
ABSTRACT
The origins of the various elements in the human antibody repertoire have been and still are subject to considerable uncertainty. Uncertainty in respect of whether the various elements have always served a specific defense function or whether they were co-opted from other organismal roles to form a crude naïve repertoire that then became more complex as combinatorial mechanisms were added. Estimates of the current size of the human antibody naïve repertoire are also widely debated with numbers anywhere from 10 million members, based on experimentally derived numbers, to in excess of one thousand trillion members or more, based on the different sequences derived from theoretical combinatorial calculations. There are questions that are relevant at both ends of this number spectrum. At the lower bound it could be questioned whether this is an insufficient repertoire size to counter all the potential antigen-bearing pathogens. At the upper bound the question is rather simpler: How can any individual interrogate such an astronomical number of antibody-bearing B cells in a timeframe that is meaningful? This review evaluates the evolutionary aspects of the adaptive immune system, the calculations that lead to the large repertoire estimates, some of the experimental evidence pointing to a more restricted repertoire whose variation appears to derive from convergent ‘structure and specificity features’, and includes a theoretical model that seems to support it. Finally, a solution that may reconcile the size difference anomaly, which is still a hot subject of debate, is suggested.
Origin of the adaptive immune system
It is currently thought that the antibody repertoire in the adaptive human immune system evolved to enable it to combat the massive number of foreign antigens derived from pathological bacteria, viruses, toxins, and the like. A critical question is whether the number of potentially dangerous pathogens on earth today has been a gradual accretion over millions of years and, pari passu, provided the driving force for the development of an adaptive immune system with an extensive antibody repertoire, or were they unconnected? The gradual accretion argument, while carrying a certain biological logic, may suffer from the teleological conundrum of the future requirements dictating the strategy of the past, particularly if the rate of pathogen diversification greatly exceeds the ability of the immune control systems to adapt by mutation at the same rate. Unless the many pathogens present today have always existed, there should be evidence of increasing ‘fitness’ of antibody diversity and associated mechanisms over time. The phylogenetic analysis of antibody genes since the divergence of the jawed vertebrates, in cartilaginous fish, bony fish, amphibians, reptiles, birds and mammals, and the absence of active recombinase activator genes RAG1 and RAG2 in jawless vertebrates suggests this to be the case.Citation1
A broader question, however, is whether repertoire diversification has run ahead of the cellular mechanisms by which such a diverse membership can be fully exploited. Marchalonis et al.Citation2 suggest that the cellular processes preexisted and were simply co-opted into the immune system, citing as an example the ancient origin of homologues of the complement system (e.g., the C3 molecule, a thioester-containing protein) in protostomes. But, analysis of actual immune responses suggests that the full antibody repertoire size is not (and probably never has been) accessed since it is far in excess of the ability of elements in the immune system in both time and space to exploit it. Current estimates of the human antibody repertoire size, although controversial, have been suggested to be ~1015 membersCitation3,Citation4 for the naïve repertoire, and as high as 1018 members based on theoretical combinatorial calculations. The 1015 estimate for the naïve B cell repertoire, the first group of antibody-bearing cells seen by an antigen, exceeds the total number of cells of all types in the body by 100 times, the total number of B cells in the body (~1011)Citation5 by 10,000 times and more importantly, the number of circulating peripheral naïve mature B-cells (~109 based on numbers of CD27−/IgD+ naïve B cellsCitation6) at any one time by a million times. While new, immature B cells are produced at the rate of ~109 per day, only a small percentage of long-lived B cells is retained in the periphery as viable mature B cells. The majority are removed as a result of ‘self-reactive’ depletion in the bone marrow, while those that enter the periphery but fail to successfully enter lymphoid follicles, or experience anergy by recognition of soluble self-antigens, have a much shorter half-life.Citation7 The dynamic diversity variation contributed by new cells that survive these pruning processes is unknown. The fundamental paradox then is that if the essentially astronomical 1015 naïve repertoire consisted of independent B cell clones it could never exist in the human body in physical terms, and, even if it were possible, it could never be accessed in a time frame that is immunologically meaningful.
The number of pathogenic species thought to be infectious for humans has been estimated at ~1400.Citation8 If we assign an estimate of 10 different accessible antigens per pathogen (probably an overestimate) and around 100 different epitopes on each antigen (this will vary considerably between different-sized antigens), this leads to the impossible conclusion that there are ~109 antibodies per epitope if the naïve repertoire is of the order of 1015 independent members. Furthermore, such a large excess of antibodies that could address each epitope in many different ways comes with a high potential cost of generating autoimmune antibodies. Marchalonis et al.Citation2 (p225) draw attention to this numbers dilemma, perhaps tangentially, by stating that in order to survive infection with new pathogens the organism’s immune system must have an ‘adequate’ set of VH/VL recognition units that continually arise throughout the life of the organism. The question then arises: what size of repertoire is sufficient or adequate and what is simply repertoire inflation, the excess of which should have been lost during natural selection given the enormous resource cost to a species of maintaining an inflated system? If, as Marchalonis et al.Citation2 (p230) also suggest, all vertebrate antibodies share two critical properties, namely epitope promiscuity, and poly-reactivity, then these properties should render an enormous repertoire expansion unnecessary.
StewartCitation9 and Marchalonis et al.Citation10 propose that the development of the antibody repertoire occurred independently of antigen-driven selection. Stewart’s hypothesis is that immunoglobulins (Ig) did not arise to fight infection at all and hangs on the notion that ‘variable region molecules’, what he calls the VRM system, existed as a critical structure in all vertebrates whose function was to control growth. There are a number of issues with Stewart’s arguments. First, it is not clear what ‘necessary’ function the VRM performed in its earliest form, necessary in the sense that a selective advantage for each of the ‘innovative elements’ of the VRM Stewart lists would have ensured their retention over time. The statement that the principal function of the VRM was to “contribute to the integration of the internal molecular environment” struggles to define anything biologically meaningful. In invertebrates, a VRM system may have been unnecessary where the processing of pathogens tends to occur ‘within’ cells rather than defending against them from the outside. Recognition elements, as part of an innate immune system that may have contained the ability to vary its response to diverse challenges, have been shown by Quenesberry, Vasta, and colleaguesCitation11 to have existed in tunicates where highly diverse lectin families may have allowed them to distinguish self from non-self. Such a function is best retained by maintenance of polymorphism – too few alleles and the distinction between self and non-self may disappear.
A provocative observation Quenesberry et al. make is that if the enormous diversity in the adaptive immune system present in the jawed vertebrates is so critical for defending against infection, how is it that invertebrates having only innate immunity mediated by molecules such as lectins, galectins, and others managed to survive throughout a much longer period of invertebrate history? A plausible answer is that the arrival of the first Ig variable region genes and the subsequent diversity mechanisms that rapidly followed would represent a ‘molecular Darwinism’ whereby the more diverse antibody system would gradually augment the lectin-based and other innate recognition systems and grow a more sophisticated acquired immunity function. The jawed vertebrates would not have needed to ‘invent’ the variable region genes, but merely duplicate them alongside natural variation over time since the V-domains of the subtype found in the receptors of T and B cells are known from both agnathans and cephalochordates, although as DzikCitation12 notes, in those species they do not rearrange. Present-day jawed vertebrates still maintain some lectins, but appear to have further evolved their pattern recognition capabilities in innate immunity by co-opting and optimizing other types of cell-surface located molecules, such as the Toll-like receptors (TLRs). These pattern recognition molecules are also found in invertebrates where they were first identified in the fruit fly, Drosophila (the toll gene). Toll was first classified by O’Neill et al.Citation13 as playing a role in pattern formation during Drosophila embryogenesis, but was later shown by LemaitreCitation14 to also play an anti-fungal defense role. In the sea urchin, more than 200 TLR genes have been identified, and they are also found in sponges. Intriguingly, plant resistance genes encoding proteins that plants use to recognize bacteria, fungi, and viruses also resemble TLRs. TravisCitation15 suggests that such ‘parasite sensors’ may have emerged even before plants and animals diverged.
DzikCitation12 further argues that the rearrangement mechanism of the lymphocyte V-domains suggests an origin from a common ancestral domain existing before the divergence of the extant gnathostome (sharks, rays) classes. For example, in the sponges, genes similar to those involved in the mammalian antibody response have been identified. A protein similar to mammalian lymphocyte-derived cytokine is up-regulated during non-self-recognition in Suberites. In Geodia, two classes of receptors with Ig-like domains have been identified, receptor tyrosine kinase (RTK) and the non-enzymic sponge adhesion molecules that contain two polymorphic Ig-like domains.Citation16 The expression of these molecules is also upregulated during grafting. Amino acid substitutions within the Ig-like domains in Geodia are restricted to “hot spots”. Close homologs of the RAG genes have been found in the sea urchinCitation17 and even in the early chordate, Amphioxus,Citation18 although the supporters of the ‘RAG arrived via a transposon’ theory suggest the sea urchin and jawed vertebrates may have received the passing transposon DNA independently. Homologs of activation induced deaminase (AID), a critical component of the variable gene hypermutation and rearrangement machinery, have only been found, however, in the gnathostomes.
Thus, while some elements of the antibody repertoire generating system have appeared relatively recently, other elements may have preexisted and been co-opted from innate immunity for their functional relevance. DzikCitation12 (p447) concludes that some of the molecules used in higher vertebrates for the innate and adaptive immune systems were already developed at the point of evolutionary splitting off of the sponges. The various mutation and selection cycles would then have evolved their ‘fitness’ for key roles in the antibody arm of acquired immunity.
In assessing the question of ‘adequacy’, if we accept the combinatorial calculations that generate the 1015−18 numbers, the theoretical repertoire is ‘more than adequate’ by a long stretch. This also raises the supplementary question of how natural selection can have missed pruning such an overly complex system with its potentially enormous drain on species resources, unless it serves a function we are not yet aware of, or is an unoptimized system still evolving or, as seems more plausible, represents the sum of individual, extensively overlapping, repertoires encoded within the total human population.
The human antibody repertoire ‘elements’
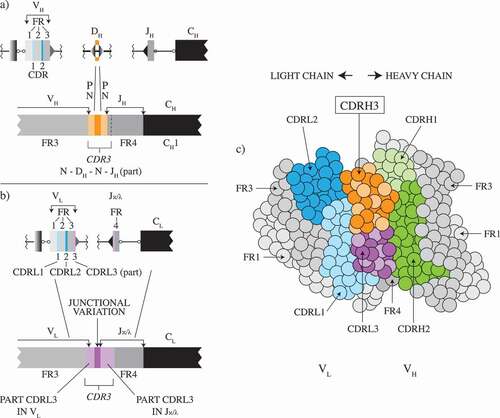
The theoretical diversity of the Ig repertoire in humans is a result of a number of different stages in the development of the B cell and of the VH and VL selection and assembly mechanisms therein. Briefly, there are 7 human VH gene families (containing 51 different VH sequences) and 16 human VL (Vκ+Vλ) gene families (containing 100 different VL sequences) that can be selected, at first sight at random. Each of the VH sequences encodes three framework regions (HFR1,2,3) and the first two complementarity-determining regions (CDRH1 and H2), while the germline VL sequences encode LFR1,2,3, the first two CDRs (CDRL1 and L2) and part of the third CDR (CDRL3). The final sequences in the VH region are assembled by the complex set of events shown in ). A similar but less complex assembly occurs to complete the VL region ()). Calculating the potential diversity available to the population of IgM-bearing peripheral B cells via the combinatorial assembly of 51 VH genes, 6 JH segments, 25 D segments for the heavy chains, and 50 Vκ genes, with 5 Jκ segments plus 50 Vλ genes, and 7 Jλ segments for the light chains, all without consideration of junctional diversity, generates a ‘core’ combinatorial diversity of 4.6 × 106 VH-VL pairs.
Figure 1. Cartoons of the combinatorial assembly process by which heavy-chain variable domains (Figure 1(a)) and light chain variable domains (Figure1(b)) are assembled in the B-cell. The relationship between the framework regions and the CDRs is shown in Figure 1(c). Figure 1(a,c) is adapted from Figure 5 in reference 4
However, on putting the DH and JH sequences together, several types of additional variation can be introduced during assembly. Nucleotides at the VH-DJ junction can be nibbled away by exonucleases and replacement nucleotides inserted (called N-nucleotide addition), thereby generating new sequences at the junction, and a similar process can occur during joining of the DH to the JH sequences. In addition, asymmetric nicking of hairpin structures at the VH-DH junction and refilling to generate blunt ends for subsequent ligation can cause sequence changes, known as P-diversity (P for palindrome), a process discovered by Tonegawa and his teamCitation19 in 1989 while studying the T-cell receptor. In some antibody sequences, exonuclease over-activity can completely obliterate the original D sequence nucleotides, or move them into a different reading frame, as shown by Darsley & ReesCitation20 in 1985, generating entirely novel sequences in the region of the heavy-chain CDR3. While such reading frame biases in the DH segment are seen in mouse heavy chains, Benechou et al.Citation21 have shown that such biases are rare in human IgM+ heavy chains (the naïve repertoire) where a single reading frame is most commonly observed, while inverted DH sequences are more or less never seen. After class switching to IgG (or IgA), B-cell biases in reading frame usage can be more pronounced accompanied by inter-individual differences. A further potential diversifying mechanism within CDRH3 involves the formation of DH-DH segments via fusion of DH regions, leading to a recombined heavy-chain VH-[DH-DH]-JH. In a study in 2012 Briney et al.Citation22 measured the frequency of such fusions in the human peripheral B cell population and obtained a figure of about 1 in 800 with a much lower frequency in memory B cells. While low frequency, the radical topographic sculpting provided by these longer lengths, not solely generated by the DH-DH mechanism, can generate useful finger-like protrusions, ideal for projecting into and binding viral surface canyons, first suggested by Webster et al.Citation23 in 1994. It is possible that such mechanisms may be focused more in the mucosal B cell repertoire with its tendency to express antibodies with longer CDR H3 sequencesCitation24 and where insult by many viral pathogens first occurs.
Two further mechanisms of diversification have been described in mouse and human tissues where the selected VH gene in a particular completed VDJ recombination can be modified. The first, described by Darlow & Stott,Citation25 was thought to occur by a mechanism similar to gene conversion, mediated by the AID enzyme. AID, undetectable in naïve resting or memory B cells,Citation26 is active during hypermutation of antibodies in germinal centers, and in the gene conversion process is proposed to mediate the transfer of homologous sequences between the rearranged VDJ gene and other VH genes. Of course, unless such interchange is restricted to functional VH regions, this mechanism could also interchange non-viable sequences from the many more VH pseudogenes present in the human genome. Examples of this form of ‘gene conversion’ in humans have been reported to occur in B cells from tonsils, synovial tissues from rheumatoid arthritis patients, and lymphomas.Citation27 (and references therein).
An additional mechanism, reviewed in a Hypothesis and Theory article by Meng et al.Citation27 in 2014, involves the replacement of the entire VH region (VHR) by a new, upstream VH gene in what Meng et al. call a VH ‘invasion’. The atypical recombination mechanism involved in this process often generates long VH CDR3s that may result in multi-specificity or even autoreactivity. Similar replacement events have been seen with light chains. Possible targets for this type of rearrangement are nonfunctional VDJ or VJ assemblies or those that require ‘receptor editing’ because of self-recognition. The frequency of such unusual modifications, their precise timing during B cell development and their overall contribution to the antibody repertoire is not yet clear.
Berek & MilsteinCitation28 estimated the diversity factor due to N- and P- junctional diversity in heavy chains to be somewhere >10 (the astronomical school put this much higher) giving a diversity of 4.6 × 107. To arrive at the 1015 number the N- and P- diversity effects, allied with other CDRH3- and CDRL3-varying mechanisms, would need to introduce a further variation factor of ~108. While such calculations are mathematically interesting, they do not take account of biological mechanisms that themselves may introduce a selection bias and reduce the effective diversity (e.g., clonal deletion in the bone marrow and peripheral lymph tissues). Khass et al.Citation29 describe a further example of bias that suggests individual DH genes are selected by evolution to preferentially generate defined categories of antigen-binding sites, likely reflecting the history of pathogen exposure to the human immune system. Once assembled, the antibody-bearing B cells at this point express the VH and VL regions in a surface-located IgM (the antigen receptor).
After a particular IgM-bearing B cell has been selected by an antigen for its ‘structural fitness’, it undergoes several changes, one of which is a switch to an IgG format. This switch, occurring in the germinal centers, also activates an internal somatic hypermutation machinery process and the spinning-off of memory B cells. The somatic hypermutations are not randomly distributed in the VH and VL regions, but are thought to be targeted to a small number of sequence motifs and are not part of the naive repertoire since they only arise after antigen selection has occurred, as discussed by Schramm & Douek.Citation30 Their role is to improve the fitness of the already selected antibodies to the antigen by climbing an affinity landscape through mutation.Citation31 The historical discovery of these various diversification mechanisms for those interested is explored elsewhere.Citation32
In referring to the astronomical (~1015) repertoire, Khass et al.Citation29 call it the ‘infinite repertoire theorem’. An example of the problem of interrogating such a vast repertoire in a reasonable time frame for the immune system to react to an infection, thus avoiding serious clinical consequences, is exposure to influenza virus. To induce a viral infection via aerosols (the most common route) Nikitin et al.Citation33 proposed that ingestion of the order of at least 3 × 103 viral particles is required. Not all ingested particles may engage in peripheral B cell contact in germinal centers since their first site of contact will likely be at mucosal barriers. But, assuming at least ~103 viral particles are involved, the B cell interrogation time can be estimated based on a productive period of each antigen-B cell interaction of one second (the approximate dissociation time for an IgM antibody-antigen complex of micromolar KD). So, 103 identical viral particles undergoing a binding event with all 109 peripheral B cells (assuming naïve to influenza infection) would take 106 seconds (~12 days) if the entire repertoire had to be screened to find a productive hit, and proportionately shorter if a hit is found more rapidly. It will be abundantly clear from even this simplistic example that an effective immune response could never be mounted if a repertoire of 1015 was required to be interrogated by an antigen in order to find a productive hit.
Recent sequencing studies by Soto et al.Citation34 of human circulating heavy chains suggested a size of about 1.1 × 107 B cell clonotypes in the naïve repertoire. In this analysis two of the three individuals whose heavy and light chains were sequenced shared between 1% and 6% of B cell heavy-chain clonotypes (0.3% shared by all three), while two of the three individuals shared 20% and 34% of λ or κ light chains, respectively, likely reflecting convergent or ‘canonical’ solutions. In another somewhat larger study, Hong et al.Citation35 analyzed the size of the expressed IgM repertoire in both neonates and healthy human adults by Illumina sequencing. Sixteen million sequences from peripheral B cells of 33 different adults (male and female) were analyzed and showed about 7 × 106 unique IgM B-cell clones. Within these clones about 9000 unique VDJ rearrangements were seen in the VH sequences, within which 3.4 × 106 unique CDRH3 sequences were found. Of particular interest was the bias of germline VH usage present in the adult repertoire, with 94.5% of VH genes arising from the VH1, VH2, VH3, and VH4 families but only 0.1% for VH5, VH6, and VH7. In the human heavy-chain repertoire 23 VH genes are in VH3, 10 in VH4 and 10 in VH1, together representing 84% of the total VH repertoire. The remaining 16% come from the VH2 (4) and VH5-7 (4) families. If VH selection is purely stochastic, Hong et al. might have expected to see an extensive spread of genes in the ratio VH3> VH4 = VH1. They did not, in line with many other observations of VH gene usage. In fact, the top VH gene frequency in the adult repertoire was from the VH4 and VH1 families, with two VH4 genes and one VH1 genes topping the table (VH4-59 > VH1-69 > VH4-54; see in Hong et al.Citation35). This suggests that calculations in which all 51 VH genes are given equal status in the combinatorial equation do not reflect the way the immune repertoire seems to operate (it is already thought that particular VH gene selection is influenced by proximity of VH families to particular JH and D segments), at least at the level of the naïve repertoire. Hong et al. also note that the extensive N-modifications at the V-D-J junctions in the adult repertoire and the impact of such junctional modifications on VH CDR3 sequences may be the “major determinants” for heavy-chain V-region diversity, a notion consistent with the views of DavisCitation36 in 2004 and Khass et al.Citation29 in 2018.
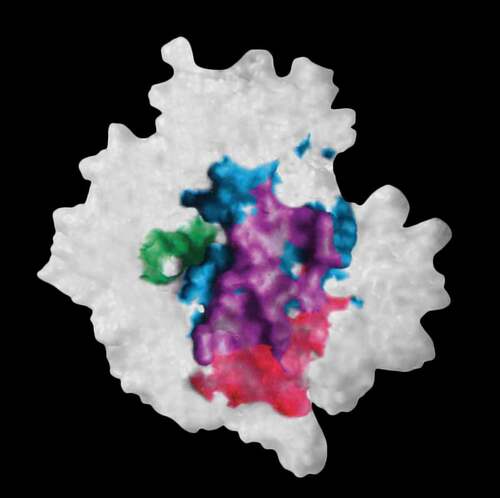
Figure 2. The molecular surface of neuraminidase showing elements of the interacting surfaces in the x-ray structures of its complex with antibody NC10 (pdb: 1NMB) and antibody NC41 (pdb: 1NCA). The epitope residues on neuraminidase common to the binding of antibodies NC10 and NC41 are shown in purple while those epitope residues only bound in NC10 are in red and for NC41 in cyan. The enzyme active site is shown in green. Reconstructed from Figure 5 in reference 58 using Adobe Illustrator
While somatic mutation can act to improve the ‘good enough’ to the ‘very good’ (increasing affinity), this does not alter the fact that the initial process of selection to find even a good enough partner is still random; finding the needle is still subject to the laws of probability. In the infinite repertoire model, there are ~108 sets of 107 IgM antibodies in the 1015 theoretical naïve repertoire. As SchroederCitation4 points out, the process of an antigen working its way through such a repertoire would also have an enormous resource (energy) cost for the organism when creating large numbers of antibodies and the cells that encode them that are unused. Other factors cautioned by Schroeder are the possibility that in a truly random process undesirable autoreactive antibodies may be generated and the lengthy time taken to find a suitable antibody would allow infections and diseases to gain hold before immune protection could occur. The infinite repertoire model seems unworkable, unless that is, the repertoire has massive redundancy such that the theoretical number of 1015 naïve antibodies encodes in reality a much smaller number of specificities, a feature that cannot be estimated solely by inspection of sequence diversity.
In a 2019 study, Briney et al.Citation3 described the sequencing of three billion heavy-chain sequences from the pooled circulating B cells from 10 individuals. The individuals were gender balanced and divided between African-American and Caucasian ethnicities. On the basis of the sequence diversity observed and the assumption that combining heavy and light chain sequences is a random process, the authors proposed that the naïve repertoire may be as large as 3 × 1015 different antibodies. They state,Citation3 (p393) however, that the dataset revealed ‘largely unique repertoires’ for each individual studied. On a practical point, the large numbers generated by high-throughput sequencing and consequent evaluation of clonotype diversity are challenging and dependent on a number of factors, such as the level of instrument sequence errors and their interpretation, as discussed recently by Vazquez-Bernat et al.Citation37 and Ohlin et al.,Citation38 and ultimately on how well the B cell repertoire members at a particular time are segregated. Hershberg and PrakCitation39 (p11) note that, when a repertoire of mutant B cells is assessed, what is actually being measured is the history of those cells that have already survived selection. The analysis by Briney et al. appears to have taken account of those post-naïve antibodies that had already undergone selection, using their hypermutation status to exclude them from the naïve repertoire calculation. Nevertheless, the repertoire estimate is based solely on ‘sequence variation’ without any consideration (understandably) of what the ‘structural repertoire’ would be, currently impossible to deduce theoretically since decoding antibody structure from sequence is barely possible. A similar calculation taking Soto et al.’s number of 1.1 × 107 heavy-chain clonotypes and adding the full diversity due to kappa and lambda light chains ((1.1 x 107 × 250) + (1.1 x 107 × 350), respectively), but without any correction for biased gene segment usage or clonal identities, leads to a maximum size of 6.6 × 109 heavy-light chain pairs, an experimentally observed repertoire close to the number of circulating B cells, but which is still around a million times smaller than the postulated 1015 number.
Is an ‘infinite’ immune repertoire required?
There seem to be two possible questions or assumptions the above issues raise. First, is the antibody repertoire in reality as large as calculations suggest and what is the experimental evidence, based on diversity of ‘structure-based specificity’ and not just ‘sequence diversity’? Second, if the repertoire is not that large, what is the lower limit of variation and how is the selection of effective (‘fit’) partner antibodies by antigens achieved in a timely and resource efficient manner? Perelsen and OsterCitation40 and Perelson and WeisbuchCitation41 approach this last question via mathematical modeling by looking at antibody–antigen interactions in terms of the ‘shape space’ of epitopes (or antigenic determinants) and the probability of a repertoire of antibodies covering the total epitope shape space. A further important aspect built into these calculations is the proportion of the theoretical repertoire that consists of antibodies reactive against self-antigens, eliminated during the processes of deletion. As De Boer & PerelsonCitation42 point out, not all self-antigens would be present during this deletion process, and the suggestion that the size of the mature repertoire is largely determined by the presence of anti-self antibodies is perplexing and raises the question of how the immune system avoids catastrophic auto-immune responses for those many self-antigens that are not accessed during deletion. In addition, loss of those anti-self antibodies in the repertoire would potentially create specificity holes that could be (and likely are) exploited by pathogens. This model of the B-cell repertoire using the notion of shape space requires that antibodies are multi-specific, that is they can bind a series of structurally related epitopes. Structural redundancy (widely reported and often referred to as poly- or oligo-reactivity) is likely to be more prevalent in antibodies triggered early in the response (e.g. IgMs) and would argue for a ‘diversity reduction factor’ that should be introduced into the combinatorial calculation.
However, it is the naïve repertoire that first meets antigen and, barring a previous encounter where memory B-cells may be present, it is this naïve repertoire’s fitness that will determine success or otherwise in eliminating the pathogen. The important basis of the Perelson model is that because each antibody can recognize all epitopes within its shape space, a finite number of antibodies can recognize an infinite number of antigens. But what is that finite number? The model estimates this by asking the question: What is the minimum number of different antibodies that will ensure an epitope will not be recognized? It turns out that if the repertoire size is ~107 different antibodies – ‘different’ here means different in specificity with an implied structure difference, not simply sequence difference – Perelson’sCitation43 model predicts that essentially all epitopes will be recognized since the probability that an epitope escapes detection is infinitely small. This is predicated of course on the assumptions that each of the 107 antibodies is different in terms of its antigen recognition surface, and that there are no significant holes in the repertoire allowing some antigens to fall through, issues that are almost impossible to resolve, theoretically or experimentally. PerelsonCitation40 notes that the expressed repertoire in mice for each of B cells and T cells is about 107, in accord with the Hong et al.Citation35 experimentally observed numbers for expressed antibodies (1.1x107), although 100x lower than the total number of circulating human B cells (109), and of the Soto et al.Citation34 extrapolated number of 6.6 × 109 antibodies. Allowing for clonal identity and sequence-based structural redundancy in the circulating repertoire, this would put the model predictions in a good place and is not inconsistent with the 1400 × 10 x 100 pathogen/antigen/epitope notion calculated earlier.
Further support for Perelsen’s ‘limited repertoire is sufficient’ model comes from the behavior of phage libraries. Early phage libraries generated antibodies with affinities (micromolar and sub-micromolar range) similar to those seen for IgM and early response IgG antibodies that would have derived from a typical naïve repertoire. In 1991 Marks et al.Citation44 prepared a phage library from peripheral human B cells with a diversity of >107 members. On screening and analysis of the library, Marks suggested that such a library could recognize any antigen, acknowledging the limited affinities obtainable. Similar results were obtained by Griffiths et al.Citation45 and others. It seems likely that, as phage libraries with greater variation in the CDRs were prepared (e.g. HuCalCitation46 libraries), they would have mimicked in their sequence space a mix of naïve-type antibodies and hypermutated antibodies, as already suggested by Griffiths et al.Citation45 (p3254)
So, how do we explain the frequently claimed proposition that the potential repertoire size is actually eight or so orders of magnitude greater than appears to be required? In this article, I maintain the evidence suggests that the theoretical antibody repertoire must be heavily redundant, and further, is in fact a ‘population repertoire’, where N individuals have different but significantly overlapping fractions, M1-n, of the available repertoire, M1-n x N. This suggests that the ‘infinite’ repertoire is not in fact infinitely large, but is in reality the sum of a large population of heavily redundant repertoires, selected and matured over millions of years and differing only where individuals have different historical pathogen experiences. Further, each individual M is able to cover Perelsen’s antigen space because of this vast redundancy, a notion not inconsistent with the observations of many of the studies cited here. The implication of this, if correct, is that the ‘specificity’ repertoire is much smaller than the repertoire calculated solely on the basis of sequence variation. One way to validate this notion is to look at the experimentally observed antibody repertoire ‘use’.
The observed diversity of antibody-antigen responses
Some of the earliest studies of antibody responses to defined antigens were carried out by Klaus RajewskyCitation47 when he was Director of the Institute for Genetics in Cologne and by Cesar Milstein at the Medical Research Council in Cambridge. Rajewsky’s analyses suggested that responses to specific antigens were restricted. As he observed:
There is evidence that certain specificities in the pre-immune repertoire are selectively expanded … This would be an efficient way to amplify the expression of selected sets of antigen binding sites whose expression is useful on the basis of … experience.Citation47 (p1092)
These are interesting comments. The words ‘useful’ and ‘experience’ conjure up a mechanistic notion that is the antithesis of a stochastic process and resonates closely with recent observations of convergence in antibody CDRH3 sequences and structural motifs for recognition of specific antigens and their epitopes, an area discussed below. It is also educational to consider some of the conclusions of Berek & MilsteinCitation28 in 1988 based on their own and others’ empirical data, summarized below:
The best available naïve repertoire (estimated at that time to be 109) has developed via natural selection while the best memory repertoire arises from antigen-driven selection;
While the memory repertoire may potentially be large, it is in fact heavily restricted since it is selected by antigen experience;
Memory cells will play a critical role in the repertoire of antigen-specific cells, but will have a negligible role in defining the total diversity of the available repertoire;
The pool of available specificities in the naïve repertoire is only a fraction of the potential repertoire and the memory pool a fraction of the available repertoire;
Somatic mutation does not contribute to the diversity of the naïve repertoire.
While these ideas are now more than 30 years old, which by the way doesn’t make them redundant, they were remarkably prescient. Around the same time, Blier & BothwellCitation48 extended the observations of Milstein, Rajewsky, and others in a study of B cell lineages that characterized specific secondary responses in the mouse. In that study, a single VH gene was present in 24 of 28 antibodies, with two similar V-lambda genes used in 25 of 28 antibodies. Bye et al.Citation49 showed in 1992 that, of 14 murine antibodies generated against the Rhesus (D) alloantigen in seven different donor mice, only two VH genes were used in the secondary response while 11 of the 14 selectively used the JH6 segment. The IgM response to this antigen was even more restricted. Ikematsu et al.Citation50 in 1993 observed a similar restricted response in anti-rabies antibodies. Of nine human anti-rabies antibodies, generated from B cells of human donors that had received the rabies vaccine, seven used the VHIII heavy-chain family. The results from these studies are consistent with Berek & Milstein’s notion that the antibody repertoire is constrained to the naïve repertoire available at the point of antigen arrival and that any expansion or diversification during the secondary response is severely restricted.
On the question of the fitness of the VH repertoire, Suarez et al.Citation51 in 2006 observed a remarkably constrained use of only a single VH gene in a transgenic mouse that contained five different human VH germ line genes. The mouse antigen response generated 30 different antibodies to different soluble proteins and whole cells by co-opting only the VH1-2 gene, along with varied D and JH segments, suggesting a possible selection process driven by the preferred complicity of CDRH3 sequences.
In 2008 Volpe & KeplerCitation52 mapped the frequency of human VH gene usage in 6500 productive heavy-chain rearrangements and more than 300 nonproductive rearrangements. Several important results of this analysis emerged. First, the frequency of the seven VH gene families found in productive rearrangement was heavily biased toward VH3 (~46%) and VH4 (~24%), with VH1 coming in third at about 18%. JH gene segment usage and D segment usage were also heavily biased, with J4 and J6, and D2 and D4 dominating the productively rearranged genes. Further, Volpe and Kepler found a statistically significant preference for certain D-J pairings, suggesting a gene proximity origin. These analyses, which were much larger than similar earlier analyses by Brezinschek et al.Citation53 but with similar conclusions, lend considerable weight to the idea that the actual size of the antibody repertoire is, in practical terms, much lower than its theoretical size, ignoring, as that calculation typically does, biases in frequencies that arise from recombination mechanisms influenced by gene segment adjacency and perhaps other recombination factors. Mark DavisCitation36 (pp239, 241) noted in 2004 that wide variations in V region vertebrate repertoires are likely to be of lesser importance than the preservation of one or more diverse CDR3 regions. He further noted that it is the diversity of heavy-chain CDR3s that drive specificity, whereas CDRH1 and CDRH2 residues are broadly cross-reactive and subject to improvement by somatic hypermutation. In a more recent review, while acknowledging the results of the 2019 study by Briney et al., Davis & BoydCitation54 (p111) comment:
It is currently unclear how diverse the Ig repertoire needs to be to provide competent humoral immunity over a human lifespan.
In 2009 Glanville et al.Citation55 isolated antibodies from the B cells of 654 human donors. The isolated cells included naïve, memory, plasma, and pre-immune cells containing somatically altered paratopes. Their conclusion was that the total diversity of the multi–individual repertoires was 3.5 × 1010. While the objective of this study was to understand how to construct an ‘adequate’ diversity into synthetic libraries, there are a number of cautionary comments that should be made. First, the VH diversity of the library was rather limited (~105) and the repertoire size was calculated by assuming random heavy chain-light chain combinations since the sequences determined were not from paired antibody chains. Second, 78% of the antibody chain sequences had between 1 and 6 amino acid mutations within the V-region encoded CDRs, reflecting somatic hypermutation (SHM) seen only after antigen stimulation, isotype switching, and movement to germinal centers where affinity maturation and spinning off of memory cells occurs (SHM in IgM B cells can occur but is rare). It is therefore likely that the diversity seen by these authors was from a combination of naïve IgM cells and B cells that were already selected, the latter the memory repertoire of Berek & Milstein. Given the immune history of the 654 individuals, the memory repertoire must have been substantial both in size and diversity. Separation of the naïve repertoire from the memory repertoire is clearly an essential requirement for establishing the real diversity of the naïve repertoire, but is a non-trivial exercise, as indicated by Lees & Shepherd.Citation56
Structural convergence
A consequence of the ‘convergence’ model of antibody fitness is that paratope structures that recognize the same or substantially overlapping epitopes can arise either by using the same or closely similar CDRH3 sequences (sequence convergence), or from using different VH, D, and JH sequences that are capable of producing the structural paratope (structural convergence) necessary to recognize the same epitope(s). In 2015 Dunand & WilsonCitation57 argued that for some antigens there exist epitopes that are ‘so unique’ (sic) there are only so many paratope structures, and hence sequences that are capable of recognizing them, calling such behavior ‘stereotyped convergence’ where individuals from diverse genetic backgrounds use highly convergent B cell receptors. Others have observed sequence (CDRH3) similarity, or convergence, but dissimilar sequences can also generate paratope complementarity. In 1994 Malby et al.Citation58 in an x-ray crystallography study described two different antibodies (NC10 and NC41) with significant CDR sequence differences that recognized ~80% of the same epitope on the surface of the influenza neuraminidase antigen. The NC10 CDRH3 was 15 residues while NC41 was 13 residues. Only four residues (all at the C-terminus of the CDR) were common between the two CDR sequences. The molecular surface of the neuraminidase monomer is shown in in which the area common to the two different antibodies is shown in purple. The two sets of CDRs produce a common complementary structure to the epitope but using different sequences. In a somewhat premonitory statement that anticipated many of the current convergence notions, Malby et al.’s parting sentence in the conclusion stated:
A fundamental principle of protein structure is that unrelated amino acid sequences may give rise to similar polypeptide folds. It is now emerging that two chemically unrelated binding sites may bind a common structure on a third protein. This is facilitated in part by the capacity of proteins to modulate their shape to achieve the topographic complementarity necessary for protein–protein interactions.Citation58 (p744)
In a later 2005 study, De Genst et al.Citation59 described an example of sequence-based convergence. They isolated two VHH antibodies from dromedaries that had been immunized with hen egg-white lysozyme (HEWL) in different locations (Morocco and Dubai) and at widely separated times (1993 and 1997, respectively). While the VH and JH regions differed between the two antibodies, the D regions were identical in sequence and structure, with both antibodies recognizing identical epitopes. Further, the two paratopes were highly complementary to their epitopes with surface correlation values (a measure of the structural complementarity between two surfaces) of 0.79 and 0.71 for the two antibodies, values close to those of many highly evolved oligomeric protein interfaces.Citation60 In addition, while CDRH1 differed between the two antibodies, both antibodies had accumulated many mutations that converged on identity in several CDRH2 positions. This cavity-protrusion feature is shown in . De Genst et al. explain the origin of the sequence convergence as a result of hypermutation:
Figure 3. The epitopes (top) and paratopes (bottom) from the crystal structures of the two VHH-HEWL (hen-egg lysozyme) complexes, D2-L19, and cAb.Lys2. The contacting regions (within 5 Å of the other protein in the complex) are colored blue. The amino acid Thr47 in the light chain is labeled and colored red and its binding pocket within the paratope is indicated with an arrow. Reproduced from ) in reference 59 with permission from CCC Market Place. The original figure was kindly supplied by the authors
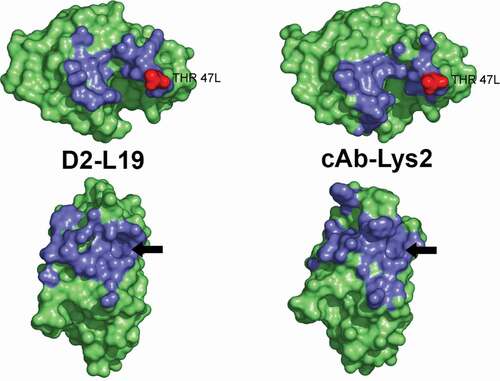
Y59A mutation occurring in both VHHs in particular plays a pivotal role for HEWL interaction because it creates a cavity between the H2 and H3 loops. The Thr-47 of HEWL (Thr-47 L) fits into this cavity and is surrounded by residues occluding solvent from this site … .Citation59 (p14117)
This example provides further support for the notion that structural, or topographic complementarity among antibodies that bind to identical or substantially similar epitopes is the driver of antibody specificity. While the sequence is important, these two studies demonstrate that structural convergence can be obtained both by closely similar and by significantly different CDR sequences. In relating these observations to the repertoire question, and essentially questioning the notion that repertoire diversity can be deduced solely be reference to sequence differences, De Genst et al. also suggest that on a structural level, acquired immunity may rely more on a well-developed somatic hypermutation machinery than on the acquisition of large primary repertoires. The generation of ‘fit’ antibodies may then begin by selecting candidate primary hits from a conservative naïve repertoire, followed by a secondary drive toward high specificity convergent solutions.
The role of V-regions and CDR3 in antigen receptor diversity
Given the memory B cell antibodies are antigen selected and optimized by somatic hypermutation that is focused mainly on the CDRH3 region, a reasonable question would be: Is the selection of specific VH germline genes and the CDRs encoded therein a critical requirement for the development of particular antigen specificities? In 2000 Xu & DavisCitation61 addressed a related question, which essentially asked: Is the CDR3 region of VH domains sufficient for most antibody specificities? Their model was a transgenic mouse with a single VH gene (VH5-51, rarely used in adult human responses) but containing 10 functional D segments, six JH segments, Cμ, and Cγ1 constant regions. The results were surprising, at least at the time. After immunizing the mice with four different protein antigens and three different haptens conjugated to protein carriers, specific IgM antibodies were obtained where, apart from one antibody, the only sequence differences seen were in the CDRH3 sequences. On further hyperimmunization, IgG antibodies were obtained containing somatic hypermutations and, for one of the antigens, exhibiting an extremely high IgG affinity (KD ~25pM). The extension of the observations in this study to other normal immune responses by these authors was that restricted V gene usage is a result of antigen-driven clonal expansion of kinetically favorable antigen receptor clones. Their conclusions also support the notion that it is the highly diverse CDRH3 regions that are dominant in encoding antigen specificity. In 2004 DavisCitation36 (p241) further developed his CDRH3 hypothesis on antibody diversity concluding that:
… the VH and VL repertoires do not make any unique contribution to specificity, but that virtually any combination can be molded by somatic mutation into a high affinity binding site provided that the ‘right’ VH CDR3 is providing the core specificity.
But, the problem with parsimonious models is that their extrapolation may not predict the full reality of an operational, in vivo repertoire at work. The ideal dataset would come from a detailed retrospective analysis of a large population of human individuals who had been subjected to a multi-antigen onslaught, such as with a multivalent vaccine. Fortunately, a number of such analyses has been made, the results from which begin to explain how the antibody repertoire works.
The central position of CDRH3 in the antibody combining site, allied with its enormous variation in length and sequences, argues for a critical role in antigen recognition. While the suggestion that CDRH3 is the focus for antigen selection, the shape of the combining site is determined by the juxtaposition of several CDRs from both the heavy and the light chain, giving rise to varied topographies, analyzed by Webster et al.Citation23 The logical construction of a naïve repertoire that provides variation for small, medium, and large antigens is then that natural selection should have ‘fixed’ members of the different topographic classes in relation to the frequency of experience by the immune system from the different molecular types of antigen assault. If this variation is represented in the naïve IgM repertoire, then fast selection of low affinity but ‘specific enough’ B cells that then enter an optimization pathway can follow. For a given class of antigen (e.g., the topographically flat surface of a protein) it is likely required that a particular combination of CDR lengths that generate such a flat paratope will be present. This may then be the first point of CDR convergence. Such a correlation was first analyzed in some detail by Vargas-Madrazo et al.Citation62 in 1995 on the basis of preferred CDR canonical classes where the class combinations for recognition of protein antigens were markedly different to those for small antigens. An intermediate ‘multi-specific’ shape class appeared to be related to CDRH3 conformation. A year later, MacCallum et al.Citation63 extended this notion and identified similar variations in antigen-dependent paratope topographies, classifying them as concave, ridged (or grooved) and planar. In a later study Zemlin et al.Citation64 compared the human and mouse repertoires with respect to CDRH3 length and amino acid composition. Their conclusions resonate with the above notion that within the naïve repertoire there are distinct paratope shape classes:
Our results suggest, however, that specific antibodies may be selected from very distinct overall repertoires … we propose that the … repertoires … differ in the array of antigen specificities and affinities, but during the response to a given antigen they obey similar rules of selection … and must therefore reflect convergent selection.Citation64 (p745)
While each B cell that selects a particular VH germline gene, where CDRH1 and CDRH2 sequences are fixed for that B cell in the naïve repertoire, other B cells using the same VH gene can have widely differing CDRH3s. If antigen recognition is largely driven by CDRH3 sequence and structure, as postulated by Khass et al.Citation29 and others, then analysis of human responses to specific antigens, such as in vaccines or after parasite infection, should provide evidence of convergence of these sequences and structures. Such memory convergence, if observed, would best be analyzed by assessing the time-dependent repertoire usage since secondary responses will contain antibodies somatically hypermutated. Such analyses have been done.
In a study by Parameswaran et al.Citation65 in 2013, convergent human antibody responses to dengue virus infection were seen, while in 2014 Jackson et al.Citation66 made the observation that convergent antibody sequence and structure may be a common feature for most if not all pathogens, a view supported by their studies on responses to influenza vaccines. The existence of a ‘public antibody repertoire’ in different individuals characterized by convergent responses to multi-antigen vaccines was also observed by Trück et al.Citation67 in 2015. In 2016 Joyce et al.Citation68 and Kallewaard et al.Citation69 analyzed the human responses to influenza vaccines and the antibody repertoire usage in different individuals. In their ‘Leading Edge Previews’ commentary on these results, Jackson and BoydCitation70 make some important observations. They note that by identifying convergent ‘classes’ of influenza-neutralizing antibodies, as in the Joyce study, diverse antibody-mediated responses of different humans can converge on similar solutions to complex targeting. They go further, stating that shared antibody structural motifs for binding viral epitopes are formed in different individuals, despite the vast diversity of antibody repertoires.Citation70 (p532)
The above examples suggest a somewhat different picture of the human antibody repertoire to that commonly expounded. In an evolving immune system that includes exposure to antigens of many different types, it seems logical that the ‘classes’ of antibody paratopes in the naïve repertoire would have been retained by natural selection on the basis of their ‘fitness’ for particular antigen topographies and size, and that the repertoire numbers in each class would reflect the frequency of assault on the immune system and the variation in shape, size, and chemistry. The mechanisms that have evolved to shoe-horn CDRH3 sequences and lengths into preferred sets rather than allowing a totally random selection, a process that would be deleterious to a naïve repertoire that can present only a few million antibodies to invading antigens, is currently unknown, but it must exist. This preference is consistent with analyses such as that of Vargas-Madrazo et al.Citation62 where, despite some 300 possible combinations of VH and VL CDR canonical structures, 87% of 381 antibody sequences analyzed used only 10 combinations.
In a more recent study, Mason et al.Citation71 constructed a library of rationally designed mutations in CDRH3 of trastuzumab, which targets human epidermal growth factor receptor 2 (HER2), and then screened for retention of binding to the HER2 receptor. Using the experimental library binding results as a training set in a neural network, a large in silico library (108 variants) was constructed from which around 106 members, with CDRH3 sequences that were intelligently varied at different positions in the CDR while maintaining only small variations in length, were predicted to be binding antibodies. From this set, 30 randomly selected variants were tested and found to bind the HER2 antigen with highly respectable affinities. While it became difficult to derive either structural or sequence-based ‘rules’ that explained the common binding specificities, this study demonstrated that many different CDRH3 sequences were capable of binding the same antigen, while clearly offering similar three-dimensional structures necessary for retaining specificity for HER2. What is apparent from such a study is that merely reciting the fact that antibodies have different sequences and that they must therefore have a different specificity contribution to the repertoire requires an understanding of how to translate sequence information into structure and specificity. Despite the application of highly sophisticated neural network methods using reliable experimental data to train the networks, Mason et al. concluded that specificity differences cannot reliably be explained (yet) at the structural level based simply on inspection of amino acid sequences.
Two further analyses have brought additional ideas that add to the body of knowledge on the determinants of antibody specificity. In 2018 D’Angelo et al.Citation72 observed that many different combinatorial rearrangements can give rise to the same heavy-chain CDR3 sequence, and that when that sequence is in the context of a specific VH-DH-JH recombination, target specificity is achieved. However, if that same CDRH3 sequence is found in the context of VH-DH recombination involving different VH genes or D segments, the target specificity is absent. The authors conclude that CDRH3 is necessary but insufficient for recognition of a specific antigenic determinant or epitope. Indeed, if CDRH3 were the sole arbiter of antibody specificity, it would be a highly convergent view of immune recognition, even allowing for the fact that the Camelidae have managed to discard the need for a light chain in their repertoire of VHH antibodies.
An explanation of how paratope specificity is encoded may be beginning to emerge from analyses such as the recent work of Akbar et al.,Citation73 whose mammoth study on 825 antibody-protein antigen complexes from 84 different antigen classes attempted to define shared paratope ‘motifs’ based on interaction surfaces involving different CDR and framework residues. The motifs derived from the data set did not cluster by antigen class but were shared across antigen classes. Further, the antigen and antibody sequences (and antibody germline V-genes) differed substantially across the different complexes. This analysis resulted in fewer than 104 interaction motifs, which the authors propose represents a substantial (50%) portion of the global paratope interaction space. This ‘structural’ space would then be more than 10 orders of magnitude smaller than the postulated global antibody sequence space. While the estimate of 104 motifs is surprisingly low, the concept aligns with Perelsen’s notion that a limited number of paratope structural motifs that is also many orders of magnitude lower than the theoretical number of sequences, can provide an interaction surface repertoire for all antigens. It also reinforces the view of D’Angelo et al.Citation72 that the contribution of a number of different CDRs (and perhaps indirectly, framework sequences) to antigen specificity is essential, notwithstanding the likelihood that CDRH3 is a critical contributor.
As a further parallel of what appears to be a ‘motif convergence’ process for antibody repertoires that may have been heavily influenced by historical assaults on the adaptive immune system, it is interesting to note that a similar selection pressure may have been operating to focus the CD4+ T-cell receptor ‘ligand’ repertoire, as suggested by Bradley and Thomas:Citation74 (p558)
… a pattern has clearly emerged: Within an epitope-specific repertoire, a portion of the responding receptors cluster closely together based on shared motifs … One is tempted to hypothesize that the presence of these clustered groups corresponds to an evolutionary focusing on particular epitopes … .
A physical model of convergent recognition supports a ‘small repertoire per epitope’
The notion of Perelson’s epitope ‘shape space’, while a stand-alone mathematically convincing model, can be examined by a practical approach that may mimic the way in which antibody-bearing B cells and antigens carry out their interaction pas de deux before converging on a subset of antibodies for further maturation. In an approach to calculate an epitope-focused antibody repertoire, a well-characterized surface region from a protein whose x-ray structure is known (human PCSK9Citation75), and to which many different antibodies have been raised,Citation76-Citation78 is taken as an example. Twelve non-contiguous amino acids (reflecting a non-linear epitope) from the surface region are examined where the amino acids are essentially in fixed positions relative to one another on the protein surface, as in most other protein epitopes. For the purposes of this purely topographic analysis, any conformational effects that may exist where residues and their neighbors may undergo small movements over time are ignored. In addition, no assumptions are made about the nature of the amino acids, other than their approximate exposed surface areas. Likewise, the capability of any of the amino acid ‘groups’ examined in mediating a thermodynamically viable binding event is not taken into account. However, and for information, in the analysis of 107 non-similar antibody-protein complexes by Kringelum et al.Citation79 (pp9,10,13) (1) none of the epitopes examined were linear epitopes; (2) epitopes were typically described as a flat oblong shape with an area of about 400Å2; and (3) the most common property of epitopes was a hydrophobic core surrounded by hydrophilic or charged perimeter (as in the dry core, wet rim model).
If the antibody paratope repertoire for such a region interacts with epitopes consisting of all possible combinations of residues 1–12, from single residue epitopes to epitopes involving all 12 residues; then, the theoretical repertoire, N, for such an epitope ensemble would be the sum of all combinations:
However, in order to arrive at the theoretical N requires all possible combinations of the 12 amino acids to be bound independently by an antibody. It is clear that not all combinations within N are available since amino acids on the surface of a protein are not free-floating ‘spheres’ that can be accessed in all discreet combinations without interference from neighboring amino acids.
shows a region of 12 amino acids on the surface of the protein, each amino acid labeled A, B, C … L. The circular and oval overlays approximate the areas of the exposed amino acid atoms.
Figure 4. A surface epitope region (red) consisting of 12 different amino acids selected from the exposed surface of the protein, PCSK9 (PDB: 2PMW). An outline of each residue side-chain is indicated by a yellow circle or oval. The epitope region is used to calculate the combinations of each unique residue set from 1 to 12 residues that could be addressed by different antibodies. The surface region is redrawn from the PDB structure (2PMW; reference 75) using Adobe Illustrator See text for explanation
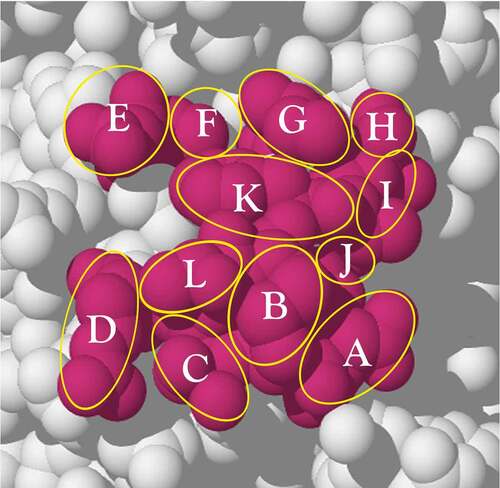
The combinatorial sum of the combinations 1, 2, 3 … 12 at a time provides an upper bound for the total number of possible combinations, but, as indicated above, the physical layout severely limits N. With no physical constraints the value of N is given by:
As a general principle for a physically valid combinatorial calculation, it is clear that for each of the defined epitope sets the amino acids within a set have either to be contiguous in order to form an interaction without interacting with others (e.g., for the 2 at a time set, I and J but not I and D), or they have no other amino acids between them, such as D and E. In addition, at least one of them will have to be on the boundary of the region (so not B alone).
In the estimates performed, since the options are constrained by the physical and topographical structure of the region, a systematic counting procedure is employed. For example, for the regions taken 3 at a time we obviously cannot have non-contiguous combinations such as AGL.
The counting procedure first involves all combinations with A in, then all without A but with B in, etc. Hence, at each stage, for example, once those with A are completed then A will not occur in any further combinations. The details of the counting procedure are shown in Appendix A. The sum of all combinations using this counting procedure is then:
This number could be considered a lower theoretical estimate for the number of antibodies with different sets of epitope residues bound, lower since it would be incorrect to gainsay all possible ways in which an antibody could address the same epitope with small variations to the sets used. It is also possible that multiple antibodies with different germline origins and different sequences could bind to the exact same sets of residues. However, based on the fact that not all sub-regions of the 12-residue set will contain an epitope profile that provides a sufficient-free energy change for productive binding (an epitope not containing a sufficiently hydrophobic core, for example), and the restricted repertoire usage observed experimentally discussed earlier, it is reasonable to expect that the upper limit ‘in practice’ will not be substantially different from this estimate and may even be lower. Taking the earlier calculation of 1400 pathogens, 10 antigens, and 100 epitopes per antigen and allowing for the existence of epitopes both larger and smaller than in our what might be called the ‘average example’, we arrive at a required repertoire size of 2.4 × 108 (171 x 1.4 × 106) different antibodies theoretically capable of addressing similarly sized epitopes across all antigens. This number sits within the range encompassed by the number of circulating IgM-bearing human B cells, ~3x109, but without any correction for specificity redundancy and clonal identities, and the postulated repertoire size from Perelsen’s calculations and other experimental observations (~107). While this topographic calculation is simply a mathematical model, it does suggest that the ‘adequate’ naive repertoire size is likely to be many, many orders of magnitude lower than the postulated number of 1015.
Conclusions
The question of how to make use of an immense sequence-based antibody diversity that is beyond the physical capacity of any individual’s immune system to access is both controversial, currently unresolved and perhaps unresolvable. As discussed, some combinatorial calculations put the naive repertoire size at >1015 members – the ‘infinite repertoire’. The functional antibody repertoire is not a single ‘entity’, but comprises a ‘naïve, IgM’ element and ‘experience-rich’ elements within the IgG bearing memory B cell population. Other theoretical estimates, based on sound mathematical ‘shape space’ considerations, propose that a naïve repertoire of about 107 different antibody ‘specificities’ (not simply sequences) may be sufficient to recognize all possible antigens. Experimental repertoire analyses largely support the shape space estimates within an order of magnitude or so. Experimental studies that have measured the clonal diversity of antibody responses to specific antigens also allow the conclusion that a naïve repertoire of around 107 B-cells may be adequate for the generation of a response to all new antigens, while the memory B cell repertoire takes care of responses to previously encountered antigens. Important contributions to the argument have also been made by transgenic animal studies in which even smaller repertoires, generated using a limited number of VH-genes coupled with the normal DH and JH segments, have been shown to be ‘adequate’. Rather than engaging in fruitless calculations on what possible number of antibodies the adaptive immune system can generate, it may be equally appropriate to reflect on the teleological conundrum that an infinite repertoire would have to have evolved for a purpose not yet required, and more importantly, that is impossible to make use of.
The observed convergence of heavy-chain CDRH3 sequences, and with it paratope structural motifs observed in many studies, supports the ‘limited but adequate’ repertoire theorem while the physical limits of how many antibodies can address a particular epitope with the likelihood of forming a viable antibody-antigen pairing in the time scale of a typical immune response, exemplified by the analysis of a 12-residue epitope with all its assumptions, suggests that the plasticity of a limited repertoire augmented by somatic hypermutation provides an adaptive immune repertoire that is fit for purpose.
A clue to the repertoire anomaly may have arisen from the study by Briney et al.Citation3 where the B cell repertoires of the 10 persons appeared to be different at the sequence level while at the same time showing the convergence of specificity. This behavior suggests that perhaps we should be describing the antibody repertoire as a ‘specificity’ repertoire whose variation is based on structural rather than sequence differences. This leads to the idea that what we could call the ‘master repertoire’ is in reality a collection in the human population of highly converged individual repertoires. If so, in the absence of any understanding of the sequence and structural redundancy among these individual repertoires whose convergence has been shaped over time by common and different antigen challenges, the frequently espoused ‘infinite’ combinatorial calculation based on sequence differences is almost certainly incorrect.
If a structure-based repertoire is the reality, acquired immunity must show some necessary elements. Many analyses of responses to particular pathogen epitopes in different individuals do show evidence of convergent antibody signatures, particularly in the sequences of the heavy-chain CDR3 that should reflect paratope ‘structural convergence’. If many different V-gene, J and D sequence combinations can produce paratopes that have the necessary ‘structural fitness’, then in reality the master sequence repertoire must be heavily ‘sequence redundant’. In that case, many combinations of CDR sequences derived from different gene combinations must be able to generate a structural paratope capable of binding a particular pathogen epitope sufficiently well to trigger the full armored response of the adaptive immune system. If this is true, the ‘meaningful repertoire’ is not inaccessibly large, which it is good news for the human population, individual repertoires within which, while exhibiting sequence differences, will contain extensively overlapping structural similarities as specificity determinants. The sequence redundancy means no individual has to explore an impossibly large sequence repertoire to be sure of finding an antibody solution to an existing or new pathogen, but can find a solution within a much more accessible structure repertoire, save for those instances where individual specificity ‘holes’ exist for genetic or developmental reasons. The key for improved therapeutic vaccine strategies may then be to identify those converged motifs that may be particularly effective for certain pathogen responses, while also cataloging the holes that some individuals may have in their historically biased anti-pathogen repertoires. Such valuable profiling could be carried out by a combination of massive comparative sequencing efforts of the sort Briney et al.,Citation3 Hong et al.,Citation35 Soto et al.,Citation34 and others have described, but on much larger numbers of individuals, followed by the much more difficult process of determining the ‘specificity motif’ redundancy within such a massive sequence space, an important start to which has been made by Krawczyk et al.Citation80
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
I would like to thank Professor Michael J. Thomas, of Auckland University Mathematics Department for help with the combinatorial epitope analysis, Professor Alan Perelsen for reviewing parts of the manuscript, Professor Gunilla Karlsson-Hedestam at the Karolinska Institute, Stockholm for valuable discussions and suggestions for improving the manuscript and Professor Laurence Hurst of the University of Bath for helpful insights (and corrections!) on the evolutionary aspects. I also thank Branddeli Ltd (UK) for help with the illustrations and gratefully acknowledge free access to the University of Bath on-line library. Finally, I thank Dr. Erwin De Genst for his kind provision of the image shown in Figure 3. I declare I have no financial interest in the publication of this paper.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- Flajnik FM, Du Pasquier L. Evolution of the immune system. In: Paul WE, editor. Fundamental immunology. 7th ed. Lippincot (PA): Williams & Wilkins; 2013. p. 67–16.
- Marchalonis JJ, Adelman MK, Schluter SF, Ramsland PA. The antibody repertoire in evolution: chance, selection, and continuity. Dev Comp Immunol. 2006;30:223–47. doi:https://doi.org/10.1016/j.dci.2005.06.011.
- Briney B, Inderbitzin A, Joyce C, Burton DR. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature. 2019;566:393–98. doi:https://doi.org/10.1038/s41586-019-0879-y.
- Schroeder HW Jr. Similarity and divergence in the development and expression of the mouse and human antibody repertoires. Dev Comp Immunol. 2006;30:119–35. doi:https://doi.org/10.1016/j.dci.2005.06.006.
- Boyd S, Joshi SA. High-throughput DNA sequencing analysis of antibody repertoires. Microbiol Spectr. 2014;2(5):AID-0017-2014. doi:https://doi.org/10.1128/microbiolspec.AID-0017-2014.
- Morbach H, Eichhorn EM, Liese JG, Girschick HJ. Reference values for B cell subpopulations from infancy to adulthood. Clin Exp Immunol. 2010;162:271–79. doi:https://doi.org/10.1111/cei.2010.162.issue-2.
- Paul WE, Ed. Fundamental Immunology. 7th ed. Lippincott (PA): Williams & Wilkins; 2013. p. Ch.8.
- Editorial. Nat Rev Microbiol. 2011;9:628. doi:https://doi.org/10.1038/nrmicro2644.
- Stewart J. Immunoglobulins did not arise in evolution to fight infection. Immunol Today. 1992;13:396–99. doi:https://doi.org/10.1016/0167-5699(92)90088-O.
- Marchalonis JJ, Kaveri S, Lacroix-Desmazes S, Kazatchkine MD. Natural recognition repertoire and the evolutionary emergence of the combinatorial immune system. Faseb J. 2002;16:842–48. doi:https://doi.org/10.1096/fj.01-0953hyp.
- Quenesberry MS, Ahmed H, Elola MT, O’Leary N, Vasta GR. Diverse lectin repertoires in tunicates mediate broad recognition and effector innate immune responses. Integr Comp Biol. 2003;43:323–30. doi:https://doi.org/10.1093/icb/43.2.323.
- Dzik JM. The ancestry and cumulative evolution of immune reactions. Acta Biochimica Polonica. 2010;57(4):443–66. doi:https://doi.org/10.18388/abp.2010_2431.
- O’Neill LAJ, Golenbock D, Bowie AG. The history of toll-like receptors —redefining innate immunity. Nat Rev Immunol. 2013;13:453–60. doi:https://doi.org/10.1038/nri3446.
- Lemaitre B. The road to toll. Nat Rev Immunol. 2004;4:521–27. doi:https://doi.org/10.1038/nri1390.
- Travis J. On the origin of the immune system. Science. 2009;324(5927):580–82. doi:https://doi.org/10.1126/science.324_580.
- Pancer Z, Skorokhod A, Blumbach B, Müller WEG. Multiple Ig-like featuring genes divergent within and among individuals of the marine sponge Geodia cydonium. Gene. 1998;207:227–33. doi:https://doi.org/10.1016/S0378-1119(97)00631-8.
- Fugmann SD. The origins of the RAG genes – from transposition to V(D)J recombination. Semin Immunol. 2010;22(1):10–16. doi:https://doi.org/10.1016/j.smim.2009.11.004.
- Zhang AN, Xu K, Deng A, Fu X, Xu A, Liu X. An amphioxus RAG1-like DNA fragment encodes a functional central domain of vertebrate core RAG1. Proc Natl Acad Sci USA. 2014;11:397–402. doi:https://doi.org/10.1073/pnas.1318843111.
- Lafaille JJ, DeCloux A, Bonneville M, Takagaki Y, Tonegawa S. Junctional sequences of T cell receptor γδ gene: implications for γδ T cell lineages and for a novel intermediate of V-(D)-J joining. Cell. 1989;53:107–15.
- Darsley M, Rees AR. Nucleotide sequences of five anti-lysozyme monoclonal antibodies. Embo J. 1985;4(2):393–98. doi:https://doi.org/10.1002/embj.1985.4.issue-2.
- Benichou J, Glanville J, Prak ETL, Azran R, Kuo TC, Pons J, Desmarais C, Tsaban L, Louzoun Y. The restricted DH gene reading frame usage in the expressed human antibody repertoire is selected based upon its amino acid content. J Immunol. 2013;190(11):5567–77. doi:https://doi.org/10.4049/jimmunol.1201929.
- Briney BS, Willis JR, Hicar MD, Thomas JW, Crowe JE. Frequency and genetic characterization of V(DD)J recombinants in the human peripheral blood antibody repertoire. Immunology. 2012;137:56–64. doi:https://doi.org/10.1111/imm.2012.137.issue-1.
- Webster DM, Henry AH, Rees AR. Antibody-antigen interactions. Curr Opin Struct Biol. 1994;4:123–29. doi:https://doi.org/10.1016/S0959-440X(94)90070-1.
- Briney BS, Willis JR, Finn JA, McKinney BA, Crowe JE. Tissue specific expressed antibody variable gene repertoires. PLoS One. 2014;9:e100839. doi:https://doi.org/10.1371/journal.pone.0100839.
- Darlow JM, Stott DI. Gene conversion in human rearranged immunoglobulin genes. Immunogenetics. 2006;58:511–22. doi:https://doi.org/10.1007/s00251-006-0113-6.
- Zan H, Casali P. Regulation of aicda expression and AID activity. Autoimmunity. 2013;46(2):83–101. doi:https://doi.org/10.3109/08916934.2012.749244.
- Meng W, Jayaraman S, Zhang B, Schwartz GW, Daber RD, Hershberg U, Garfall AL, Carlson CS, Luning Prak ET. Trials and tribulations with VH replacement. Front Immunol. 2014;5(10):1–12. doi:https://doi.org/10.3389/fimmu.2014.00010.
- Berek C, Milstein C. The dynamic nature of the antibody repertoire. Immunol Rev. 1988;105:5–26. doi:https://doi.org/10.1111/imr.1988.105.issue-1.
- Khass M, Vale AM, Burrows PD, Schroeder HW. The sequences encoded by immunoglobulin diversity (DH) gene segments play key roles in controlling B-cell development, antigen-binding site diversity, and antibody production. Immunol Rev. 2018;284:106–19. doi:https://doi.org/10.1111/imr.2018.284.issue-1.
- Schramm CA, Douek DC. Beyond hot spots: biases in antibody somatic hypermutation and implications for vaccine design. Front Immunol. 2018;9:1876. doi:https://doi.org/10.3389/fimmu.2018.01876.
- Voigt CA, Kauffman S, Wang Z. Rational evolutionary design: the theory of in vitro protein evolution. Adv Protein Chem. 2000;55:79–160.
- Rees AR. The antibody molecule: from antitoxins to therapeutic antibodies. Oxford, UK: Oxford University Press; 2014. p. 238–56.
- Nikitin N, Petrova E, Trifonova E, Karpova O. Influenza virus aerosols in the air and their infectiousness. Adv Virol. 2014;2014:859090.
- Soto C, Bombardi RG, Branchizio A, Kose N, Matta P, Sevy AM, Sinkovits RS, Gilchuk P, Finn JA, Crowe JE, et al. High frequency of shared clonotypes in human B cell receptor repertoires. Nature. 2019;566:398–402. doi:https://doi.org/10.1038/s41586-019-0934-8.
- Hong B, Wu Y, Li W, Wang X, Wen Y, Jiang S, Dimitrov DS, Ying T. In-depth analysis of human neonatal and adult IgM antibody repertoires. Front Immunol. 2018;9:128. doi:https://doi.org/10.3389/fimmu.2018.00128.
- Davis MM. The evolutionary and structural ‘logic’ of antigen receptor diversity. Semin Immunol. 2004;16:239–43. doi:https://doi.org/10.1016/j.smim.2004.08.003.
- Vazquez-Bernat N, Corcoran M, Hardt U, Kaduk M, Phad GE, Martin M, Karlsson Hedestam GB. High-quality library preparation for NGS-based immunoglobulin germline gene inference and repertoire expression analysis. Front Immunol. 2019;10:660. doi:https://doi.org/10.3389/fimmu.2019.00660.
- Ohlin M, Scheepers C, Corcoran M, Lees WD, Busse CE, Bagnara D, Thörnqvist L, Bürckert J-P, Jackson KJL, Ralph D, et al. Inferred allelic variants of immunoglobulin receptor genes: a system for their evaluation, documentation, and naming. Front Immunol. 2019;10:435. doi:https://doi.org/10.3389/fimmu.2019.00435.
- Hershberg U, Prak ELT. The analysis of clonal expansions in normal and autoimmune B cell repertoires. Phil Trans R Soc B. 2015;370:20140239. doi:https://doi.org/10.1098/rstb.2014.0239.
- Perelson AS, Oster GF. Theoretical studies on clonal selection: minimal antibody repertoire size and reliability of self non-self discrimination. J Theor Biol. 1979;81:645–70. doi:https://doi.org/10.1016/0022-5193(79)90275-3.
- Perelson A, Weisbuch G. Immunology for physicists. Rev Mod Phys. 1997 October;69(4):1219–68. doi:https://doi.org/10.1103/RevModPhys.69.1219.
- De Boer RJ, Perelson A. How diverse should the immune system be?. Proc R Soc Lond B. 1993;252:171–75.
- Perelson AS. AMS Josiah Willard gibbs lecture “immunology for mathematicians”. Baltimore (MD): Joint Mathematics Meetings (JMM); 2019 January 16. https://www.youtube.com/watch?v=R3xvxxLbk_0.
- Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization human antibodies from V-gene libraries displayed on phage. J Mol Biol. 1991;222:581–97. doi:https://doi.org/10.1016/0022-2836(91)90498-U.
- Griffiths AD, Williams SC, Hartley O, Tomlinson IM, Waterhouse P, Crosby WL, Kontermann RE, Jones PT, Low NM, Allison TJ, et al. Isolation of high affinity human antibodies directly from large synthetic repertoires. Embo J. 1994;13(14):3245–60. doi:https://doi.org/10.1002/embj.1994.13.issue-14.
- Knappik A, Ge L, Honegger A, Pack P, Fischer M, Wellnhofer G, Hoess A, Wölle J, Plückthun A, Virnekäs B, et al. Fully synthetic Human Combinatorial Antibody Libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Biol. 2000;296:57–86. doi:https://doi.org/10.1006/jmbi.1999.3444.
- Rajewsky K, Forster I, Cumano A. Evolutionary and somatic selection of the antibody repertoire in the mouse. Science. 1987;238:1088–94. doi:https://doi.org/10.1126/science.3317826.
- Blier PR, Bothwell A. A limited number of B cell lineages generates the heterogeneity of a secondary immune response. J Immunol. 1987;139:3996–4006.
- Bye JM, Carter C, Cui Y, Gorick BD, Songsivilai S, Winter G, Hughes-Jones NC, Marks JD. Germline variable region gene segment derivation of human monoclonal anti-Rh(D) antibodies. J Clin Invest. 1992;90:2481–90. doi:https://doi.org/10.1172/JCI116140.
- Ikematsu H, Harindranath N, Ueki Y, Notkins AL, Casali P. Clonal analysis of a human antibody response. J Immunol. 1993;150:1325–37.
- Suarez E, Magadan S, Sanjuan I, Valladares M, Molina A, Gambon F, Diazespada F, Gonzalezfernandez A. Rearrangement of only one human IGHV gene is sufficient to generate a wide repertoire of antigen specific antibody responses in transgenic mice. Mol Immunol. 2006;43:1827–35. doi:https://doi.org/10.1016/j.molimm.2005.10.015.
- Volpe JM, Kepler TB. Large-scale analysis of human heavy chain V(D)J recombination patterns. Immunome Res. 2008;4:3. doi:https://doi.org/10.1186/1745-7580-4-3.
- Brezinschek HP, Brezinschek RI, Lipsky PE. Analysis of the heavy chain repertoire of human peripheral B cells using single-cell polymerase chain reaction. J Immunol. 1995;155:190–202.
- Davis MM, Boyd SD. Recent progress in the analysis of ab T cell and B cell receptor repertoires. Curr Opin Immunol. 2019;59:109–14. doi:https://doi.org/10.1016/j.coi.2019.05.012.
- Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, Ni I, Mei L, Sundar PD, Day GMR, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci USA. 2009;106(48):20216–21. doi:https://doi.org/10.1073/pnas.0909775106.
- Lees WD, Shepherd AJ. Studying antibody repertoires with next-generation sequencing. In: Keith JM, editor. Bioinformatics Volume II: structure, Function, and applications. Second ed. 2017. p. 257–70
- Dunand CJ, Wilson PC. Restricted, canonical, stereotyped and convergent immunoglobulin responses. Phil Trans R Soc B. 2015;370:20140238. doi:https://doi.org/10.1098/rstb.2014.0238.
- Malby RL, Tulip WR, Harley VR, McKimm-Breschkin JL, Laver WG, Webster RG, Colman PM. The structure of a complex between the NC10 antibody and influenza virus neuraminidase and comparison with the overlapping binding site of the NC41 antibody. Structure. 1994;2:733–46. doi:https://doi.org/10.1016/S0969-2126(00)00074-5.
- De Genst E, Silence K, Ghahroudi MA, Decanniere K, Loris R, Kinne J, Wyns L, Muyldermans S. Strong in vivo maturation compensates for structurally restricted H3 loops in antibody repertoires. J Biol Chem. 2005;280(14):14114–21. doi:https://doi.org/10.1074/jbc.M413011200.
- Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–50. doi:https://doi.org/10.1006/jmbi.1993.1648.
- Xu JL, Davis MM. Diversity in the CDR3 region of VH is sufficient for most antibody specificities. Immunity. 2000;13:37–45. doi:https://doi.org/10.1016/S1074-7613(00)00006-6.
- Vargas-Madrazo E, Lara-Ochoa F, Carlos Almagro J. Canonical structure repertoire of the antigen-binding site of immunoglobulins suggests strong geometrical restrictions associated to the mechanism of immune recognition. J Mol Biol. 1995;254:497–504. doi:https://doi.org/10.1006/jmbi.1995.0633.
- MacCallum RM, Martin ACR, Thornton JM. Antibody-antigen interactions: contact analysis and binding site topography. J Mol Biol. 1996;262:732–45. doi:https://doi.org/10.1006/jmbi.1996.0548.
- Zemlin M, Klinger M, Link J, Zemlin C, Bauer K, Engler JA, Schroeder HW, Kirkham PM. Expressed murine and human CDR-H3 Intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. J Mol Biol. 2003;334:733–49. doi:https://doi.org/10.1016/j.jmb.2003.10.007.
- Parameswaran P, Liu Y, Roskin K, Jackson KL, Dixit V, Lee J-Y, Artiles KL, Zompi S, Vargas M, Simen B, et al. Convergent antibody signatures in human dengue. Cell Host Microbe. 2013;13:691–700. doi:https://doi.org/10.1016/j.chom.2013.05.008.
- Jackson KL, Liu Y, Roskin K, Glanville J, Hoh R, Seo K, Marshall E, Gurley T, Moody M, Haynes B, et al. Human responses to influenza vaccination show seroconversion signatures and convergent antibody rearrangements. Cell Host Microbe. 2014;16:105–14. doi:https://doi.org/10.1016/j.chom.2014.05.013.
- Trück J, Ramasamy MN, Galson JD, Rance R, Parkhill J, Lunter G, Pollard AJ, Kelly DF. Identification of antigen-specific B cell receptor sequences using public repertoire analysis. J Immunol. 2015;194(1):252–61. doi:https://doi.org/10.4049/jimmunol.1401405.
- Joyce MG, Wheatley AK, Thomas PV, Chuang G-Y, Soto C, Bailer RT, Druz A, Georgiev IS, Gillespie RA, Kanekiyo M, et al. Vaccine-induced antibodies that neutralize group 1 and group 2 influenza A viruses. Cell. 2016;166:609–23. doi:https://doi.org/10.1016/j.cell.2016.06.043.
- Kallewaard NL, Corti D, Collins PJ, Neu U, McAuliffe JM, Benjamin E, Wachter-Rosati L, Palmer-Hill FJ, Yuan AQ, Walker PA, et al. Structure and function analysis of an antibody recognizing all influenza A subtypes. Cell. 2016;166:596–608. doi:https://doi.org/10.1016/j.cell.2016.05.073.
- Jackson KJL, Boyd SD. Broadening horizons: new antibodies against influenza. Cell. 2016;166:532–33. doi:https://doi.org/10.1016/j.cell.2016.07.023.
- Mason DM, Friedensohn S, Weber CR, Jordi C, Wagner B, Meng S, Gainza P, Correia B, Reddy ST. Deep learning enables therapeutic antibody optimization in mammalian cells by deciphering high-dimensional protein sequence space. bioRxiv. 2019. doi:https://doi.org/10.1101/617860.
- D’Angelo S, Ferrara F, Naranjo L, Erasmus MF, Hraber P, Bradbury ARM. Many routes to an antibody heavy-chain CDR3: necessary yet insufficient, for specific binding. Front Immunol. 2018;9:Article 395, 1–12. doi:https://doi.org/10.3389/fimmu.2018.00395.
- Akbar R, Robert PA, Pavlović M, Jeliazkov JR, Snapkov I, Slabodkin A, Weber CR, Scheffer L, Miho E, Haff IH, et al. A compact vocabulary of paratope-epitope interactions enables predictability of antibody-antigen binding. bioRxiv (Cold Spring Harbor). 2019. doi:https://doi.org/10.1101/759498.
- Bradley P, Thomas PG. Using T cell receptor repertoires to understand the principles of adaptive immune recognition. Annu Rev Immunol. 2019;37:547–70. doi:https://doi.org/10.1146/annurev-immunol-042718-041757.
- Piper DE, Jackson S, Liu Q, Romanow WG, Shetterly S, Thibault ST, Shan B, Walker NPC. The crystal structure of PCSK9: a regulator of plasma LDL-cholesterol. Structure. 2007;15:545–52. doi:https://doi.org/10.1016/j.str.2007.04.004.
- Chan JCY, Piper DE, Cao Q, Liu D, King C, Wang W, Tang J, Liu Q, Higbee J, Xia Z, et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci. 2009;106(24):9820–25. doi:https://doi.org/10.1073/pnas.0903849106.
- Liang H, Chaparro-Riggers J, Strop P, Geng T, Sutton JE, Tsai D, Bai L, Abdiche Y, Dilley J, Yu J, et al. Proprotein convertase substilisin/kexin type 9 antagonism reduces low-density lipoprotein cholesterol in statin-treated hypercholesterolemic nonhuman primates. J Pharmacol Exp Ther. 2012;340(2):228–36. doi:https://doi.org/10.1124/jpet.111.187419.
- Ni YG, Di Marco S, Condra JH, Peterson LB, Wang W, Wang F, Pandit S, Hammond HA, Rosa R, Cummings RT, et al. A PCSK9-binding antibody that structurally mimics the EGF(A) domain of LDL-receptor reduces LDL cholesterol in vivo. J Lipid Res. 2011;52:78–86. doi:https://doi.org/10.1194/jlr.M011445.
- Kringelum JV, Nielsen M, Padkjær SB, Lund O. Structural analysis of B-cell epitopes in antibody: protein complexes. Mol Immunol. 2013;53(1–2):24–34. doi:https://doi.org/10.1016/j.molimm.2012.06.001.
- Krawczyk K, Kelm S, Kovaltsuk A, Galson JD, Kelly D, Trück J, Regep C, Leem J, Wong WK, Nowak J, et al. Structurally mapping antibody repertoires. Front Immunol. 2018;9:1698. doi:https://doi.org/10.3389/fimmu.2018.01698
Appendix A
Regions 1 at a time
Expected: A, C, D, E, G, H, I
Maximum total of 6 out of 12
Regions 2 at a time
Expected: AB, AC, AI (example of exception to general contiguous rule), AJ, CD, DE (example of exception to general contiguous rule), DL, EF, FG, GH, HI, IJ
Maximum total of 12 out of 66
Regions 3 at a time
Expected: ABC, ABJ, ACD, AIJ, BCD, CDL, DEF, DEL, EFG, EFK, FGH, GHI, HIJ, IJK
Possible: BCL, FGK, HIK, AIH, DKL
Maximum total of 19 out of 220
Regions 4 at a time
Expected: ABCD, ABCL, ABIJ, AHIJ, AIJK, BCDL, CDEL, CDKL, DEFK, DEKL, EFGH, EFGK, FGHI, FGHK, GHIJ, GHIK, HIJK
Possible: ABJK, AGHI, BDKL, ACDL
Maximum total of 21 out of 495
Regions 5 at a time
Expected: ABCDL, ABCIJ, ABCJL, ABHIJ, ABIJK, BCDEL, BCDKL, CDEKL, DEFGK, DEFKL, EFGHI, EFGHK, FGHIK, GHIJK,
Possible: ABIJL, ABCKL, ABGIJK
Maximum total of 17 out of 792
Regions 6 at a time
Expected: ABCDEL, ABCDJL, ABCDKL, ABCIJL, ABCIJK, ABHIJK, ABIJKL, BCDEKL, BCDJKL, BEFGKL, BGHIJK, CDEFKL, DEFGHK, DEFGKL, EFGHIK, EFGHKL, FGHIJK,
Possible: BCDFKL, EFGIKL, FGHIKL
Maximum total of 20 out of 924
Regions 7 at a time
Expected: ABCDEKL, ABCDJKL, ABCDFKL, ABCHIJK, ABCIJKL, ABGHIJK, ABHIJKL, AFGHIJK, BCDEFKL, BCDFJKL, BGHIJKL, CDEFGKL, DEFGHKL, DEFGHIK, EFGHIJK, EFGHIKL,
Possible: ABCDIJL, ABCGKJL, BCDIJKL
Maximum total of 19 out of 792
Regions 8 at a time
Expected: ABCDEFKL, ABCDEJKL, ABCDFJKL, ABCDIJKL, ABCGIJKL, ABCHIJKL, ABCGHIJK, ABFGHIJK, ABGHIJKL, BCDEFGKL, BCDEFJKL, BCDFGJKL, BDEFGHKL, BFGHIJKL, BEFGHIJK, CDEFGHKL, DEFGHIKL,
Possible: BDEFGJKL, BEFGHJKL, BEFGHIKL, EFGHIJKL
Maximum total of 21 out of 495
Regions 9 at a time
Expected: ABCDEFGKL, ABCDEFJKL, ABCDGIJKL, ABCDHIJKL, ABCFGIJKL, ABCGHIJKL, ABFGHIJKL, ABEFGHIJK, BCDEFGHKL, BCDFGIJKL, BEFGHIJKL
Possible: ABCDFGJKL, BCDEFGJKL, BCEFGIJKL, BCFGHIJKL, CDEFGHIKL, DEFGHIJKL
Maximum total of 17 out of 220
Regions 10 at a time
Expected: ABCDEFIJKL, ABCDFGIJKL, ABCDGHIJKL, ABCEFGIJKL, ABCFGHIJKL, ABEFGHIJKL, BCDEFGHIKL, BCDEFGIJKL, BDEFGHIJKL
Possible: ABCDEFGJKL, ABCDFGHJKL, BCDEFGHJKL, BCDFGHIJKL
Maximum total of 13 out of 66
Regions 11 at a time
Expected: ABCDEFGIJKL, ABCDFGHIJKL, ABCEFGHIJKL, ABDEFGIHJKL, BCDEFGIHJKL,
Maximum total of 5 out of 12
Regions 12 at a time
Maximum total of 1 out of 1
____________________________________________________________________________________________
Total of ‘Expected plus Possible’ = 6 + 12 + 19 + 21 + 17 + 20 + 19 + 21 + 17 + 13 + 5 + 1 = 171