
ABSTRACT
Gene therapy approaches now allow for the production of therapeutic antibodies by healthy or cancerous human tissues directly in vivo, and, with an increasing number of gene delivery methods available, the cell type for expression can be chosen. Yet, little is known about the biophysical changes introduced by expressing antibodies from producer cells or tissues targeted by gene therapy approaches, nor about the consequences for the type of glycosylation. The effects of different glycosylation on therapeutic antibodies have been well studied by controlling their glycan compositions in non-human mammalian production cells, i.e., Chinese hamster ovary cells. Therefore, we investigated the glycosylation state of clinically approved antibodies secreted from cancer tissues frequently targeted by in vivo gene therapy, using native mass spectrometry and glycoproteomics. We found that antibody sialylation and fucosylation depended on the producer tissue and the antibody isotype, allowing us to identify optimal producer cell types according to the desired mode of action of the antibody. Furthermore, we discovered that high amounts (>20%) of non-glycosylated antibodies were produced in cells sensitive to the action of the produced antibodies. Different glycosylation in different producer cells can translate into an altered potency of in-vivo produced antibodies, depending on the desired mode of action, and can affect their serum half-lives. These results increase our knowledge about antibodies produced from cells targeted by gene therapy, enabling development of improved cancer gene therapy vectors that can include in vivo glycoengineering of expressed antibodies to optimize their efficacies, depending on the desired mode of action.
Introduction
The use of monoclonal antibodies (mAbs) for the treatment of cancer has been one of the most successful therapeutic strategies for the treatment of both hematologic malignancies and solid tumors. To date, the US Food and Drug Administration and European Medicines Agency have approved over 80 therapeutic antibodies, with over 570 being currently in various stages of clinical trials.Citation1 The majority of these therapeutic antibodies belong to the immunoglobulin class G (IgG), and of those, the majority to IgG1, as this subclass can elicit cytotoxicity on target cells and has the most prolonged serum half-life via neonatal Fc receptor (FcRn)-mediated recycling.Citation2 Alternatively, antibody therapeutics whose function would be impaired by target cell depletion, such as checkpoint inhibitors that bind to T-cells, are often developed as IgG4s that elucidate low Fcγ-receptor activation.
IgG half-life and immune effector cell activation are highly influenced by their glycosylation pattern.Citation3 Antibody glycosylation represents a complex post-translational protein modification that includes a cascade of different trimming and extension steps. In IgG1, it occurs via N-linked glycosylation at asparagine 297 in the crystallizable fragment (Fc) of the IgG heavy chain.
Briefly, fucosylation of the proximal N-acetylglucosamine (GlcNAc) reduces antibody binding to the activating FcγRIIIa receptor on natural killer (NK) cells, a subset of peripheral (5–10%) and splenic monocytes as well as macrophages by steric hindrance, resulting in a reduction of antibody-dependent cell-mediated cytotoxicity (ADCC).Citation4 In contrast, the lack of antibody fucosylation causes increased IgG binding to the low-affinity FcγRIIIb on granulocytes (neutrophils, eosinophils and basophils), which leads to repressed ADCC responses and increased phagocytosis selectively in this cell population.Citation5 Furthermore, terminal galactose or mannose negatively influences the circulating half-life of the antibody, although the importance of this effect is still debated.Citation6 Sialylation of the terminal galactose suppresses antibody-mediated immune responses by: 1) reducing ADCC and antibody-dependent cellular phagocytosis (ADCP) via a decreased affinity to FcγRIIIa; 2) binding to macrophage-presented Siglec-7/9, or by binding to T cell presented Siglec-15; and 3) by impairing complement-dependent cytotoxicity (CDC).Citation7–Citation10 Conversely, antibody sialylation increases the circulating half-life by preventing terminal galactose from binding to the hepatocytic asialoglycoprotein receptor.Citation11 Therefore, depending on the intended biological activity of the antibody, different glycoforms of the antibody may be desired.
Antibody glycosylation is not uniform, resulting in a heterogeneous mixture of different glycoforms. The distribution of glycoforms depends on the producer cell,Citation12 i.e., whether (natural) antibodies are expressed from plasma cells in vivo, or produced ex vivo as recombinant therapeutics, which is typically done in Chinese hamster ovary (CHO) cell lines, giving rise to high yields with facile cultivation.
The glycosylation pattern of CHO-produced therapeutic antibodies can nowadays be modified by glycoengineering. Antagonistic antibodies, such as the anti-HER2 antibody trastuzumab, profit from an increased ADCC, which is achieved by decreased fucosylation.Citation13–Citation15 Conversely, for T-cell-binding checkpoint inhibitors like nivolumab, an ADCC response would lead to T cell depletion and therefore counteract the intended T cell activation. Consequently, a glycosylation pattern with reduced ADCC response and increased circulating half-life, i.e., by increasing fucosylation and sialylation, could be beneficial.Citation16,Citation17
Gene therapy approaches are currently being developed with an increasing number of strategies, utilizing both viral and non-viral vectors, for the delivery of genes encoding therapeutics, including antibodies or antibody fragments. Such in vivo expressed antibodies can, in principle, be produced in a variety of different organs and cell types.Citation18 The in-vivo expression of the anti-human epidermal growth factor receptor 2 (HER2) antibody trastuzumab, for example, has shown promising results in cancer gene therapy.Citation19 Despite the increasing number of gene therapy studies targeting cancer cells, the glycosylation state of antibodies expressed from cancer cells has not been investigated yet. Considering the extensive knowledge about antibody glycosylation in CHO or other ex vivo producer cells and the previously reported altered glycosylation of cancer cell surface receptors,Citation20 it can be presumed that antibodies produced from cancer cells might also have altered glycosylation patterns. However, detailed data about the cancer cell-induced glycosylation pattern of expressed and secreted antibodies are required to lay the foundation for improved cancer gene therapy approaches.
For this reason, we address in this study four key aspects: 1) A comparison of the glycosylation of antibodies produced from target cells for cancer gene therapy with those from established ex vivo producer cells; 2) the influence of an autocrine (growth inhibitory) effect of the expressed antibody on antibody glycosylation; 3) a correlation between antibody isotypes and cancer cell induced glycosylation; and 4) the effect of the gene delivery vectors (viral vs. non-viral) on antibody glycosylation. Therefore, we investigated the glycosylation pattern of two different clinically approved antibodies, trastuzumab (Herceptin®, humanized IgG1κ) and nivolumab (OPDIVO®, IgG4κ). We investigated them upon expression from cancer cells with a variable range of HER2-dependence (), as well as from fibroblasts, which can be a major stromal component of many cancers. Trastuzumab can induce direct cell growth inhibition but not an apoptotic effect in HER2-sensitive cells; thus, HER2-sensitive producer cell lines can be inhibited through autocrine effects. In contrast, nivolumab, an anti-programmed cell death protein 1 (PD1) antibody, has no reported binding to tumor cells.Citation21 Furthermore, antibodies were expressed upon gene delivery with DNA transfection and via different viral gene delivery vectors in cells that promote or do not promote viral replication, to investigate whether active viral replication can affect the glycosylation patterns of the secreted antibodies.
Table 1. Overview of investigated mAb producer cell lines.
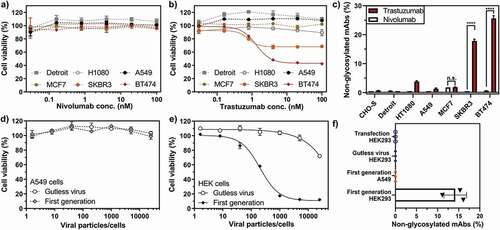
Figure 1. Loss of antibody glycosylation induced by the antagonistic autocrine effect of the produced antibody and virus replication. Growth inhibitory effect of nivolumab (a) or trastuzumab (b) on non-HER2 expressing (A549 and Detroit 551) and HER2-positive (MCF7, SKBR3 and BT474) cells, normalized to untreated cells. SKBR3 and BT474 cells showed a significant decrease in cell viability upon trastuzumab addition (continuous lines), while the cell viability of the remaining cell lines was not influenced by trastuzumab (dotted lines). c) Quantification of non-glycosylated antibody species of nivolumab (white bars) or trastuzumab (red bars) by peptide mass analysis. Only trastuzumab-responsive BT474 and SKBR3 cells produced elevated levels of non-glycosylated or half-glycosylated antibody species. The cell viability of A549 (d) or HEK293 (e) cells treated with a nivolumab-encoding first-generation (filled circles) or gutless adenoviral vector (empty circles) normalized to untreated cells. f) Non-glycosylated antibody forms could only be observed under conditions promoting viral replication (i.e., from the first-generation virus in HEK293 cells). Cell viability was obtained from three separate cell populations. Peptide mass spectrometry data represent three individual consecutive measurements. Error bars represent SEM. Data comparison was done with a two-way ANOVA test, p > 0.01 (**) and p < 0.0001 (****) are indicated.
Recent innovations in state-of-the-art hybrid mass spectrometry (MS) approaches, combining high-resolution native MS and peptide-centric MS, have enabled comprehensive characterization of biopharmaceuticals, including the measurement of structural micro- and macro-heterogeneity of protein proteoforms. Making use of such methods, it is now possible to characterize in-depth various therapeutic and serum glycoproteins.Citation22–Citation24 We have now made use of these methods to characterize for the first time the glycosylation and corresponding biological effects of different antibodies produced in different cell types and cell lines and by different expression systems. This knowledge will help to further design, develop and optimize complex biologics produced in vivo, both with regards to finding the best production site, as well as defining glycoengineering that may be warranted for the intended mode of action.
Results
Homogenous amino acid composition of produced antibodies:
To produce the antibodies in the desired host-cells, we encoded both heavy and light chains of the same antibody under the control of a single promoter, using a furin site followed by a self-cleaving 2A site (), to provide approximately equimolar expression of both chains. To test whether homogeneous backbone production with no residual side products is obtained, we tested the homogeneity and purity of three different 2A site variants (), which originated from three different viruses, namely foot-and-mouth disease virus (F2A), porcine teschovirus-1 (P2A) and thosea asigna virus (T2A). The heavy chain is linked at its C-terminus to a furin site, followed by the 2A site, followed by the signal sequence of the light chain (, Sup. Fig. 1 and 2). Briefly, ribosome skipping leads to a cleavage between the 2A sequence and the light chain signal sequence, followed by removal of the 2A sequence by furin, followed by carboxypeptidase trimming of the four basic amino acids on the C-terminus of the heavy chain.
Figure 2. Optimization of homogeneous production of nivolumab. a) Construct used for expression. The heavy and light chain (dark and light blue) of the expressed antibodies are expressed under the control of a CMV promotor with a 2A site to facilitate equimolar expression. The heavy and light chain are getting separated by a ribosomal skipping mechanism initiated by a 2A site (black). In order to remove residual 2A site residues at the C-terminus of the heavy chain, a furin site (yellow) was encoded upstream of the 2A site. Cleavage sites are depicted in the protein sequence by black arrows. Following cleavage, the signal sequence is translocated and processed, and the residual basic residues of the furin site removed by carboxypeptidases, resulting in unmodified heavy and light chain molecules. The full antibody sequence of trastuzumab and nivolumab are shown in Sup. Fig. 1 and 2. Three 2A sites (F2A, P2A and T2A) were tested to achieve homogenous antibody backbone masses. b) Mass analysis of the PNGase F deglycosylated antibodies. Nearly complete processing of nivolumab is revealed for the P2A construct, as evidenced by the identical mass as observed for the commercial Opdivo. In contrast, the T2A construct shows the presence of minor antibody species with incomplete carboxypeptidase cleavage. In all cases, some low abundant species were detected, representing the clipping of glycine from the C-terminus of the heavy chain. The glycosylation state is shown of nivolumab (c) or trastuzumab (d) expressed from CHO-S cells, transfected with naked DNA or infected with a replication-deficient adenoviral vector. Very similar glycosylation patterns are observed by quantitative glycopeptide analyses. Peptide mass spectrometry data represent three individual consecutive measurements (n = 3).
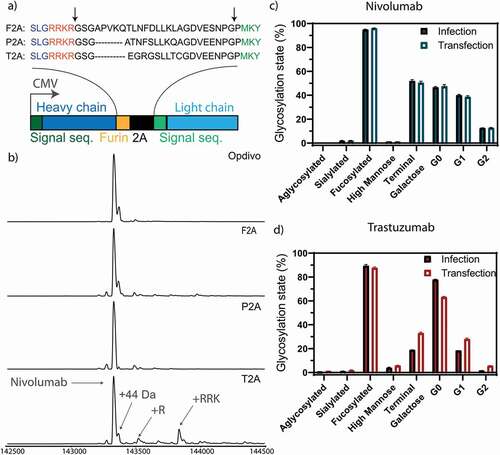
Upon transient expression and Protein A purification of antibodies produced by HEK293-F cells, the P2A variants showed the most homogenous production of the antibody backbone (). Similar results were obtained for the F2A site, while some heterogeneity was observed for the T2A site. As a side note, C-terminal extensions (incomplete carboxypeptidase trimming) observed when using the T2A site have often been identified in recombinantly produced antibodies as a result of stress introduced by the high-level production.Citation25 However, even in this case, despite increasing the heterogeneity of the produced recombinant antibody, the effect of this subpopulation on antibody structure, thermal stability, pharmacokinetics as well as antigen and FcRn binding of this antibody subpopulation is currently considered negligible.Citation25 Nevertheless, we decided to use the P2A construct in further studies, as it showed the most homogeneous processing toward the desired molecular species upon transient expression in HEK293-F cells, which is desired for MS analysis. For trastuzumab, the F2A construct yielded the most homogenous backbone and highest yields upon transient expression, comparable to nivolumab. Both the P2A construct of nivolumab and the F2A construct of trastuzumab resulted in monodisperse proteins with a single major species of the correct size, without any detectable degradation or aggregation (Sup. Fig. 3).
Virus production and targeted cancer cell infection
Both antibodies were encoded on a first-generation adenoviral vector: trastuzumab as an F2A construct and nivolumab as a P2A construct. Negative staining and transmission electron microscopy (TEM) were used to confirm the intact structure of purified virions (Sup. Fig. 4a). Both antibodies produced from CHO-S cells, either transduced with non-replicating viral vectors or by transfection with naked DNA, showed nearly identical glycosylation patterns in native mass analysis (Sup. Fig. 5) and in complementary quantitative glycopeptide analysis (), demonstrating that the viral gene delivery per se, in the absence of active virus replication, does not introduce major changes in the glycosylation state of CHO-expressed antibodies. Trastuzumab from transfected CHO cells showed 15% less agalactosylated, 10% more mono- and 5% more fully galactosylated antibodies than trastuzumab produced from virus-infected cells (). Nevertheless, it should be emphasized that batch-to-batch variations in galactosylation of commercial Herceptin® have been observed and are accepted in the GMP production of the antibody.Citation26 Moreover, changes in the glycosylation between antibody produced from transfected or infected cells were not found for nivolumab (), and so we believe these findings on galactosylation cannot be generalized.
In order to infect HER2-positive cancer cells (MCF7, SKBR3, and BT474) with high efficiency, the purified viruses were retargeted using a previously designed trimeric adaptor molecule,Citation27,Citation28 which redirects the natural tropism of the adenovirus from the coxsackie adenovirus receptor (CAR) to HER2. All other cell lines (CHO-S, Detroit 551, HT1080, and A549) were infected with a naked virus via the natural transduction mechanism that employs CAR binding. A list of all investigated infected cell lines is given in . The transduction efficiency (Sup. Fig. 4b) and the amount of cell-expressed antibody (Sup. Fig. 4c) were similar for both antibody-encoding viruses, except for MCF7, which produced less nivolumab than trastuzumab. Furthermore, trastuzumab-sensitive cells (BT474 and SKBR3 cells) generally produced less antibody than fibroblasts (Detroit) or human non-cancer (HEK293) producer cells (Sup. Fig. 4c). In line with previous findings,Citation29 infection of non-HER2 expressing cancer cells (i.e., A549 cells) could be achieved by infecting cells with non-retargeted adenoviral vectors.
Autocrine effects of produced antibodies lead to growth inhibition and concurrent production of non-glycosylated forms
We acquired high-resolution native mass spectra for all antibodies produced from the different cell lines and, in parallel, performed (glyco)peptide-centric analysis of their glycosylation patterns. The native MS spectra provide an overview of the proteoform profiles of all antibodies analyzed, which we represent by annotated zero-charge deconvoluted spectra and by a list of annotated proteoforms (Sup. Fig. 6 and Sup. Table 2). Complementary to the native MS data, quantitative MS analysis of the IgG glycopeptides (Sup. Fig. 7), generated by proteolytic digestion, provided in-depth glycosylation profiles, from which we derived the relative abundances of all validated glycoforms (Sup. Table 3). Suggested compositions of the glycan structures were obtained from manual inspection of selected MS/MS spectra, which are all depicted in Sup. Data 1 and 2.
Our data revealed that, in addition to the significant differences in the quantity and composition of antibody glycosylation caused by the different origin of the producer cell line, we detected substantial portions (over 20%) of non-glycosylated and a smaller portion (over 2%) of half-glycosylated trastuzumab (), when trastuzumab was expressed from the trastuzumab-responsive cell lines SKBR3 and BT474 (). In sharp contrast, HER2-negative cell lines (A549, CHO-S, Detroit, HT1080), which are not responsive to trastuzumab (), showed negligible amounts of non-glycosylated antibodies. For confirmation of this finding, the non-glycosylated antibody species were also quantified by peptide-centric MS () and were found to be in a good agreement with the native MS results (Sup. Fig. 8). MCF7 cells, which express lower levels of HER2 and are non-responsive to trastuzumab, also showed no significantly elevated levels of non-glycosylated antibodies. Notably, the amount of non-glycosylated trastuzumab correlated with HER2 sensitivity () and HER2 transcription levels (Sup. Fig. 9). This leads us to the conclusion that the signaling inhibition with ensuing growth inhibition by trastuzumab upon binding to HER2 is the biological course for non-glycosylated antibodies. Since we did not find any correlation with the amount of FcγR/FcRn on the producer cancer cells (Sup. Fig. 9), we can exclude Fc binding of trastuzumab to cancer cells as a possible cause.
Figure 3. Non-glycosylated trastuzumab is exclusively produced from HER2-addicted cells. Zero-charge deconvoluted native mass spectra of the intact trastuzumab (a) and nivolumab (b) produced from Detroit and BT474 cell lines. The spectra are separated into three sections, highlighting the mass range of non-glycosylated (red), half-glycosylated (blue), and fully glycosylated forms (gray). c) Trastuzumab expressed from HER2-addicted cells (BT474 and SKBR3) led to the production of a substantial amount of non-glycosylated or half-glycosylated antibodies, while trastuzumab expressed from non-HER2 addicted cells resulted in only fully glycosylated antibodies. d) As a control, nivolumab showed neither non- nor half-glycosylated variants regardless of HER2 addiction of the producer cell line.
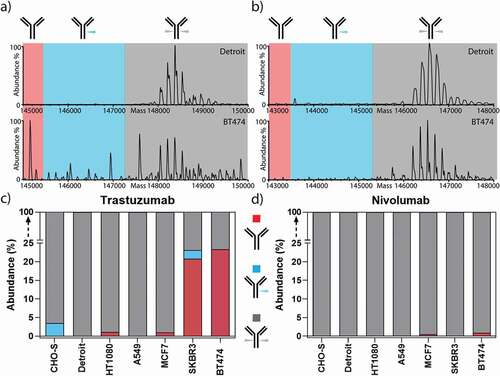
In line with this hypothesis, the expression of nivolumab, which does not bind to HER2, did not impair the growth or metabolism of the tested cell lines (), and did not reveal any incomplete antibody glycosylation (). Therefore, highly elevated levels of non-glycosylated antibody species are restricted exclusively to trastuzumab expression from HER2-sensitive cell lines, and thus this correlates with the antagonistic action of the antibody on its producer cell line.
As a side note, although we optimized different 2A cleavage constructs using a human cell line (HEK293-F) before investigating cancer cell infection, a low abundance (typically around 10%) of incomplete furin-cleaved antibodies were observed in some cell lines (Detroit 551, HT1080, SKBR3, and BT474), most likely caused by a lower expression of furin in these cell lines, compared to HEK293 cells.Citation30
Active viral replication also leads to non-glycosylated antibody forms
To further investigate the observed correlation between inhibition of cell growth and antibody glycosylation, we performed experiments to induce cellular stress by viral replication. We found that non-glycosylated antibody forms were only observed in conditions that facilitate virus-mediated cell lysis, i.e., in HEK293 cells, which provide adenoviral genes (E1A and E1B) in trans for virus particle amplification ().Citation31 For this purpose, we expressed nivolumab, which does not have any autocrine effects on the producing cell lines (here A549 and HEK293), from either a first-generation adenovirus (replication-competent in HEK293 cells) or gutless adenoviral vectors (rendered replication-deficient due to the lack of the viral genome).Citation32 Replication of the first-generation virus in HEK293 cells led to cell growth arrest (), whereas the gutless adenovirus only inhibited proliferation under very high multiplicities of infection (MOIs). Gutless virus-mediated growth inhibition is most likely caused by very low residual amounts of helper virus (which is replication-competent in HEK293 cells), remaining from the production, as has been previously described.Citation32 In A549 cells, which lack the E1 gene and thus do not facilitate the replication of E1-deleted adenoviral vectors, neither first-generation nor gutless virus showed any growth inhibitory effect (). Accordingly, non-glycosylated nivolumab forms were only observed from HEK293 cells infected with first-generation adenovirus (). HEK293 cells transduced with a gutless viral vector or transfected with naked DNA of the antibody plasmids, as well as A549 cells transduced with a first-generation vector, did not lead to incomplete glycosylation ().
Unique glycosylation pattern of cancer cells
As the next step, we performed an in-depth analysis of the cancer cell-mediated glycosylation patterns by combining native MS and glycopeptide analysis to further investigate changes in the composition of the fully glycosylated antibody forms. Overall, we found that the microheterogeneity of the glycosylation profile of the expressed antibody differs with respect to the origin of the producer cell line, quantitatively presented in for trastuzumab expressed from representative cell lines. More specifically, we observed an increased amount of N-acetylneuraminic acid (Neu5Ac) sialylation – further only referred to as sialylation – for both nivolumab and trastuzumab expressed from the various cancer cells, compared to a negligible amount of sialylated antibodies expressed from a classical antibody producer (CHO-S) cell line or a fibroblast (Detroit 551) cell line (). Sialyation levels varied in between the cancer cell lines used, ranging from marginal (i.e., 5% in MCF7 and 7% in BT474) to moderate (i.e., 17% in A549) and high (30–45% in SKBR3) levels. The cancer cells we used belonged to several different cancer types, such as breast adenocarcinomas (MCF7, SKBR3, BT474), fibroblast sarcoma (HT1080) and non-small-cell lung cancer (A549). Although previous studies had shown high amounts of a sialylated therapeutic produced from HT1080 cells,Citation12 we could not detect elevated levels of sialylated antibody expressed from HT1080 cells in our study. Of note, we used HT1080 cells that were previously modified to express the fibroblast activation protein (FAP) for an increased adenovirus infection. Observed elevated levels of antibody sialylation correlated with the upregulation of the α2-6 sialic acid transferase ST6GAL1 mRNA (Sup. Fig. 10) and protein levels () in these cancer cell lines. Also, in line with our findings, orthogonal expression of ST6GAL1 in CHO-S cells has been described to increase antibody sialylation.Citation16 No immune-stimulating, non-endogenous forms of sialic acid N-glycolyl-neuraminic acid (Neu5Gc) were detected on the cancer cell-expressed antibodies.
Figure 4. Glycosylation profile of trastuzumab expressed in a representative set of cell lines. Quantitative comparison of glycosylation patterns based on mass spectrometric analysis of tryptic glycopeptides derived from trastuzumab produced by CHO-S, fibroblast (Detroit 551), and two cancer cell lines (A549 and SKBR3). Overall differences in the antibody glycosylation are expressed in bar charts showing a comparison of three functionally relevant glycan types among different cell lines. Changes in the microheterogeneity are shown by the different coloring of the segments in the stacked columns, representing different glycan compositions, and by depicting some of the most abundant glycans next to the corresponding segment. Of note, some glycoforms appear in both fucosylated and sialylated classes, as they contain both sialic acid and fucose. Antibodies expressed from tumor cells showed substantial differences in microheterogeneity in comparison to antibody glycosylation from non-cancer cells, represented by the table under the chart summarizing the number of glycoforms found for each cell line. Peptide mass spectrometry data represent three individual consecutive measurements.
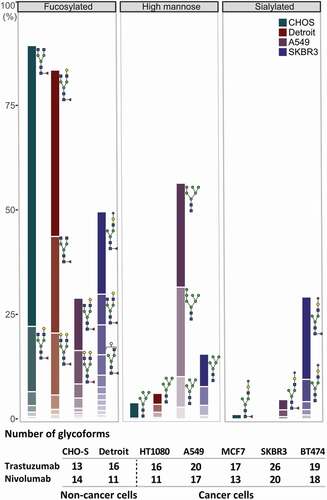
Figure 5. Key glycosylation characteristics of cancer cell-produced antibodies. Antibody glycosylation was quantified by MS analysis of tryptic digests derived from corresponding IgG products. a) Exclusively cancer cells produced antibodies with elevated levels of sialylation. b) The α2-6 sialic acid transferase ST6GAL1 was elevated in cancer cell lines as detected by Western blot, whereas no ST6GAL1 was detected in non-cancer cell lines. c) While the amount of fucosylated nivolumab was consistently high in all cell lines, fucosylation was substantially reduced for trastuzumab produced in several cancer cell lines, independent of HER2 expression. d) Expressed antibodies consist of a mixture of different glycoforms, i.e., strongly ADCC-promoting, with a non-fucosylated glycan (green), weakly ADCC-promoting, fucosylated (gray) or ADCC-impaired, non-glycosylated (red) antibodies. e) A FcγRIIIa-activation assay was used to elucidate the potential effect of cancer cell expressed antibodies on their ADCC response. Trastuzumab binding to HER2-positive cells (SKBR3) leads to the induction of luciferase in a Jurkat detection cell line upon FcγRIIIa-activation. f) FcγRIIIa-activation of trastuzumab expressed from different cell lines as compared to Herceptin is shown. Opdivo, which is a non-HER2 binding IgG4, functions as a negative control, as this isotype has impaired FcγRIIIa binding. FcγRIIIa activation data was obtained from three separate cell populations. Peptide mass spectrometry data represent three individual consecutive measurements. Error bars represent SEM. Data comparison was done with a two-way ANOVA test, p < 0.01 (**) and p < 0.0001 (****) are indicated.
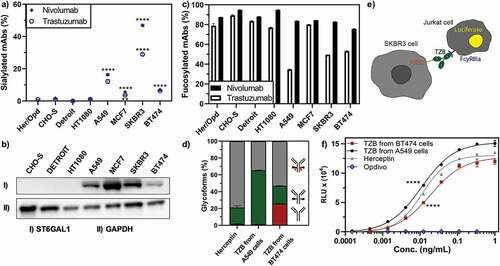
The remaining antibody glycosylation forms we analyzed (fucosylated, as shown in , galactosylated (Sup. Fig. 11) or high-mannose forms (Sup. Fig. 11)) showed no general cancer-cell specific patterns, while particular cancer cells did show very interesting glycoforms. Of note, A549 cells led to a higher amount of high-mannosylated glycoforms (Sup. Fig. 11) and, more importantly, an exceptionally low amount of fucosylated forms for the IgG1 antibody trastuzumab (). This decrease of fucosylation translated to an increased FcγRIIIa binding and activation, detected here via a fluorescence readout assay (), and thus to increased FcRγIIIa engagement of the A549-expressed trastuzumab compared to the clinically used Herceptin (), predicting an increased ADCC response. Notably, this assay does not account for competition of foreign Fc molecules preoccupying the FcRγIIIa receptor, thus in vivo results might differ. Conversely, when trastuzumab was expressed in HER2-addicted (BT474) cells, due to the high amounts of non-glycosylated () and sialylated () antibody species, FcγRIIIa binding, and thus the potential ADCC responses, were lower than when trastuzumab was expressed from HER2-negative cancer (A549) cells or compared to the commercial Herceptin produced in CHO cells ().
Discussion
In this study, we determined whether and how glycosylation patterns of antibodies are affected when they are produced in human tumor cells or fibroblast cells as a prerequisite for designing and optimizing targeted human gene therapy. We compare these results to those from standard producer cell lines (CHO and HEK293); thereby, we benchmarked these glycosylation profiles against their corresponding clinically approved CHO-produced therapeutics, with which properties of different glycoforms have been well established. We focused our work on two well-studied, clinically relevant antibodies of two different classes and modes of action (trastuzumab, an IgG1 where ADCC is desired, and nivolumab, an IgG4 where ADCC is undesired).
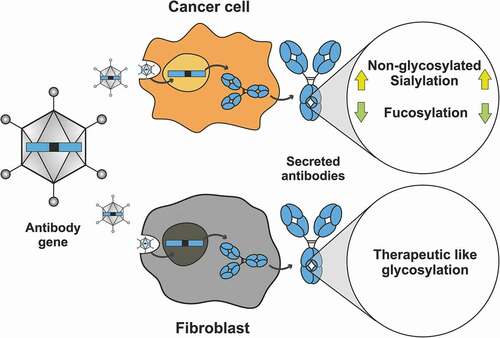
In summary (), we observed three critical variations in the glycosylation of the antibodies at N297: 1) elevated, yet heterogeneous levels of sialylated antibodies produced by the tumor cells; 2) decreased fucosylation of trastuzumab, but not nivolumab, produced in the A549 tumor cell line; and 3) the presence of non-glycosylated antibodies when the cell line is sensitive to the antibody, or when it is put under stress by viral replication, such as by infection with a replication-competent virus. Here, we will discuss the potential implications of these observations for the future developments of cancer gene therapy as these properties relate to Fc receptor affinities, antibody effector functions, serum half-lives and immunogenicity.
Figure 6. Summary: Expression in different host cells influences antibody glycosylation, potentially enhancing activity. Here we expressed two clinically approved antibodies (trastuzumab and nivolumab) in various cancer and non-cancerous cell lines upon gene delivery by a viral vector. The cancer cell lines induced specific glycosylation patterns different from the commercially produced therapeutic antibody, whereas the fibroblast cell line showed more therapeutic-like glycosylation with regards to sialyation and fucosylation. Hypersialylation, as reported to occur on surface receptors of cancer cell lines, is also reflected by the antibodies that are secreted from these cells, and can be linked to the enhanced expression of the sialic acid transferase ST6GAL1 in the tested cancer cell lines. Several cancer cell lines produced and secreted IgGs harboring substantially less fucosylation, which we showed can lead to improved FcγRIIIa binding and thus increased ADCC. Cancer cells sensitive to the antibody they secrete do often respond by the production of non-glycosylated antibody molecules. Thus, when antibodies are produced in vivo, they have to be targeted by viral vectors to the optimal producer cells, which has now become possible. Furthermore, such strategies could further benefit from glycoform engineering, potentially further increasing their therapeutic efficacy.
We revealed that N297 antibody sialylation was elevated for both trastuzumab (IgG1) and nivolumab (IgG4) in all adenocarcinoma and non-small-cell lung cancer lines. These levels also did not significantly vary by antibody class (IgG1 vs. IgG4) when produced by the same cell line, except in the case of SKBR3 cells, which produced nivolumab (IgG4) with significantly higher sialylation levels than observed for trastuzumab (IgG1). We hypothesize that this is not intrinsic to the antibody class itself, but rather due to the autocrine effects of the produced trastuzumab, leading to a negative regulation and overall loss of antibody glycosylation ().
Furthermore, our data indicate that the varying degrees of elevated sialylation across tumor cell lines could be a result of differential upregulation of the α2-6 sialic acid transferase, ST6GAL1, in these tumor cell lines, shown by us via protein levels () and by others via mRNA levels.Citation33 In line with these findings, it has been reported to be sufficient to overexpress human ST6GAL1 during antibody production in CHO cells to induce increased amounts of antibody sialylation.Citation16
Cancer immunotherapeutics like checkpoint blockade antibodies (e.g., anti-PD1) or costimulatory agonistic antibodies (e.g., anti-4-1BB) have been engineered to have impaired ADCC/CDC function to prevent depletion of activated effector cells. Their local production by tumor cells may thus be an attractive design goal, since increased sialylation may already occur naturally in some tumor lines and may otherwise be achieved by ST6GAL1 overexpression. It will have to be assessed, however, whether the thereby obtained extended circulating half-lifeCitation11 and the reduced ADCC responseCitation34 of sialylated antibodies can outperform T cell inactivation by sialic acid-activated Siglec-15Citation9 and NK cell silencing by sialic-activated Siglec-7, −9.Citation8
For many anti-cancer therapies, however, reduced ADCC can be an undesirable effect and reduce antibody efficacy. Furthermore, once bound to the tumor cell, the anti-inflammatory properties of sialic acid could have other consequences. For example, sialylated antagonistic antibodies against tumor receptors that bind directly to tumor cells, such as trastuzumab, might cover the tumor with an additional anti-inflammatory signal that would silence tumor-infiltrating lymphocytes and NK cells instead of activating them.
Fucosylation can also negatively impact ADCC by reducing affinity for FcγRIIIa. Nonetheless, in contrast to sialylation, therapeutic producer cell lines (i.e., CHO) commonly produce antibodies with fucosylated glycans (80–100%).Citation35 One cancer cell line used in our study, i.e., A549, produced significantly lower levels of fucosylation, which indeed translated to increased FcγRIIIa binding and activation compared to the commercial drug Herceptin, produced from CHO cells, and may thus potentially lead to an increased ADCC response. It should be noted that recently several therapeutic anti-tumor antibodies have been glycoengineered to decrease or eliminate fucosylation, showing increased ADCC and promising therapeutic effects surpassing those of the corresponding conventional CHO-produced commercial products.Citation35
Interestingly, reduced fucosylation was only found for the IgG1 (trastuzumab) expressed from a variety of cancer cell lines (A549, SKBR3 and BT474) cells, but not for the IgG4 (nivolumab), which showed normal levels of fucosylation. Differences in the fucosylation state of antibodies may be related to antibody class in addition to the cell line from which they are expressed, as this has been previously described also for IgG1 and IgG4 antibodies expressed from the same cell line.Citation36
Our final key finding was that, in addition to the host cell line and the antibody class, the specificity of the produced antibodies, i.e., antibody-induced autocrine effects on the producer cell line, had significant effects on antibody glycosylation. Native MS analysis revealed that cancer cell lines that were sensitive to the antibodies they produced resulted in significant levels of antibodies lacking glycosylation in one heavy chain (half-glycosylated forms) or both heavy chains (fully non-glycosylated). The loss of glycosylation was exclusively observed in cell lines sensitive to the expressed antagonist trastuzumab, leading to a measurable reduction of FcγRIIIa binding and activation, by the cancer cell-expressed antibodies in comparison to the commercial therapeutic, Herceptin (). We hypothesize that the cellular stress by the signaling inhibition caused by the autocrine effects of the antibody leads to a negative feedback loop where cells release more non-glycosylated antibodies via the secretory pathway or via partial cell lysis.
Similar to the autocrine effect of the produced therapeutic, cellular stress induced by viral replication-mediated cell lysis also enhanced the formation of non-glycosylated antibodies (). In contrast, in the absence of signaling inhibition by the secreted product, no detectable non-glycosylated antibodies were produced from non-replicative vectors or following DNA transfection. Furthermore, CHO cells transfected with DNA or infected with non-replicative vectors showed identical sialylation and fucosylation of the produced antibodies, with some changes only observed in the galactosylation of trastuzumab among the produced antibodies. We thus conclude that, at least in CHO cells, gene delivery by non-replicative viral vectors has no overall impact on the glycosylation pattern compared to DNA transfection. However, further studies will be required to evaluate whether this conclusion is applicable also to cancer and fibroblast cells.
In contrast, our finding that replicative vectors showed increased amounts of non-glycosylated antibodies under conditions that allow viral replication may highlight a limitation of oncolytic viruses: antibodies expressed in vivo from cancer cells infected by a replicative oncolytic virus may lead to decreased therapeutic efficacy because aglycosylated antibodies lack Fc engagement, which is a limitation for antagonistic antibodies that function primarily through ADCC.
In this context, it would be of interest to determine whether the in vitro observed tendency of non-glycosylated antibodies to more easily aggregate and to have lower thermal stability also has a significant limiting influence on the effector function of non-glycosylated antibodies produced by oncolytic viruses.Citation25
The glycosylation pattern of endogenous, B cell-produced antibodies is not static but highly regulated, based on genetics, diet and even diseases,Citation37,Citation38 which indicates that the glycosylation pattern of cancer-cell-produced antibodies in vivo might be even more heterogeneous due to the tumor cells’ complex interactions with the tumor microenvironment, disease progress and responses to therapies than we have addressed here. Gene therapy approaches offer a unique opportunity to adapt and control the glycosylation pattern of in vivo expressed antibodies by employing previously developed antibody glycoengineering strategiesCitation39-Citation42 in vivo, to optimize the physiological functions of the antibody. In fact, in vivo antibody-mediated sialylation has recently shown promising result in anti-inflammatory diseases,Citation43 potentially paving the way for a new field of in vivo glycoengineering. Depending on the desired mode of action, coexpression of sialidasesCitation44 or sialyl transferases,Citation16 decreased fucosylation by GnT-III (β-1,4-mannosyl-glycoprotein 4-β-N-acetylglucosaminyltransferase),Citation42 which induces a bisecting GlcNAc preventing fucosylation due to steric clashes, or RMD (GDP-6-deoxy-D-lyxo-4-hexulose reductase),Citation45 which limits the substrate supply for antibody fucosylation, or, alternatively, increased fucosylation by fucosyl transferases are accessible through appropriate vector design.
In conclusion, the analysis of factors influencing the glycosylation of antibodies produced in various cancer cell lines and fibroblasts, as demonstrated here, will guide the further development of gene therapy for production of therapeutic proteins in vivo.
Materials and methods
Chemicals, antibodies, and cell lines
All chemicals, antibodies, and cell lines used in this study are described in the Supplementary Information.
Antibody design
The heavy and light chains of the humanized IgG1κ antibody, trastuzumab (Sup. Fig. 1), and the human IgG4κ antibody, nivolumab (Sup. Fig. 2), were synthesized from protein sequences of the commercially available therapeutic (Herceptin®, Genentech) and IMGT accession No. 9623, respectively. Signal sequences H7 and L1 were chosen for trastuzumab heavy and light chain, while signal sequences H5 and L2 were chosen for nivolumab to achieve antibody secretion.Citation46 Antibody heavy and light chains were separated by the foot-and-mouth disease virus 2A self-processing peptide (F2A).Citation47 The 2A sequences are preceded by an N-terminal furin cleavage site RKRR to remove excess 2A amino acids from the antibody heavy chain allowing the trimming of the remaining RKRR by carboxypeptidase. The F2A sequences were later exchanged with the P2A or T2A sequence using Gibson assembly (New England Biolabs).
The antibody genes were subcloned into the pShuttle plasmid, where they could be used directly for transient transfection experiments. Further, pShuttle plasmids were assembled with a modified pAdEasy-1 plasmid, carrying a mutation in the hypervariable region 7 of the hexon for first-generation virus production, as described in the AdEasy Adenoviral Vector System kit manual (Agilent Cat. No 240009). For cloning gutless viruses, the expression cassettes from the pShuttle constructs were subcloned directly into the backbone plasmid pC4HSu from Microbix© using standard cloning techniques.
Virus production
First-generation adenoviruses were made as described in the AdEasy Adenoviral Vector System kit‘s manufacturer’s instructions, using HEK293 cells as a packaging cell line. The nivolumab gutless virus was amplified using the cell line 116 as described.Citation48 All viral constructs were purified after production with two consecutive cesium chloride density gradients and stored at −80°C in 20 mM HEPES pH 8.1, 150 mM NaCl, 1 mM MgCl2, 10% glycerol upon usage.
Antibody production
Adherent cells were adapted to DMEM medium containing IgG-depleted serum (PAN Biotech, Cat# P30-2802) for two passages before infection. The day prior to infection, adherent cells were seeded in 5 × 15 cm dishes to reach a confluence of 90% the following day. The density of the suspension cell lines was adjusted to 2 × 106 (HEK293-F) and 4 × 106 (CHO-S) cells/mL on the day of infection. Adherent cells were infected with one infectious particle of untreated adenovirus per cell, except for HER2-expressing cells (i.e., MCF7, BT474, SKBR3), which were infected via HER2-retargeted adenoviral vectors as described previously.Citation27,Citation28 Suspension cells were infected with ten infectious particles per cell of untreated adenovirus. Infectious particles were determined in A549 cells as described in the supplementary part. Cell supernatants were harvested 5, 10- and 15-days post-infection for adherent cells and 7 days post-infection for suspension cells.
Antibody purification
Cell supernatants were filtered through a 0.2 µm filter, loaded onto a prepacked 1 ml GE Protein A column (Cat#: 17040201), washed with 10 column volumes of phosphate-buffered saline (PBS), and antibodies were eluted with 0.1 M citric acid, pH 2.5. Samples with detectable absorbance at 280 nm were pooled, dialyzed against PBS, and stored at −80°C until usage.
Western blotting
ST6Gal1 and GAPDH expression was analyzed by first separating the protein content of the cell lysate on an SDS gel followed by protein transfer to a PVDF membrane and staining by anti ST6GAL1 and GAPDH antibodies. The detailed procedure and antibodies that were used can be found in the supplementary part.
Size exclusion chromatography
Purified antibodies were analyzed by size exclusion chromatography coupled to multi-angle light scattering (SEC-MALS) under non-reducing conditions, following de-glycosylation with PNGase F (NEB cat. no. P0704S). Antibodies (30 µg each) were loaded onto a BEH 500 UPHLC column and separated with a flow rate of 0.3 mL/min in PBS running buffer. The eluted protein was detected by 280 nm absorbance and the molecular mass determined by a three-angle light scattering detector (µDAWN).
Sample preparation for native MS
Purified protein solutions (containing 20 µg of the protein) were buffer-exchanged into 150 mM aqueous ammonium acetate (pH 7.5) by ultrafiltration (vivaspin500, Sartorius Stedim Biotech, Germany) using a 10 kDa cutoff filter, and the protein concentration was adjusted to 2–3 µM prior to native MS analysis. The same batch of samples was used to investigate the backbone mass heterogeneity of all IgG samples. The samples were treated with five units of PNGase F (Roche, IN, USA), and incubated at room temperature overnight. Following incubation, IgG samples were buffer-exchanged once more into 150 mM ammonium acetate (pH 7.5) and immediately analyzed by native MS.
Native MS analysis
Approximately 2–3 µM of protein was directly sprayed into a modified Exactive Plus Orbitrap instrument with extended mass range (EMR) (Thermo Fisher Scientific, Bremen) using a standard m/z range of 150–15,000. The voltage offsets on transport multi-poles and ion lenses were manually tuned to achieve optimal transmission of protein ions at elevated m/z. Nitrogen was used in the higher-energy collisional dissociation cell at a gas pressure of 6–8 × 10−10 bar. The following MS parameters were used: spray voltage 1.2–1.3 V, source fragmentation, and collision energy were varied from 20–100 V to achieve optimal desolvation, source temperature 250°C, and resolution (at m/z 200) 35,000. The instrument was mass calibrated in the 500–5,000 m/z range using CsI clusters, as described previously.Citation49
Proteolytic digestion for glycopeptide-centric analysis
5 µg aliquots of selected IgG samples were brought to 100 mM ammonium bicarbonate (pH 8.5) to achieve a concentration of 1 mg/ml. The samples were denatured with 1% sodium deoxycholate (SDC) and reduced with 5 mM TCEP. This mixture was incubated for 4 hours at 37°C with LysC (enzyme:protein ratio of 1:75 w/w), followed by overnight incubation at 37°C with trypsin (1:100 w/w). Hereafter, the SDC was precipitated by bringing the samples to 1% trifluoroacetic acid and centrifugation at maximum speed for 10 min. The supernatant was collected for subsequent desalting by an Oasis µElution HLB 96-well plate (Waters, Wexford, Ireland) positioned on a vacuum manifold. All desalted proteolytic digests containing modified glycopeptides were dried with a SpeedVac apparatus and reconstituted in 40 µL of 0.1% formic acid prior to liquid chromatography (LC)-MS and MS/MS analysis.
For native MS analysis, proteins were buffer-exchanged into 150 mM aqueous ammonium acetate (pH 7.5), and analyzed on a modified Q-exactive Plus Orbitrap mass spectrometer with extended mass range (EMR) (Thermo Fisher Scientific). The raw native MS spectra were deconvoluted to zero-charge spectra by Intact Mass softwareCitation50 (Protein Metrics, CA, USA). We annotated the spectra by use of an in-house developed R script for semi-automated peak annotation as described previously.Citation51 For LC-MS and MS/MS analysis, we prepared Lys-C and tryptic digests from all protein samples. Resulting peptide mixtures were desalted, dried, and reconstituted in formic acid before MS analysis. All peptides were separated and analyzed using an Agilent 1290 Infinity HPLC system (Agilent Technologies) coupled on-line to an Orbitrap Q exactive HF mass spectrometer (Thermo Fisher Scientific). The data were processed by the software Byonic and Skyline and eventually manually curated. A detailed description of all data analysis can be found in the Supplementary Materials.
Cell viability assay
After four days of antibody or virus incubation with the respective cell line, antibody-induced growth inhibition was measured by a NeoFroxx XTT assay (Cat# 1167TT000) according to the manufacturer’s protocol. Due to different proliferation rates, cells were seeded with adjusted cell densities the day prior to treatment (i.e., 4,000 cells/well for Detroit, 3,200 cells/well for BT474, 2,500 cells/well for MCF7 and 2,000 cells/well for SKBR3, HEK293 and A549) in 96-well plates. Signal intensity was measured at 463 nm absorbance, referenced to 670 nm absorbance.
Antibody FcRγIIIa binding quantification
Due to the cell growth-arresting effect of trastuzumab on HER2-expressing cells, a signaling-based assay from Promega (Cat# G7010) was used to determine the intensity of the FcRγIIIa-mediated response. For this purpose, 5,000 SKBR3 cells were incubated per well with a target-to-effector cell ratio of 1:15, and the FcRγIIIa-induced luciferase signal was detected after 30 minutes of exposure of the cells with Bio-Glo™ Luciferase Assay Substrate using an exposure time of 10 seconds on a Tristar Multimode Reader LB 942 (Berthold).
Disclosure of Potential Conflicts of Interest
The authors have no known competing financial interests or personal relationships that could have appeared to influence the work reported here.
Author contributions
D.B., A.P. and S.N.S designed the project; S.N.S. cloned the plasmids for first-generation adenoviral vectors; D.B. cloned all plasmids for gutless adenoviral vectors, produced all viral constructs, generated and purified all antibodies from respective cell lines, performed size exclusion and cell-based analysis of purified antibodies; V.F. measured native, and peptide mass spectrometry; V.F. and A.J.R.H. analyzed mass spectrometry data; S.N.S. designed antibody expression constructs, performed western blots and ELISAs; D.B., S.N.S and V.F. coordinated the project; A.P. and A.J.R.H supervised the project; D.B., V.F. and S.N.S with the help of A.P. and A.J.R.H wrote the paper.
Supplemental Material
Download Zip (112.6 MB)Acknowledgments
We acknowledge Gery Barmettler and the Center for Microscopy and Image Analysis (ZMB) at the University of Zürich for assistance with TEM, Markus Schmid (University of Zurich) for assistance with virus production, Birgit Dreier (University of Zurich) and Sebastian Globe (Wyatt Technologies Europe) for assistance with the multi-angle light scattering (SEC-MALS) analysis and Prof. Dr. Uwe Zangemeister (University of Bern), Dr. Med. Oliver Gautschi (Luzerner Kantonsspital) and Dr. med. Thilo Zander (Luzerner Kantonsspital) for providing and coordinating the donation of Opdivo©. This research is supported by the SNF Sinergia grant 170929 (to AP), National Cancer Institute of the National Institutes of Health under Award number F32CA189372 (to SNS), and the UZH Forschungskredit 2017 ID 3761 (to DB). VF and AJRH acknowledge support from the Netherlands Organization for Scientific Research (NWO) through the NWO TOP-Punt Grant 718.015.003 and the and EU Horizon 2020 program INFRAIA project Epic-XS (Project 823839).
Supplementary material
Supplemental data for this article can be accessed on the here.
Additional information
Funding
References
- Kaplon H, Reichert JM. Antibodies to watch in 2019. MAbs. 2019;11:219–13. doi:https://doi.org/10.1080/19420862.2018.1556465.
- Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I–related receptor FcRn. Annu Rev Immunol. 2000;18:739–66. doi:https://doi.org/10.1146/annurev.immunol.18.1.739.
- Jennewein MF, Alter G. The immunoregulatory roles of antibody glycosylation. Trends Immunol. 2017;38:358–72. doi:https://doi.org/10.1016/j.it.2017.02.004.
- Chung S, Quarmby V, Gao X, Ying Y, Lin L, Reed C, Fong C, Lau W, Qiu Z, Shen A, et al. Quantitative evaluation of fucose reducing effects in a humanized antibody on Fcγ receptor binding and antibody-dependent cell-mediated cytotoxicity activities. MAbs. 2012;4:326–40. doi:https://doi.org/10.4161/mabs.19941.
- Treffers LW, Van Houdt M, Bruggeman CW, Heineke MH, Zhao XW, Van Der Heijden J, Nagelkerke S, Verkuijlen P, Geissler J, Lissenberg-Thunnissen S, et al. FcγRIIIb restricts antibody-dependent destruction of cancer cells by human neutrophils. Front Immunol. 2018;9:3124. doi:https://doi.org/10.3389/fimmu.2018.03124.
- Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6:576–88. doi:https://doi.org/10.1002/psp4.12224.
- Li T, DiLillo DJ, Bournazos S, Giddens JP, Ravetch JV, Wang LX. Modulating IgG effector function by Fc glycan engineering. Proc Natl Acad Sci U S A. 2017;114:3485–90. doi:https://doi.org/10.1073/pnas.1702173114.
- Jandus C, Boligan K, Chijioke O, Liu H, Dahlhaus M, Démoulins T, Schneider C, Wehrli M, Hunger RH, Baerlocher GM, et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J Clin Invest. 2014;124:1810–20. doi:https://doi.org/10.1172/JCI65899.
- Wang J, Sun J, Liu LN, Flies DB, Nie X, Toki M, Zhang J, Song C, Zarr M, Zhou X, et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med. 2019;25:656–66. doi:https://doi.org/10.1038/s41591-019-0374-x.
- Quast I, Keller CW, Maurer MA, Giddens JP, Tackenberg B, Wang LX, Münz C, Nimmerjahn F, Dalakas MC, Lünemann JD. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J Clin Invest. 2015;125:4160–70. doi:https://doi.org/10.1172/JCI82695.
- Park EI, Manzella SM, Baenziger JU. Rapid clearance of sialylated glycoproteins by the asialoglycoprotein receptor. J Biol Chem. 2003;278:4597–602. doi:https://doi.org/10.1074/jbc.M210612200.
- Goh JB, Ng SK. Impact of host cell line choice on glycan profile. Crit Rev Biotechnol. 2018;38:851–67. doi:https://doi.org/10.1080/07388551.2017.1416577.
- Junttila TT, Parsons K, Olsson C, Lu Y, Xin Y, Theriault J, Crocker L, Pabonan O, Baginski T, Meng G, et al. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res. 2010;70:4481–89. doi:https://doi.org/10.1158/0008-5472.CAN-09-3704.
- Schuster M, Umana P, Ferrara C, Brünker P, Gerdes C, Waxenecker G, Wiederkum S, Schwager C, Loibner H, Himmler G, et al. Improved effector functions of a therapeutic monoclonal Lewis Y-specific antibody by glycoform engineering. Cancer Res. 2005;65:7934–41. doi:https://doi.org/10.1158/0008-5472.CAN-04-4212.
- Malphettes L, Freyvert Y, Chang J, Liu P, Chan E, Miller J, Zhou Z, Nguyen T, Tsai C, Snowden AW, et al. Highly efficient deletion of FUT8 in CHO cell lines using zinc-finger nucleases yields cells that produce completely nonfucosylated antibodies. Biotechnol Bioeng. 2010;106:774–83. doi:https://doi.org/10.1002/bit.22751.
- Lin N, Mascarenhas J, Sealover NR, George HJ, Brooks J, Kayser KJ, Gau B, Yasa I, Azadi P, Archer-Hartmann S. Chinese hamster ovary (CHO) host cell engineering to increase sialylation of recombinant therapeutic proteins by modulating sialyltransferase expression. Biotechnol Prog. 2015;31:334–46. doi:https://doi.org/10.1002/btpr.2038.
- Du J, Meledeo MA, Wang Z, Khanna HS, Paruchuri VDP, Yarema KJ. Metabolic glycoengineering: sialic acid and beyond. Glycobiology. 2009;19:1382–401. doi:https://doi.org/10.1093/glycob/cwp115.
- Hollevoet K, Declerck PJ. State of play and clinical prospects of antibody gene transfer. J Transl Med. 2017;15:131. doi:https://doi.org/10.1186/s12967-017-1234-4.
- Jiang M, Shi W, Zhang Q, Wang X, Guo M, Cui Z, Su C, Yang Q, Li Y, Sham J et al. Gene therapy using adenovirus-mediated full-length anti-HER-2 antibody for HER-2 overexpression cancers. Clin Cancer Res. 2006;12:6179–85. doi:https://doi.org/10.1158/1078-0432.ccr-06-0746.
- Rodrigues E, Macauley MS. Hypersialylation in cancer: modulation of inflammation and therapeutic opportunities. Cancers (Basel). 2018;10:207. doi:https://doi.org/10.3390/cancers10060207.
- Brockhoff G, Heckel B, Schmidt-Brecken E, Plander M, Hofstaedter F, Vollmann A, Diermeier S. Differential impact of Cetuximab, Pertuzumab and Trastuzumab on BT474 and SK-BR-3 breast cancer cell proliferation. Cell Prolif. 2007;40:488–507. doi:https://doi.org/10.1111/j.1365-2184.2007.00449.x.
- Yang Y, Liu F, Franc V, Halim LA, Schellekens H, Heck AJR. Hybrid mass spectrometry approaches in glycoprotein analysis and their usage in scoring biosimilarity. Nat Commun. 2016;7:13397. doi:https://doi.org/10.1038/ncomms13397.
- Franc V, Zhu J, Heck AJR. Comprehensive proteoform characterization of plasma complement component C8αβγ by hybrid mass spectrometry approaches. J Am Soc Mass Spectrom. 2018;29:1099–110. doi:https://doi.org/10.1007/s13361-018-1901-6.
- Čaval T, Tian W, Yang Z, Clausen H, Heck AJR. Direct quality control of glycoengineered erythropoietin variants. Nat Commun. 2018;9:3342. doi:https://doi.org/10.1038/s41467-018-05536-3.
- Liu H, Ponniah G, Zhang H, Nowak C, Neil A, Gonzalez-Lopez N, Patel R, Cheng G, Kita AZ, Andrien B. In vitro and in vivo modifications of recombinant and human IgG antibodies. MAbs. 2014;6:1145–54. doi:https://doi.org/10.4161/mabs.29883.
- Kim S, Song J, Park S, Ham S, Paek K, Kang M, Chae Y, Seo H, Kim H, Flores M. Drifts in ADCC-related quality attributes of Herceptin®: impact on development of a trastuzumab biosimilar. MAbs. 2017;9:704–14. doi:https://doi.org/10.1080/19420862.2017.1305530.
- Dreier B, Honegger A, Hess C, Nagy-Davidescu G, Mittl P, Grütter MG, Belousova N, Mikheeva G, Krasnykh V, Plückthun A. Development of a generic adenovirus delivery system based on structure-guided design of bispecific trimeric DARPin adapters. Proc Natl Acad Sci U S A. 2013;110:E869–77. doi:https://doi.org/10.1073/pnas.1213653110.
- Schmid M, Ernst P, Honegger A, Suomalainen M, Zimmermann M, Braun L, Stauffer S, Thom C, Dreier B, Eibauer M, et al. Adenoviral vector with shield and adapter increases tumor specificity and escapes liver and immune control. Nat Commun. 2018;9:450. doi:https://doi.org/10.1038/s41467-017-02707-6.
- Lyons M, Onion D, Green NK, Aslan K, Rajaratnam R, Bazan-Peregrino M, Phipps S, Hale S, Mautner V, Semour LW, et al. Adenovirus type 5 interactions with human blood cells may compromise systemic delivery. Mol Ther. 2006;14:118–28. doi:https://doi.org/10.1016/j.ymthe.2006.01.003.
- Petryszak R, Keays M, Tang Y, Fonseca N, Barrera E, Burdett T, Füllgrabe A, Fuentes A, Jupp S, Koskinen S, et al. Expression Atlas update - an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2016;44:D746–D752. doi:https://doi.org/10.1093/nar/gkv1045.
- Jager L, Hausl MA, Rauschhuber C, Wolf NM, Kay MA, Ehrhardt A. A rapid protocol for construction and production of high-capacity adenoviral vectors. Nat Protoc. 2007;2:1236–47. doi:https://doi.org/10.1038/nprot.2009.4.
- Rosewell A, Vetrini F, Ng P. Helper-dependent adenoviral vectors. J Genet Syndr Gene Ther. 2011;Suppl 5:1. doi:https://doi.org/10.4172/2157-7412.s5-001.
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–07. doi:https://doi.org/10.1038/nature11003.
- Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–73. doi:https://doi.org/10.1126/science.1129594.
- Pereira NA, Chan KF, Lin PC, Song Z. The “less-is-more” in therapeutic antibodies: afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs. 2018;10:693–711. doi:https://doi.org/10.1080/19420862.2018.1466767.
- Bruggeman CW, Dekkers G, Bentlage AEH, Treffers LW, Nagelkerke SQ, Lissenberg-Thunnissen S, Koeleman CAM, Wuhrer M, van den Berg TK, Rispens T, et al. Enhanced effector functions due to antibody defucosylation depend on the effector cell Fcγ receptor profile. J Immunol. 2017;199:204–11. doi:https://doi.org/10.4049/jimmunol.1700116.
- Ackerman ME, Crispin M, Yu X, Baruah K, Boesch AWN, Harvey DJ, Dugast AS, Heinzen EL, Eran A, Choi I, et al. Natural variation in Fc glycosylation of HIV-specific antibodies impacts antiviral activity. J Clin Invest. 2013;123:2183–92. doi:https://doi.org/10.1172/JCI65708.
- Mahan AE, Jennewein MF, Suscovich T, Dionne K, Tedesco J, Chung AW, Streeck H, Pau M, Schuitemaker H, Francis D, et al. Antigen-specific antibody glycosylation is regulated via vaccination. PLoS Pathog. 2016;12:e1005456. doi:https://doi.org/10.1371/journal.ppat.1005456.
- Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the “high-hanging fruit.”. Nat Rev Drug Discov. 2018;17:197–223. doi:https://doi.org/10.1038/nrd.2017.227.
- Jung ST, Reddy ST, Kang TH, Borrok MJ, Sandlie I, Tucker PW, Georgiou G. Aglycosylated IgG variants expressed in bacteria that selectively bind FcγRI potentiate tumor cell killing by monocyte-dendritic cells. Proc Natl Acad Sci U S A. 2010;107:604–09. doi:https://doi.org/10.1073/pnas.0908590107.
- Chen TF, Sazinsky S, Houde D, DiLillo DJ, Bird J, Li KK, Cheng GT, Qiu H, Enge JR, Ravetch JV, et al. Engineering aglycosylated IgG variants with wild-type or improved binding affinity to human Fc gamma RIIa and Fc gamma RIIIAs. J Mol Biol. 2017;429:2528–41. doi:https://doi.org/10.1016/j.jmb.2017.07.001.
- Ferrara C, Brünker P, Suter T, Moser S, Püntener U, Umaña P. Modulation of therapeutic antibody effector functions by glycosylation engineering: influence of Golgi enzyme localization domain and co-expression of heterologous β1, 4-N-acetylglucosaminyltransferase III and Golgi α-mannosidase II. Biotechnol Bioeng. 2006;93:851–61. doi:https://doi.org/10.1002/bit.20777.
- Pagan JD, Kitaoka M, Anthony RM. Engineered sialylation of pathogenic antibodies in vivo attenuates autoimmune disease. Cell. 2018;172:564–77. doi:https://doi.org/10.1016/j.cell.2017.11.041.
- Naso MF, Tam SH, Scallon BJ, Raju TS. Engineering host cell lines to reduce terminal sialylation of secreted antibodies. MAbs. 2010;2:519–27. doi:https://doi.org/10.4161/mabs.2.5.13078.
- Von Horsten HH, Ogorek C, Blanchard V, Demmler C, Giese C, Winkler K, Kaup M, Berger M, Jordan I, Sandig V, et al. Production of non-fucosylated antibodies by co-expression of heterologous GDP-6-deoxy-d-lyxo-4-hexulose reductase. Glycobiology. 2010;20:1607–18. doi:https://doi.org/10.1093/glycob/cwq109.
- Haryadi R, Ho S, Kok YJ, Pu HZ, Zheng L, Pereira NA, Li B, Bi X, Goh L, Yang Y, et al. Optimization of heavy chain and light chain signal peptides for high level expression of therapeutic antibodies in CHO cells. PLoS One. 2015;10:1–16. doi:https://doi.org/10.1371/journal.pone.0116878.
- Chng J, Wang T, Nian R, Lau A, Hoi KM, Ho SCL, Gagnon P, Bi X, Yang Y. Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. MAbs. 2015;7:403–12. doi:https://doi.org/10.1080/19420862.2015.1008351.
- Palmer D, Ng P. Improved system for helper-dependent adenoviral vector production. Mol Ther. 2003;8:846–52. doi:https://doi.org/10.1016/j.ymthe.2003.08.014.
- Rosati S, Yang Y, Barendregt A, Heck AJR. Detailed mass analysis of structural heterogeneity in monoclonal antibodies using native mass spectrometry. Nat Protoc. 2014;9:967–76. doi:https://doi.org/10.1038/nprot.2014.057.
- Bern M, Caval T, Kil YS, Tang W, Becker C, Carlson E, Kletter D, Sen KI, Galy N, Hagemans D, et al. Parsimonious charge deconvolution for native mass spectrometry. J Proteome Res. 2018;17:1216–26. doi:https://doi.org/10.1021/acs.jproteome.7b00839.
- Lin Y-H, Zhu J, Meijer S, Franc V, Heck AJR. Glycoproteogenomics: A frequent gene polymorphism affects the glycosylation pattern of the human serum fetuin/α-2-HS-glycoprotein. Mol Cell Proteomics. 2019;18:1479–90. doi:https://doi.org/10.1074/mcp.ra119.001411.