
ABSTRACT
Conditionally active COBRA™ (COnditional Bispecific Redirected Activation) T cell engagers are engineered to overcome the limitations of inherently active first-generation T cell engagers, which are unable to discern between tumor and healthy tissues. Designed to be administered as prodrugs, COBRAs target cell surface antigens upon administration, but engage T cells only after they are activated within the tumor microenvironment (TME). This allows COBRAs to be preferentially turned on in tumors while safely remaining inactive in healthy tissue. Here, we describe the development of the COBRA design and the characterization of these conditionally active T cell engagers. Upon administration COBRAs are engineered to bind to tumor-associated antigens (TAAs) and serum albumin (to extend their half-life in circulation), but are inhibited from interacting with the T cell receptor complex signaling molecule CD3. In the TME, a matrix metalloproteinase (MMP)-mediated linker cleavage event occurs within the COBRA construct, which rearranges the molecule, allowing it to co-engage TAAs and CD3, thereby activating T cells against the tumor. COBRAs are conditionally activated through cleavage with MMP9, and once active are highly potent, displaying sub-pM EC50s in T cell killing assays. Studies in tumor-bearing mice demonstrate COBRA administration completely regresses established solid tumor xenografts. These results strongly support the further characterization of the novel COBRA design in preclinical development studies.
Introduction
Monoclonal antibodies have transformed the field of cancer therapeutics through their ability to precisely target specific proteins on the surface of tumor cells. Efforts to increase the activity of these drugs have included engineering antibody binding sites to increase affinity to their target antigens, as well as conjugation of antibodies to potent cytotoxins for targeted delivery to tumor cells.Citation1−Citation7 In addition to their selective recognition of individual proteins on tumor cells, antibodies can also induce immune-mediated destruction of these cells.Citation8-Citation11 Engineering antibodies to enhance their ability to recruit a cytotoxic immune response has proven to be an effective strategy for increasing their potency.
Bispecific antibodies that engage T cells possess two distinct binding sites, one specifically targets an antigen on the surface of the tumor cell and the other binds an activating receptor on the surface of a T cell. The first therapeutic molecules of this type, called bispecific T cell engagers, have proven to be very potent in directing cytotoxic T cell responses to specific target cells both in vitro and in vivo.Citation12-Citation15 When the selected target antigen is only expressed on tumor cells and non-essential normal cells, this strategy has produced very potent therapeutic molecules like blinatumomab, which targets the CD19 antigen on the surface of certain B cell leukemias (and normal B cells).Citation16-Citation18
The application of this strategy to the treatment of solid tumors has been more challenging because most solid tumor antigens are also expressed on essential normal cells.Citation19-Citation23 Compounding this issue is the need to deliver higher dose levels to patients with solid tumors due to the high interstitial pressure and poor penetration of therapeutics into the tumors.Citation24-Citation26 Thus, it may not be possible to deliver doses that would be efficacious in patients with cancer if the therapeutic also targets cytotoxic T cell responses to normal cells expressing solid tumor antigens. Strategies to address this problem have included designing bispecific T cell engaging prodrugs that can be selectively activated in the tumor micro-environment (TME). Several physiological features of the TME set it apart from most normal tissues. For example, disorganized tissue growth in a tumor can lead to an anoxic environment with a lower extracellular pH.Citation27-Citation32 In addition, rapid cell division in a tumor is often associated with increased extracellular matrix remodeling and increased proteolysis.Citation33,Citation34 Due to dysregulated gene expression, tumors also frequently co-express cell-surface antigens that are not co-expressed on normal tissues. To exploit these characteristics of the TME, antibodies have been engineered to bind preferentially at low pH or to bind only in the presence of aptamers activated under hypoxic conditions.Citation35-Citation37 Other groups have designed protease-activated, bispecific T cell engagers either using masks attached via protease-cleavable linkers to block one or both of the antibody binding sites or employing protein domain complementation and Boolean logic gating to ensure bispecific activation only in the presence of two different tumor antigens.Citation38-Citation40
The approach taken in this study was to separate the active domains of the T cell binding site from each other such that CD3 binding by the prodrug is impaired, and to use the functional tumor-targeting domains in the bispecific to accumulate these molecules on the surface of tumor cells. These prodrugs can then be selectively activated in the protease-rich TME and rearrange to become active T cell engagers while remaining as inactive prodrugs in normal tissues, which have less proteolytic activity. Linkers sensitive to matrix metalloproteases (MMPs), especially MMP2 and MMP9, were chosen as the activation switches in these molecules due to the ubiquitous expression of these proteases in tumors and their tight regulation in normal tissues.Citation41-Citation44 Another important consideration in the design of these molecules was to minimize the loss of potency in the underlying T cell bispecific while maximizing the conditionality of the prodrug. Finally, the serum half-life of the activated drug was designed to be reduced relative to the prodrug in order to broaden the therapeutic index of the resulting molecules.
Results
Hemi-COBRA structure and MOA
To generate conditional T cell engagers, the two variable domains of an active anti-CD3ε single-chain variable fragment (scFv) were placed into two separate molecules termed hemi-COBRAs. Each of these active domains (Vh or Vl) was attached to a C-terminal complementary inactivated variable domain (Vli or Vhi) connected by a protease-cleavable scFv linker to stabilize the protein. A tumor-targeting single domain antibody (sdAb) was added to the N-terminus of each active anti-CD3 variable domain and an anti-human serum albumin (HSA) sdAb was added to the C-terminus of each inactive anti-CD3 domain. Each of the molecules in a complementary pair was designed to have an extended serum half-life due to its C-terminal anti-HSA sdAb and bind a target antigen on a tumor cell using its N-terminal sdAb. The ability of the anti-HSA sdAb to extend the serum half-life of protein molecules to which it is linked is well established.Citation45,Citation46 Once bound to a cellular target the hemi-COBRAs were designed not to bind CD3 and activate T cells until proteolytic cleavage of the scFv linker. Upon cleavage, the inactive anti-CD3 scFvs can subsequently domain exchange to create an active CD3 binding site tethered to the surface of the tumor cell (Supplemental Figure 1). In addition, having lost the downstream HSA binding sdAb, these active molecules should have a reduced serum half-life should they escape the TME or form in the periphery. Since the TME is known to be enriched for a subset of activated proteases relative to normal tissues, activation of hemi-COBRAs by these proteases should favor T cell-mediated tumor killing over T cell-mediated toxicity in normal tissues.
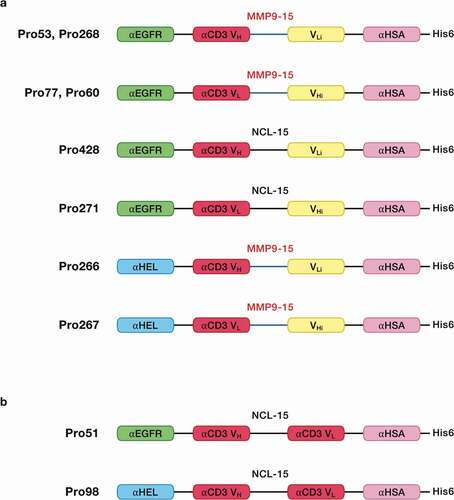
To test this strategy, complementary pairs of hemi-COBRAs were produced (). The first pair of hemi-COBRAs (Pro268, Pro60) used an anti-epidermal growth factor receptor (anti-EGFR) sdAb as the tumor-targeting domain. The inactive anti-CD3 domains (Vhi, Vli) were created by substituting part or all of the normal CDR2 region with the Flag sequence (DYKDDDDK). The inactive anti-CD3 domains were attached to their complimentary active anti-CD3 domains using MMP2/9 cleavable scFv linkers (15aa long). The non-cleavable hemi-COBRA pair (Pro428, Pro271) had the same structure, but the MMP2/9 cleavable scFv linkers were replaced with standard, non-cleavable scFv linkers. The non-tumor-targeting hemi-COBRA pair (Pro266, Pro267) used an anti-hen egg lysozyme (αHEL) sdAb in place of the anti-EGFR sdAbs in Pro268 and Pro60. A final pair of hemi-COBRAs (Pro53, Pro77) used more conservative germline reversion mutations to create the inactive anti-CD3 domains (GL3, GL). Pro51 was constructed as a T cell-engaging positive control using the anti-EGFR sdAb linked to a fully active CD3 binding scFv followed by an anti-HSA sdAb. Pro98 was constructed as a non-tumor binding negative control by substituting the anti-HEL sdAb for the tumor-targeting anti-EGFR sdAb in Pro51 ().
Figure 1. Molecular schematics.
In vitro activity of complementary Hemi-COBRA pairs
Initial studies tested the binding of the uncleaved molecules to EGFR-expressing HT-29 cells and CD3-expressing Jurkat cells (Supplemental Figure 2). As expected, the molecules carrying the anti-EGFR sdAb bound to HT-29 tumor cells, whereas the molecules with the anti-HEL sdAb did not. Likewise, the two control molecules with active anti-CD3-binding scFvs bound to the Jurkat T cell line, but the hemi-COBRAs did not. Each of the molecules was tested for its binding affinity to human EGFR, human CD3ε and HSA via biolayer interferometry (Octet) (Supplemental Figure 3a-j). All molecules possessing the anti-EGFR sdAb bound to human EGFR with a KD from 1–3 nM. None of the molecules with an anti-HEL sdAb showed detectable binding to EGFR. None of the hemi-COBRAs showed binding to human CD3ε, whereas the control molecules with an active anti-CD3 scFv bound CD3ε with an affinity close to 5 nM. Each of the intact molecules bound HSA with an affinity of around 10 nM.
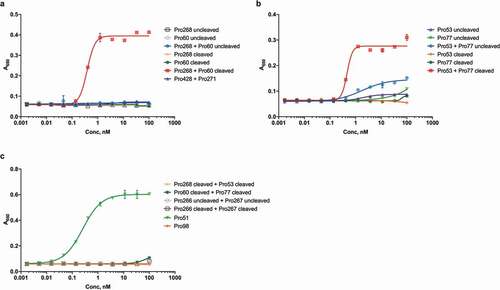
To test the activity of cleaved hemi-COBRAs, the molecules were treated with purified human MMP9 and cleavage of the scFv linker was assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (). Hemi-COBRAs with the MMP9 cleavable linker were cleaved predominantly into two protein fragments of the expected sizes, whereas the hemi-COBRAs bearing the non-cleavable linkers were not affected by the MMP9 treatment. The ability of complementary hemi-COBRAs to create CD3 binding sites following MMP9 activation was demonstrated using a sandwich enzyme-linked immunosorbent assay (ELISA). Hemi-COBRAs or controls were bound to immobilized human EGFR, after which the binding of human CD3ε was assessed. Cleaved Pro268 only showed CD3ε binding when it was mixed with cleaved Pro60 (). Neither hemi-COBRA alone (cleaved or uncleaved) showed CD3ε binding, and the mixture of the two uncleaved molecules failed to create CD3ε binding sites. The Pro53 + Pro77 hemi-COBRA pair showed similar activity, although the inactivation of the anti-CD3 domains was less complete than seen with the Flag inactivated domains. When mixed, uncleaved Pro53 + Pro77 showed a low level of CD3ε binding that was substantially increased upon MMP9 cleavage (). Neither molecule showed CD3ε binding on its own, either cleaved or uncleaved. The need for complementary, activated hemi-COBRAs was shown by mixing cleaved Pro268 with cleaved Pro53 and cleaved Pro60 with cleaved Pro77. Neither of these mixtures contain complementary active anti-CD3 (Vh + Vl) and neither mixture showed significant CD3ε binding ().
Figure 2. Cleaved vs uncleaved hemi-COBRAs – SDS-PAGE.
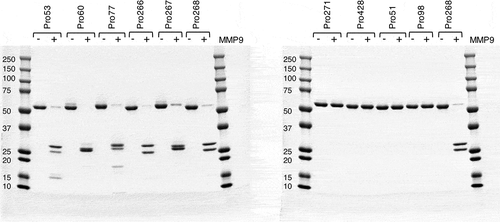
Figure 3. Sandwich ELISA of intact and cleaved hemi-COBRAs.
Similar results were achieved using a sandwich fluorescence-activated cell sorting (FACS) assay on EGFR-expressing HT-29 cells. CD3ε binding sites were created on the surface of these EGFR-expressing cells when cleaved Pro268 + Pro60 were bound to the cells, but not when the uncleaved molecules were bound (). Similarly, cleaved Pro53 created a substantial number of CD3ε binding sites when mixed with cleaved Pro77, even though the uncleaved mixture showed no CD3ε binding (). also shows that while the complementary hemi-COBRA pairs demonstrate conditional binding to CD3ε, they have gained this conditionality by sacrificing some potency relative to a constitutive T cell engager like Pro51.
Figure 4. Sandwich FACS of intact and cleaved hemi-COBRAs.

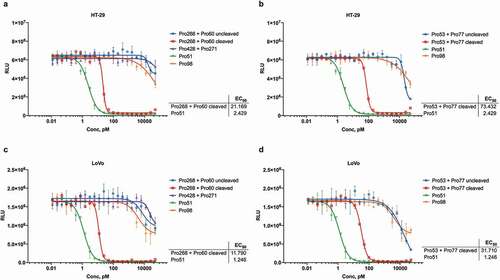
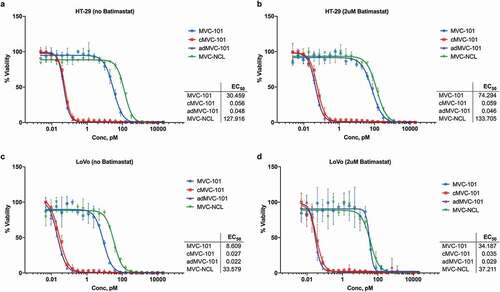
The EGFR-binding hemi-COBRA pair, Pro268 + Pro60, was then tested for its ability to induce primary human T cells to lyse HT-29 cells in vitro in a T cell-dependent cellular cytotoxicity (TDCC) assay. Only after MMP9 cleavage was the pair able to efficiently induce TDCC activity. The uncleaved hemi-COBRA pair showed at least 500-fold less potency in this assay and had similar activity to the non-tumor-targeting control and the non-cleavable hemi-COBRA pair (). Comparable results were seen with the hemi-COBRA pair carrying the germline inactivated anti-CD3 domains (Pro53/Pro77 in ). In both cases the activated hemi-COBRA pairs showed reduced potency compared to the fully active control (Pro51). Analogous results were generated using LoVo cells as the EGFR-expressing target cell line (,). Comparing the potencies of the cleaved hemi-COBRA pairs in TDCC assays on HT-29 cells, 72 pM for Pro268 + Pro60 and 22 pM for Pro53 + Pro77, with their ability to generate CD3-binding sites on these cells as measured by FACS demonstrates that efficient T cell killing is engendered at a very low level of receptor occupancy.
Figure 5. Hemi-COBRA TDCC.
Structure and MOA of full-length COBRAs
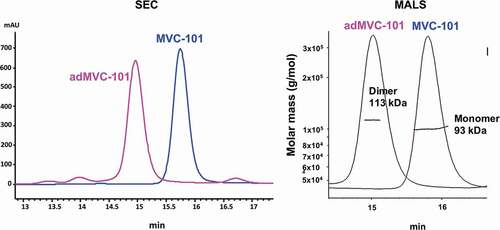
Having shown that the separated active anti-CD3 heavy chain and light chain domains can assemble on the surface of tumor cells to promote T cell killing, we attempted to design a therapeutic that would combine the two hemi-COBRAs into a single molecule with similar properties as a hemi-COBRA pair. This was done by creating constrained active and inactive anti-CD3 scFvs with short linkers (8aa) that prevented the heavy chain and light chain variable domains from pairing. The full-length COBRA (MVC-101) was designed such that the constrained, active anti-CD3 domains were flanked on both sides by tumor-targeting sdAbs at the N-terminus of the molecule. The MMP9 cleavable linker was used to separate this fragment from the Flag-inactivated anti-CD3 domains, followed by the anti-HSA sdAb at the C-terminus of the molecule (). Following cleavage by MMP9, two active fragments could dimerize on the tumor cell surface to create two CD3 binding sites with four target binding sdAbs, as diagrammed in Supplemental . This model is supported by size exclusion chromatography combined with multi-angle light scattering (SEC-MALS) showing that expression of MVC-101 produces a single peak with a molecular weight of 93 kDa, as expected for a monomeric protein folded into a single isoform. In contrast, the isolated active fragment (adMVC101) produces a dominant peak with a molecular weight of 113 kDa, which is consistent with a homodimeric active domain ().
Figure 6. Schematics of full-length COBRAs.
Figure 7. SEC-MALS analysis of MVC-101 and adMVC-101.
In vitro activity of full-length COBRAs
The affinities of the binding sites within the full-length COBRAs for their target antigens were measured by Octet. The two anti-EGFR sdAbs in MVC-101 appeared to bind in a concerted fashion to human EGFR, producing an apparent affinity of 330 pM, as compared to the 3 nM binding affinity observed with the individual hemi-COBRAs. Importantly, binding to human CD3ε was not detected (Supplemental Figure 5a). Similar results were obtained with the non-cleavable control, MVC-NCL (Supplemental Figure 5b). In contrast, the recombinantly expressed active dimer, adMVC-101, showed even stronger binding to human EGFR with an apparent affinity 54 pM and an apparent affinity for human CD3ε of 58 pM (Supplemental Figure 5c). The binding affinities of MVC-101, MVC-NCL, and adMVC-101 to non-human primate antigens, rhesus EGFR, cynomolgus CD3ε, and cynomolgus serum albumin were within 3-fold of their affinities to the respective human antigens (Supplemental Figure 5d-f). Neither MVC-101 nor MVC-NCL showed detectable binding to mouse EGFR or mouse CD3ε, but did bind mouse serum albumin, which is expected to promote half-life extension in mouse models (Supplemental Figure 5g,h).
Cleavage of MVC-101 with either MMP2 or MMP9 produced the expected size fragments on an SDS-PAGE gel, whereas MVC-NCL was unaffected by MMP2 or MMP9 (Supplemental Figure 6). Interestingly, MVC-101 did not rapidly dissociate into its component parts in solution following cleavage with MMP9, as shown by size exclusion chromatography (Supplemental Figure 7). However, the cleavage fragments do dissociate slowly at 4°C (data not shown).
The MVC-101 monomer can simultaneously bind both EGFR and HSA as shown by SEC analysis (Supplemental Figure 8a). The MMP9 cleavage site remains accessible to MMP9 after the complex is formed as evident in the SDS PAGE gel of the COBRA treated with MMP9 in the presence of HSA and EGFR (Supplemental Figure 8b). Thus, the protein folds in such a manner that efficient target binding and protease cleavage are maintained.
The full-length COBRAs were evaluated for their ability to bind EGFR-expressing cells via FACS. All molecules bound HT-29 cells and LoVo cells in a dose-dependent manner (Supplemental Figure 9a). When the cell-bound, full-length COBRAs were tested for CD3ε binding, only the active dimer, adMVC-101, showed significant binding (supplemental Figure 9b).
The potencies of these molecules were measured in TDCC assays against EGFR-expressing cells. Using HT-29 cells as targets, the potency of pre-cleaved MVC-101 was similar to the potency of the adMVC-101 dimer. These data support the hypothesis that pre-cleaved MVC-101 efficiently rearranged to produce the active dimer on the surface of target cells. Uncleaved MVC-101 showed significantly less potency than the pre-cleaved MVC-101, but higher activity than the non-cleavable control, MVC-NCL (). The TDCC activity of uncleaved MVC-101 was reduced by the addition of the specific MMP inhibitor batimastat to the defined serum-free TDCC media, demonstrating that MVC-101 was being activated by MMP activity generated by the cells during the assay (). Similar results were obtained when LoVo was used as the target cell line in the assay ().
Figure 8. TDCC with full-length COBRAs.
Pharmacokinetics and COBRA-mediated regression of established solid tumors in mice
Next, the stability of MVC-101 was evaluated in vivo. To measure the pharmacokinetics of COBRAs in mice, MVC-101 and MVC-NCL were dosed intravenously (i.v.) at 500 µg/kg. The plasma exposure of MVC-101 and MVC-NCL were found to be very similar, with a serum half-life of 18 hours for MVC-101 and 22 hours for MVC-NCL (Supplemental Figure 10). This demonstrates that inclusion of the MMP9 cleavable linker in MVC-101 did not substantially affect its serum stability. In addition, the PK assay measures only full-length prodrug and not the active dimer, suggesting that little cleavage of MVC-101 occurred in the blood. Given that the affinity of the anti-HSA sdAb to HSA is about 10-fold greater than its affinity to mouse serum albumin, the ability of the anti-albumin domain to extend the half-life of COBRAs in human serum may be even greater.
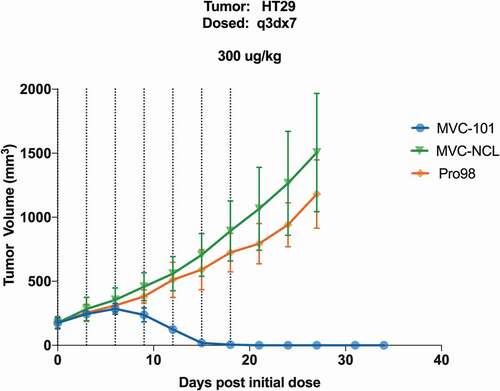
To test the ability of MVC-101 to be activated in vivo and regress established solid tumors in mice, the COBRAs were administered every three days for a total of seven doses to NSG mice bearing established HT-29 tumors. Primary human T cells were injected via a single i.v. injection upon COBRA dose initiation. In animals dosed with the non-cleavable control, MVC-NCL, at 300 µg/kg, the tumors grew at a consistent rate over the course of the study, similar to that of the negative control molecule, the HEL-targeted Pro98. In contrast, in animals dosed with MVC-101 at 300 μg/kg, complete tumor regressions were observed (). These experiments demonstrate that proteolytic cleavage of COBRAs is necessary to achieve tumor regressions in mice and that MVC-101 can be efficiently activated in vivo to produce complete tumor regressions at very modest doses.
Figure 9. MVC-101 regresses established solid tumors in mice.
Discussion
Bispecific T cell engagers have proven very effective at generating potent immune responses toward cells expressing specific target antigens. The challenge in developing these molecules as therapeutics for the treatment of solid tumors is to selectively direct their activity toward tumor cells and away from essential normal cells. Complementary pairs of hemi-COBRAs can achieve this selectivity by efficiently binding to cells expressing the target antigen, and only rearranging to become active T cell engagers after cleavage by proteases that are enriched in the tumor microenvironment. This selective rearrangement favors the accumulation of active CD3 binding domains on tumor cells since they are tethered to the cell surface via their target-binding sdAbs. This intermolecular rearrangement also releases the inactive anti-CD3 scFv domains and the half-life extension elements from the cell surface since they are no longer connected to the tumor-targeting domain, effectively making this an irreversible reaction.
The hemi-COBRA strategy achieves its primary goal of focusing T cell killing to the protease-rich TME, but it also creates several complications for the development of a therapeutic. Achieving similar concentrations of the two hemi-COBRAs on the surface of the tumor cells can be challenging unless the pharmacokinetic and pharmacodynamic properties of the two molecules are very similar in each patient. These features can be difficult to engineer. In addition, the conditional activity of the hemi-COBRAs is achieved at the expense of their potency, so larger doses would need to be administered to achieve the desired efficacy. Finally, the production and development of two proteins as a single therapeutic dramatically increases the manufacturing and regulatory complexity of the final product.
Combining the conditional activity and tumor targeting of a hemi-COBRA pair into a single molecule addresses these challenges. The full-length COBRA uses a linker that is sensitive to proteases that are enriched in the TME to separate an active fragment containing two tumor-targeting sdAbs and the constrained active anti-CD3 domains from the inactivating fragment that contains the constrained inactive anti-CD3 domains and the half-life extension element (HLE). This sequence arrangement produces a protein that stably folds into a structure that binds to the tumor target with high avidity but does not bind to the CD3 complex on T cells. Cleavage of this prodrug in vitro with a tumor-associated protease, MMP9, increases its potency in a TDCC assay by more than 500-fold compared to the non-cleavable control, demonstrating the conditional nature of the prodrug. The full-length COBRAs also have increased molar potency compared to the hemi-COBRA pair due to avidity of the two target-binding sdAbs in the active domain.
When injected into mice harboring primary human T cells, COBRAs with MMP2/9 cleavable linkers completely regress established human tumors at modest doses. In contrast, COBRAs with non-cleavable linkers do not induce tumor regression. This demonstrates that the prodrug is efficiently processed in vivo to create active and targeted T cell engagers. Since the anti-EGFR sdAbs do not cross-react to murine EGFR, the selectivity of COBRA activation in tumor tissues as compared to normal tissues cannot be estimated. Experiments employing mouse cross-reactive COBRA constructs are currently underway to determine the degree of selectivity afforded by the MMP cleavable linker. Separating the active T cell-engaging fragments from their HLEs after protease cleavage also reduces the serum half-life of the active domains and limits their ability to migrate to normal tissues and cause on-target, off-tumor toxicity.
Since the two tumor-targeting sdAbs in the active fragment bind to their antigen in a concerted fashion, another potentially useful feature of the full-length COBRA format that has not been explored in this study is the ability to target two different tumor antigens with the same COBRA. Selecting a pair of tumor antigens that are expressed on the same tumor, but not co-expressed on the same normal cells may allow the construction of COBRAs with an improved safety profile. To achieve this goal, the affinities of tumor-targeting sdAbs could be engineered to be low so as not to target normal cells expressing either antigen individually, but rely on the avidity of the COBRA to tumor cells co-expressing both antigens to selectively drive their destruction.
Alternatively, COBRAs that target two different tumor antigens could address the problem of acquired resistance due to the heterogeneous nature of protein expression within human tumors.Citation48-Citation53 To address this problem, COBRAs would contain two tumor-targeting sdAbs, each of which is able to induce potent T cell engagement alone. Such molecules would exert anti-tumor activity on tumor cells that express either antigen, and, as a result, would be expected to have activity on a broader range of tumors cells than COBRAs that target a single antigen. This strategy may be especially effective when the two antigens are overexpressed on tumors relative to normal tissues, which would reduce the potential toxicity of a COBRA targeting two tumor antigens. This approach would be very valuable for directing T cell killing to variant tumor cells that have lost expression of either target antigen and for reducing tumor relapse in the situation where expression of either or both antigens is heterogeneous on cells within the tumor.
Materials and methods
Cells
Cell lines were purchased from ATCC: HT-29 colorectal cell line, LoVo colorectal cell line, and Jurkat T cell leukemia cell line. Cells were propagated in the media recommended by the vendor, supplemented with 10% fetal bovine serum (FBS). Human pan-T cells were isolated from fresh leukopaks (Stemcell Technologies, #70500) using an immunomagnetic negative selection kit (Stemcell Technologies, 17951). Cells were analyzed on FACS for purity and viability and frozen into single use vials.
Recombinant proteins
COBRA molecules and recombinant proteins generated at Maverick were produced via transient transfection using the Expi-293 system (ThermoFisher Scientific) and were purified by Protein A or Ni-affinity chromatography using standard techniques. A fusion protein containing the extracellular domain of human EGFR (amino acids 26–645), a Factor Xa cleavage site, and human IgG1 Fc was expressed in mammalian cells and purified by standard Protein A purification. Subsequently, the fusion protein was cleaved with Factor Xa (New England Biolabs), and the Fc was removed with Protein A sepharose to yield monomeric recombinant EGFR protein that was used in the sandwich ELISA. Biotinylated recombinant CD3ε was purchased from Acrobiosystems and Streptavidin-AF647 was purchased from ThermoFisher Scientific.
Cleavage of hemi-COBRA molecules
MMP9 (R&D Systems) was activated with 1 mM APMA (Sigma) overnight at 37°C, after which the activated enzyme was incubated with hemi-COBRA molecules at a 1:200 molar ratio overnight at room temperature. Cleavage was confirmed by subjecting approximately 1 µg of cleaved and uncleaved molecules to SDS-PAGE and staining with InstantBlue Coomassie stain (Expedeon).
Sandwich ELISA
Recombinant EGFR protein was immobilized on Immulon 4HBX plates (VWR) at 1 µg/mL in 0.2 M carbonate buffer, pH 9, after which the plates were blocked with SuperblockTM (ThermoFisher Scientific). After washing the plates several times with phosphate-buffered saline (PBS) containing 0.05% Tween 20 (PBS/T), uncleaved or cleaved hemi-COBRAs, diluted in PBS/T, were then added to the plates and incubated for 2 hours. The plates were then washed with PBS/T, after which biotinylated recombinant CD3ε (0.5 µg/mL in PBS/T) was added for 2 hours, followed by another wash in PBS/T and a 1 hour incubation with streptavidin-HRP (ThermoFisher Scientific), diluted 1:10,000 in PBS/T. The plates were washed in PBS/T and were developed with TMB (VWR), followed by the addition of 650 nm stop solution (VWR). The plates were read on a SpectraMax M2 (Molecular Devices) at 650 nm, and the data were analyzed using GraphPad Prism version 7.05 for Windows (GraphPad Software).
Sandwich FACS
HT-29 cells were removed from the flask with 20 mM ethylenediaminetetraacetic acid (EDTA) in PBS, washed once with RPMI+10% FBS and once with PBS+10% FBS, after which the cells were resuspended in a solution of hemi-COBRAs, diluted in PBS+1% FBS. Cells were incubated for 30 minutes on ice, after which unbound hemi-COBRAs were removed by several washes in PBS+1% FBS. Cells were then incubated with biotinylated recombinant CD3ε (100 nM in PBS+1% FBS) for 30 minutes on ice, washed in PBS+1% FBS, then incubated for 30 minutes on ice with streptavidin-AF647, diluted to 2 µg/mL in PBS+1% FBS. Cells were washed several times in PBS+1% FBS, after which flow cytometry was performed on a CytoFlex LX (Beckman Coulter). The data were analyzed using Flow Jo (BD Biosciences) and GraphPad Prism version 7.05 for Windows (GraphPad Software).
TDCC assays
Tumor cell lines (HT-29 and LoVo) were engineered to constitutively express firefly luciferase. Cells were removed from the flask with TrypLE Express, centrifuged and resuspended in culture medium. The culture medium for hemi-COBRA TDCC assays was RPMI + 10% FBS (Gibco); for TDCC assays using full-length COBRAs, all steps were performed in defined AIM-V media (Gibco) with or without the addition of 2 µM batimastat (Selleckchem). Purified human pan T cells were also thawed, centrifuged, and resuspended in culture medium. Tumor cells and T cells were counted and mixed together at a ratio of 1:10, respectively. The co-culture of cells was then added to the wells of a 384-well assay plate. Serial dilutions of COBRAs were prepared separately in culture medium and transferred to the assay plate. Cells were incubated at 37°C for 48 hours, after which luciferase levels were measured using an Envision luminometer (Perkin Elmer) by adding an equal volume of SteadyGlo (Promega). Data were analyzed using GraphPad Prism version 8.3.1 for MacOS (GraphPad Software).
In vivo efficacy and PK
NSG mice (The Jackson Laboratory) were implanted with tumor cell lines subcutaneously. Human T cells were isolated from leukopak via negative selection (StemCell Technologies) and expanded utilizing G-Rex technology (Wilson Wolf) in combination with T cell expansion/activation beads (Miltenyi). Once tumor growth was established, mice were randomized into groups (N = 6) based on tumor volume, expanded human T cells were implanted i.v. and test articles were dosed as indicated. Tumor volume was assessed by caliper measurement. To assess pharmacokinetics, test articles were administered i.v. into NOD-SCID mice (The Jackson Laboratory) and blood was collected and processed to plasma as indicated. Each dosing group included 3 animals. Plasma concentration was determined by MSD assay using anti-idiotype antibody as capture, and anti-His detection.
Abbreviations
COBRA Conditional Bispecific Redirected ActivationTME Tumor microenvironmentTAA Tumor-associated antigensMMP Matrix metalloproteinasescFv Single-chain variable fragmentsdAb Single domain antibodyCD3 Cluster of differentiation 3HSA Human serum albuminEGFR Epidermal growth factor receptorHEL Hen egg lysozymeSDS-PAGE Sodium dodecyl sulfate-polyacrylamide gel electrophoresisELISA Enzyme-linked immunosorbent assayTDCC T cell-dependent cellular cytotoxicity assayFACS Fluorescence-activated cell sortingSEC Size exclusion chromatographyMALS Multi-angle light scatteringPK PharmacokineticsHLE Half-life extension elementi.v. IntravenouslyFBS Fetal bovine serumAPMA 4-Aminophenylmercuric acetatePBS Phosphate-buffered salineEDTA Ethylenediaminetetraacetic acid
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download Zip (23.2 MB)Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- Yu B, Liu D. Antibody-drug conjugates in clinical trials for lymphoid malignancies and multiple myeloma. J Hematol Oncol. 2019;12:94. doi:https://doi.org/10.1186/s13045-019-0786-6.
- Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, Forero-Torres A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. NE J Med. 2010;363:1812–11. doi:https://doi.org/10.1056/NEJMoa1002965.
- Tabasinezhad M, Talebkhan Y, Wenzel W, Rahimi H, Omidinia E, Mahboudi F. Trends in therapeutic antibody affinity maturation: from in-vitro towards next-generation sequencing approaches. Immunol Lett. 2019;212:106–13. doi:https://doi.org/10.1016/j.imlet.2019.06.009.
- Peters C, Brown S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Bioscience Rep. 2015;35(4):e00225. doi:https://doi.org/10.1042/BSR20150089.
- Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, Wu HC. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27:1. doi:https://doi.org/10.1186/s12929-019-0592-z.
- Lo KM, Leger O, Hock B. Antibody Engineering. Microbiol Spect. 2014;2:AID–0007. doi:https://doi.org/10.1128/microbiolspec.AID-0007-12.
- Shim H. Bispecific antibodies and antibody-drug conjugates for cancer therapy: technological considerations. Biomolecules. 2020;10:360. doi:https://doi.org/10.3390/biom10030360.
- Varchetta S, Gibelli N, Oliviero B, Nardini E, Gennari R, Gatti G, Silva LS, Villani L, Tagliabue E, Menard S, et al. Elements related to heterogeneity of antibody-dependent cell cytotoxicity in patients under trastuzumab therapy for primary operable breast cancer overexpressing Her2. Cancer Res. 2007;67(24):11991–99. doi:https://doi.org/10.1158/0008-5472.CAN-07-2068.
- Nimmerjahn F, Ravetch JV. Antibody-mediated modulation of immune responses. Immunol Rev. 2010;236:265–75. doi:https://doi.org/10.1111/j.1600-065X.2010.00910.x.
- Golay J, Zaffaroni L, Vaccari T, Lazzari M, Borleri GM, Bernasconi S, Tedesco F, Rambaldi A, Introna M. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood. 2000;95:3900–08. doi:https://doi.org/10.1182/blood.V95.12.3900.
- Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–58. doi:https://doi.org/10.1182/blood.V99.3.754.
- Jitschin R, Saul D, Braun M, Tohumeken S, Volkl S, Kischel R, Lutteropp M, Dos Santos C, Mackensen A, Mougiakakos D. CD33/CD3-bispecific T-cell engaging (BiTE) antibody construct targets monocytic AML myeloid-derived suppressor cells. J Immunother Cancer. 2018;6:116. doi:https://doi.org/10.1186/s40425-018-0432-9.
- Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93:290–96. doi:https://doi.org/10.1038/icb.2014.93.
- Fu M, He Q, Guo Z, Zhou X, Li H, Zhao L, Tang H, Zhou X, Zhu H, Shen G, et al. Therapeutic Bispecific T-Cell Engager Antibody Targeting the Transferrin Receptor. Front Immunol. 2019;10:1396. doi:https://doi.org/10.3389/fimmu.2019.01396.
- Dreier T, Baeuerle PA, Fichtner I, Grun M, Schlereth B, Lorenczewski G, Kufer P, Lutterbuse R, Reithmuller G, Gjorstrup P, et al. T cell costimulus-independent and very efficacious inhibition of tumor growth in mice bearing subcutaneous or leukemic human B cell lymphoma xenografts by a CD19-/CD3- bispecific single-chain antibody construct. J Immunol (Baltimore, Md. 1950;2003(170):4397–402.
- Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104. doi:https://doi.org/10.1186/s13045-015-0195-4.
- Goebeler ME, Bargou R. Blinatumomab: a CD19/CD3 bispecific T cell engager (BiTE) with unique anti-tumor efficacy. Leuk Lymphoma. 2016;57:1021–32. doi:https://doi.org/10.3109/10428194.2016.1161185.
- Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science (New York, NY). 2008;321:974–77. doi:https://doi.org/10.1126/science.1158545.
- Zhang S, Zhang HS, Cordon-Cardo C, Ragupathi G, Livingston PO. Selection of tumor antigens as targets for immune attack using immunohistochemistry: protein antigens. Clin Cancer Res. 1998;4:2669–76.
- Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res. 1997;3:81–85.
- Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mole Ther. 2011;19:620–26. doi:https://doi.org/10.1038/mt.2010.272.
- Palmer DC, Chan CC, Gattinoni L, Wrzesinski C, Paulos CM, Hinrichs CS, Powell DJ, Klebanoff CA, Finkelstein SE, Fariss RN, et al. Effective tumor treatment targeting a melanoma/melanocyte-associated antigen triggers severe ocular autoimmunity. Proc Natl Acad Sci U S A. 2008;105:8061–66. doi:https://doi.org/10.1073/pnas.0710929105.
- Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–37. doi:https://doi.org/10.1158/1078-0432.CCR-09-0737.
- Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev. 2008;60:1421–34. doi:https://doi.org/10.1016/j.addr.2008.04.012.
- Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–92. doi:https://doi.org/10.1038/nrc1893.
- Jain RK. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 1990;50:814s–9s.
- Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11:671–77. doi:https://doi.org/10.1038/nrc3110.
- Stubbs M, McSheehy PM, Griffiths JR, Bashford CL. Causes and consequences of tumour acidity and implications for treatment. Mol Med Today. 2000;6:15–19. doi:https://doi.org/10.1016/S1357-4310(99)01615-9.
- Pillai SR, Damaghi M, Marunaka Y, Spugnini EP, Fais S, Gillies RJ. Causes, consequences, and therapy of tumors acidosis. Cancer Metastasis Rev. 2019;38:205–22. doi:https://doi.org/10.1007/s10555-019-09792-7.
- Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckland, NZ). 2015;3:83–92.
- Moulder JE, Rockwell S. Tumor hypoxia: its impact on cancer therapy. Cancer Metastasis Rev. 1987;5:313–41. doi:https://doi.org/10.1007/BF00055376.
- Engin K, Leeper DB, Cater JR, Thistlethwaite AJ, Tupchong L, McFarlane JD. Extracellular pH distribution in human tumours. Int J Hyperther. 1995;11:211–16. doi:https://doi.org/10.3109/02656739509022457.
- Rakashanda S, Rana F, Rafiq S, Masood A, Amin S. Role of proteases in cancer: A review. Biotechnol Mole Biol Rev. 2012;7:90–101. doi:https://doi.org/10.5897/BMBR11.027.
- DeClerck YA, Mercurio AM, Stack MS, Chapman HA, Zutter MM, Muschel RJ, Raz A, Matrisian LM, Sloane BF, Noel A, et al. Proteases, extracellular matrix, and cancer: a workshop of the path B study section. Am J Pathol. 2004;164:1131–39. doi:https://doi.org/10.1016/S0002-9440(10)63200-2.
- Zhou F, Fu T, Huang Q, Kuai H, Mo L, Liu H, Wang Q, Peng Y, Han D, Zhao Z, et al. Hypoxia-activated PEGylated conditional aptamer/antibody for cancer imaging with improved specificity. J Am Chem Soc. 2019;141:18421–27. doi:https://doi.org/10.1021/jacs.9b05063.
- Sharp L, Chang C, Frey G, Liu H, Xing C, Wang J, Walls M, Wheeler C, Ben Y, Boyle WJ, et al. Potent CAB CTLA4 antibody to reduce immune side effects and toxicities associated with single-agent and combination cancer immuno therapies. Cancer Res. 2019;79:2708.
- Chang C, Frey G, Sharp L, Boyle WJ, Wang J, Xing C, Liu H, Wheeler C, Walls M, Short JM. Novel conditionally active biologic (CAB) antibody targeting EpCAM demonstrates anti-tumor efficacy in vivo. Cancer Res. 2019;79:356.
- Minogue E, Millar D, Chuan Y, Zhang S, Grauwet K, Guo M, Langenbucher A, Benes CH, Heather J, Minshull J, et al. Redirecting T-cells against AML in a multidimensional targeting space using T-cell engaging antibody circuits (TEAC). Blood. 2019;134:2653. doi:https://doi.org/10.1182/blood-2019-127634.
- Boustany LM, Wong L, White CW, Diep L, Huang Y, Liu S, Richardson JH, Kavanaugh WM, Irving BA. EGFR-CD3 bispecific probody therapeutic induces tumor regressions and increases maximum tolerated dose >60-fold in preclinical studies. Mole Cancer Ther. 2018;17:A164–A.
- Ellerman D. Bispecific T-cell engagers: towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods (San Diego, Calif). 2019;154:102–17. doi:https://doi.org/10.1016/j.ymeth.2018.10.026.
- Waas ET, Lomme RM, DeGroot J, Wobbes T, Hendriks T. Tissue levels of active matrix metalloproteinase-2 and −9 in colorectal cancer. British J Cancer. 2002;86:1876–83. doi:https://doi.org/10.1038/sj.bjc.6600366.
- Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi:https://doi.org/10.1016/j.cell.2010.03.015.
- Kato K, Hara A, Kuno T, Kitaori N, Huilan Z, Mori H, Toida M, Shibata T. Matrix metalloproteinases 2 and 9 in oral squamous cell carcinomas: manifestation and localization of their activity. J Cancer Res Clin Oncol. 2005;131(6):340–46. doi:https://doi.org/10.1007/s00432-004-0654-8.
- Edwards JG, McLaren J, Jones JL, Waller DA, O’Byrne KJ. Matrix metalloproteinases 2 and 9 (gelatinases A and B) expression in malignant mesothelioma and benign pleura. Br J Cancer. 2003;88:1553–59. doi:https://doi.org/10.1038/sj.bjc.6600920.
- Sven H, Ingrid O, Judith B, Maria S-M. Pre-clinical intravenous serum pharmacokinetics of albumin binding and non-half-life extended nanobodies. Antibodies. 2015;4:141–56. doi:https://doi.org/10.3390/antib4030141.
- Andersen JT, Dalhus B, Cameron J, Daba MB, Plumridge A, Evans L, Brennan SO, Gunnarsen KS, Bjoras M, Sleep D, et al. Structure-based mutagenesis reveals the albumin-binding site of the neonatal Fc receptor. Nat Comm. 2012;3:610. doi:https://doi.org/10.1038/ncomms1607.
- Igawa T, Tsunoda H, Kikuchi Y, Yoshida M, Tanaka M, Koga A, Sekimori Y, Orita T, Aso Y, Hattori K, et al. VH/VL interface engineering to promote selective expression and inhibit conformational isomerization of thrombopoietin receptor agonist single-chain diabody. Protein Eng Des Sel. 2010;23:667–77. doi:https://doi.org/10.1093/protein/gzq034.
- Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, Seth S, Chow CW, Cao Y, Gumbs C, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science (New York, NY). 2014;346:256–59. doi:https://doi.org/10.1126/science.1256930.
- Yuan Y. Spatial heterogeneity in the tumor microenvironment. Cold Spring Harbor Perspect Med. 2016;6:a026583. doi:https://doi.org/10.1101/cshperspect.a026583.
- Ramon y Cajal S, Sese M, Capdevila C, Aasen T, De Mattos-Arruda L, Diaz-Cano SJ, Hernández-Losa J, Castellví J. Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mole Med (Berlin, Germany). 2020;98:161–77. doi:https://doi.org/10.1007/s00109-020-01874-2.
- Ojha J, Secreto C, Rabe K, Ayres-Silva J, Tschumper R, Dyke DV, Slager S, Fonseca R, Shanafelt T, Kay N, et al. Monoclonal B-cell lymphocytosis is characterized by mutations in CLL putative driver genes and clonal heterogeneity many years before disease progression. Leukemia. 2014;28(12):2395–98. doi:https://doi.org/10.1038/leu.2014.226.
- Caswell-Jin JL, McNamara K, Reiter JG, Sun R, Hu Z, Ma Z, Ding J, Suarez CJ, Tilk S, Raghavendra A, et al. Clonal replacement and heterogeneity in breast tumors treated with neoadjuvant HER2-targeted therapy. Nat Comm. 2019;10(1):657. doi:https://doi.org/10.1038/s41467-019-08593-4.
- Aleksakhina SN, Kashyap A, Imyanitov EN. Mechanisms of acquired tumor drug resistance. Biochim Et Biophys Acta Rev Cancer. 2019;1872:188310. doi:https://doi.org/10.1016/j.bbcan.2019.188310.