
ABSTRACT
Ebola virus (EBOV) can cause severe hemorrhagic fever in humans, and no approved treatment is currently available. Although several antibodies have achieved good protection in animal models, the potential emerging isolates of ebolavirus and the unknown effects of experimental antibodies in humans underscore the need to develop additional antibodies to address the threat of Ebola. Here, we isolated a series of memory B cell-derived monoclonal antibodies from healthy Chinese adults vaccinated with Ad5-EBOV. These antibodies were encoded by diverse germline genes and had high levels of somatic hypermutation. Most antibodies were cross-reactive and could bind at least two ebolavirus glycoproteins (GPs). Seven neutralizing antibodies were identified using HIV-EBOV GP-Luc pseudovirus, and they effectively neutralized authentic EBOV. In particular, monoclonal antibody 2G1 exhibited potent cross-neutralization against HIV-EBOV/SUDV/BDBV GP-Luc bearing different ebolavirus GPs. We used truncated GPs, competition assays, and software prediction to analyze seven neutralizing antibodies, which bound four different epitopes on GP. Importantly, three of these antibodies provided complete protection in mice when administered one day post-infection. Our study expands the list of candidate antibodies and the options for successfully treating ebolavirus infection.
Introduction
Ebola virus is a highly dangerous single-stranded negative-sense RNA virus of the Filoviridae family. It can cause severe Ebola virus disease (EVD) in both human and non-human primates, with mortality rates of up to 90%.Citation1 Ebola virus (EBOV) belongs to the Ebolavirus genus, which also includes Bundibugyo virus (BDBV), Sudan virus (SUDV), Reston virus (RESTV), and Taï Forest virus (TAFV). Bombali virus (BOMV), which was isolated from small tailless bats in 2018, represents a potential new addition to the genus.Citation2,Citation3 Since the discovery of EBOV 44 years ago, ebolaviruses have reemerged in the human population 28 times.Citation4 EBOV, the most deadly ebolavirus, has caused as many as 18 outbreaks, including the 2013–2016 epidemic in West Africa and the ongoing epidemic in the Republic of the Congo. The 2013–2016 Ebola epidemic in West Africa resulted in 28,616 cases and 11,310 deaths; it also marked the first occurrence of ebolavirus infection outside Africa.Citation5,Citation6 The fatality rate of EBOV (40%~90%) is higher than that of SUDV (36%~65%) and BDBV (25%~36%).Citation7 TAFV has a high mortality rate in chimpanzee populations, but only one serious non-lethal case has been reported in humans.Citation8 RESTV appears to be asymptomatic in humans,Citation9 and it remains unclear if the newly discovered BOMV causes disease in animals or humans.
In spite of impressive progress toward EVD treatment,Citation10 no drug has been approved thus far. A single surface glycoprotein (GP) mediates adhesion and invasion of ebolavirus, and is the key target for designing vaccines and entry inhibitors.Citation10,Citation11 Long-term persistence of specific antibodies has been observed in animals and humans surviving EVD, suggesting their potential for therapy. Numerous GP-targeting monoclonal antibodies (mAbs) or mAb cocktails have been developed in recent years. Among them, ZMapp,Citation12 MIL77E,Citation13 mAb114,Citation14 and REGN-EB3,Citation15 have proven highly protective in non-human primates. In August 2018, another epidemic of EBOV broke out in the Democratic Republic of the Congo and rapidly grew into the second largest filovirus outbreak in history. During the epidemic, four main investigational therapies, ZMapp, mAb114, REGN-EB3, and the small molecule remdesivir (GS-5734)Citation16 were approved for emergency use by the World Health Organization. Recently reported preliminary data show that the mortality rates of patients treated with REGN-EB3 and mAb114 were reduced from 67% to 29% and 34%, respectively, which is more than the reduction achieved by ZMapp (49%) and remdesivir (53%).Citation17 These exciting results further prove the prospect of mAbs as a treatment for EVD.
However, the antibodies or cocktails mentioned above are only specific for EBOV. SUDV and BDBV pose a similarly great threat to human life and health. The conserved GP sequence and structure of ebolavirus makes it possible to screen broadly protective antibodies. Recently, several mAbs targeting conserved epitopes, such as FVM04,Citation18 ADI-15878/15742,Citation19 CA45,Citation20 and EBOV520,Citation21 have been reported. These antibodies could neutralize at least two ebolaviruses both in vitro and in vivo, offering a potential optimal solution against filovirus infection.
Isolation of human mAbs from EVD survivors is a useful strategy. However, EBOV can exist in survivors for a long period of time,Citation22 increasing the risk of potential exposure to virus when working with human blood samples. Recently, a recombinant adenovirus type-5 vector-based vaccine (Ad5-EBOV) carrying the GP of the EBOV variant Makona-C15 showed good immunogenicity and elicited high levels of GP-specific antibody responses in clinical trials,Citation23,Citation24 providing a suitable alternative resource for isolating GP-specific antibodies. Here, we isolated a number of mAbs from memory B cells of humans vaccinated with Ad5-EBOV.Citation23 Seven EBOV-neutralizing antibodies (nAbs) were identified, and a broadly cross-reactive mAb potently neutralized EBOV, BDBV, and SUDV GP-mediated infections in vitro. Three of these neutralizing mAbs completely protected mice in the challenge model of EVD when administered one day after exposure to EBOV. Our study provides a strategy for screening EBOV mAbs from healthy people immunized with vaccines. The identified protective antibodies further expand the list of candidate EVD therapies and, therefore, the options for post-exposure treatment of ebolavirus infections. Furthermore, a broadly neutralizing mAb can potentially serve as a prospective pan-ebolavirus therapeutic antibody.
Results
Preparation of truncated EBOV GPs
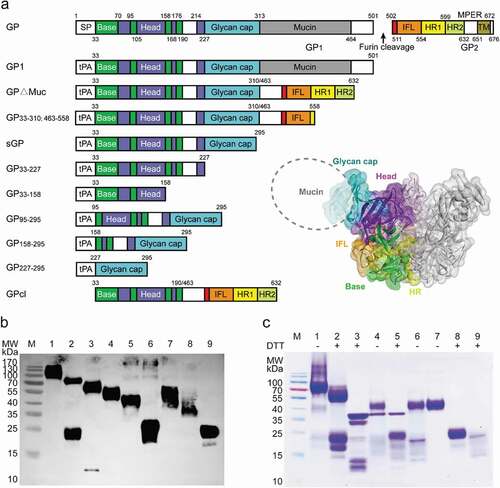
GP exists on the surface of the virus membrane in the form of a trimer, and each monomer is composed of two subunits, GP1 and GP2. GP1 mediates adhesion to the cell membrane and binding to the virus receptor, whereas GP2 mediates membrane fusion between the virus and endosome following a conformational change.Citation25,Citation26 To sort GP-specific memory B cells and analyze antibody-binding regions, we produced a panel of truncated EBOV GPs by deleting previously described GP domains.Citation26 The panel consisted of GP1, mucin domain-deleted GP (GP∆Muc), secreted GP (sGP), as well as several new forms: GP33-310; 463–558, resulting from a removal of the heptad repeat region on GP∆Muc backbone; GP33-227, comprising a truncated sGP without the glycan cap (GC) domain; GP33-158 comprising most of the base and head domains; GP95-295, an sGP variant without the base; GP158-295, comprising a further truncation of GP95-295, with the removal of the head region; GP227-295, comprising only the GC domain (). These truncated GPs were well expressed in soluble form in a mammalian expression system, and the proteins were purified through Ni-NTA affinity purification. Likely due to protein glycosylation, the apparent molecular weights of the GP truncations observed on Western blots were larger than the predicted ones (). Studies have shown that cleavage of GP∆Muc using thermolysin in vitro could simulate the processing of GP by cathepsin in vivo, resulting in a cleaved GP (GPcl) similar to that observed during natural infection.Citation27-Citation29 We digested GP∆Muc with thermolysin in phosphate-buffered saline (PBS, pH7.4) and then isolated GPcl by size-exclusion chromatography (). We verified the main types of truncated GPs (GPdTM, GP1, sGP, GP∆Muc and GPcl) using mAbs of MIL77E (one GC binder, two Base binders) by ELISA. Consistent with the epitopes previously reported,Citation30 MIL77-3 bound GPdTM, GP1, and sGP, while MIL77-1 and MIL77-2 bound GPdTM, GP∆Muc, and GPcl.
Figure 1. Generation of truncated EBOV GPs. (a) Design of truncated EBOV GPs. GP includes the signal peptide (SP), Base, Head, glycan cap (GC), mucin-like domain (Mucin), internal fusion loop (IFL), heptad repeat 1 (HR1), heptad repeat 2 (HR2), and transmembrane (TM) domain. The same color-coding is used also for a three-dimensional model of GP (PDB ID: 3CSY). (b) Expi293 expression supernatants of truncated EBOV GPs were identified using anti-6× His tag pAb-HRP by western blotting. Lane 1: GP133-501; 2: GP∆Muc33-310; 463–632; 3: GP33-310; 463–558; 4: sGP33-295; 5: GP33-227; 6: GP33-158; 7: GP95-295; 8: GP158-295; 9: GPcl. (c) Soluble GP∆Muc was digested by thermolysin to generate GPcl. Lanes 1–2: GP∆Muc; 3: Thermolysin; 4, 5: GP∆Muc incubated with thermolysin; 6: GP∆Muc-thermolysin mixture retained by a 50-kDa cutoff filter; 7, 8: GPcl collected by a Superdex 200 Increase 10/300 GL column; 9: Removed impurities. Symbols “+”, “-” indicate reduction or not with dithiothreitol (DTT)
Isolation of ebolavirus GP-specific antibodies
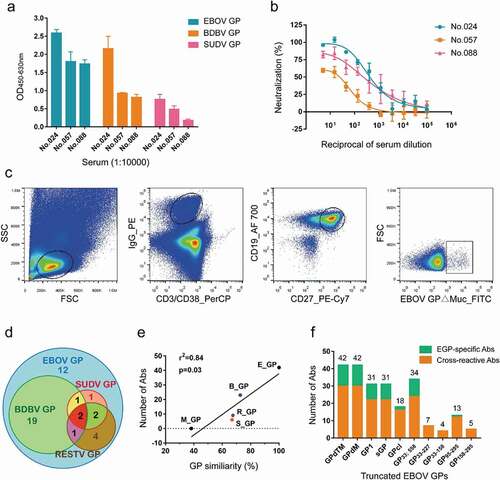
The Ad5-EBOV vaccine has been shown to elicit a strong antibody response against EBOV GP in a recent clinical trial.Citation23,Citation24 To isolate GP-specific mAbs from vaccinated participants, serum from vaccinated donors collected 28 days after the second vaccination was examined for binding activity against recombinant EBOV, BDBV, and SUDV GPs lacking their transmembrane domains (GPdTM). ELISA revealed that sera from three vaccinees exhibited cross-reactivity against all three GPs ( and Figure S1a). Moreover, all three sera could neutralize HIV-EBOV GP-Luc, an HIV-based pseudovirus carrying EBOV GP and expressing luciferase reporter, in vitro (). To obtain high-affinity antibodies derived from memory B cells, EBOV GP∆Muc was labeled with fluorescein isothiocyanate (FITC) and CD3−/CD38−/IgG+/CD19+/CD27+/GP∆Muc+ GP-specific memory B cells were isolated from the subjects’ peripheral blood mononuclear cells (PBMCs) (). A total of 358 GP-specific memory B cells were sorted, accounting for about 1% of mIgG-type memory B cells. Paired light and heavy chain variable region genes were amplified from single memory B cells by single-cell reverse-transcription PCR (RT-PCR) and nested PCR. This procedure yielded 161 VH genes (45.0%), 176 Vκ genes (49.2%), and 105 Vλ genes (29.3%), of which 133 pairs (79 κ chains and 54 λ chains) were successfully matched (37.2% success rate).
Figure 2. Isolation of GP-specific monoclonal antibodies. (a) Binding capacity of the serum of vaccine-immunized subjects # 024, 057, and 088 to EBOV GP, BDBV GP, and SUDV GP. Values represent the difference in optical density (OD) between sera (1:10,000) on day 28 post-boost immunization and day 0 from the same donor. See also Figure S1a. (b) Neutralizing capacity of the serum of vaccine-immunized subjects # 024, 057, and 088 against pseudotyped HIV-EBOV GP-Luc. Data on the curve represent the difference in neutralization ability between sera on day 28 post-boost immunization and day 0 from the same donor. (c) Sorting of CD3−/CD38−/IgG+/CD19+/CD27+/GP∆Muc+ single memory B cells obtained from PBMCs one month post-boost immunization to identify GP-specific mAbs. (d) Number of specific or cross-reactive antibodies identified using the supernatants of Ig genes linear expression cassettes. See also Figure S1b. (e) Correlation between GP sequence similarity to EBOV GP and number of binding antibodies. (f) Number of antibodies binding to different truncated EBOV GPs determined by ELISA using 293 T supernatants. See also Figure S1b
The light and heavy chain variable region genes of the antibodies were constructed into full-length linear expression cassettes,Citation31 and then co-transfected into 293 T cells. EBOV/SUDV/BDBV/RESTV/Marburg virus (MARV) GPdTM and several truncated EBOV GPs were used to analyze the specificity, cross-reactivity, and binding region of the antibodies in 293 T expression supernatant. Finally, 42 EBOV GP∆Muc-binding antibodies (40.6%) were screened from the above 133 pairs of antibody genes. The relatively weak binding of these antibodies to GPdTM (Figure S1b) might be related to binding affinity, epitope exposure, and other factors.
Binding profiles of GP-specific antibodies
Interestingly, the selected EBOV GP-binding antibodies showed unexpectedly high cross-activity, with 30/42 (71.4%) binding at least two types of ebolavirus GPs ( and Figure S1b). Specifically, 24 mAbs bound two ebolavirus GPs, four mAbs (2F4, 5D7, 5G11, and 9G11) bound three GPs, and two mAbs (2G1 and 9D10) bound all four ebolavirus GPs. The majority of the cross-reactive mAbs bound BDBV GP (23/30), six bound SUDV GP, and nine bound RESTV GP, which is consistent with the similarities of their protein sequences (EBOV vs BDBV, 72.7% amino acid similarity; EBOV vs SUDV, 66.9%; EBOV vs RESTV 67.5%). None of the mAbs cross-reacted to MARV GP, likely due to the low amino acid similarity between MARV GP and EBOV GP (38.2%). There was a significant positive correlation between the number of GP-cross-reactive antibodies and protein sequence similarity to EBOV GP (p = .03, r2 = 0.84) ().
We next attempted to map the target regions of the mAbs on EBOV GP using purified GP truncations ( and Figure S1b): both GP1 and sGP (31); GPcl (18); GP33-310; 463–558 (34); GP33-227 (7); GP33-158 (4); GP95-295 (13); and GP158-295 (5). The mAbs differentially bound to the truncated GPs, suggesting diverse epitopes. All 31 GP1-binding mAbs also bound sGP, suggesting that their epitopes were mainly located within the first 295 amino acids (aa) of GP. Twenty-two of the GP1-binding antibodies failed to bind GPcl, indicating that their epitopes were possibly lost or changed after thermolysin cleavage. Nine of the 18 GPcl-binding antibodies also boundGP1, suggesting the recognition of a common sequence on GP1 (approximately 33 ~ 190 aa). The other nine GPcl-binding mAbs could bind to all truncated forms (GPdTM, GP∆Muc, and GPcl) containing GP2 subunit and did not bind to any of the truncated forms with GP2 deletion, implying that GP2 was essential for binding. Two antibodies failed to bind either GP1 or GPcl, suggesting that their epitopes might be dependent on the whole structure of GP or GP∆Muc. Some mAbs bound to nearly all truncated GPs, implying recognition of a common region of these truncated GPs, and their epitopes were exposed and maintained. The accessibility of the truncated GPs to some of the mAbs suggested that the GP mutants maintained such epitopes; however, lack of binding did not necessarily imply the absence of mAb epitopes. There are two possible reasons for the latter: 1) some key amino acids for antibody binding might have been missing and the remaining structure was not sufficient to support antibody binding; or 2) the truncated GP could not maintain the same conformation as native GP.
Diversity in gene usage and high levels of somatic hypermutation
The variable regions of the heavy (VH) and light (VL) chains of the isolated mAbs were sequenced and analyzed to determine germline gene distribution, complementarity-determining region (CDR) sequences, nucleotide and amino acid mutations. The mAbs were revealed to originate from a variety of germline genes and possess unique sequences (). The heavy chains were encoded by 18 different VH germline genes derived from V1, V3, and V4 variable gene families, with V3 being the most abundant, followed by V4 and V1 (). The kappa (κ) light chains were encoded by V1, V2, and V3 variable gene families and contained 10 different Vκ germline genes, whereas the lambda (λ) light chains were encoded by 10 different Vλ germline genes derived from V1, V2, and V4 variable gene families. Some germline genes exhibited high frequency of usage, including HV3-11 (6), HV3-48 (4), and HV3-15 (4) of VH; κV1-39 (10) and κV3-20 (9) of Vκ; as well as λV2-14 (5) and λV1-40 (4) of Vλ (Figure S2). Remarkably, all 10 mAbs with κV1-39 (the most used gene) exhibited cross-reactivity, whereas only two of the nine mAbs with κV3-20 bound at least two kinds of ebolavirus GPs.
Figure 3. Analysis of EBOV GP-specific antibody sequences. See also Figure S2. (a) Antibodies with the same VH germline genes were grouped and their CDR3 s are listed. Bold letters indicate identical or conserved amino acids, whereas gray letters denote amino acids with different characteristics. (b) Heavy (top) and light chain (κ/λ, bottom) variable region gene family distributions of GP-specific antibodies. (c) Number of nucleotide (Nt) and amino acids (AA) mutations in VH and VL genes. (d) CDR length of VH and VL genes. Mean ± SD are shown in red. (e) Multiple alignment of VH, Vκ, and Vλ of mAbs depicted by WebLogo 3. CDR regions are highlighted
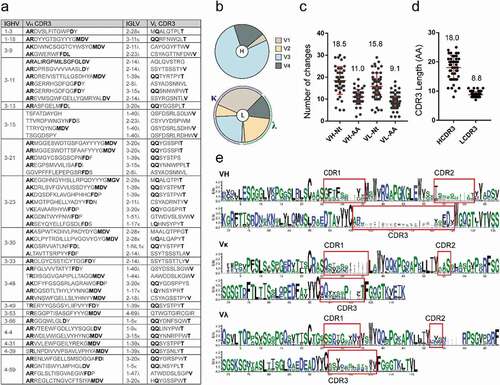
The variable region of mAbs exhibited high levels of somatic hypermutation, with an average of 18.5 nucleotide and 11 amino acid mutations for VH, and 15.8 and 9.1, respectively, for VL (). A large difference was observed between the VH genes and germline genes of the mAbs, which might result from the high frequency of mutations and their gradual accumulation in somatic cells during affinity maturation under antigen selection. The average CDR3 lengths of VH and VL were 18.0 and 8.8 amino acids, respectively (). The VH or VL sequence logo plots revealed numerous differences in the variable regions, particularly in the CDRs. Interestingly, some sites in the CDR of germline genes showed certain preference and conservation during somatic hypermutation, such as “D” at position 124 of HCDR3 and “S” at position 56 of κCDR2 ().
Seven mAbs potently neutralize Ebola virus
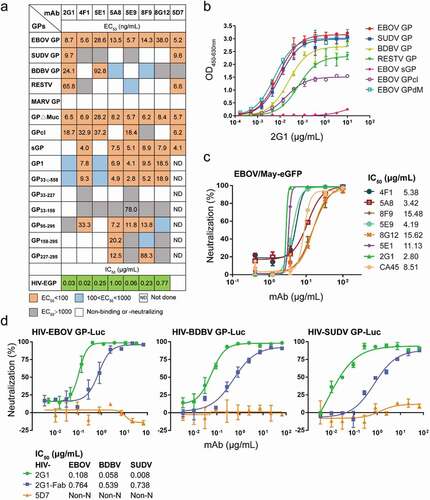
We prepared mAbs in a mammalian expression system and tested their binding activity to various GPs. All mAbs showed strong binding to EBOV GP, and the half-maximal effective concentration (EC50) ranged from 5 to 50 ng/mL by ELISA (). The cross-reactivity of mAbs was consistent with that observed during the screening process. Some mAbs, such as 2G1 and 5D7, presented a wide cross-reactivity to ebolavirus GPs. The EC50 values of mAb 2G1 to EBOV/SUDV/BDBV/RESTV GP were 8.7, 9.7, 24.1, and 65.8 ng/mL, respectively (,). Similarly, mAb 5D7 bound EBOV/SUDV/RESTV GP with EC50 values of 5.2, 9.6, and 6.6 ng/mL, respectively. As expected, no mAb could effectively bind MARV GP. Seven mAbs, including five GP1-specific mAbs and two GP2-related mAbs, exhibited potent neutralizing activity against pseudotyped HIV-EBOV GP-Luc, with a half-maximal inhibitory concentration (IC50) ranging from 0.02 to 1.00 μg/mL (Figure S4a). Similarly, the seven mAbs were able to neutralize authentic EBOV/May-eGFP, and four of them exhibited stronger neutralization than CA45 (). Notably, 2G1, the most cross-reactive mAb, effectively neutralized HIV-EBOV/SUDV/BDBV GP-Luc (), whereas another pan-ebolavirus mAb, 5D7, could not neutralize any of the three pseudotyped viruses.
Figure 4. Profiles of antibody binding and neutralizing abilities. (a) EC50 and IC50 values of the identified antibodies binding to Ebola virus GPs, truncated EBOV GPs, and HIV-EBOV. See also Figure S3 and Figure S4a. (b) Binding curves of mAb 2G1 to ebolavirus GPs or truncated EBOV GPs. (c) Neutralization of authentic EBOV/May-eGFP by nAbs; corresponding IC50 values are shown. (d) Neutralization of cross-reactive mAbs or Fab to HIV-EBOV/BDBV/SUDV GP-Luc; corresponding IC50 values are shown. Binding and neutralization assays were performed in duplicate or triplicate
Seven nAbs recognize four distinct areas of GP
The neutralizing antibodies (nAbs) were further analyzed using truncated GPs to determine their approximate binding regions ( and Figure S3). For example, mAb 5A8 bound to GP95–295, GP158–295, and GP227–295, so its epitope was predicted to be located within aa 227–295, the only region shared by all three of those truncated GPs. We similarly inferred the binding regions for 2G1 (GP2-related), 4F1 (95–190 aa), 5E1 (GP2-related), 5E9 (95–190 aa), 8F9 (227–295 aa), and 8G12 (190–295 aa) (Figure S5).
We further analyzed the epitopes of the seven nAbs by competitive binding ELISA (). MIL77-1/2/3, derived from the component mAbs of ZMapp, were used as contrasts.Citation13,Citation30 Biotinylated antibodies at their EC50 were incubated with 100-fold excess competitors, and competitiveness was defined by the ratio of biotinylated antibody binding in the presence of competitors versus an irrelevant mAb. MAbs were considered competing for the same site if the competition value was <30, they were deemed non-competing if the value was >60, and competition was considered intermediate if the value was between 30 and 60. Accordingly, mAbs were roughly divided into five groups: groups 1 and 2 interacted with GP1, whereas groups 3, 4, and 5 interacted with the GP2 subunit. All mAbs in group 1 could compete with MIL77-3 (T270, K272) binding to the GC, suggesting that the epitopes of these mAbs were spatially close to that of MIL77-3. However, MIL77-3 could not completely block the binding of most mAbs from group 1, which might be related to the location and angle of antibody binding. MIL77-3 binds almost vertically to the top of the flexible GC,Citation30 probably leaving room for other mAbs to bind. The five nAbs in group 1 could be further divided into three subgroups: 4F1 and 5E9, 8F9 and 5A8, and 8G12. The mAbs in each of the first two subgroups competed with each other, but there was no competition between subgroups, indicating that the epitopes of the two subgroups did not overlap. The subgroup 8G12 was similar to MIL77-3 and was easily outcompeted by mAbs of the other two subgroups. 2G1 and 5E1, two nAbs binding to the GP2 subunit, belonged to group 3 and group 5, respectively. The epitopes of these two groups were different from those of MIL77-1 (C511, N550, G553, and C556) and MIL77-2 (C511, D552, and C556)Citation30 in group 4. Overall, seven nAbs targeted mainly four diverse areas on GP, suggesting their potential for cocktail therapies.
Figure 5. Competition assays and epitope prediction. (a) Competition assays. Numbers represent the percentage binding of biotinylated mAbs in the presence of a competitor versus an irrelevant mAb. Values <30 (white numbers in black grids) indicate mAbs with an identical or close epitope; values >60 (white boxes with black numbers) indicate noncompetitive mAbs; values in the 30%~60% range (black numbers in gray grids) indicate intermediate binding ability. MIL77-1/2/3 are abbreviated as M-1/2/3. G1 to G5 represent five groups with different binding areas (red squares); seven nAbs are highlighted by green squares. (b) Mapping of predicted critical amino acids of nAbs on a GP model (PDB ID: 5KEL). See also Figure S5
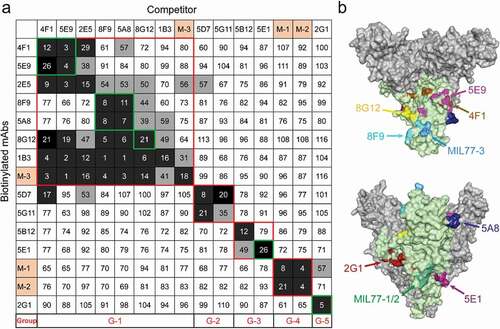
We built an antigen-binding fragment (Fab) binding model of nAbs and docked it with EBOV GP (PDB ID: 5KEL) using a ZDOCK method to generate the top 2000 poses in the GP-Fab complex clusters ranked by ZRANK score. We then applied an optimized residue contact frequency (RCF) algorithm to analyze the top 101 conformations and obtain the score of each amino acid residue.Citation32 The amino acid residues ranked among the top 10 or 20 scores (Figure S5) and located in the region previously delineated by truncated GPs were considered critical sites for binding. Finally, we mapped the possible epitopes of the seven nAbs on the GP trimer ().
Thermolysin cleavage affects nAbs binding to GP
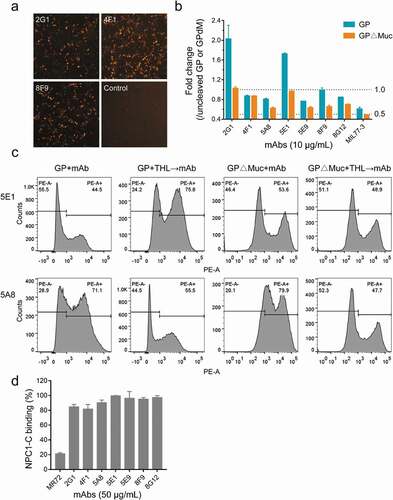
To determine whether the presence of GP in a soluble or membrane-anchored form, or in a cleaved or non-cleaved form, affected mAb binding, we displayed EBOV GP or GP∆Muc on the cell surface and then examined the changes in mAb binding to GPs with or without thermolysin digestion. Phycoerythrin (PE)-labeled anti-human IgG secondary antibodies stained BHK-T7 cells expressing GP (), indicating that the mAbs could bind natural GP trimers. We further displayed GP or GP∆Muc on the surface of 293 T cells and quantitatively analyzed the changes in mAb binding after thermolysin digestion using flow cytometric analysis (,, Figure S6, and Figure S7). Although all mAbs bound GP, GP2-related mAbs showed a lower positive signal (32.3% and 44.5%) than GP1-binding mAbs (63.1% to 73.5%). On the contrary, 2G1 showed even stronger activity than other mAbs in the presence of soluble GP by ELISA. This difference might be explained by the fact that after the GP was directionally anchored into the cell membrane, the epitopes of the five GP1-related mAbs were better exposed and more accessible, whereas those of GP2-related mAbs were more easily obscured by the mucin-like domain or disturbed by the membrane. This conjecture is supported by the analysis of cell surface-displayed GP∆Muc and thermolysin digestion. For surface-displayed GP∆Muc, positive binding between cells and GP1 mAbs increased by 8.9%, whereas that of 2G1 and 5E1 increased by 47.4% and 20.4%, respectively. The binding of mAbs to GPs changed in unexpected ways after thermolysin cleavage. For example, MR191, a MARV GP-specific antibody, which could not bind GP and GP∆Muc of EBOV, was found capable of binding EBOV GPcl.Citation33 The binding of MR191 to thermolysin-digested GP or GP∆Muc increased by 4.1-fold and 15.5-fold (Figure S6 and Figure S7), respectively, suggesting that the GPcl-like structure was generated after digestion. For 2G1 and 5E1, the number of positive cells with cleaved GP increased by 2.1-fold and 1.7-fold, respectively, and GP∆Muc binding remained unchanged after thermolysin digestion. However, thermolysin digestion greatly affected the binding of GP1-related mAbs to GP or GP∆Muc, and the percentage of positive cells decreased by 1-40% and 13-52%, respectively. These results indicate that the cleavage of GP in late endosomes might be either beneficial or harmless for GP2-related mAbs, but detrimental for some GP1-binding mAbs.
Figure 6. Influence of GP form and thermolysin cleavage on mAbs binding. (a) Binding of mAbs to EBOV GP displayed on the surface of BHK-T7 cells. Images were acquired under a 4× objective on a Cytation imaging reader. (b) and (c) Binding of mAbs to uncleaved or cleaved EBOV GP/GP∆Muc displayed on the surface of 293 T cells and analyzed using flow cytometry. Fold change in mAb binding after thermolysin cleavage was calculated by comparing the percentage of PE-positive cells with or without thermolysin (THL) digestion. See also Figure S6 and Figure S7. (d) NPC1-C binding inhibition. See also Figure S4b
Inhibition of NPC1-C binding to GPcl
The interaction between GPcl and C domain of Niemann-Pick C1 (NPC1-C) has been identified as a critical process for ebolavirus entry.Citation34 To test whether the seven nAbs could block the binding of GPcl to NPC1-C, we pre-incubated mAbs with GPcl, and then added biotinylated NPC1-C. MR72, a MARV GP-specific mAb, was reported to block the binding of EBOV GPcl to NPC1-C.Citation21,Citation35 We confirmed the blocking activity of MR72 in our assays, but none of the nAbs affected the binding of NPC1-C to GPcl ( and Figure S4b). This suggests that our nAbs may play a neutralizing role by inhibiting cathepsin cleavage, affecting GP allostery or other mechanisms.
Therapeutic protection of mice by neutralizing antibodies
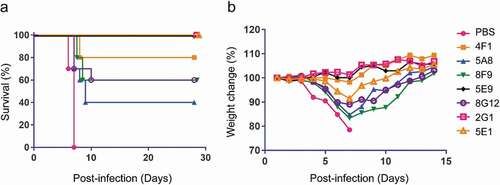
Finally, we evaluated the in vivo protection of nAbs in a mouse model (). Groups (n = 10) of BALB/c mice were challenged intraperitoneally (i.p.) with 1000 × median lethal dose (LD50) of mouse-adapted EBOV (MA-EBOV) variant Mayinga, and then treated with PBS or 100 μg of each nAb one day post-infection. All mice treated with PBS died within seven days. However, 3/7 nAbs (2G1, 5E1, and 5E9) provided complete protection while the remaining nAbs afforded 40% to 80% survival. Moreover, no weight loss was observed in mice treated with highly protective nAbs ().
Figure 7. Survival and weight change of mice treated with EBOV mAbs. (a) BALB/c mice (n = 10 per group) were inoculated with mouse-adapted EBOV, treated with PBS or 100 μg nAbs at one day post-infection, and monitored for 28 days. (b) Weight change of mice after virus challenge and mAbs administration. Mice were monitored for 14 days
Discussion
EBOV has caused dozens of outbreaks, but the scale of the last two outbreaks has far exceeded the previous ones. Indeed, EBOV, and related viruses, continue to pose a threat to global public health, and they continue to be prime candidates for the development of biological weapons.Citation36 Meanwhile, new EBOV-like filoviruses are still being discovered.Citation2,Citation37 All these factors underscore the importance of developing preventive and therapeutic agents. Although the vaccine Ervebo (Merck) was recently approved by the FDA to prevent EVD caused by EBOV in adults,Citation38 there remains no clinically approved post-exposure therapeutic. Antibodies have good pharmacokinetics, specificity, and tolerability, and represent promising potential therapeutics for a number of viral diseases, including those caused by filoviruses. Indeed, two antibody treatments, REGN-EB3 and mAb114, have recently been shown to significantly improve patient survival in a clinical trial undertaken during the ongoing EBOV outbreak in the Democratic Republic of the Congo,Citation39 highlighting the potential for antibodies to be effective treatments against filoviruses.
Here, we isolated a set of antibodies derived from memory B cells of Ad5-EBOV Phase 1 clinical trial participants. Vaccines expressing EBOV GP can stimulate antibodies cross-reactive to multiple ebolavirus GPs, but with relatively low ratio.Citation40 Interestingly, a large portion of our mAbs (30/42) cross-reacted to a few other ebolavirus GPs, with most of them recognizing two GPs. The cross-reactivity among the two-GP binding mAbs is species-dependent, meaning the closer the phylogenetic relationship, the larger the number of cross-reactive mAbs. As a result, most two-GP cross-reactive mAbs are EBOV/BDBV double positive, followed by EBOV/RESTV and EBOV/SUDV double positive. As expected, no cross-reactivity to MARV GP was observed.
To define the target regions of the mAbs on EBOV GP, we analyzed their binding profiles against a panel of truncated EBOV GPs. The mAbs recognized diverse regions, and some could bind all types of truncated GPs, indicating that some conserved structure was maintained on these truncated types. A series of antibodies derived from human donors, who received ChAd3 EBOZ and were boosted with MVA-BN Filo, have shown lower levels of somatic mutations, with an average of five VH amino acid changes compared to germline genes.Citation40 Our mAbs showed higher somatic hypermutation levels with an average of 11 VH amino acid mutations, which suggests a promising immunization strategy or an optimized marker choice for isolating antibodies undergone high levels of affinity maturation.
As expected, all mAbs showed strong binding to EBOV GP, with EC50 values ranging from 5 to 50 ng/mL (0.03 to 0.3 nM) by ELISA. We screened seven potent nAbs using HIV-EBOV GP-Luc pseudotyped virus and verified the effectiveness of the procedure with authentic EBOV. In particular, a broad-binding spectrum nAb, 2G1, effectively neutralized EBOV, SUDV, and BDBV pseudotypes. We analyzed the epitopes of the seven nAbs through competition assays and found four main targets. Epitopes of five nAbs were located on sGP and close to the non-neutralizing antibody MIL77-3; whereas binding of the other two nAbs likely requires the GP2 subunit, but is in regions different from the epitopes of MIL77-1 and −2 in GP base domain.
Previously, a systematic analysis with 171 EBOV antibodies showed that neutralization in vitro and immune effector functions (IEFs) were the most predictive of protection in animals.Citation41 Epitopes of a large number of neutralizing antibodies remain on cleaved GP, while antibodies with the strongest IEFs usually bind to the top of the GP trimer away from the viral membrane. In this study, four GPcl-binding nAbs, 2G1, 4F1, 5E1, and 5E9, provided the best protection in mice (three at 100% and one at 80%). They also displayed good protective efficacy when administered at two days post infection (70% ~ 100%, data not shown). Three nAbs, 5A8, 8F9, and 8G12, failed to bind GPcl and exhibited relatively lower protection (two at 60% and one at 40%). Given that our mAbs were incorporated the same constant region during the screening process, the IEFs should be similar for five nAbs (5E9, 4F1, 8F9, 8G12, and 5A8) bound to the top of GP1. However, their protection in mice ranged from 100% to 40%, suggesting that GPcl-related nAbs are highly correlated with protection in vivo. Furthermore, because of the protective effect mediated by IEFs, antibody performance might be further improved by optimizing Fc structure and functions.
A cocktail of antibodies is considered necessary and effective to treat ebolavirus infections.Citation42 However, it is not clear which combinations of epitopes or protection mechanisms are the most beneficial. ZMapp consists of a non-neutralizing antibody and two competing nAbs,Citation12,Citation30 which may not be an ideal cocktail strategy. By contrast, REGN-EB3 consists of three nAbs targeting different epitopes.Citation15 This difference may partly explain the different protection afforded to patients. Initially, it was thought that the large amount of soluble sGP expressed in the body during natural infection might act as a decoy,Citation43 resulting in fewer antibodies available against the virus. However, a recent study has shown that sGP-binding antibodies do not appear to adversely affect protection.Citation41 Although this “apparent negative effect” has not been proven experimentally or therapeutically, it may be related to the need for high doses of antibody or cocktail. Furthermore, we observed that GP1-binding nAbs bound more efficiently to surface-displayed GP, but were also more susceptible to thermolysin cleavage. In contrast, the binding of GP2-related nAbs to membrane GP was relatively less efficient, but was less-sensitive to thermolysin. Our seven nAbs were isolated from healthy individuals and recognize four different epitopes located on GP1 or GP2. Although we endeavored to delineate the possible epitopes of seven nAbs based on truncated GPs and computer simulations, the key amino acids enabling each mAb binding need to be further validated through site-directed mutations or analysis of the antibody-GP complex structure. In addition, because the neutralization mechanism and synergy effects of these antibodies remain unclear, it remains to be determined how to choose the nAbs for an effective cocktail.
In summary, we isolated a number of ebolavirus cross-reactive antibodies with unique sequences, high levels of somatic hypermutation, and diverse epitopes from Ad5-EBOV-immunized clinical donors. Seven neutralizing antibodies were identified, and three that targeted different epitopes could confer complete protection against EBOV in a mouse model, indicating their potential as therapeutic candidates or cocktail components for EVD treatment. In particular, a pan-ebolavirus mAb, 2G1, may provide broad resistance to ebolavirus infections.
Materials and methods
Ethics statement
Human blood samples used in this study were from the Ad5-EBOV (Ebola virus/H sapiens-wt/GIN/2014/Makona-C15) vaccine trial conducted in China (Clinical Trials registration number, NCT02326194).Citation23,Citation24 The ethical approval for this study was issued by the Institutional Review Board of the Jiangsu Provincial Center of Disease Control and Prevention. All participants provided written informed consent. The study was conducted following the 1975 Declaration of Helsinki and Good Clinical Practice guidelines.
The animal experiments in this study were performed in the containment level 4 (CL4) facility at the Canadian Science Center for Human and Animal Health (Winnipeg, Canada), part of the Public Health Agency of Canada’s National Microbiology Laboratory (NML). All work was approved by the institutional animal care committee in compliance with guidelines from the Canadian Council on Animal Care (CCAC).
Production of proteins
EBOV/SUDV/BDBV/RESTV/MARV rGPdTM (0501-016/0502-015/0505-015/0504-015/0506-015) was purchased from IBT BIOSERVICES. The sequences of truncated EBOV GPs, including GP133-501, GP∆Muc 33–310; 463–632, GP33-310; 463–558, sGP33-295, GP33-227, GP33-158, GP95-295, and GP158-295, were amplified from a codon-optimized full-length GP of the Makona-C15 strain (GenBank: KJ660346.2). The original signal peptide (SP, 1–32 aa) of GP was replaced by the first 23 aa of tissue-type plasminogen activator (t-PA, GenBank: AAA60111.1) with one site mutation (P22 N), and a 6× His tag was added at the C-terminus of truncated GPs for identification and purification. The sequences were digested by EcoRI and HindIII (R3101 and R0104; New England Biolabs) and then cloned into pcDNA3.4 plasmids. Truncated GPs were expressed in the ExpiFectamine™ 293 system (A14524; Gibco) following the manufacturer’s instructions. Briefly, 30 μg of GP-bearing plasmid and 80 μL of transfection reagent were diluted each in 1.5 mL Opti-MEM I Reduced Serum Medium (31985–062; Gibco) and then incubated at room temperature for 5 min before mixing them together. After a 20-min incubation, the mixture was added to a shaker flask containing 7.5 × 107 Expi293 F™ cells in 25.5 mL expression medium. After 16 h, 150 μL of enhancer 1 and 1.5 mL of enhancer 2 were added to each flask. Cells were cultured at 37°C, 125 rpm in 8% CO2, and harvested at 72 ~ 96 h post-transfection. The culture supernatant was collected by centrifugation at 3000 × g for 10 min, and then filtered through 0.2-μm syringe filters (4612; PALL). About 1 to 5 μL of each GP supernatant was identified by western blotting using an anti-6× His tag pAb-horseradish peroxidase (HRP) antibody (Ab1187; Abcam). A HisTrap HP column (17524801; GE Healthcare) loaded with binding buffer (20 mM sodium phosphate, 20 mM imidazole, 0.5 M NaCl, pH 7.4) and elution buffer (20 mM sodium phosphate, 500 mM imidazole, 0.5 M NaCl, pH 7.4) was employed for the purification of GPs. Purified GPs were buffer-exchanged into PBS and then stored at −80°C after quantification with the BCA Protein Assay Kit (23227; Pierce, Thermo Fisher Scientific).
To generate GPcl, GP∆Muc was digested by thermolysin as previously described with a minor modification.Citation27,Citation44 Thermolysin (T7902; Sigma-Aldrich) was added to a solution of 2 mg/mL GP∆Muc in PBS at a final concentration of 0.5 mg/mL and incubated for 1 h at 37°C. The reaction was stopped by phosphoramidon at a final concentration of 0.5 mM, and then the buffer was exchanged for PBS using an Amicon Ultra 0.5 mL-50 kDa cutoff centrifugal filter unit (UFC5050; Merck). The concentrated solution was filtered through 0.20-μm microfilters (SLLGR04NL; Merck) and purified on a Superdex 200 Increase 10/300 GL gel column (28990944; GE Healthcare).
The cDNAs encoding domain C (374–620 aa) of human NPC1 (GenBank: NM_000271.4) were synthesized (Sangon Biotech) and then cloned into a pET32a vector by NdeI and XhoI (R0111 and R0146; New England Biolabs). NPC1-C was expressed in the Escherichia coli BL21 strain (CB105-02; TIANGEN Biotech) and the inclusion bodies were dialyzed and renatured in PBS. A soluble form of NPC1-C was also prepared in the ExpiFectamine™ 293 system using the pcDNA3.4 vector as described above.
Packaging of ebolavirus GP-pseudotyped recombinant viruses
Full-length genes of wild-type EBOV GP (GenBank: KJ660346.2), BDBV GP (GenBank: YP_003815435.1), and SUDV GP (GenBank: NC_006432.1) were synthesized and constructed into pDC316 vectors using EcoRI and HindIII. 293 T cells were seeded into T75 flasks (0030711122; Eppendorf) and pseudovirus packaging was conducted when cells reached 70%~80% density. A total of 22 μg of plasmids (pNL4-3.Luc.R-E- and GP at a mass ratio of 5:1) was mixed with 40 μL of TurboFect reagent (R0531; Thermo Fisher Scientific) in 2 mL Opti-MEM medium. The mixture of DNA-reagent was added to flasks after incubation for 15 min at room temperature, and cells were cultured at 37°C, 5% CO2 for 24 h. The supernatant was centrifuged at 800 × g for 5 min, filtered through a 0.45-μm filter, and stored at −80°C. The GP of packaged HIV-EBOV GP-Luc virus was identified by western blotting using MIL77-3, a previously reported antibody specific to EBOV GP. Luciferase activity was detected with the Luciferase Assay System (E1501; Promega) and expressed as relative luciferase units to determine the dilution used in neutralization assays. All operations involving the use of pseudoviruses were carried out under biosafety level 2 conditions.
Labeling of GP∆Muc with FITC
About 400 μg of GP∆Muc was prepared in 200 μL of fresh 0.1 M sodium carbonate buffer (pH 9.0) before conjugation. The solution was mixed with 20 μL FITC (F4274; Sigma-Aldrich) at 1 mg/mL, and then incubated in the dark for 8 h at 4°C. NH4Cl was added to a final concentration of 50 mM and incubated for 2 h at 4°C to stop the reaction. Conjugated GP∆Muc was buffer-exchanged into PBS using a centrifugal filter unit until the filtrate was colorless. The FITC-labeled GP∆Muc was stored away from light at 4°C.
Isolation of memory B cells and single-cell PCR
PBMCs were isolated from clinical subjects one month after booster immunization during the Phase 1 clinical trial of Ad5-EBOV, and frozen in liquid nitrogen. PBMCs were thawed and centrifuged at 800 × g for 5 min to remove the storage solution. Cells were washed twice using 2 mL FPBS (PBS containing 2% (v/v) fetal bovine serum (FBS)) and resuspended in 100 μL FPBS. Memory B cell sorting was performed following a previously described method with some modifications.Citation45,Citation46 Briefly, 1 × 106 cells were incubated with fluorescently-conjugated antibodies against CD3 (PerCP, SP34-2; BD Biosciences), CD19 (APC-AF 700, J3-119; BD Biosciences), CD27 (PE-Cy7, 1A4CD27; Beckman-Coulter), CD38 (PerCP, HIT2; Biolegend), and IgG (PE, G18-145; BD Biosciences) according to the manufacturers’ instructions. GP∆Muc-FITC was also used in the fluorescence cocktail at 10 μg/mL to identify antigen-specific memory B cells. The cells were washed with FPBS after 1 h of staining at 4°C, and then filtered through a 40-μm cell strainer. GP∆Muc-specific memory B cells were sorted on a SH800 S flow cell sorter (SONY) to isolate single IgG+CD3−CD38−CD19+CD27+GP∆Muc+ cells. Single cells were sorted into 96-well PCR plates (MLL9601; Bio-Rad) containing 20 μL RNase-free water (GI201-01; TransGen) and 20 U of RNasin inhibitors (N2115; Promega) in each well. After cell sorting, RT-PCR was immediately carried out using the SuperScript™ III First-Strand Synthesis System (18080051; Invitrogen, Thermo Fisher Scientific), and followed by a nested PCR using TransStart Taq DNA polymerase (AP141; TransGen) to amplify cDNAs encoding VH and VL of antibodies from single cells.Citation47,Citation48 VH or VL genes were then constructed into linear cassettes containing a CMV promoter, Ig leader fragments, CH or CL of IgG1, and poly-A tail to obtain full-length Ig heavy and light chains as previously described.Citation31
Quick expression of antibodies using linear cassettes
Human embryonic kidney HEK293 T cells (ATCC® CRL-11268; ATCC) were seeded into 96-well culture plates at 2 × 104 cells/well 24 h before transfection and cultured in 200 μL Dulbecco’s Modified Eagle Medium (DMEM) (C11995500; Gibco) supplemented with 10% FBS, 100 mg/mL streptomycin, and 100 I.U./mL penicillin at 37°C and 5% CO2. Linear DNA vectors expressing genes of heavy and light chains (0.15 μg each) were mixed with 0.4 μL TurboFect transfection reagent in 20 μL Opti-MEM medium and incubated at room temperature for 15 min. After transfection, cells were cultured at 37°C, 5% CO2 for 36 h and the supernatant was collected to screen for specific antibodies.
ELISA
To screen serum samples from human vaccines, EBOV, BDBV or SUDV GPdTM proteins were coated onto microplates (9018; Corning) at 1 μg/mL and incubated overnight at 4°C. On the following day, the plates were washed three times and blocked with 2% (w/v) bovine serum albumin (BSA) in PBS containing 0.1% (v/v) Tween-20 (PBST) for 1 h at 37°C. After three washes, serial 3-fold dilutions of the pre-vaccination serum (V0) or the serum on day 28 post-2nd vaccination (V10) of vaccines # 024, 057 or 088 were added (100 μL/well) and incubated for 1 h at 37°C, followed by three washes. Plates were washed and a 1:10,000 dilution of goat anti-human IgG Fc-HRP (Ab97225; Abcam) diluted in PBST containing 0.1% (w/v) BSA was added for 1 h at 37°C. After a final wash, plates were incubated with 100 μL of 3,3ʹ,5,5ʹ-tetramethylbenzidine substrate (PR1200; Solarbio) for 6 min at room temperature, followed by addition of 50 μL stop solution. The optical density at a dual-wavelength of 450 nm and 630 nm was read on a Spectramax 190 reader (Molecular Devices).
To screen for GP-specific antibodies, different GPs were coated onto 96-well microplates at 1 μg/mL. After blocking at 37°C for 1 h, plates were washed before adding 100 μL of collected supernatant described above and then incubated at 37°C for 1 h. The same procedures described above were applied thereafter. The cutoff value was defined as twice the OD value of the negative control.
To assay the binding activity of purified mAbs, microplates were coated with GPs at 1 μg/mL and incubated with purified antibodies in serial dilutions starting at 10 μg/mL. Optical density was detected using goat anti-human IgG Fc-HRP at 450/630 nm and transformed to fit a four-parameter curve. The EC50 of each antibody was calculated using GraphPad Prism 7 software.
Sequence analysis of GP-binding antibodies
The nested PCR products of positive clones identified by ELISA were sequenced (Sangon Biotech) and then gene family usage, nucleotide mutations, as well as amino acids changes of variable regions were analyzed using IMGT/V-Quest (http://www.imgt.org/IMGT_vquest). Phylogeny trees were built using MEGA version 7.0 (https://megasoftware.net/) based on ClustalW alignment and the neighbor-joining method. Diversity and conservation of variable regions were depicted with WebLogo 3 (http://weblogo.threeplusone.com/create.cgi).
Generation of antibodies and fabs
To produce antibodies, the linear cassettes of heavy or light chains were cleaved by EcoRI and NotI (R3101, R3189; New England Biolabs), and ligated into the pcDNA3.4 vector by T4 DNA Ligase (M0202; New England Biolabs). Cloned plasmids were aligned with sequences of nested PCR fragments by Vector NTI (Thermo Fisher Scientific) and extracted using the PureYieldTM plasmid miniprep system (A1222; Promega). Heavy and light chains (15 μg each) were used for expression in Expi293 F™ cells as described above. Antibodies were purified on a HiTrap rProtein A column (17507901; GE Healthcare) with PBS (pH 7.4) for equilibrium, 0.1 M glycine (pH 3.0) for elution, and 1 M Tris for neutralization (pH 9.0). The antibody concentration was determined by a BCA Protein Assay Kit after buffer-exchange into PBS, and the antibodies were stored at −80°C.
The constant regions of IgG1 were added after the synthesis of VH or VL genes of MR72/191,Citation33 and antibodies were expressed and prepared in the same way as described above.
The Fab was generated as previously described.Citation49 Briefly, the fragment containing VH and CH1 domains with a 6× His tag at the C-terminus (Fd-His6) was amplified from full-length heavy chain, and then cloned into the pcDNA3.4 vector. The pcDNA3.4-Fd-His6 and full-length light chain were co-transfected into Expi293 F™ cells, and the Fab was purified on a HisTrap HP column.
Competition ELISA
Antibodies were labeled with EZ-Link Sulfo-NHS-LC-Biotin (21435; Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, 200 μg antibodies were incubated with a 20-fold molar excess of biotin at room temperature for 1 h. Biotinylated antibodies were buffer-exchanged several times to remove excess biotin, and the concentration was determined using NanoVue Plus (GE Healthcare) at 280 nm.
Purified GP∆Muc was coated onto microplates at 1 μg/mL and incubated overnight at 4°C. After blocking, biotinylated antibodies at a final EC50 calculated as above were mixed with 5 μg/mL of competitors (about 100-fold molar excess), and the mixture was added to coated plates, followed by incubation for 1 h at 37°C. An antibody specific to anthrax protective antigen, 8A7,Citation48 was used as an irrelevant competitor. Biotinylated antibodies bound to GP∆Muc were detected using HRP-conjugated streptavidin (SNN1004; Thermo Fisher Scientific) at 450/630 nm. The percentage of bound biotinylated antibodies was calculated by comparing the absorbance value in the presence of competitors to that in the presence of an irrelevant control. Antibodies were considered competing for the same or close epitope if the percentage of bound detecting mAb was <30%. Antibodies were assumed to bind to different sites if the percentage value was >60%. A group of intermediate competitive antibodies was identified if their percentage value was 30%~60%.
Neutralization of ebolavirus GP-pseudotyped HIV
To detect neutralizing activity in human blood, sera of donor vaccines were diluted in DMEM at a starting ratio of 1:5, followed by 3-fold serial dilutions. Next, 50 μL of diluted sera were incubated with an equal volume of HIV-EBOV GP-Luc (luciferase units were adjusted to 20,000 ~ 100,000 in the absence of mAbs) for 1 h at 37°C. Then, 2 × 104 293 T cells in 100 μL DMEM were added to the virus-antibody mixtures. After infection at 37°C, 5% CO2 for 36 h, medium was removed, and cells were incubated with 50 μL lysis buffer (E1531; Promega) for 10 min at room temperature. A 20-μL volume of cell lysate was added to 96-well white assay plates (3599; Costar, Corning) and light intensity was read immediately on a GloMax 96 Microplates Luminometer (Promega) after addition of 50 μL luciferase assay reagent to each well. The neutralizing ability of mAbs was calculated by comparing the light intensity of wells in the presence of mAbs to that of wells containing virus only, and the IC50 was calculated by fitting to a four-parameter curve using GraphPad Prism 7 software.
To examine neutralizing activity of purified mAbs or Fabs, 50 μL antibodies or Fabs in serial dilutions starting at 100 μg/mL were incubated with 50 μL diluted HIV pseudotype ebolavirus in 96-well plates at 37°C for 1 h, followed by the same procedures described above.
EBOV/May-eGFP neutralization assay
Neutralizing activity of the mAbs against authentic EBOV was assessed in the NML CL4 laboratory. Basically, mAbs in serial dilutions were pre-incubated with EBOV/May-eGFP at a final multiplicity of infection of 0.05 for 1 h at 37°C before applying to green monkey Vero E6 cells (ATCC) in 96-well plates for 1 h at 37°C. The inocula were then removed, and cells were maintained in fresh DMEM containing 2% (v/v) bovine growth serum for 3 to 4 days at 37°C, 5% CO2. GFP intensities were recorded using a Synergy HTX Multi-Mode Microplate Reader (BioTek).
Prediction of antibody-antigen complex structure
The structure of the mAb Fab-GP complex was predicted using a method based on our previous work.Citation32 Briefly, a homologous constructed model of mAb Fab and a structure of EBOV GP (PDB ID: 5KEL) were used for docking with a Dock Proteins protocol (ZDOCK) in Discovery Studio 4.5 (https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/). Approximately 54,000 poses of antigen-antibody complexes were generated and followed by evaluation with a ZRANK scoring function. The top 101 poses with the highest ZRANK score were used to calculate the RCF of Fabs and GP.
Cell imaging
Hamster BHK-T7 cells were seeded into 24-well plates at 1.5 × 105 cells/well and cultured in DMEM at 37°C, 5% CO2. Cells were transfected with 1 μg PDC316-EBOV GPfull-length when cell density reached 80%. After incubation for 24 h at 37°C and 5% CO2, medium was removed, and cells were incubated with 5 μg/mL mAbs in fresh medium at 37°C for 1 h. Cells were washed twice and incubated with 5 μL PE-conjugated mouse anti-human IgG (555787; BD Biosciences) in 200 μL fresh medium at 37°C for 1 h. After a final wash, the fluorescent images were captured under a 4× objective in the red channel (586 nm, 647 nm) using a Cytation imaging reader (BioTek).
Binding of mAbs to cell surface-displayed GPs
To display GP or GP∆Muc on the membrane surface, 20 μg PDC316-EBOV GP or GP∆Muc containing full-length GP2 was transfected into 293 T cells cultured in T75 flasks. Cells were cultured at 37°C, 5% CO2 for 24 h, medium was removed, and cells were washed twice with 10 mL FPBS by centrifugation at 500 × g for 5 min and 4°C. For mAbs binding only, ~5 × 105 cells in 100 μL FPBS were incubated with 10 μg/mL mAbs at 37°C, 5% CO2 for 1 h. For thermolysin cleavage followed by mAbs binding, ~5 × 105 cells were incubated with 0.25 mg/mL thermolysin at 37°C, 5% CO2 for 1 h and washed before incubation with 10 μg/mL mAbs. Cells were washed and incubated with 10 μL PE-conjugated mouse anti-human IgG at room temperature for 1 h. Unbound PE-conjugated antibody was removed by a final wash, and cells were resuspended in 200 μL FPBS. Stained cells were measured using a FACSCanto II flow cytometer (BD Biosciences), 50,000 events of each test were recorded and then analyzed using FlowJo V10 software. A GC-specific mAb, an irrelevant antibody, as well as an EBOV GPcl-binding mAb rather than GP- or GP∆Muc-binding mAbs served as positive or negative controls in this experiment.
Inhibition of NPC1-C binding
Plates were coated overnight with 1 μg/mL GPcl at 4°C. After blocking, plates were incubated with mAbs in serial dilutions starting at 50 μg/mL for 30 min at 37°C. Plates were washed and incubated with 5 μg/mL biotinylated NPC1-C at 37°C for 30 min. Bound NPC1-C was detected at 450/630 nm using HRP-conjugated streptavidin. Inhibition of NPC1-C binding was calculated by comparing the optical density value in the presence of mAbs to that in the presence of an irrelevant control. MR72, a previously reported competitive mAb against NPC1-C, was used as a positive control.
Protection in MA-EBOV-challenged mice
Briefly, 4-8-week-old female BALB/c mice (Charles River, Canada) were housed in microisolator cages in the NML CL4 laboratory. Groups of BALB/c mice (n = 10) were challenged with 1000-fold LD50 of MA-EBOV Mayinga (GenBank: AF499101) via the IP route, and intraperitoneally treated with 100 μg of individual mAbs or the same volume of PBS 1 day post challenge. Mice were monitored daily for signs of disease and weight changes for 14 days, and observed for another 14 days. Moribund mice were humanely euthanized according to the CCAC guidelines. All mice were euthanized at the end of the study.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download MS Word (3.4 MB)Acknowledgments
We thank Dr. Feng Jiannan for providing MIL77-1/2/3; thank Kevin Terniery, Alix Albietz and VTS staff at NML for assistance with animal care and technical support.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- World Health Organization. Ebola virus disease; 2019. [accessed 2019 Dec 18]. https://www.who.int/en/news-room/fact-sheets/detail/ebola-virus-disease
- Goldstein T, Anthony SJ, Gbakima A, Bird BH, Bangura J, Tremeau-Bravard A, Belaganahalli MN, Wells HL, Dhanota JK, Liang E, et al. The discovery of Bombali virus adds further support for bats as hosts of ebolaviruses. Nat Microbiol. 2018;3:1084–15. doi:https://doi.org/10.1038/s41564-018-0227-2.
- Forbes KM, Webala PW, Jaaskelainen AJ, Abdurahman S, Ogola J, Masika MM, Kivisto I, Alburkat H, Plyusnin I, Levanov L, et al. Bombali virus in mops condylurus Bat, Kenya. Emerg Infect Dis. 2019;25. doi:https://doi.org/10.3201/eid2505.181666.
- Centers for Disease Control and Prevention. Ebola virus outbreaks by species and size, since 1976; 2019. [accessed 2019 Dec 20]. https://www.cdc.gov/vhf/ebola/history/distribution-map.html
- Parra JM, Salmeron OJ, Velasco M. The first case of Ebola virus disease acquired outside Africa. N Engl J Med. 2014;371:2439–40. doi:https://doi.org/10.1056/NEJMc1412662.
- Liddell AM, Davey RT Jr., Mehta AK, Varkey JB, Kraft CS, Tseggay GK, Badidi O, Faust AC, Brown KV, Suffredini AF, et al. Characteristics and clinical management of a cluster of 3 patients with Ebola virus disease, including the first domestically acquired cases in the United States. Ann Intern Med. 2015;163:81–90. doi:https://doi.org/10.7326/M15-0530.
- Centers for Disease Control and Prevention (CDC). History and outbreaks of Ebola virus disease; 2018. [accessed 2019 Dec 20]. https://www.cdc.gov/vhf/ebola/history/distribution-map.html
- Le Guenno B, Formenty P, Wyers M, Gounon P, Walker F, Boesch C. Isolation and partial characterisation of a new strain of Ebola virus. Lancet. 1995;345:1271–74. doi:https://doi.org/10.1016/S0140-6736(95)90925-7.
- Burk R, Bollinger L, Johnson JC, Wada J, Radoshitzky SR, Palacios G, Bavari S, Jahrling PB, Kuhn JH. Neglected filoviruses. FEMS Microbiol Rev. 2016;40:494–519. doi:https://doi.org/10.1093/femsre/fuw010.
- Cross RW, Mire CE, Feldmann H, Geisbert TW. Post-exposure treatments for Ebola and Marburg virus infections. Nat Rev Drug Discovery. 2018;17:413–34. doi:https://doi.org/10.1038/nrd.2017.251.
- Hoenen T, Groseth A, Feldmann H. Therapeutic strategies to target the Ebola virus life cycle. Nat Rev Microbiol. 2019;17:593–606. doi:https://doi.org/10.1038/s41579-019-0233-2.
- Qiu X, Wong G, Audet J, Bello A, Fernando L, Alimonti JB, Fausther-Bovendo H, Wei H, Aviles J, Hiatt E, et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature. 2014;514:47–53. doi:https://doi.org/10.1038/nature13777.
- Qiu X, Audet J, Lv M, He S, Wong G, Wei H, Luo L, Fernando L, Kroeker A, Fausther Bovendo H, et al. Two-mAb cocktail protects macaques against the Makona variant of Ebola virus. Sci Transl Med. 2016;8:329ra33. doi:https://doi.org/10.1126/scitranslmed.aad9875.
- Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, Ploquin A, Doria-Rose NA, Staupe RP, Bailey M, et al. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science. 2016;351:1339–42. doi:https://doi.org/10.1126/science.aad5224.
- Pascal KE, Dudgeon D, Trefry JC, Anantpadma M, Sakurai Y, CD M, Turner HL, Fairhurst J, Torres M, Rafique A, et al. Development of clinical-stage human monoclonal antibodies that treat advanced Ebola virus disease in nonhuman primates. J Infect Dis. 2018;218:S612–S26. doi:https://doi.org/10.1093/infdis/jiy285.
- Warren TK, Jordan R, Lo MK, Ray AS, Mackman RL, Soloveva V, Siegel D, Perron M, Bannister R, Hui HC, et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. 2016;531:381–85. doi:https://doi.org/10.1038/nature17180.
- New antibodies best ZMapp in Ebola trial. Nat Biotechnol. 2019;37:1105. doi:https://doi.org/10.1038/s41587-019-0284-y.
- Keck ZY, Enterlein SG, Howell KA, Vu H, Shulenin S, Warfield KL, Froude JW, Araghi N, Douglas R, Biggins J, et al. Macaque monoclonal antibodies targeting novel conserved epitopes within filovirus glycoprotein. J Virol. 2016;90:279–91. doi:https://doi.org/10.1128/JVI.02172-15.
- Bornholdt ZA, Turner HL, Murin CD, Li W, Sok D, Souders CA, Piper AE, Goff A, Shamblin JD, Wollen SE, et al. Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science. 2016;351:1078–83. doi:https://doi.org/10.1126/science.aad5788.
- Zhao X, Howell KA, He S, Brannan JM, Wec AZ, Davidson E, Turner HL, Chiang CI, Lei L, Fels JM, et al. Immunization-elicited broadly protective antibody reveals ebolavirus fusion loop as a site of vulnerability. Cell. 2017;169:891–904 e15. doi:https://doi.org/10.1016/j.cell.2017.04.038.
- Gilchuk P, Kuzmina N, Ilinykh PA, Huang K, Gunn BM, Bryan A, Davidson E, Doranz BJ, Turner HL, Fusco ML, et al. Multifunctional pan-ebolavirus antibody recognizes a site of broad vulnerability on the Ebolavirus glycoprotein. Immunity. 2018;49:363–74 e10. doi:https://doi.org/10.1016/j.immuni.2018.06.018.
- Fischer WA, Brown J, Wohl DA, Loftis AJ, Tozay S, Reeves E, Pewu K, Gorvego G, Quellie S, Cunningham CK, et al. Ebola virus ribonucleic acid detection in semen more than two years after resolution of acute ebola virus infection. Open Forum Infect Dis. 2017;4:ofx155. doi:https://doi.org/10.1093/ofid/ofx155.
- Zhu F-C, Hou L-H, Li J-X, Wu S-P, Liu P, Zhang G-R, Hu Y-M, Meng F-Y, Xu -J-J, Tang R, et al. Safety and immunogenicity of a novel recombinant adenovirus type-5 vector-based Ebola vaccine in healthy adults in China: preliminary report of a randomised, double-blind, placebo-controlled, phase 1 trial. The Lancet. 2015; 385:2272–79. doi:https://doi.org/10.1016/s0140-6736(15)60553-0
- Zhu F-C, Wurie AH, Hou L-H, Liang Q, Li Y-H, Russell JBW, Wu S-P, Li J-X, Hu Y-M, Guo Q, et al. Safety and immunogenicity of a recombinant adenovirus type-5 vector-based Ebola vaccine in healthy adults in Sierra Leone: a single-centre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2017;389:621–28. doi:https://doi.org/10.1016/s0140-6736(16)32617-4.
- Takada A, Robison C, Goto H, Sanchez A, Murti KG, Whitt MA, Kawaoka Y. A system for functional analysis of Ebola virus glycoprotein. Proc Natl Acad Sci U S A. 1997;94:14764–69. doi:https://doi.org/10.1073/pnas.94.26.14764.
- Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008;454:177–82. doi:https://doi.org/10.1038/nature07082.
- Ng M, Ndungo E, Jangra RK, Cai Y, Postnikova E, Radoshitzky SR, Dye JM, Ramirez de Arellano E, Negredo A, Palacios G, et al. Cell entry by a novel European filovirus requires host endosomal cysteine proteases and Niemann-Pick C1. Virology. 2014;468-470:637–46. doi:https://doi.org/10.1016/j.virol.2014.08.019.
- Wang H, Shi Y, Song J, Qi J, Lu G, Yan J, Gao GF. Ebola viral glycoprotein bound to its endosomal receptor Niemann-Pick C1. Cell. 2016;164:258–68. doi:https://doi.org/10.1016/j.cell.2015.12.044.
- Zhang Q, Gui M, Niu X, He S, Wang R, Feng Y, Kroeker A, Zuo Y, Wang H, Wang Y, et al. Potent neutralizing monoclonal antibodies against Ebola virus infection. Sci Rep. 2016;6:25856. doi:https://doi.org/10.1038/srep25856.
- Davidson E, Bryan C, Fong RH, Barnes T, Pfaff JM, Mabila M, Rucker JB, Doranz BJ. Mechanism of binding to ebola virus glycoprotein by the ZMapp, ZMAb, and MB-003 cocktail antibodies. J Virol. 2015;89:10982–92. doi:https://doi.org/10.1128/JVI.01490-15.
- Liao HX, Levesque MC, Nagel A, Dixon A, Zhang R, Walter E, Parks R, Whitesides J, Marshall DJ, Hwang KK, et al. High-throughput isolation of immunoglobulin genes from single human B cells and expression as monoclonal antibodies. J Virol Methods. 2009;158:171–79. doi:https://doi.org/10.1016/j.jviromet.2009.02.014.
- Liu W, Ren J, Zhang J, Song X, Liu S, Chi X, Chen Y, Wen Z, Li J, Chen W. Identification and characterization of a neutralizing monoclonal antibody that provides complete protection against Yersinia pestis. PLoS One. 2017;12:e0177012. doi:https://doi.org/10.1371/journal.pone.0177012.
- Flyak AI, Ilinykh PA, Murin CD, Garron T, Shen X, Fusco ML, Hashiguchi T, Bornholdt ZA, Slaughter JC, Sapparapu G, et al. Mechanism of human antibody-mediated neutralization of Marburg virus. Cell. 2015;160:893–903. doi:https://doi.org/10.1016/j.cell.2015.01.031.
- Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, et al. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. Embo J. 2012;31:1947–60. doi:https://doi.org/10.1038/emboj.2012.53.
- Wec AZ, Herbert AS, Murin CD, Nyakatura EK, Abelson DM, Fels JM, He S, James RM, de La Vega M-A, Zhu W, et al. Antibodies from a human survivor define sites of vulnerability for broad protection against Ebolaviruses. Cell. 2017;169:878–90.e15. doi:https://doi.org/10.1016/j.cell.2017.04.037.
- Borio L, Inglesby T, Peters CJ, Schmaljohn AL, Hughes JM, Jahrling PB, Ksiazek T, Johnson KM, Meyerhoff A, O’Toole T, et al. Hemorrhagic fever viruses as biological weapons: medical and public health management. Jama. 2002;287:2391–405. doi:https://doi.org/10.1001/jama.287.18.2391.
- Yang XL, Tan CW, Anderson DE, Jiang RD, Li B, Zhang W, Zhu Y, Lim XF, Zhou P, Liu XL, et al. Characterization of a filovirus (Mengla virus) from Rousettus bats in China. Nat Microbiol. 2019;4:390–95. doi:https://doi.org/10.1038/s41564-018-0328-y.
- U.S. Food and Drug Administration. First FDA-approved vaccine for the prevention of Ebola virus disease, marking a critical milestone in public health preparedness and response; 2019. [accessed 2019 Dec 22]. https://www.fda.gov/news-events/press-announcements/first-fda-approved-vaccine-prevention-ebola-virus-disease-marking-critical-milestone-public-health?utm_campaign=121919_PR_First%20FDA-approved%20vaccine%20for%20the%20prevention%20of%20Ebola%20virus%20disease&utm_medium=email&utm_source=Eloqua
- Ehrhardt SA, Zehner M, Krahling V, Cohen-Dvashi H, Kreer C, Elad N, Gruell H, Ercanoglu MS, Schommers P, Gieselmann L, et al. Polyclonal and convergent antibody response to Ebola virus vaccine rVSV-ZEBOV. Nat Med. 2019;25:1589–600. doi:https://doi.org/10.1038/s41591-019-0602-4.
- Rijal P, Elias SC, Machado SR, Xiao J, Schimanski L, O’Dowd V, Baker T, Barry E, Mendelsohn SC, Cherry CJ, et al. Therapeutic monoclonal antibodies for Ebola virus infection derived from vaccinated humans. Cell Rep. 2019;27:172–86 e7. doi:https://doi.org/10.1016/j.celrep.2019.03.020.
- Saphire EO, Schendel SL, Fusco ML, Gangavarapu K, Gunn BM, Wec AZ, Halfmann PJ, Brannan JM, Herbert AS, Qiu X, et al. Systematic analysis of monoclonal antibodies against Ebola virus GP defines features that contribute to protection. Cell. 2018;174:938–52.e13. doi:https://doi.org/10.1016/j.cell.2018.07.033.
- Saphire EO, Aman MJ. Feverish quest for ebola immunotherapy: straight or cocktail? Trends Microbiol. 2016;24:684–86. doi:https://doi.org/10.1016/j.tim.2016.05.008.
- de La Vega MA, Wong G, Kobinger GP, Qiu X. The multiple roles of sGP in Ebola pathogenesis. Viral Immunol. 2015;28:3–9. doi:https://doi.org/10.1089/vim.2014.0068.
- Hashiguchi T, Fusco ML, Bornholdt ZA, Lee JE, Flyak AI, Matsuoka R, Kohda D, Yanagi Y, Hammel M, Crowe JE Jr., et al. Structural basis for Marburg virus neutralization by a cross-reactive human antibody. Cell. 2015;160:904–12. doi:https://doi.org/10.1016/j.cell.2015.01.041.
- Wang Y, Sundling C, Wilson R, O’Dell S, Chen Y, Dai K, Phad GE, Zhu J, Xiao Y, Mascola JR, et al. High-resolution longitudinal study of HIV-1 Env vaccine-elicited B cell responses to the virus primary receptor binding site reveals affinity maturation and clonal persistence. J Immunol. 2016;196:3729–43. doi:https://doi.org/10.4049/jimmunol.1502543.
- Sholukh AM, Mukhtar MM, Humbert M, Essono SS, Watkins JD, Vyas HK, Shanmuganathan V, Hemashettar G, Kahn M, Hu SL, et al. Isolation of monoclonal antibodies with predetermined conformational epitope specificity. PLoS One. 2012;7:e38943. doi:https://doi.org/10.1371/journal.pone.0038943.
- Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R, Wilson PC. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc. 2009;4:372–84. doi:https://doi.org/10.1038/nprot.2009.3.
- Chi X, Li J, Liu W, Wang X, Yin K, Liu J, Zai X, Li L, Song X, Zhang J, et al. Generation and characterization of human monoclonal antibodies targeting anthrax protective antigen following vaccination with a recombinant protective antigen vaccine. Clin Vaccine Immunol. 2015;22:553–60. doi:https://doi.org/10.1128/CVI.00792-14.
- Zhao Y, Gutshall L, Jiang H, Baker A, Beil E, Obmolova G, Carton J, Taudte S, Amegadzie B. Two routes for production and purification of Fab fragments in biopharmaceutical discovery research: papain digestion of mAb and transient expression in mammalian cells. Protein Expr Purif. 2009;67:182–89. doi:https://doi.org/10.1016/j.pep.2009.04.012.