal antitumor effects (maximal TGI) were observed on Day 13. Thus, the data collected after Day 13 are not appropriate to evaluate anti-PD1 efficacy. But on Day 9 and 13, a statistical difference (P < .05, compared with isotype control) was still observed both with the high (10 mg/kg) and middle dose (5 mg/kg) group of HX008, while less significant difference was observed in the nivolumab-treated group.
In contrast to the Mixeno model, the results of the HuGEMM model were fairly explicit. Administration of HX008 and pembrolizumab showed a similar trend of dramatic reduction in tumor growth, among which HX008 at 5 mg/kg showed the most effective inhibition, with tumor growth reduced by 93% on Day 13 post dose (P < .001). Notably, when taking individual response into account, we found that the groups dosed with HX008 at 5 mg/kg and 10 mg/kg both exhibited an extraordinary complete tumor remission rate of 62.5% (five of eight mice), which is higher than that of the pembrolizumab control group (37.5%, three of eight mice). Collectively, these results revealed potent antitumor response of HCC827- and MC38-bearing mice that received HX008, with comparable activity to the approved drugs. The absence of severe weight loss or deaths suggested that HX008 was well tolerated in rodents. Moreover, the comparable antitumor effect of the middle dose (5 mg/kg) and high dose (10 mg/kg) suggested that HX008 might work at a moderate dose, consistent with the inference in cell-based assays.
In the single-dose PK study, administration of HX008 to cynomolgus monkeys at 1 mg/kg, 3 mg/kg and 10 mg/kg was well tolerated without obvious weight loss or unexpected deaths. Given the fact that PK of HX008 was affected by ADA, the PK of HX008 in cynomolgus monkeys was assessed based on initial exposure up to 9 days post dose, by which time ADA had not taken effect.Citation45 It has been demonstrated that ADA responses in animals are not predictive of responses in humans.Citation46,Citation47 HX008 is a humanized antibody, the immunogenicity of which may be different between monkeys and humans. In the Phase 1a clinical trial of HX008, after multiple administrations (treatment cycles varied between patients because of withdrawal or discontinuation), the incidence rate of ADA was 1/30, the longest cycle of which included 19 administrations of HX008 up to13 months, and no > grade 4 treatment-related adverse events were induced by the drug. Notably, the half-life of HX008 at 1 mg/kg and 10 mg/kg (23.0 and 23.5 days) is longer than that of nivolumab (15.0 to 21.0 days) and pembrolizumab (16.0 to 18.1 days) at the same dose reported in clinical trials conducted in Asian patients,Citation48–Citation51 indicating an overall PK benefit of HX008 in human.
During the development of HX008, we engineered its Fc domain with a TPA mutation, the mutated residues of which had been reported to enhance pH-dependent FcRn binding, and several combinations of the three mutations have demonstrated a 2 ~ 4 fold improvement of PK in primates.Citation23,Citation26,Citation52 The data from the PK study in cynomolgus monkeys and Phase 1a clinical trial provided a favorable PK evaluation for HX008, but the direct correlation between the engineered Fc and in vivo PK remains unclear and requires further investigation. More studies will be done to reveal the relationship of modified Fc-mediated FcRn binding with in vivo disposition by directly comparing the wildtype and TPA variants of HX008, and further clinical investigation is ongoing in Phase 1b (CTR20191676) and Phase 2 (NCT03704246) trials.
Taken together, our study demonstrates that HX008 has similar binding properties for human PD-1 and possesses comparable ability to promote T-cell function when compared with nivolumab. Negligible binding to FcγRIIIa and C1q can be predictive of reduced ADCC and CDC activity associated with its IgG4 backbone. In vivo studies validated the therapeutic potential of HX008 with significant antitumor response to multiple cancer cells and favorable PK properties. On the whole, our preclinical data lay the foundation for further clinical investigations of HX008, and suggest that it is a suitable drug candidate for cancer immunotherapy.
Materials and methods
Cell lines and reagents
CHO-PDL1-CD3L and Jurkat-PD-1-NFAT cell lines were obtained from National Institutes for Food and Drug Control (China), cultured in DMEM/F12 (11330–032, Gibco) or RPMI-1640 (11875–093, Gibco) medium containing 10% fetal bovine serum (FBS, Gibco), 1% NEAA (11140–050, Gibco) and 100 μg/mL penicillin-streptomycin (SV30010, Hyclone), and selected with hygromycin B (400052, Merck Millipore) and puromycin (P8230, Solarbio), respectively. HCC827 and MC38 cell lines were purchased from American Type Culture Collection (ATCC) and cultured in RPMI-1640 (Gibco) supplemented with 10% FBS (Gibco).
Antibody generation
To prepare the mAbs, Balb/C mice (Charles River) were immunized with recombinant human PD-1 protein (produced in house). Splenocyte-derived hybridomas producing mAbs reactive to PD-1 were screened by ELISA. The generation and humanization of the murine mAb was performed as described previously.Citation53 Briefly, total RNA was isolated from hybridoma cells producing the anti-PD-1 mAbs. Variable region genes of the antibody were obtained by degenerate polymerase chain reaction and ligated into the mouse IgG light chain and heavy chain constant regions. After activity validation of the recombinant antibody, the humanized antibody was constructed by 1) using a human IgG4 format containing an S228P (Ser228 to Pro228) mutation to prevent the IgG4 arm exchange, and 2) replacing the murine framework region with a human counterpart. Activity was tested after every modification. The S254T/V308P/N434A (TPA) variants were produced using the method of QuikChange and were confirmed by DNA sequencing. Antibodies were expressed in Chinese hamster ovary stable cell line and purified with protein G affinity chromatography.
Binding activity assays
PD-L1 and PD-L2 blocking ELISA
The potency of HX008 to block the PD-1 engagement of PD-L1 and PD-L2 was evaluated in a competitive ELISA. PD-1-hFc protein (PD-1 protein with human IgG1Fc, Akesobio) was incubated in 96-well plates at 4°C overnight. Three-fold serial dilutions of HX008 and nivolumab protein (4.12 ~ 3000 ng/mL, Bristol-Meyers Squibb, Lot. AAE6383) were applied to duplicate wells in the presence of 0.3 μg/mL PD-L1-mFc (containing a mouse IgG2aFc). After one-hour incubation, the bound competitive ligands were detected by horseradish peroxidase (HRP)-conjugated goat anti-mouse antibodies (Jackson ImmunoResearch, Cat. 115-035-062) and developed with 3,3′,5,5′-Tetramethylbenzidine (TMB, Biosharp, Cat. Amresco0759) substrates. The IC50 (half maximal inhibitory concentration) of antibodies was determined by fitting the absorbance-concentration data to a four-parameter regression model. PD-L2 inhibition ELISA was performed in the same set-up with higher ligand concentration (1 μg/mL PD-L2).
Binding kinetics of HX008 to FcγRIIIa and Clq
Binding kinetics of the HX008 to FcγRIIIa and Clq were tested using Octet system based on BLI technology. In the binding assay of FcγRIIIa, 1 μg/mL biotin-labeled FcγRIIIa (produced in house) was immobilized on streptavidin-coated biosensor for 300 s to reach a saturation level. HX008 was added with serial concentrations initiating from 125 nM and contacted for 120 s, followed by 180 s dissociation. Binding kinetics to C1q were determined by coating 100 μg/mL HX008 on an anti-human Fab-CH1 2nd Generation (FAB2G) Biosensor for 300 s. The captured antibodies were then detected by loading C1q protein (fitzgerald, Cat. 32R-AC049) of serial dilutions ranging from 3.13 to 200 nM, allowing the proteins to associate for 120 s and dissociate for 180 s. All BLI experiments were performed on the Octet instrument (ForteBio, Inc.) in phosphate-buffered saline (PBS) buffer, and all kinetics data were collected and analyzed using ForteBio Data Analysis 7.0 software.
In vitro functional assays
Stimulation of cytokine secretion in MLR
The MLR assays were used to evaluate and compare the capacity of HX008 and nivolumab to stimulate the release of IL-2 and IFNγ by T lymphocytes, details being described as follows. PBMC were isolated from fresh blood samples of healthy donors using ficoll density gradient separation. All blood samples were taken after the donors provided written informed consent for the donation. After a 4-h incubation at 37°C, adherent cells were cultured in supplemented RPMI medium containing 10% FBS together with human granulocyte-macrophage colony-stimulating factor (peprotech, Cat. 300–03) and IL-4 (peprotech, Cat. 200–04) to obtain immature dendritic cells (iDCs). Maturation of iDCs was accomplished with tumor necrosis factor (peprotech, Cat. 300-01A) stimulation. Then, the mature DCs were co-cultured with allogeneic T cells from another healthy donor in the presence of a titration of antibodies. After incubating for 5 days, the secretion levels of IL-2 and IFNγ in the cell supernatant were determined using commercially available ELISA kits in accordance with the manufacturer’s instructions (Dakewe).
Reporter gene assay to measure the biological function of HX008 on T cells
The ability of HX008 to improve T-cell functions via inhibition of PD-1/PD-L1 signaling was determined using two engineered cell lines: 1) CHO-PDL1-CD3L expressing PD-L1 and anti-CD3 single-chain antibody fragment, and 2) Jurkat-PD-1-NFAT expressing PD-1 and the luciferase gene under the control of the NFAT response elements. The experiments were carried out as described previously.Citation54 CHO-PD-L1-CD3L cells were seeded in 96-well plate and cultured for 16 ± 2 h prior to the addition of serially diluted HX008 and nivolumab. Then, the cultures were mixed with Jurkat-PD-1-NFAT cells at a ratio of 1:2 and incubated at 37°C, 5% CO2 for 6 h. The relative luciferase intensity was revealed with luminescent substrate and detected by EnVision multimode plate reader (Perkin Elmer). The data were analyzed with a four-parameter regression model.
In vivo activity studies
In vivo studies in mice were contracted to Crown Bioscience Inc., and the PK study in cynomolgus monkeys was contracted to WestChina-Frontier PharmaTech Co. Ltd. All animal experiments in this study were carried out in accordance with the Guide for the Care and Use of Medical Laboratory Animals (Ministry of Health, People’s Republic of China), and were approved and overseen by the Institutional Animal Care and Use Committee.
Antitumor effect of HX008 in HCC827 (human non-small cell lung cancer) MiXeno model
An adoptive transfer of human PBMCs into NSG mice (Model Animal Research Center of Nanjing University) following tumor cells inoculation was used to establish a MiXeno model and investigate the antitumor effect of HX008 in HCC827 (human non-small cell lung cancer). 5 × 106 HCC827 cells were inoculated subcutaneously in the right flank of 40 NSG mice (32 mice plus 8 in surplus) on Day 0. When the mean tumor volume reached 66 mm3 after 6 days of tumor cell inoculation (Day 6), 32 mice were evenly divided into 4 groups according to the tumor volume. Then, PBMCs re-suspended in PBS were injected into the tail vein of NSG mice. Mice of groups 1 ~ 4 were given i.v. administration of HX008 5 mg/kg and 10 mg/kg (test group), nivolumab 5 mg/kg (positive group, Bristol-Myers Squibb Company) and isotype antibody human IgG4 5mg/kg (control group, produced by Crown Bioscience (Taicang) Inc), and each dose was administered on Day 6, 9, 13, 16, 19 and 22 after inoculation, six administrations in total. Efficacy and safety were evaluated according to the tumor growth inhibition value based on relative tumor volume (TGIRTV), animal body weight changes and death rate.
Antitumor effect of HX008 in HuGEMM implanted with syngenic MC38 mouse tumor
The HuGEMM model (Model Animal Research Center of Nanjing University) developed by replacing the extracellular region of C57BL/6 mouse PD-1 gene with a human counterpart enabled us to test the antitumor activity of HX008 directly on mouse MC38 tumor. MC38 cells were subcutaneously inoculated in the right flank of the tested mice (1 × 106 cells/mouse). When the average tumor volume reached about 134 mm3, mice were randomly divided into four experimental groups according to the tumor volume (8 mice per group). Mice of four groups received tail i.v. administration of HX008 (5 mg/kg and 10 mg/kg, test groups), pembrolizumab (10 mg/kg, positive control group, Merck) and isotype antibody human IgG4 (5 mg/kg, negative control group, produced by Crown Bioscience (Taicang) Inc) twice weekly for a total of 6 doses. Efficacy and safety evaluation were accomplished based on tumor growth inhibition value-based TGIRTV, animal body weight changes, and death rate.
Pharmacokinetics in cynomolgus monkeys
In a single-dose PK study, 24 cynomolgus monkeys (Macaca fascicularis) were randomly divided into four groups according to body weight: a high dose group of nivolumab (10 mg/kg), and low, middle, and high dose group of HX008 (1, 3, 10 mg/kg). Each group has six monkeys (three males and three females per group) and received a single i.v. administration. The whole blood was collected and processed for serum pre-dose, and at 5, 30 min and 1, 2, 4, 8, 24, 48, 144, 216 h post-dose. Concentrations of nivolumab and HX008 in cynomolgus monkey serum were determined with standard curve plotted by ELISA, and related PK parameters were calculated using Phoenix WinNonlin (Pharsight) 6.4.
The concentration of HX008 in the serum of cynomolgus monkeys was tested via ELISA. PD1-mFc was coated on the plate overnight followed by washing and antibody incubation. Then, HRP-conjugated goat anti-human IgG was added and detection signals were produced by TMB substrate. Finally, the reaction was stopped by sulfuric acid and detected at 450 nm. The color density of reaction is associated with HX008 concentration. Repeatability, precision, and accuracy had been validated for the method.
Anti-HX008 antibodies in the serum of cynomolgus monkeys are tested with Bridging-ELISA method. HX008 should be coated on the plate first. Positive controls (rabbit polyclonal anti-HX008 antibodies, HanX Biopharmaceuticals, produced in house) were added, followed by incubation with biotin-labeled HX008 (HX008-Biotin). Captured HX008-biotin was then detected by HRP-conjugated streptavidin. Reaction was revealed by TMB, stopped with 0.5 M H2SO4, and read at wavelength of 450 nm. Absorbance is proportional to concentrations of positive control samples or ADA in study samples.
Brief description of clinical design
This study was an open-label, single-arm Phase 1a clinical trial (CTR20180125) conducted in Chinese patients with advanced solid tumors. The study was designed to investigate the safety and tolerability, PK, immunogenicity, and anti-tumor activity of HX008 monotherapy. HX008 was administered as a 60-min i.v. infusion at a dose of 1 mg/kg, 3 mg/kg, and 10 mg/kg or a fixed dose of 200 mg/patient every 3 weeks, with a 6, 8, 6, 10-patient cohort, respectively. Serum samples were collected at 1 h pre-administration and 0, 2, 8, 24, 72, 168, 366 h post-administration and the single-dose PK was evaluated on Day 21 after administration.
The concentration of HX008 in human serum sample is tested using the Bridging-ECLIA (Electrochemiluminescent Immunoassay) method of MSD. Biotinylated PD-1-his was captured on the MSD SA plate. Then test samples, standard, and quality control were added into the plate. After washing excess antibodies, sulfo-tag-conjugated goat anti-human IgG4 antibody were added to form the complex of biotin-PD-1 – HX008 – Sulfo-Tag-Mouse anti-human IgG4 antibody. Finally, the reaction was developed by MSD Read Buffer and read on MSD instruments. The results were processed by fitting the data to a five-parameter model, the weight coefficient of which is 1/Y2.
The analytical method for detecting ADA in human blood sample is based on the Bridging-ECLIA method of MSD. The assay is briefly described as follows. The working solution of capture reagent (biotinylated drug), detection reagent (sulfo-tag conjugated drug), and neutralizing reagent was added to the polypropylene plate, and then the test sample, verification sample or internal control after acidolysis were added to the plate. After a complete reaction in the dark with shaking, the sample mixture was transferred to a blocked MSD Streptavidin Gold Plate. The Drug-ADA-Drug complex bound to the MSD SA Plate after incubation in dark with shaking. Finally, the reaction was developed by MSD Read Buffer T and read on the MESO QUICKPLEX SQ120. The signal value was directly proportional to the concentration of ADA. The highest tolerance of drug for the method can reach up to 200.0 µg/mL, 50.0 µg/mL, and 20.0 µg/mL under the conditions of 1000 ng/mL, 200 ng/mL, and 100 ng/mL ADA (molar ratio is about 1: 200, 1: 250, 1: 200), respectively.
The human clinical study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice, and the study protocol and all amendments were approved by the institutional ethics committees. Each patient provided written informed consent.
Statistical analysis
The potency of activity assay is expressed as the ratio of EC50. A four-parameter model was used to fit a dose–response curve, through which the EC50 of drugs was determined. All error bars represent the mean ± SD or SEM as indicated. In vitro assays were performed in triplicate. For the in vivo assays, the significance was determined using one-way ANOVA with the Dunn-Sidak post hoc test. The value for which P < .05, 0.01 or 0.001 was considered statistically significant, and was marked by one to three asterisks, respectively.
Disclosure of Potential Conflicts of Interest
Faming Zhang, Ying Huang, and Gan Xi have ownership interest (including patents) in HanX Biopharmaceuticals Inc. The remaining authors have declared that they have no financial conflict of interest with regard to this work.
Supplemental Material
Download Zip (11.1 MB)Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Iwai Y, Hamanishi J, Chamoto K, Honjo T. Cancer immunotherapies targeting the PD-1 signaling pathway. J Biomed Sci. 2017;24:26. doi:https://doi.org/10.1186/s12929-017-0329-9.
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–12. doi:https://doi.org/10.1056/NEJMoa1305133.
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi:https://doi.org/10.1056/NEJMoa1200694.
- Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. Embo J. 1992;11:3887–95. doi:https://doi.org/10.1002/j.1460-2075.1992.tb05481.x.
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34. doi:https://doi.org/10.1084/jem.192.7.1027.
- Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8:765–72. doi:https://doi.org/10.1093/intimm/8.5.765.
- Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–44. doi:https://doi.org/10.1182/blood-2008-12-195792.
- Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42. doi:https://doi.org/10.1111/j.1600-065X.2010.00923.x.
- Nishimura H1, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–51. doi:https://doi.org/10.1016/S1074-7613(00)80089-8.
- Nishimura H. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–22. doi:https://doi.org/10.1126/science.291.5502.319.
- Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, Collins M, Honjo T, Freeman GJ, Carreno BM. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol. 2002;32:634–43. doi:https://doi.org/10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9.
- Peggs KS, Quezada SA, Allison JP. Cell intrinsic mechanisms of T-cell inhibition and application to cancer therapy. Immunol Rev. 2008;224:141–65. doi:https://doi.org/10.1111/imr.2008.224.issue-1.
- Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, Greenfield EA, Freeman GJ. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–66. doi:https://doi.org/10.4049/jimmunol.170.3.1257.
- Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–75. doi:https://doi.org/10.1200/JCO.2009.26.7609.
- Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, Powderly JD, Heist RS, Carvajal RD, Jackman DM, et al. Overall survival and long-term safety of nivolumab (Anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2015;33:2004–12. doi:https://doi.org/10.1200/JCO.2014.58.3708.
- Kwok G, Yau TC, Chiu JW, Tse E, Kwong YL. Pembrolizumab (Keytruda). Hum Vaccin Immunother. 2016;12:2777–89. doi:https://doi.org/10.1080/21645515.2016.1199310.
- Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy-inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res. 2012;18:6580–87. doi:https://doi.org/10.1158/1078-0432.CCR-12-1362.
- Jefferis R, Lund J. Interaction sites on human IgG-Fc for FcgammaR: current models. Immunol Lett. 2002;82:57–65. doi:https://doi.org/10.1016/S0165-2478(02)00019-6.
- Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daëron M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–25. doi:https://doi.org/10.1182/blood-2008-09-179754.
- Jefferis R. Isotype and glycoform selection for antibody therapeutics. Arch Biochem Biophys. 2012;526:159–66. doi:https://doi.org/10.1016/j.abb.2012.03.021.
- Raghavan M, Bonagura VR, Morrison SL, Bjorkman PJ. Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry. 1995;34:14649–57. doi:https://doi.org/10.1021/bi00045a005.
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276:6591–604. doi:https://doi.org/10.1074/jbc.M009483200.
- Yeung YA, Leabman MK, Marvin JS, Qiu J, Adams CW, Lien S, Starovasnik MA, Lowman HB. Engineering human IgG1 affinity to human neonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J Immunol. 2009;182:7663–71. doi:https://doi.org/10.4049/jimmunol.0804182.
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–25. doi:https://doi.org/10.1038/nri2155.
- Ober RJ, Martinez C, Vaccaro C, Zhou J, Ward ES. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J Immunol. 2004;172:2021–29. doi:https://doi.org/10.4049/jimmunol.172.4.2021.
- Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. 2006;281:23514–24. doi:https://doi.org/10.1074/jbc.M604292200.
- Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res. 2013;19:5626–35. doi:https://doi.org/10.1158/1078-0432.CCR-13-0545.
- Li QX, Feuer G, Ouyang X, An X. Experimental animal modeling for immuno-oncology. Pharmacol Ther. 2017;173:34–46. doi:https://doi.org/10.1016/j.pharmthera.2017.02.002.
- Watanabe Y, Takahashi T, Okajima A, Shiokawa M, Ishii N, Katano I, Ito R, Ito M, Minegishi M, Minegishi N, et al. The analysis of the functions of human B and T cells in humanized NOD/shi-scid/gammac(null) (NOG) mice (hu-HSC NOG mice). Int Immunol. 2009;21:843–58. doi:https://doi.org/10.1093/intimm/dxp050.
- Traggiai E. Development of a human adaptive immune system in cord blood cell transplanted mice. Science. 2004;304:104–07. doi:https://doi.org/10.1126/science.1093933.
- Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100:3175–82. doi:https://doi.org/10.1182/blood-2001-12-0207.
- Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, Watanabe T, Akashi K, Shultz LD, Harada M. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood. 2005;106:1565–73. doi:https://doi.org/10.1182/blood-2005-02-0516.
- Wang Z, Cai B, Chen GANG, Liu J, An X, Wang Z, Ouyang D, Wery J-P, Liu J, Dong X, et al. HuGEMM-h/mPD1 mouse models for assessing anti-human PD1 therapeutics [abstract]. Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; 2015 Nov 5–9; Boston, MA. Philadelphia (PA): AACR; Mol Cancer Ther. 2015;14:Abstractnr A11
- Lute KD, May KF Jr, Lu P, Zhang H, Kocak E, Mosinger B, Wolford C, Phillips G, Caligiuri MA, Zheng P, et al. Human CTLA4 knock-in mice unravel the quantitative link between tumor immunity and autoimmunity induced by anti-CTLA-4 antibodies. Blood. 2005;106:3127–33. doi:https://doi.org/10.1182/blood-2005-06-2298.
- Burova E, Hermann A, Waite J, Potocky T, Lai V, Hong S, Liu M, Allbritton O, Woodruff A, Wu Q, et al. Characterization of the Anti-PD-1 antibody REGN2810 and its antitumor activity in human PD-1 knock-in mice. Mol Cancer Ther. 2017;16:861–70. doi:https://doi.org/10.1158/1535-7163.MCT-16-0665.
- Lin S, Huang G, Cheng L, Li Z, Xiao Y, Deng Q, Jiang Y, Li B, Lin S, Wang S, et al. Establishment of peripheral blood mononuclear cell-derived humanized lung cancer mouse models for studying efficacy of PD-L1/PD-1 targeted immunotherapy. MAbs. 2018;10:1301–11. doi:https://doi.org/10.1080/19420862.2018.1518948.
- Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, Garcia C, Wu Y, Kuhne M, Srinivasan M, et al. In Vitro characterization of the Anti-PD-1 antibody nivolumab, BMS-936558, and In Vivo toxicology in non-human primates. Cancer Immunol Res. 2014;2:846–56. doi:https://doi.org/10.1158/2326-6066.CIR-14-0040.
- Guma M, Firestein GS. IgG4-related diseases. Best Pract Res Clin Rheumatol. 2012;26:425–38. doi:https://doi.org/10.1016/j.berh.2012.07.001.
- Scapin G, Yang X, Prosise WW, McCoy M, Reichert P, Johnston JM, Kashi RS, Strickland C. Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat Struct Mol Biol. 2015;22:953–58. doi:https://doi.org/10.1038/nsmb.3129.
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–67. doi:https://doi.org/10.1038/nature14011.
- van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martínez-Martínez P, Vermeulen E, den Bleker TH, Wiegman L, Vink T, Aarden LA, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317:1554–57. doi:https://doi.org/10.1126/science.1144603.
- Bowles JA, Wang SY, Link BK, Allan B, Beuerlein G, Campbell MA, Marquis D, Ondek B, Wooldridge JE, Smith BJ, et al. Anti-CD20 monoclonal antibody with enhanced affinity for CD16 activates NK cells at lower concentrations and more effectively than rituximab. Blood. 2006;108:2648–54. doi:https://doi.org/10.1182/blood-2006-04-020057.
- Stein R, Qu Z, Chen S, Solis D, Hansen HJ, Goldenberg DM. Characterization of a humanized IgG4 anti-HLA-DR monoclonal antibody that lacks effector cell functions but retains direct antilymphoma activity and increases the potency of rituximab. Blood. 2006;108:2736–44. doi:https://doi.org/10.1182/blood-2006-04-017921.
- Zhang J, Qiu J, Qiao M, Shi Q. 14 Mixeno mouse models for in vivo evaluation of anti-human cancer immunotherapeutics. Eur J Cancer. 2014;50:11. doi:https://doi.org/10.1016/S0959-8049(14)70140-6.
- Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. Aaps J. 2012;14:296–302. doi:https://doi.org/10.1208/s12248-012-9340-y.
- van Meer PJ, Kooijman M, Brinks V, Gispen-de Wied CC, Silva-Lima B, Moors EH, Schellekens H. Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. MAbs. 2013;5:810–16. doi:https://doi.org/10.4161/mabs.25234.
- Bugelski PJ, Treacy G. Predictive power of preclinical studies in animals for the immunogenicity of recombinant therapeutic proteins in humans. Curr Opin Mol Ther. 2004;6:10–16.
- Shimizu T, Seto T, Hirai F, Takenoyama M, Nosaki K, Tsurutani J, Kaneda H, Iwasa T, Kawakami H, Noguchi K, et al. Phase 1 study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in Japanese patients with advanced solid tumors. Invest New Drugs. 2016;34:347–54. doi:https://doi.org/10.1007/s10637-016-0347-6.
- Yamamoto N, Nokihara H, Yamada Y, Shibata T, Tamura Y, Seki Y, Honda K, Tanabe Y, Wakui H, Tamura T. Phase I study of Nivolumab, an anti-PD-1 antibody, in patients with malignant solid tumors. Invest New Drugs. 2017;35:207–16. doi:https://doi.org/10.1007/s10637-016-0411-2.
- Lee KW, Lee DH, Kang JH, Park JO, Kim SH, Hong YS, Kim ST, Oh DY, Bang YJ. Phase I pharmacokinetic study of nivolumab in Korean patients with advanced solid tumors. Oncologist. 2018;23:155–e17. doi:https://doi.org/10.1634/theoncologist.2017-0528.
- ClinicalTrials.gov. An open-label, randomized phase I study investigating safety, tolerability, pharmacokinetics, and efficacy of pembrolizumab (MK-3475) in Chinese subjects with non-small-cell lung cancer. Accessed 7 October 2019. https://clinicaltrials.gov/ct2/show/results/NCT02835690.
- Datta-Mannan A, Chow CK, Dickinson C, Driver D, Lu J, Witcher DR, Wroblewski VJ. FcRn affinity-pharmacokinetic relationship of five human IgG4 antibodies engineered for improved in vitro FcRn binding properties in cynomolgus monkeys. Drug Metab Dispos. 2012;40:1545–55. doi:https://doi.org/10.1124/dmd.112.045864.
- Presta LG, Chen H, O’Connor SJ, Chisholm V, Meng YG, Krummen L, Winkler M, Ferrara N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–99.
- Wang L, Yu C, Yang Y, Gao K, Wang J. Development of a robust reporter gene assay to measure the bioactivity of anti-PD-1/anti-PD-L1 therapeutic antibodies. J Pharm Biomed Anal. 2017;145:447–53. doi:https://doi.org/10.1016/j.jpba.2017.05.011.