
ABSTRACT
The role of brain-derived neurotrophic factor (BDNF) signaling in chronic pain has been well documented. Given the important central role of BDNF in long term plasticity and memory, we sought to engineer a high affinity, peripherally-restricted monoclonal antibody against BDNF to modulate pain. BDNF shares 100% sequence homology across human and rodents; thus, we selected chickens as an alternative immune host for initial antibody generation. Here, we describe the affinity optimization of complementarity-determining region-grafted, chicken-derived R3bH01, an anti-BDNF antibody specifically blocking the TrkB receptor interaction. Antibody optimization led to the identification of B30, which has a > 300-fold improvement in affinity based on BIAcore, an 800-fold improvement in potency in a cell-based pERK assay and demonstrates exquisite selectivity over related neurotrophins. Affinity improvements measured in vitro translated to in vivo pharmacological activity, with B30 demonstrating a 30-fold improvement in potency over parental R3bH01 in a peripheral nerve injury model. We further demonstrate that peripheral BDNF plays a role in maintaining the plasticity of sensory neurons following nerve damage, with B30 reversing neuron hyperexcitability associated with heat and mechanical stimuli in a dose-dependent fashion. In summary, our data demonstrate that effective sequestration of BDNF via a high affinity neutralizing antibody has potential utility in modulating the pathophysiological mechanisms that drive chronic pain states.
Introduction
Neurotrophins represent a family of structurally-related proteins that have diverse biological functions. During development, neurotrophins critically regulate processes underlying cell survival and neuronal differentiation. In the adult nervous system, neurotrophin signaling is required for maintenance of synaptic function and plasticity.Citation1-Citation3 Damage to peripheral nerves triggers changes in the expression of ion channels, neuropeptides, growth factors, cytokines and chemokines that ultimately culminates in enhanced excitability of the peripheral and central nervous system.Citation4 Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin family that is upregulated following peripheral nerve injury.Citation5-Citation10 Following synthesis in small- to medium-sized dorsal root ganglion (DRG) neurones,Citation9 BDNF is anterogradely transported to the dorsal horn where it is released upon noxious stimulation in an activity-dependent manner.Citation11 The physiological actions of BDNF are mediated via interaction with two receptors, the high affinity tyrosine receptor kinase B (TrkB) and p75NTR, also known as low-affinity nerve growth factor receptor (LNGFR). BDNF engagement of TrkB results in receptor dimerization, leading to autophosphorylation of tyrosine residues in the cytoplasmic domain. This results in the presentation of docking sites for adapter proteins containing phosphotyrosine-binding (PTB) or src-homology-2 (SH2) motifs that couple the receptor to multiple intracellular signaling cascades, including Ras/ERK (extracellular signal–regulated kinase), PI3 K (phosphatidylinositol-3-kinase) and PLC-γ (phospholipase C γ).Citation1,Citation2 p75NTR, on the other hand, is a member of the tumor necrosis receptor superfamily that binds all members of the neurotrophin family with similar affinities. Activation of p75NTR may potentiate Trk signaling and furthermore promote the discrimination of Trk receptors for their preferred neurotrophin ligands.Citation12,Citation13
BDNF has been documented to be a key driver of central sensitization in the spinal cord, and accumulating evidence suggests that microglial-derived BDNF promotes long-term hypersensitivity and aberrant nociceptive processing through mechanisms involving spinal disinhibition.Citation14 In support of this, sequestration of spinal BDNF using the scavenging molecule TrkB-Fc attenuated pain related behavior in models of peripheral nerve damage.Citation15-Citation17 TrkB-Fc is a fusion protein that comprises the Fc domain of human IgG1 and the ectodomain of TrkB, and it has been used as a tool to specifically antagonize BDNF activity. To date, many studies have focused on the central role of BDNF using local administration of TrkB-Fc. However, few studies have investigated how peripherally-derived BDNF affects sensory processing under chronic pain conditions. In human, dysregulated BDNF levels have been documented in joint diseases, peripheral nerve damage, intervertebral disc degeneration, bladder cancer and chronic pancreatitis,Citation18-Citation24 with some reports demonstrating correlations between peripheral BDNF levels and pain or disease severity.Citation25-Citation28 Agents that specifically recognize BDNF, and inhibit BDNF signaling through its receptor, may provide a novel approach for the treatment of certain chronic pain conditions. However, a key consideration in the design of a potential therapeutic is the restriction of drug access to the brain to ensure there is minimal interference of BDNF function in the CNS. Given the key role of this mediator in long-term synaptic plasticity, learning and memory, disruption of this pathway could lead to undesirable cognitive effects.
Recent technological advances in antibody engineering have led to a growing interest in the development of monoclonal antibodies for therapeutic clinical applications. Monoclonal antibodies offer high specific binding to the target with little off-target activity, a long half-life (>20 days) and limited CNS penetration based on molecular weight. The therapeutic potential of an anti-NGF monoclonal antibody has been demonstrated in patients with advanced osteoarthritis and chronic low back pain, where it was shown to provide long-term analgesic efficacy.Citation29-Citation32 Given the role of BDNF in chronic pain, we sought to engineer an antibody that would preferentially block BDNF signaling with high affinity and specificity. The starting point for optimization was a BDNF monoclonal antibody, R3bH01, which was derived using a combination of in vivo chicken immunization and in vitro phage display.Citation33 Here, we describe the affinity optimization of a complementarity determining region (CDR)-grafted variant of R3bH01. Optimization led to the identification of B30, which has a > 300-fold improvement in affinity based on BIAcore, an 800-fold improvement in potency in a cell based pERK assay and demonstrates exquisite selectivity over related neurotrophins. To explore whether the affinity and potency improvements measured in vitro translated to in vivo pharmacological activity, we compared the parental R3bH01 to optimized B30 in a rat model of peripheral nerve injury. B30 demonstrated a return to baseline in the Kv current suppression model at 0.03 mg/kg compared to the 1 mg/kg required for the parental antibody. We further demonstrate that peripheral BDNF plays a role in maintaining the plasticity of sensory neurons following nerve damage, with B30 reversing neuron hyperexcitability associated with heat and mechanical stimuli in a dose-dependent fashion. In summary, our data demonstrate that effective sequestration of BDNF via a high affinity neutralizing antibody has potential utility in modulating the pathophysiological mechanisms that drive chronic pain states.
Results
Discovery, humanization & optimization of an anti-BDNF antibody
In vivo immunization of chickens was coupled with in vitro phage display to derive a panel of neutralizing anti-BDNF antibodies (workflow summarized in supplementary Figure 1a and methods described by Finlay et alCitation34). Clone R3bH01 was chosen for humanization based on BDNF binding (supplementary Figure 1b) and TrkB neutralization (supplementary Figure 1c). A classical CDR-grafting approach was taken, grafting the chicken-derived CDRs onto well-behaved acceptor human frameworks VH3-23;Vλ3-19.Citation35,Citation36 A single back-mutation, L46 T, was required in the VL FW1 region to retain the functionality of the parental chicken-human chimeric IgG1. This particular back-mutation has been previously described during humanization of a chicken anti-IL12 antibody.Citation35 The resulting humanized H01 was shown to maintain affinity for BDNF using an epitope competition assay (supplementary Figure 2a) and Biacore kinetics (supplementary Figure 2b).
Figure 1. Workflow for anti-BDNF antibody affinity maturation
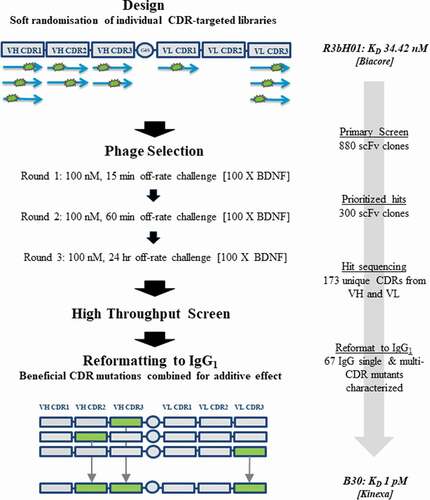
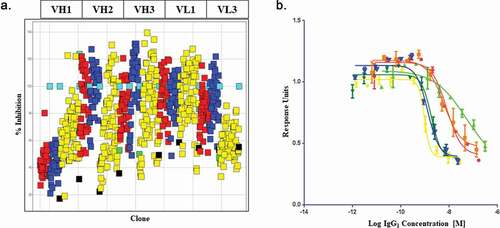
The approach taken to optimization is summarized in . CDR mutagenesis was achieved using a soft randomization approach that aims to incorporate approximately 50% wild-type and 50% any other amino acid at each position across all CDRs except VL-CDR2. Mutational libraries were assessed by sequence verification of approximately 96 clones per library. In our experience, this primer-based method of mutagenesis generally leads to wild-type representation ranging from 40% to 70% at each amino acid position. Sequencing quality control for all 5 targeted CDRs is included in supplementary Figure 3. Libraries were rescued using helper phage M13K07 and phage pools were subjected to off-rate selection in order to drive affinity. Selection outputs were expressed as single-chain variable fragments (scFv) in periplasmic fractions and assessed for performance against the parental R3bH01 and humanized H01 in a competition homogeneous time resolved fluorescence (HTRF) assay (). Top-performing clones from the competition enzyme-linked immunosorbent assay (ELISA) were reformatted into full-length IgG1 in a combinatorial fashion, mixing mutant VH and VL chains, in addition to combining mutations across multiple CDRs. IgGs were prioritized for potency in the pERK TrkB signaling assay (). Cumulative data identified B30 as the top performing high affinity variant and this was brought forward for further characterization. Given the extensive surface positive charge and high pI of BDNF, lead clones were also assessed for nonspecific binding to several related neurotrophins and unrelated chemokines with high pI and positive charge patches. Representative data is shown in supplementary Figure 4.
Figure 2. High-throughput screening of soft mutagenesis phage libraries for affinity-improved anti-BDNF antibodies
Affinity determination of BDNF antibodies by surface plasmon resonance and KinExA analysis
Surface plasmon resonance (SPR) was used to determine the binding kinetics of B30 compared to the parental chimeric R3bH01 antibody. Low densities of human BDNF were directly immobilized onto a Biacore sensor chip to reduce nonspecific binding and to minimize avidity and rebinding effects. A dilution series of the R3bH01 and affinity-optimized B30 were flowed over the sensor chip surface, providing dose-dependent responses (). The dissociation phase for the highest concentration of antibody was extended to 4000 sec to achieve a signal decay minimum of 5%.Citation37 The kinetic rate constants were determined and the apparent KD values are reported in .
Table 1. Comparison of parental chicken R3bH01 to humanized & affinity optimized B30
Figure 3. Demonstration of affinity improvement of B30 over parental R3bH01
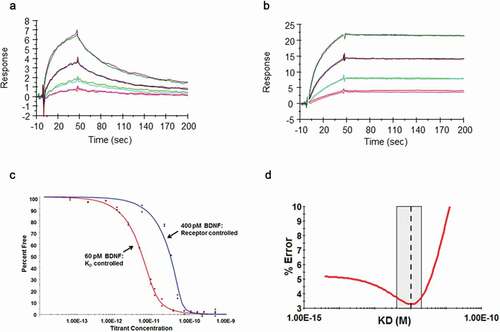
However, the off-rate for B30 was reaching the limits of sensitivity of the BIAcore T200, and a solution phase kinetic exclusion assay was subsequently used to determine the equilibrium dissociation constant for the antigen-binding fragment (Fab) of B30 binding to human BDNF.Citation38 Two fixed concentrations of BDNF were used, which aimed to provide KD and active binding concentration (ABC)-controlled curves to permit a global N-curve analysis of the sensorgram data (). The ABCs used were 60 pM for a KD controlled curve and 400 pM for a receptor-controlled curve, incorporating 2 binding sites for each BDNF homodimer. A wide range of Fab concentrations, two replicates for each concentration, yielded several data points between 0 and 100% free human BDNF, indicating assay reproducibility and confirming equilibrium. A 95% confidence interval of KD values and % error are shown in .
The SPR apparent KD value for B30 was determined to be 120 pM, indicating a 286-fold improvement over R3bH01, with off-rate being the main contributor to this effect. The KinExA-determined equilibrium dissociation rate constant for Fab B30 was 1 pM. This difference may be explained by the potential effect on binding functionality by directly immobilizing BDNF or solid surface matrix effects.Citation39 A number of attempts were made to reverse the orientation of the SPR assay, but, as a result of the high pI of BDNF, this resulted in a significant charge-based ligand interaction with the CM5 dextran chip. In addition to this, although the SPR assay was carried out at a very fast flow rate and elevated temperature (37°C), it was still operating close to the limits of sensitivity of the SPR method.
Affinity optimized B30 is more effective than R3bH01 at reversing injury induced ion channel plasticity in rat dorsal root ganglion neurons
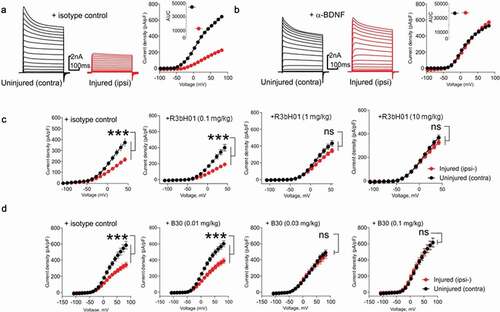
A phenomenon associated with peripheral nerve damage is a change in the expression pattern of ion channels,Citation40 which contributes to aberrant sensory processing. Voltage gated potassium (Kv) channels comprise a diverse family of ion channels that intrinsically govern resting membrane potential, firing frequency and shape action potential repolarization in excitable cells.Citation41 Dysregulation of Kv channels and a consequent reduction in the Kv current have been hypothesized to underlie neuronal hyperexcitability in injury states.Citation42-Citation45 A previous report has demonstrated that this may, in part, be mediated through elevated BDNF levels as a result of injury.Citation42 To evaluate whether altered BDNF signaling drives Kv channel plasticity, we investigated the effects of peripheral BDNF sequestration in animals sustaining peripheral nerve injury using systemic administration of anti-BDNF antibodies chimeric R3bH01 & B30. Animals were given a single injection of R3bH01, B30 or hIgG1 negative control 4 to 7 days prior to electrophysiological studies. Consistent with previous reports, spinal nerve ligation (SNL) led to a dramatic attenuation of the native Kv current density in injured L5/L6 DRG neurones compared to those on the contralateral (uninjured) side in the same animal ( & ). Recordings made in animals treated with R3bH01, the parental anti-BDNF molecule, revealed dose-dependent reversal of these deficits (injury-induced Kv current suppression), with significant efficacy achieved at 1 and 10 mg/kg (). These data support findings that the antibody, through specific binding of BDNF, modulates sensory neurone function and alters the injury phenotype of sensory neurones. Based on the improved potency and binding affinity of B30, we explored a lower dose range of this antibody (0.01 to 0.1 mg/kg) in the same model. B30 produced full reversal of Kv current deficits at 0.03 and 0.1 mg/kg, while the 0.01 mg/kg dose remained ineffective (). Improvement in antibody potency achieved via affinity maturation translated to enhanced pharmacological activity in vivo.
Figure 4. Affinity of anti-BDNF antibodies correlates to potency of neuropathy-induced Kv current suppression
B30 reverses neuronal hyperexcitability in a rat model of peripheral nerve injury
Having demonstrated the ability to reverse Kv current plasticity in injured DRG neurons, we next examined the impact of B30 on primary afferent excitability using an ex vivo skin nerve preparation.Citation46,Citation47 A key feature of peripheral nerve damage is peripheral sensitization, a phenomenon characterized by reduced activation thresholds, enhanced firing activity to evoked inputs and increased background activity in primary afferent fibers.Citation48
The pharmacological activity of B30 was evaluated in nerve injured rats (SNL) at 3 weeks post-surgery. Recordings were made from the tibial nerve, which was dissected free along with the associated glaborous skin. Modulation of thermal hypersensitivity was investigated using two heat stimulation protocols that evoked a different firing pattern between ipsilateral and the contralateral nerve preparations in SNL rats. Delivery of a noxious heat stimulus (36-52°C over 50 seconds, at 0.4°C/second) onto the center of the receptive field evoked robust afferent firing in both ipsilateral and contralateral preparations. A second heat ramp (36 − 48°C over 50 seconds, at 0.2°C/s) elicited lower levels of firing (<600 spikes in 50 seconds) in the absence of injury in the contralateral preparation, whilst the same stimulus elicited a greater firing response (>1000 spikes) in preparations taken from the ipsilateral side. This is interpreted to be a manifestation of peripheral sensitization, and this pattern of activity was observed in animals treated with hIgG1 negative control (). The thermal hypersensitivity resulting from nerve injury was dose dependently reversed by B30, with both doses (0.03 mg/kg and 0.1 mg/kg) significantly reducing excitability in the skin-nerve preparation (). Similarly, activity evoked by the noxious heat ramp was significantly reduced at the 0.1 mg/kg dose (). These data support the potential utility of B30 in reversing mechanisms underlying peripheral nerve hyper-excitability following peripheral nerve damage.
Figure 5. B30 reduces thermal hyperalgesia in the tibial nerve of SNL injured rats
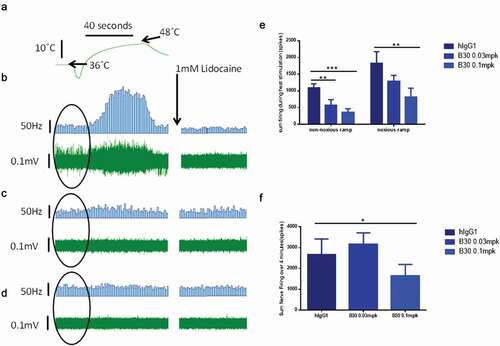
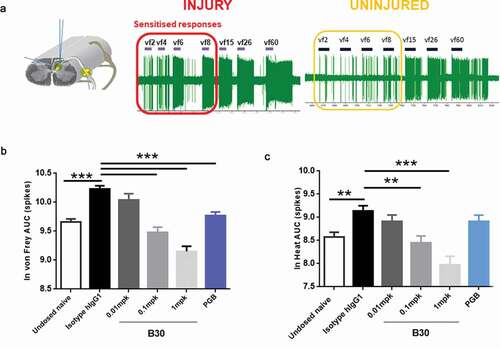
To further explore whether the reversal of neuronal plasticity seen in in vitro and ex vivo preparations can also be demonstrated in vivo, spinal cord recordings were conducted in nerve injured animals at 3 weeks post tibial nerve injury. Single dorsal horn recordings were made from ipsilateral wide dynamic range (WDR) neurones 4–7 days following dosing with B30 (0.01, 0.1, 1 mg/kg), negative isotype control or pregabalin. A mechanical and/or heat response function was constructed by quantification of neuronal activity to graded von Frey filaments (2 to 60 g) or heat (32 to 50 degrees) applied to the peripheral receptive field (). The area under the curve (AUC) of the stimulus response curve is shown in & . Evidence for spinal cord hyperactivity was observed in neurones receiving input from the injured hindpaw; these neurones displayed exaggerated firing activity to low intensity mechanical punctate stimuli. This hyperexcitability was attenuated in animals receiving pregabalin over 5 days (twice daily, 15 mg/kg). Similarly, a single injection of B30 dose dependently attenuated the hypersensitivity to mechanical punctate stimuli and heat in dorsal horn neurons. At the same dose (0.1 mg/kg) that B30 showed reversal of the Kv current deficit and heat sensitization in peripheral nerves, spinal cord excitability was restored back to uninjured control levels (see undosed naïve group in ). This is in contrast to animals treated with the negative control (hIgG1), where the sensitized phenotype was still present. In these animals, the magnitude of firing to peripheral inputs was overall greater compared to that seen in uninjured naïve animals.
Figure 6. B30 reduces spinal cord hyperexcitability in spinal dorsal horn (DH) neurones of nerve injured animals
Taken together, our data supports a role for peripheral BDNF in the mechanisms underlying neuronal hyperexcitability following peripheral nerve injury. Both the parental BDNF antibody (R3bH01) and the affinity-matured antibody B30 specifically bind BDNF and neutralize the neurotrophin’s activity in vivo. We provide evidence that through affinity maturation, the potency of the B30 molecule was greatly enhanced, resulting in in vivo efficacy at much lower doses compared to the parental R3bH01 antibody.
Discussion
Here, we describe the successful generation of a high affinity anti-BDNF molecule that displays potent in vivo pharmacological activity and reverses key features of injury-induced plasticity in sensory neurones. To our knowledge, this is the first report of an anti-BDNF molecule that binds BDNF with high affinity, potently neutralizes BDNF activity in vivo and shows selectivity in binding over related neurotrophins and similarly charged chemokines. While a previous study demonstrated that local administration of anti-BDNF promoted nerve fiber integrity in a rat nerve transection model, this study used a polyclonal antibody which has not been fully characterized for BDNF specificity or functional activity.Citation16
Mature BDNF is 100% conserved across mammals, including rat and mouse.Citation49 Due to the high homology, traditional antibody generation methods such as mouse hybridoma approaches provide a challenge in breaking tolerance. Based on the phylogenetic distance between chickens and mammals, we used an in vivo avian immunization approach,Citation34 coupled with in vitro phage display of the generated chicken immune repertoire. This led to the identification of R3bH01. The desired pM affinity, however, was not achieved directly from the library, and an optimization campaign was required post-humanization, which led us to clone B30. Given the extent of engineering ultimately required, alternative methods could be considered for this target, and we have successfully used large naïve human antibody libraries coupled with optimization via phage display to achieve pM affinities in the past.Citation38 B30 demonstrated pM affinity for BDNF, potent neutralization of TrkB signaling in cell-based assays and retained exquisite selectivity for BDNF over related neurotrophins. We demonstrate that the improvement in in vitro binding affinity achieved through affinity maturation correlates with increased potency observed in vivo. Efficacy was observed in the Kv electrophysiological assay at doses as low as 0.03 mg/kg for B30 with 1 mg/kg of R3bH01 required to achieve the same level of protection.
A key finding in the present study is that systemic administration of BDNF antibody reverses hyperexcitability and injury-associated processes that likely contribute to abnormal processing in damaged sensory neurones. In addition to their developmental role in governing cell growth, differentiation and neuronal survival, neurotrophins function to regulate synaptic plasticity in the adult nervous system. Through both transcriptional and posttranslational mechanisms, this class of molecules exert structural and functional modifications of synaptic connections and profoundly alter neuronal excitability in sensory neurones.Citation2,Citation50
Reduced expression of voltage-gated potassium channels and functional deficits in channel activity have been described across many injury models.Citation40-Citation42,Citation44,Citation51-Citation55 One mechanism contributing to these changes is an alteration in BDNF/TrkB signaling, and pharmacological blockade of this pathway has been shown to reverse Kv channel plasticity in culture.Citation42 Kv channels represent a large family of ion channels that critically regulate aspects of action potential firing, including its initiation, propagation, duration and firing frequency.Citation53,Citation56 The composition of the Kv channel subtype, the density and distribution pattern along the axon are crucial determinants controlling axonal excitability.Citation57 As these channels function to suppress excitability in peripheral nerves, reorganization in their expression pattern or pharmacological blockade are expected to result in abnormal excitability and promote ectopic firing.Citation55,Citation57,Citation58
Using administration of a highly selective monoclonal antibody, we provide novel evidence that sustained BDNF suppression over a period of several days reverses phenotypes that are commonly associated with nerve injury. Evidence for hyperexcitability is observed at multiple levels of the neuraxis, including the peripheral nerve, DRG, spinal cord and supraspinal regions of the brain. Decreased activation thresholds, ongoing activity and enhanced firing to peripheral inputs are all hallmark features of peripheral and central sensitization. The ability to bring about long-term excitability changes through modulation of synaptic transmission and connectivity suggests that neurotrophins may be responsible for promoting the chronicity of the pain state following injury. This, in turn, could drive abnormal pain symptoms that typically include ongoing pain, allodynia and hyperalgesia. Our findings suggest that the excitability of the peripheral pain pathway can be restored to physiological levels by peripheral sequestration of BDNF through mechanisms that partly involve Kv channel activity. Targeting dysregulated BDNF levels using a highly specific antibody molecule therefore offers a novel approach to the treatment of certain chronic pain conditions, particularly where pain is driven through abnormal activity in the injured nervous system. Further studies are required to evaluate whether anti-BDNF therapy can be beneficial in reversing neurological imbalances in diseases such as neuropathic pain.
Materials and methods
Isolation and humanization of lead chicken clone
Human BDNF (Peprotech, Catalog Number 450–02) was used to immunize 2 chickens and an scFv phage library was subsequently constructed from spleen and bone marrow post-immunization using methods previously described.Citation34 R3bH01 was identified as the lead clone using the Pathhunter pERK assay and was subsequently humanized using a classical CDR grafting approach as previously described.Citation36 Sequence alignment identified VH3-23 and Vλ3-19 as the closest human germline framework sequences and these were used as acceptors for the R3bH01 CDRs (Kabat definitionCitation59). Humanized grafts were tested for maintenance of functional activity using an ELISA-based competition assay against biotinylated chimeric R3bH01.
Competition ELISA
NUNC Maxisorp plates (384-well) were coated with 20 µL BDNF protein (Peprotech, Catalog 450–02) at a concentration of 1 µg/mL in phosphate-buffered saline (PBS) pH 7.4, overnight with shaking. After overnight incubation, the plates were washed five times with 200 µL PBS containing 0.02% (v/v) Tween and blocked for 1 hour using 1% Casein/PBS (v/v) (Thermo Scientific, Catalog 37528). Anti-BDNF antibodies were titrated in casein. Titrated antibodies were mixed with biotinylated chimeric R3bH01 and then applied to immobilized BDNF for 1 hour. Plates were washed as above and a 1/1,2000 dilution of horseradish peroxidase (HRP) conjugated streptavidin (Thermo Scientific, Catalog N100) was added for 1 hour. Plates were washed and developed by addition of 25 µL 3,3′,5,5′-Tetramethylbenzidine (TMB, Thermo Scientific, Catalog 34029) substrate and subsequently stopped with 25 µL 0.18 M phosphoric acid (Fisher Scientific). Plates were processed on the EnVision MultiLabel Plate Reader (Perkin-Elmer) with absorbance at 450 nm.
Affinity optimization library construction and selection
Mutational scFv libraries based on humanized H01 were constructed, rescued and selected as previously described.Citation60 In summary, 11 H01 mutagenic soft-randomization sub-libraries were constructed by splice-overlap-extension (SOE) PCR, with mutations being introduced into VH-CDR1, VH-CDR2, VH-CDR3, VL-CDR1 and VL-CDR3. Different numbers of mutagenic primers were required for each CDR based on loop length. This is represented schematically in , and primer sequences are provided in Supplementary Table 1. VL-CDR2 was not targeted in our mutational strategy based on observations from previous in-house optimization campaigns, indicating that this CDR is rarely a driver for affinity gains. SOE-PCR products were restriction digested, purified and ligated into the phagemid vector pWRIL-1. Libraries were transformed into E.coli TG1 electro-competent cells. Post sub-library titration, sub-libraries from across the length of each CDR were pooled in equimolar amounts, resulting in five CDR libraries that were selected and screened separately.
For phage display selections, a standard stepwise technique was applied.Citation61 NUNC immunotubes were coated with 100 nM BDNF in PBS, pH 7.4. A stepwise strategy was used where off-rate selection pressure with a 100-fold excess of BDNF in solution (10μ M) was applied for 15 min, 1 h and overnight from rounds 1 to 3, respectively. To avoid charge enrichment, de-selection was employed in all rounds of selection using calf thymus histones (Sigma Aldrich, 20 µg/mL).
High-throughput screening of mutants via HTRF
HTRF screening was performed as described previously.Citation61 A high-throughput scFv competition HTRF assay was established to facilitate screening for affinity improved clones while maintaining the R3bH01 epitope. Parental R3bH01 was labeled with europium cryptate using a cryptate labeling kit, according to the manufacturer’s instructions (CisBio, Catalog 62EUSUEA). The final assay mixture consisted of in-house biotinylated BDNF (Peprotech, 1 nM), europium cryptate labeled-R3bH01 (1/1,000 dilution), SA-XL665 (CisBio, Catalog 610SAXLF) (1/2,000) and periplasmic extract containing the scFv of interest in a total reaction volume of 20 µL in 1x assay buffer (CisBio, Catalog 61DB9RDF). Reactions were allowed to proceed for 3 hours at room temperature and plates were subsequently read on the EnVision MultiLabel Plate Reader (Perkin-Elmer) with excitation at 340 nm and two emission readings at 615 nm (measuring input donor fluorescence from R3bH01 europium cryptate) and 665 nm (measuring output acceptor fluorescence from SAXL665). All readings were initially calculated as a ratio of fluorescence at 665/615 and then expressed as % inhibition compared to the negative control, an unrelated scFv periprep sample. Prioritized hits were sequenced, reformatted to full-length IgG1 and purified at small-scale for further characterization as described previously.Citation62
Biacore affinity determination
SPR was used to characterize the binding kinetics of chimeric R3bH01, humanized H01 and affinity optimized antibody B30 against human BDNF (Sino Biological Inc., Catalog 50240-MNAS). A low density of BDNF was amine coupled onto a carboxymethylated dextran sensor chip surface (CM5, GE Healthcare) using a Biacore T200 instrument (GE Healthcare). Three-fold serial dilutions of antibodies, chimeric R3bH01, H01 and B30, ranging from 243 nM to 9 nM were prepared in HBS-EP+ buffer (0.01 M HEPES, 0.15 M NaCl, 3 mM EDTA, and 0.05% (v/v) surfactant P20, pH7.4). Two replicate samples at each concentration were injected at a flow rate of 100 μL/min for a 47 sec association step and either a 300 or 4000 sec dissociation. The human BDNF surface was regenerated with 3 pulses, 30 sec each, of 10 mM glycine, pH 1.5 at a flow rate of 50 μL/min and then equilibrated with a single 30 sec pulse of HBS-EP+ buffer at a flow rate of 50 μL/min. Original SPR experiments for chimeric R3bH01 and humanized H01 were performed, at least in duplicate, at 25°C with a data collection rate of 1 Hz and HBS-EP+ used as both the sample and running buffer. The resulting sensorgrams were double referencedCitation63 with buffer injections and an activated and quenched flow cell surface. Rate constants were determined by applying a 1:1 Langmuir binding model using the T200 evaluation software v1.0, subsequently determining the equilibrium dissociation constant with the equation KD = kd/ka. Given the much higher affinity of the clones derived from the optimization campaign, SPR for these clones was performed under the same conditions except at an elevated temperature of 37°C.
KinExA affinity determination
The solution KD value for the affinity-optimized B30 Fab against human BDNF was determined using a Sapidyne KinExA 3200 instrument. In this kinetic exclusion assay, the percentage of unbound, or free, human BDNF was measured from several equilibrated solutions containing a dilution of affinity-optimized B30 Fab combined with a fixed concentration of human BDNF antigen. Specifically, B30 Fab ranging in concentration from 2 nM to 343 fM was incubated with human BDNF, either at 60 pM or 400 pM overnight at room temperature (RT) to attain equilibrium. These fixed concentrations of human BDNF were near the SPR-determined KD value for B30 and provided measurable signal above background. Polymethylmethacrylate beads coated with antibody B30 at 30 μg/mL for two hours at room temperature, blocked with 10 mg/mL bovine serum albumin (BSA), and further diluted with PBS, were used to capture the unbound human BDNF from the equilibrated Fab antigen solutions. The captured free human BDNF was detected with the non-competing anti-BDNF mouse mAb848 (R&D Systems, Catalog MAB848) at 5 μg/mL followed by the Alexa Fluor® 647 anti-mouse IgG antibody (Jackson ImmunoResearch, Catalog 115-605-062) at 0.5 μg/mL. Human BDNF without Fab and buffer alone were also evaluated to determine the maximum signal and level of nonspecific binding (NSB), respectively. These multiple curve data were globally fit using N-curve analysis and Sapidyne evaluation software version 3.6.8 with Fab as the concentration reference to identify the best fit KD value.
Pathhunter pERK assay
U2OS cells expressing TrkB+p75NTR (DiscoverX, Catalog 93–0463 C3) were plated overnight in minimum essential medium (Life Technologies) with 0.5% horse serum (Life Technologies, Catalog 26050088). On the day of the assay, R3bH01, and test anti-BDNF antibodies were serially diluted 1:3 in PBS to create a 10-point concentration response curve. 10 μL of the serial diluted samples were added to the cells and incubated for 1 hour at 37°C, before the addition of 10 μL of 1.8 nM BDNF (Peprotech, Catalog Number 450–02) in PBS with 0.25% BSA to each well (BDNF final assay concentration (FAC): 150 pM). The plate was incubated for 30 min at room temperature before media removal and the addition of Cellul’erk lysis buffer (Cisbio, Catalog 64KL1FDF). The plate was stored at −80°C overnight. After thawing, lysates were transferred to a 384-well Proxiplate (Perkin Elmer, Catalog 6008260), Cellul’erk HTRF reagents were added and incubated as per kit instructions (Cisbio, Catalog 64NRKPET) before reading on an Envision plate reader (Perkin Elmer).
Surgery for peripheral nerve injury
All experimental procedures were conducted in accordance with the UK Animals ActCitation64 and followed the guidelines under the International Association for the Study of Pain.Citation65 Male Sprague Dawley rats (170–190 g) were subjected to 2 models of peripheral nerve injury, SNL and tibial nerve transection (TNT).
All animals were allowed to acclimatize to the animal facilities for at least 5 days prior to surgery. All surgery procedures were conducted under sterile conditions in a dedicated surgery suite. The back of the animal (SNL) or the left hindleg (TNT) was shaved and swabbed with Pevedine (Sigma Aldrich) and Chlorhexidine solution (Sigma Aldrich).
For ligation of L5/6 spinal nerves, animals were placed under general anesthesia (isoflurane, Sigma Aldrich, 5% induction, 2% maintenance) and the left paraspinal muscles were separated from spinous processes at L4-S2 levels. The L6 transverse process was removed to allow identification of L4-6 spinal nerves. L5 and L6 spinal nerves were isolated and tightly ligated with 6–0 silk thread (Mersilk). For TNT, the tibial nerve was carefully exposed at the point of trifurcation of the sciatic nerve, following which it was tightly ligated with 6–0 silk and transected distally to the ligature.
Hemostasis was confirmed and muscles were closed in layers using Vicryl (5–0) suture. Skin was closed with sterile surgical clips. Animals were placed in a heated recovery box until fully awake and transferred to a clean homecage. Animals were given wet mash for the first 5 days and daily post-operative observations made for 7 days post-surgery.
In vitro Kv recordings
Three weeks post-surgery, animals were randomized to receive dosing of R3bH01 (0.1, 1 or 10 mg/kg), B30 (0.01, 0.03 or 0.1 mg/kg) or hIgG1 isotype control (0.1 or 10 mg/kg). All electrophysiological studies were fully blinded. 4–7 days post injection, animals were sacrificed and ipsilateral and contralateral L5/6 DRG neurones were dissected for subsequent in vitro electrophysiological recordings. L5 and L6 DRG from the same side were pooled and dissociated. DRGs were digested in medium containing collagenase and subsequently incubated in medium containing trypsin. Following washing and trituration, the dissociated cells were centrifuged, re-suspended and plated on glass coverslips. All recordings were made on the same day as the dissociation. Voltage-clamp recordings were performed from a Vhold of −90 mV and then stepped to +60 mV in 10 mV increments. The delayed-rectifier currents (Ik quantified at the end of the test pulse) were quantified in subsequent analyses. All measured currents were normalized to the cell’s size as measured by cellular capacitance resulting in current densities (pA/pF).
Skin nerve recordings
All animals were randomly allocated to the treatment group and the study was blinded. Dosing was carried out by a separate experimenter to that conducting the electrophysiological study. 3 weeks post-surgery, animals were dosed (dosing volume 1 ml/kg) with a single injection of B30 (0.03 or 0.1 mg/kg) or hIgG1 isotype control (hIgG1, 0.1 mg/kg). 4–7 days post injection of the antibody, animals were culled using a schedule 1 method. Both legs were shaved, removed and the tibial nerve, along with the associated glaborous skin, was dissected free as described previously.Citation46 The skin was placed, glaborous side down, in a recording chamber that was continually superfused (10 mL/min; 36 ± 1°C) with carbonated (95% O2, 5% CO2) modified Krebs’ solution (in mM: NaCl 107, KCl 3.48, NaHCO3 26.2, MgSO4 (7H2O) 0.69, NaH2PO4 1.67, Na-gluconate 9.64, sucrose 7.6, Glucose 5.5, CaCl2 1.53). Extracellular recordings were made from the distal end of a bundle from the tibial nerve using a borosilicate glass suction electrode. Following a 60 min acclimatization period, a heat stimulus consisting of hot Krebs was flowed onto the skin over 50 sec. The heat was delivered either via a slow ramp (36-48°C in 50 sec, at 0.2°C/s) or via a fast ramp where temperature rose more rapidly (36-52°C in 50 sec, at 0.4°C/s). The two ramps were delivered 15 min apart and at least 2 sets of baseline readings were taken for each preparation. Data were captured and analyzed by a CED 1401 interface coupled to a computer with Spike 2 software (Cambridge Electronic Designs). At the end of the experiment, 1 mM lidocaine (Sigma Aldrich) was superfused onto the preparation for 15 min to inhibit all physiological activity.
In vivo electrophysiology
All recordings were made in animals that underwent surgery for TNT 2–3 weeks prior to the study. Animals were given a single subcutaneous injection of B30 (0.01, 0.1, 1 mg/kg, dosing volume 1 mL/kg) and animals were prepared for electrophysiological studies 4–7 days later. Studies were run in a blinded manner, and separate experimenters conducted the animal dosing and electrophysiological studies. In a separate group of animals, animals sustaining tibial nerve injury rats were orally dosed with pregabalin (7.5 mg/kg, twice daily). On the day of the recording, animals were anesthetized with isoflurane (5% induction, 1.5–2% maintenance) and the jugular vein and carotid artery were cannulated to enable drug administration and blood sampling, respectively. Body temperature was monitored by a rectal probe and maintained at 37°C using a heating blanket. Animals treated with B30 or isotype control received infusion of saline (1 mL/kg/h) while animals treated with pregabalin received pregabalin infusion (0.75 mg/kg/h) throughout the length of the study to maintain the drug at steady state. Animals were placed in a stereotaxic frame and a laminectomy was performed to expose the region of the hindpaw receiving afferent input from the hindpaw region of the rat. Cell searching commenced around 1 hour following the start of intravenous infusion and recordings were made from single WDR neurones in the ipsilateral dorsal horn that were responsive to a range of modalities, including brush, mechanical punctate and heat. On identification of a single unit, the neuronal receptive field was probed to determine the most responsive region of the paw. Neurones that were sampled in the study were those that displayed robust response to brush stimulation (>100 spikes), showed sustained firing to noxious pinch applied using an arterial clip and responded (>40 spikes) to von Frey hair stimulation with 26 g hair. To quantify the stimulus response curve to mechanical and heat stimulation, the center of the receptive field was probed with graded von Frey hairs (2, 4, 6, 8, 15, 26, 60 g) or water jet (22, 35, 40, 43, 45, 48 and 50°C) applied over 10 sec per stimulus. For thermal stimulation, a water jet was used to apply a constant stream of water at set temperatures onto the center of the receptive field. The data was captured by Spike 2 software and analyzed to establish spike firing frequency. The AUC of the stimulus response curve (mechanical or heat) was calculated for each cell and an ANOVA was used to compare the difference between treatment groups.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Declaration of conflict of interest
All authors are past or present employees of Pfizer.
Supplemental Material
Download MS Word (670.1 KB)Acknowledgments
The authors acknowledge Ying Sun and Stefano Gulla for derivation of the original chicken antibodies. The authors also thank Weijun Ma, Xiaotian Zhong, Khetemenee Lam, Kelvin Kerns, Angela Boisvert, Richard Zollner, Aaron D’Antona & Stephane Olland for protein expression and purification support.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- Pezet S. Neurotrophins and pain. Biol Aujourdhui. 2014;208:21–13. doi:https://doi.org/10.1051/jbio/2014002.
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–64. doi:https://doi.org/10.1098/rstb.2006.1894.
- Chao MV. Neurotrophin receptors: a window into neuronal differentiation. Neuron. 1992;9:583–93. doi:https://doi.org/10.1016/0896-6273(92)90023-7.
- von Hehn CA, Baron R, Woolf CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron. 2012;73:638–52. doi:https://doi.org/10.1016/j.neuron.2012.02.008.
- Verge VM, Gratto KA, Karchewski LA, Richardson PM. Neurotrophins and nerve injury in the adult. Philos Trans R Soc Lond B Biol Sci. 1996;351:423–30.
- Li W, Cai WQ, Li CR. Repair of spinal cord injury by neural stem cells modified with BDNF gene in rats. Neurosci Bull. 2006;22:34–40.
- Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21:4891–900. doi:https://doi.org/10.1523/JNEUROSCI.21-13-04891.2001.
- Castel D, Sabbag I, Brenner O, Meilin S. Peripheral neuritis trauma in pigs: a neuropathic pain model. J Pain. 2016;17:36–49. doi:https://doi.org/10.1016/j.jpain.2015.09.011.
- Luo XG, Rush RA, Zhou XF. Ultrastructural localization of brain-derived neurotrophic factor in rat primary sensory neurons. Neurosci Res. 2001;39:377–84. doi:https://doi.org/10.1016/S0168-0102(00)00238-8.
- Michael GJ, Averill S, Shortland PJ, Yan Q, Priestley JV. Axotomy results in major changes in BDNF expression by dorsal root ganglion cells: BDNF expression in large trkB and trkC cells, in pericellular baskets, and in projections to deep dorsal horn and dorsal column nuclei. Eur J Neurosci. 1999;11:3539–51. doi:https://doi.org/10.1046/j.1460-9568.1999.00767.x.
- Pezet S, Malcangio M, Lever IJ, Perkinton MS, Thompson SW. Noxious stimulation induces Trk receptor and downstream ERK phosphorylation in spinal dorsal horn. Mol Cell Neurosci. 2002;21:684–95. doi:https://doi.org/10.1006/mcne.2002.1205.
- Bibel M, Hoppe E, Barde YA. Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. Embo J. 1999;18:616–22. doi:https://doi.org/10.1093/emboj/18.3.616.
- Chao MV, Hempstead BL. p75 and Trk: a two-receptor system. Trends Neurosci. 1995;18:321–26. doi:https://doi.org/10.1016/0166-2236(95)93922-K.
- Ferrini F, De Koninck Y. Microglia control neuronal network excitability via BDNF signalling. Neural Plast. 2013;2013:429815. doi:https://doi.org/10.1155/2013/429815.
- Sanna MD, Ghelardini C, Galeotti N. Blockade of the spinal BDNF-activated JNK pathway prevents the development of antiretroviral-induced neuropathic pain. Neuropharmacology. 2016;105:543–52. doi:https://doi.org/10.1016/j.neuropharm.2016.02.016.
- Valverde Guevara YM, Yoshikawa H, Saito I, Maeda T, Seo K. Effect of local application of an antibody against brain-derived neurotrophic factor on neuroma formation after transection of the inferior alveolar nerve in the rat. Neuroreport. 2014;25:1069–74. doi:https://doi.org/10.1097/WNR.0000000000000231.
- Zhang X, Wang J, Zhou Q, Xu Y, Pu S, Wu J, Xue Y, Tian Y, Lu J, Jiang W, et al. Brain-derived neurotrophic factor-activated astrocytes produce mechanical allodynia in neuropathic pain. Neuroscience. 2011;199:452–60. doi:https://doi.org/10.1016/j.neuroscience.2011.10.017.
- Grimsholm O, Guo Y, Ny T, Forsgren S. Expression patterns of neurotrophins and neurotrophin receptors in articular chondrocytes and inflammatory infiltrates in knee joint arthritis. Cells Tissues Organs. 2008;188:299–309. doi:https://doi.org/10.1159/000121432.
- Rihl M, Kruithof E, Barthel C, De Keyser F, Veys EM. Involvement of neurotrophins and their receptors in spondyloarthritis synovitis: relation to inflammation and response to treatment. Ann Rheum Dis. 2005;64:1542–49. doi:https://doi.org/10.1136/ard.2004.032599.
- Sobue G, Yamamoto M, Doyu M, Li M, Yasuda T, Mitsuma T. Expression of mRNAs for neurotrophins (NGF, BDNF, and NT-3) and their receptors (p75NGFR, trk, trkB, and trkC) in human peripheral neuropathies. Neurochem Res. 1998;23:821–29. doi:https://doi.org/10.1023/A:1022434209787.
- Wang DD, Tian T, Dong Q, Xu XF, Yu H. Transcriptome profiling analysis of the mechanisms underlying the BDNF Val66Met polymorphism induced dysfunctions of the central nervous system. Hippocampus. 2014;24:65–78. doi:https://doi.org/10.1002/hipo.22204.
- Weidler C, Holzer C, Harbuz M, Hofbauer R, Angele P. Low density of sympathetic nerve fibres and increased density of brain derived neurotrophic factor positive cells in RA synovium. Ann Rheum Dis. 2005;64:13–20. doi:https://doi.org/10.1136/ard.2003.016154.
- Navone SE, Marfia G, Canzi L, Ciusani E, Canazza A. Expression of neural and neurotrophic markers in nucleus pulposus cells isolated from degenerated intervertebral disc. J Orthop Res. 2012;30:1470–77. doi:https://doi.org/10.1002/jor.22098.
- Purmessur D, Freemont AJ, Hoyland JA. Expression and regulation of neurotrophins in the nondegenerate and degenerate human intervertebral disc. Arthritis Res Ther. 2008;10:R99. doi:https://doi.org/10.1186/ar2487.
- Zhu ZW, Friess H, Wang L, Zimmermann A, Buchler MW. Brain-derived neurotrophic factor (BDNF) is upregulated and associated with pain in chronic pancreatitis. Dig Dis Sci. 2001;46:1633–39. doi:https://doi.org/10.1023/A:1010684916863.
- Yu L, Dan QQ, Li JT, Zhang YH. [Block effects of BDNF antibody on MEK expression in lung of rats with brain ischemia]. Sichuan Da Xue Xue Bao Yi Xue Ban. 2012;43:926–29.
- Lopez-Perez AE, Nurgali K, Abalo R. Painful neurotrophins and their role in visceral pain. Behav Pharmacol. 2018;29:120–39. doi:https://doi.org/10.1097/FBP.0000000000000386.
- Luo C, Zhong XL, Zhou FH, Li JY, Zhou P, Xu JM, Song B, Li C-Q, Zhou X-F, Dai R-P, et al. Peripheral brain derived neurotrophic factor precursor regulates pain as an inflammatory mediator. Sci Rep. 2016;6:27171. doi:https://doi.org/10.1038/srep27171.
- Kivitz AJ, Gimbel JS, Bramson C, Nemeth MA, Keller DS, Brown MT, West CR, Verburg KM. Efficacy and safety of tanezumab versus naproxen in the treatment of chronic low back pain. Pain. 2013;154:1009–21. doi:https://doi.org/10.1016/j.pain.2013.03.006.
- Gimbel JS, Kivitz AJ, Bramson C, Nemeth MA, Keller DS, Brown MT, West CR, Verburg KM. Long-term safety and effectiveness of tanezumab as treatment for chronic low back pain. Pain. 2014;155:1793–801. doi:https://doi.org/10.1016/j.pain.2014.06.004.
- Lane NE, Schnitzer TJ, Birbara CA, Mokhtarani M, Shelton DL, Smith MD, Brown MT. Tanezumab for the treatment of pain from osteoarthritis of the knee. N Engl J Med. 2010;363:1521–31. doi:https://doi.org/10.1056/NEJMoa0901510.
- Kan SL, Li Y, Ning GZ, Yuan ZF, Chen LX, Bi MC, Sun J-C, Feng S-Q. Tanezumab for patients with osteoarthritis of the knee: a meta-analysis. PLoS One. 2016;11:e0157105. doi:https://doi.org/10.1371/journal.pone.0157105.
- Finlay WJ, Bloom L, Grant J, Franklin E, Shuilleabhain DN, Cunningham O. Phage display: a powerful technology for the generation of high-specificity affinity reagents from alternative immune sources. Methods Mol Biol. 2017;1485:85–99.
- Finlay WJ, Bloom L, Cunningham O. Optimized generation of high-affinity, high-specificity single-chain Fv antibodies from multiantigen immunized chickens. Methods Mol Biol. 2011;681:383–401.
- Tsurushita N, Park M, Pakabunto K, Ong K, Avdalovic A, Fu H, Jia A, Vásquez M, Kumar S. Humanization of a chicken anti-IL-12 monoclonal antibody. J Immunol Methods. 2004;295:9–19. doi:https://doi.org/10.1016/j.jim.2004.08.018.
- Nishibori N, Horiuchi H, Furusawa S, Matsuda H. Humanization of chicken monoclonal antibody using phage-display system. Mol Immunol. 2006;43:634–42. doi:https://doi.org/10.1016/j.molimm.2005.04.002.
- Katsamba PS, Navratilova I, Calderon-Cacia M, Fan L, Thornton K, Zhu M, Bos TV, Forte C, Friend D, Laird-Offringa I. Kinetic analysis of a high-affinity antibody/antigen interaction performed by multiple Biacore users. Anal Biochem. 2006;352:208–21. doi:https://doi.org/10.1016/j.ab.2006.01.034.
- Darling RJ, Brault PA. Kinetic exclusion assay technology: characterization of molecular interactions. Assay Drug Dev Technol. 2004;2:647–57. doi:https://doi.org/10.1089/adt.2004.2.647.
- Drake AW, Tang ML, Papalia GA, Landes G, Haak-Frendscho M, Klakamp SL. Biacore surface matrix effects on the binding kinetics and affinity of an antigen/antibody complex. Anal Biochem. 2012;429:58–69. doi:https://doi.org/10.1016/j.ab.2012.06.024.
- Kim DS, Choi JO, Rim HD, Cho HJ. Downregulation of voltage-gated potassium channel alpha gene expression in dorsal root ganglia following chronic constriction injury of the rat sciatic nerve. Brain Res Mol Brain Res. 2002;105:146–52. doi:https://doi.org/10.1016/S0169-328X(02)00388-1.
- Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, Trimmer JS. Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci U S A. 2001;98:13373–78. doi:https://doi.org/10.1073/pnas.231376298.
- Cao XH, Byun HS, Chen SR, Cai YQ, Pan HL. Reduction in voltage-gated K+ channel activity in primary sensory neurons in painful diabetic neuropathy: role of brain-derived neurotrophic factor. J Neurochem. 2010;114:1460–75. doi:https://doi.org/10.1111/j.1471-4159.2010.06863.x.
- Takeda M, Tsuboi Y, Kitagawa J, Nakagawa K, Iwata K, Matsumoto S. Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol Pain. 2011;7:5. doi:https://doi.org/10.1186/1744-8069-7-5.
- Xiao Y, Wu Y, Zhao B, Xia Z. Decreased voltage-gated potassium currents in rat dorsal root ganglion neurons after chronic constriction injury. Neuroreport. 2016;27:104–09. doi:https://doi.org/10.1097/WNR.0000000000000505.
- Yang EK, Takimoto K, Hayashi Y, de Groat WC, Yoshimura N. Altered expression of potassium channel subunit mRNA and alpha-dendrotoxin sensitivity of potassium currents in rat dorsal root ganglion neurons after axotomy. Neuroscience. 2004;123:867–74. doi:https://doi.org/10.1016/j.neuroscience.2003.11.014.
- Rong W, Hillsley K, Davis JB, Hicks G, Winchester WJ, Grundy D. Jejunal afferent nerve sensitivity in wild-type and TRPV1 knockout mice. J Physiol. 2004;560:867–81. doi:https://doi.org/10.1113/jphysiol.2004.071746.
- Du J, Koltzenburg M, Carlton SM. Glutamate-induced excitation and sensitization of nociceptors in rat glabrous skin. Pain. 2001;89:187–98. doi:https://doi.org/10.1016/S0304-3959(00)00362-6.
- Shim B, Kim DW, Kim BH, Nam TS, Leem JW, Chung JM. Mechanical and heat sensitization of cutaneous nociceptors in rats with experimental peripheral neuropathy. Neuroscience. 2005;132:193–201. doi:https://doi.org/10.1016/j.neuroscience.2004.12.036.
- Maisonpierre PC, Le Beau MM, Espinosa R 3rd, Ip NY, Belluscio L, de la Monte SM, Squinto S, Furth ME, Yancopoulos GD. Human and rat brain-derived neurotrophic factor and neurotrophin-3: gene structures, distributions, and chromosomal localizations. Genomics. 1991;10:558–68. doi:https://doi.org/10.1016/0888-7543(91)90436-I.
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–38. doi:https://doi.org/10.1146/annurev.neuro.29.051605.112929.
- Ishikawa K, Tanaka M, Black JA, Waxman SG. Changes in expression of voltage-gated potassium channels in dorsal root ganglion neurons following axotomy. Muscle Nerve. 1999;22:502–07. doi:https://doi.org/10.1002/(SICI)1097-4598(199904)22:4<502::AID-MUS12>3.0.CO;2-K.
- Li Q, Wanderling S, Paduch M, Medovoy D, Singharoy A, McGreevy R, Villalba-Galea CA, Hulse RE, Roux B, Schulten K. Structural mechanism of voltage-dependent gating in an isolated voltage-sensing domain. Nat Struct Mol Biol. 2014;21:244–52. doi:https://doi.org/10.1038/nsmb.2768.
- Tsantoulas C, Zhu L, Yip P, Grist J, Michael GJ, McMahon SB. Kv2 dysfunction after peripheral axotomy enhances sensory neuron responsiveness to sustained input. Exp Neurol. 2014;251:115–26. doi:https://doi.org/10.1016/j.expneurol.2013.11.011.
- Park SY, Choi JY, Kim RU, Lee YS, Cho HJ, Kim DS. Downregulation of voltage-gated potassium channel alpha gene expression by axotomy and neurotrophins in rat dorsal root ganglia. Mol Cells. 2003;16:256–59.
- Tsantoulas C, Zhu L, Shaifta Y, Grist J, Ward JP. Sensory neuron downregulation of the Kv9.1 potassium channel subunit mediates neuropathic pain following nerve injury. J Neurosci. 2012;32:17502–13. doi:https://doi.org/10.1523/JNEUROSCI.3561-12.2012.
- Pongs O. Regulation of excitability by potassium channels. Results Probl Cell Differ. 2008;44:145–61.
- Calvo M, Richards N, Schmid AB, Barroso A, Zhu L, Ivulic D, Zhu N, Anwandter P, Bhat MA, Court FA, et al. Altered potassium channel distribution and composition in myelinated axons suppresses hyperexcitability following injury. Elife. 2016;5:e12661. doi:https://doi.org/10.7554/eLife.12661.
- Nagado T, Arimura K, Sonoda Y, Kurono A, Horikiri Y, Kameyama A, Kameyama M, Pongs O, Osame M. Potassium current suppression in patients with peripheral nerve hyperexcitability. Brain. 1999;122(Pt 11):2057–66. doi:https://doi.org/10.1093/brain/122.11.2057.
- Kabat EA, Wu T T, Bilofsky H, Resources NIoHDoR, Health NIo. sequences of immunoglobulin chains: tabulation and analysis of amino acid sequences of precursors, V-regions, C-regions, J-chain and BP-microglobulins. Department of Health, Education, and Welfare, Public Health Service, National Institutes of Health, 1979
- Franklin E, Cunningham O, Fennell B. Parallel evolution of antibody affinity and thermal stability for optimal biotherapeutic development. Methods Mol Biol. 2018;1827:457–77.
- Fennell BJ, McDonnell B, Tam AS, Chang L, Steven J, Broadbent ID, Gao H, Kieras E, Alley J, Luxenberg D. CDR-restricted engineering of native human scFvs creates highly stable and soluble bifunctional antibodies for subcutaneous delivery. mAbs. 2013;5:882–95. doi:https://doi.org/10.4161/mabs.26201.
- Shih HH, Tu C, Cao W, Klein A, Ramsey R, Fennell BJ, Lambert M, Ní Shúilleabháin D, Autin B, Kouranova E, et al. An ultra-specific avian antibody to phosphorylated tau protein reveals a unique mechanism for phosphoepitope recognition. J Biol Chem. 2012;287:44425–34. doi:https://doi.org/10.1074/jbc.M112.415935.
- Myszka DG. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Curr Opin Biotechnol. 1997;8:50–57. doi:https://doi.org/10.1016/S0958-1669(97)80157-7.
- The Animals HC. scientific procedures) Act 1986. Lancet. 1986;2:32–33. doi:https://doi.org/10.1016/s0140-6736(86)92571-7.
- Zimmermann M. Ethical considerations in relation to pain in animal experimentation. Acta Physiol Scand Suppl. 1986;554:221–33.