
ABSTRACT
The long circulating half-life and inherently bivalent architecture of IgGs provide an ideal vehicle for presenting otherwise short-lived G-protein-coupled receptor agonists in a format that enables avidity-driven enhancement of potency. Here, we describe the site-specific conjugation of a dual agonist peptide (an oxyntomodulin variant engineered for potency and in vivo stability) to the complementarity-determining regions (CDRs) of an immunologically silent IgG4. A cysteine-containing heavy chain CDR3 variant was identified that provided clean conjugation to a bromoacetylated peptide without interference from any of the endogenous mAb cysteine residues. The resulting mAb-peptide homodimer has high potency at both target receptors (glucagon receptor, GCGR, and glucagon-like peptide 1 receptor, GLP-1R) driven by an increase in receptor avidity provided by the spatially defined presentation of the peptides. Interestingly, the avidity effects are different at the two target receptors. A single dose of the long-acting peptide conjugate robustly inhibited food intake and decreased body weight in insulin resistant diet-induced obese mice, in addition to ameliorating glucose intolerance. Inhibition of food intake and decrease in body weight was also seen in overweight cynomolgus monkeys. The weight loss resulting from dosing with the bivalently conjugated dual agonist was significantly greater than for the monomeric analog, clearly demonstrating translation of the measured in vitro avidity to in vivo pharmacology.
Introduction
The incretin hormone glucagon-like peptide 1 (GLP1), and activation of its receptor (GLP-1R), have an array of beneficial effects in glucose metabolism and energy balance in humans, including stimulation of glucose-dependent insulin secretion,Citation1 as well as inhibiting glucagon secretion,Citation2 gastric emptying,Citation3 and food intake.Citation4 As such, numerous GLP-1R agonists requiring daily (e.g., liraglutide and exenatide) or weekly (e.g., albiglutide, dulaglutide, and semaglutide) injections to treat type 2 diabetes mellitus (T2DM) and/or obesity have been approved. It has also been shown that activation of the glucagon receptor (GCGR) with a 1 mg dose of glucagon before each meal for two weeks in normal humans significantly decreased both food intake and body weight, albeit at increased risk of hyperglycemia.Citation5
The potential beneficial effects of activating both receptors have been investigated with oxyntomodulin, the endogenous incretin dual agonist of both receptors. Acute infusions or injections of oxyntomodulin before meals for four weeks inhibited food intake in normalCitation6,Citation7 and obeseCitation8 volunteers. Oxyntomodulin or infusions of glucagon (with or without GLP1) also increased energy expenditure in overweight or obeseCitation9 and healthyCitation10,Citation11 humans (although another studyCitation7 showed no such impact on energy expenditure). Acute infusions of the native peptide in overweight or obese patients with or without T2DM significantly increased insulin secretion and attenuated glycemic excursions during a graded glucose infusion.Citation12
All these endogenous peptide hormones have very short half-lives, making them impractical as therapeutics. Their circulating half-lives are limited both by proteolytic liabilities from dipeptidyl peptidase IV (DPP4)Citation13 and neutral endopeptidase,Citation14 as well as by rapid renal filtration.Citation15 Incorporation of both native and non-natural amino acids has been successful in stabilizing peptides commensurate with once-daily administration. The issue of renal filtration has been addressed by preparing conjugates that have strong binding to circulating serum albumin via lipidCitation16,Citation17 or cholesterylCitation18 augmentation, or that have increased steric bulk due to attachment of polyethylene glycol.Citation19 Half-life extension approaches that combine once-weekly dosing with a minimal peak to trough ratio may achieve a superior profile by allowing for better tolerability (less gastrointestinal distress) and improved patient compliance.Citation20
Chemically and biochemically modified GLP-1R agonist peptides with extended half-lives enabled by the incorporation of non-natural amino acids such as liraglutide and semaglutide have been approved for use in T2DM patients. In addition, two fully recombinant GLP-1R single agonist fusion proteins have been approved: 1) dulaglutide, which is a fusion of two GLP1 peptides to the Fc of an antibody; and 2) albiglutide, which is a GLP1 fusion to human serum albumin (HSA). Both exploit neonatal Fc receptor (FcRn) recycling to extend their half-lives. Covalently modified IgG1 for chronic administration of GLP-1R agonists has been implemented as CovX-BodiesCitation21,Citation22 with some success, demonstrating that intact immunoglobins have potential as stable scaffolds with extended half-lives on which to precisely and simultaneously pose multiple functional moietiesCitation23 with the promise of increased potency resulting from polyvalent avidity.Citation24
Here, we describe the chemical conjugation of a metabolically stable oxyntomodulin analog (OXM) to an immunologically silent mAb. Potential effector function of the IgG was suppressed by using an IgG4-PAA isotypeCitation25,Citation26 and inherent complementarity-determining region (CDR) activity was mitigated by using predominantly germline CDR sequences. A cysteine residue introduced into the heavy chain (HC) CDR3 loop provides the site of conjugation. The specific CDR3 variant was selected for its propensity to undergo facile conjugation to a maleimide probe. This unique cysteine residue was used to site-specifically conjugate the mAb to a proteolytically stable dual agonist peptide bearing a bromoacetamide group resulting in mAb-OXM conjugates 1 and 2 ().
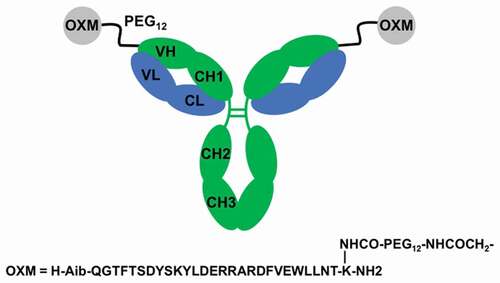
Figure 1. Schematic illustration of mAb-peptide homodimeric conjugate 2. mAb HC shown in green, LC in blue. OXM peptide shown as gray circles. The variable and constant regions of the mAb are labeled. For monomeric conjugate 1, one of the OXM peptides was replaced by acetamide.
Both monomeric (1) and dimeric (2) thioether conjugates were prepared to establish the effects of bivalent avidity on both in vitro receptor agonism and in vivo pharmacology. We show that the expected avidity of the homodimeric conjugate is much greater at GCGR than at GLP-1R in vitro, and that this translates to more profound agonism at GCGR in vivo, driving significantly more pronounced anorectic and weight loss effects when 2 is dosed in diet-induced obese (DIO) mice than the monomeric version 1. These weight loss effects were also observed in lean rats and overweight cynomolgus monkeys. The mAb-OXM conjugates have a half-life in cynomolgus monkeys commensurate with once weekly dosing in humans.
Results
GCGR/GLP-1R dual agonist peptide synthesis
The amino acid sequence of oxyntomodulin is predominantly that of glucagon, with a cationic C-terminal extension. We adopted Santoprete’s substitution of the serine at position 2 for aminoisobutyrate to increase in vitro GLP1 R agonism together with resistance to DPP4 proteolysis.Citation27 The helical topology of the peptide was further stabilized by introducing a bifurcated salt bridge from Q20 R to S16E and Q24E, and a potential oxidation liability was mitigated by a M27 L substitution. A short oligoethylene glycol spacer was incorporated between the mAb and the peptide to ensure unhindered access of the peptide to GLP-1 and glucagon receptors. The spacer also confers favorable aqueous solubility that facilitates the conjugation chemistry. Attachment of the peptide-spacer to the mAb was accomplished by introducing a reactive bromoacetamide group at the distal end of the glycol spacer. The proximal end of the spacer was attached via the sidechain of K30 of the OXM peptide ().
Selection and engineering of mAb cysteine variants
The antibody light chain variable region (VL) and the antibody heavy chain variable region (VH) were selected as the starting variable regions from which to engineer a mAb enabled for peptide conjugation.Citation28
VL:EIVLTQSPATLSLSPGERATLSCRASQSVSSYLAWYQQKPGQAPRLLIYDASNRATGIPARFSGSGSGTDFTLTISSLEPEDFAVYYCQQRSNWPLTFGQGTKVEIK
VH:EVQLLESGGGLVQPGGSLRLSCAASGFTFSSYAMSWVRQAPGKGLEWVSAISGSGGSTYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKYDGIYGELDFWGQGTLVTVSS
VL is composed completely of human Ig germline V gene sequences. The CDR3 of VH (underscored) is the only segment not composed of human germline V gene sequences and is identical to the CDR3 of an anti-human CCL2 antibody (US 20100074886 A1). Variants of VH containing a single cysteine substitution at select CDR residues across all three CDRs of the V region were designed, generated, and cloned into a mammalian host expression vector as complete heavy chains with a human IgG1 constant region. The VH/VL antigen-binding fragments crystal structure (5i1a.pdb) was used to aid in selection of CDR residues most suited for conjugation as cysteine variants. Variants were also constructed with additional glycine residues flanking the introduced cysteine residue to increase accessibility for conjugation. Similar variants of VL were designed and generated as complete light chains with a human kappa constant region. A total of 24 expression constructs of VH single cysteine variants and 22 expression constructs of VL single cysteine variants were generated. Five initial HC cysteine variants were found to express in good yields as monomeric proteins. Analytical mass determination indicated two disulfide adducts per mAb of LC, glutathione, or (predominantly) cysteine at the site engineered for conjugation, as well as clipping of the HC C-terminal lysine residue, which is commonly seen in recombinantly produced mAbs. To prepare the variant mAbs for conjugation, disulfide adducts were removed by tris(2-carboxyethyl)phosphine (TCEP) reduction whereby reaction time and reagent concentrations were optimized to maintain the native disulfide bonds within the mAb. Both TCEP reduction and conjugation efficiency to an OXM-maleimide test peptide differed between mAb variants as demonstrated by qualitative estimation of the relative percentage of conjugation reaction products. Variants for which reduction of disulfide adducts could not be achieved cleanly without disruption of the mAb were not pursued further. The greatest efficiency, as measured by the greatest percentage of conjugated product, was observed with the HC I102 C point mutant. Insertion of flanking glycines to increase solvent presentation of the cysteine sidechain was not advantageous. Little or no conjugation was observed with Y103 C variants with or without flanking glycines. The I102 C variant was expressed from transiently transfected ExpiCHO-S cells as an IgG4 PAA variant at 200 mg/L of cell culture supernatant.
The linkage resulting from maleimide conjugation is known to be potentially reversible, so bromoacetamide conjugation chemistry, which produces a more stable linkage, was adopted and implemented successfully to prepare 1 and 2, antibodies containing either 1 or 2 equivalents of peptide, respectively.
Synthesis and characterization of mAb-OXM 1 and mAb-OXM2 2
Both conjugates were prepared using excess lyophilized OXM peptide (1.5 equivalents and 7.6 equivalents vs mAb for 1 and 2, respectively) at relatively high protein concentration (5–10 mg/mL was found to be optimal). Key to the success of the conjugation was the selective reduction of the cysteine at the conjugation site, requiring only 1.5 equivalents of TCEP per site. Performing the reduction at lower pH (5–6) prevented the inter-mAb oxidation and disulfide shuffling that are problematic at higher pH. In addition to the lower peptide stoichiometry, a shorter reaction time was optimal for obtaining good yields of 1. In this case the unreacted cysteine residue was capped by addition of excess iodoacetamide in situ, again with no unwanted additional adducts.
Conjugates were characterized as sharp single peaks on analytical size exclusion chromatography (>95% monomer Figs S6 and S8), and measurement of intact mass by liquid chromatography-electrospray ionization mass spectrometry (LC ESI-MS) showed the correct molecular weights for G0 F/G1 F glycoforms (Figs S5 and S7). Peptide mapping of 2 showed that the OXM peptides were conjugated exclusively at the HC I102 C cysteine residue and that the peptide:antibody ratio was 2.0 (Figs S9 and S10). No free thiol was detectable in the purified conjugates. Thermal unfolding of the conjugates followed the same trajectory with the same melting transitions as the unconjugated mAb (Fig S11).
Residual native CDR binding was assessed by surface plasmon resonance (SPR). While the carrier mAb was selected for lack of specific antigen binding, the most likely antigen that this mAb might bind, if any, is human CCL2 based on the origin of the VH CDR3.Citation29 Conjugates were surface immobilized using anti-Fc capture. A commercially available anti-CCL2 mouse mAb served as a positive control and two nonspecific human antibodies, anti-RSV-F (B21 M) and anti-B7H3, served as negative controls. Recombinant human CCL2 accumulation was seen with the positive control, but neither the negative controls nor with 2, confirming that the peptide-mAb conjugate lacks human CCL2 binding (Fig S12).
Ex vivo plasma stability and In vivo pharmacokinetics of 2
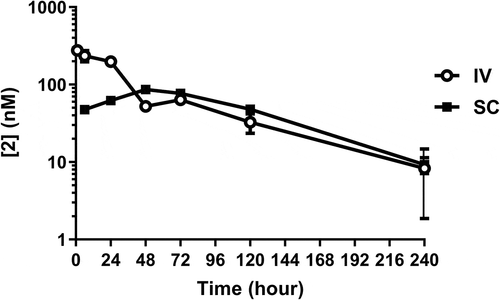
Conjugate 2 was stable in human plasma, with over 80% of bioactive compound remaining after 1 week (). The pharmacokinetic (PK) parameters of 2 are shown in . The half-lives in DIO mice, Tg32 mice, rats, and cynomolgus monkeys () were 62.0, 45.6, 38.5 ± 12.8, and 51.9 ± 6.2 hours, respectively.
Table 1. Pharmacokinetic parameters of 2 in DIO mice, Tg32 mice, SD rats, and cynomolgus monkeys. Exposures were measured by LCMS determination of intact mass. Mouse values were calculated from pooled samples.
Figure 2. Ex vivo human plasma stability of 2 over 1 week (filled circles). Percentage remaining is measured by functional assay at recombinant GLP1 receptors. The negative control (open circles) is a bioconjugate of a different oxyntomodulin variant known to be unstable in human plasma.
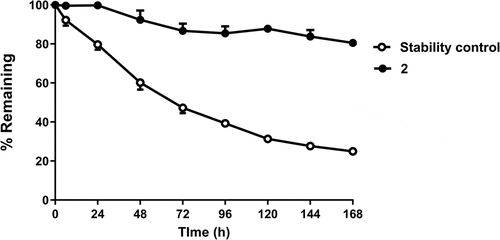
Figure 3. Pharmacokinetics of 2 in lean cynomolgus monkeys over 10 days. Exposures are shown for intravenous (IV) and subcutaneous (SC) administration. Exposures were measured by LCMS determination of intact mass.
In vitro activities of 2 at GLP-1Rand GCGR
Conjugate 2 is a GLP-1R agonist with in vitro potencies similar or better than native oxyntomodulin in all species tested (). The in vitro potencies of conjugate 2 at GCGR were also similar or better than native oxyntomodulin except at the cynomolgus GCGR receptor. The design criteria for the long-acting dual agonist were equivalent potency at human GCGR and 5- to 6-fold greater potency at human GLP1 R compared to native oxyntomodulin, criteria that were fully met in 2. The avidity effect of the dimeric conjugate was demonstrated by comparing 2 to 1, the monomeric version in which the same mAb is conjugated to a single OXM peptide. At the human GCGR, 2 was 19.5-fold more potent than 1, while the enhancement was 29.6-fold at the mouse GCGR. The difference between the monomeric and dimeric conjugates was much less pronounced at the GLP1 receptors; a 1.5-fold improvement at the mouse receptor and 2.7-fold at the human. This trend in relative potencies is reflected in the binding affinities (). In competition with GLP1 at human GLP-1R, the monomer showed 8.1-fold less receptor affinity (Ki = 19.44 ± 3.93 nM) than the dimer (Ki = 2.39 ± 0.55 nM).
Table 2. In vitro potencies, mean ± SEM, of 1, 2 and native oxyntomodulin (OXM) on cells expressing (a) GLP-1R and (b) GCGR of various species.
Table 3. Inhibition constants, mean ± SEM, of 1, 2 and GLP1 vs radiolabeled GLP1. Human GLP-1R membranes were incubated with 0.3 nM Citation1Citation25I-GLP1.
Glucose tolerance in DIO mice – single dose study with 2
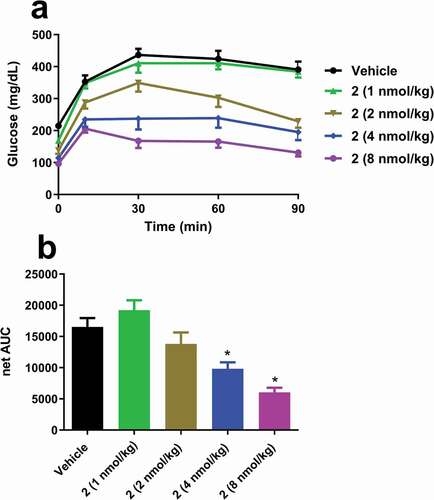
DIO mice were dosed subcutaneously (SC) with 2 at 1, 2, 4 and 8 nmol/kg. Eighteen hours later they underwent a 6 hour fast before undergoing an intraperitoneal glucose tolerance test (IPGTT). Before the IPGTT, 2 significantly decreased fasting blood glucose () but had no effect on fasting plasma insulin (data not shown). Administration of 2 at 4 and 8 nmol/kg significantly improved the glucose excursion in these insulin resistant mice, though an increase in insulin was not captured at 10 minutes. 2 at 4 and 8 nmol/kg significantly increased terminal plasma fibroblast growth factor 21 (FGF21) (2.1 ± 0.4 and 3.5 ± 0.4, respectively versus 0.4 ± 0.1 ng/mL in vehicle).
Figure 4. Single dose effects of 2 on (a) glucose excursions and (b) net AUC in insulin-resistant DIO mice. Groups were compared using One-way ANOVA, followed by Tukey’s multiple comparison post-hoc test (*p < .05, versus Vehicle).
Food intake and energy metabolism in DIO mice – single and repeat dosing studies with 2
The effects of a single SC dose of 2 on food intake and body weight were investigated in DIO mice (Fig S13). After a single dose, 2 significantly inhibited food intake over the first two days at all doses tested (1, 2, 4, or 8 nmol/kg). Five hour fasted glucose was significantly decreased by all doses (169 ± 11, 168 ± 10, 136 ± 5, and 93 ± 5 mg/dL in 1, 2, 4, and 8 nmol/kg, respectively) relative to vehicle (210 ± 10 mg/dL). Five hour fasted insulin was significantly decreased by 2–4 nmol/kg (2.0 ± 0.2, 1.8 ± 0.2, 1.5 ± 0.1, and 0.3 ± 0.1 ng/mL in 1, 2, 4, and 8 nmol/kg, respectively) relative to vehicle (2.9 ± 0.3 ng/mL). A significant decrease in body weight was maintained over 3 days in a dose-dependent manner. These trends were also evident in Sprague-Dawley rats (though fasting glucose and insulin did not change; data not shown), though overall potency of 2 was reduced compared to DIO mice (Fig. S14).
Sub-chronic efficacy of 2 was assessed by SC dosing every 3 days for 9 days in DIO mice (). At lower doses (1 or 2 nmol/kg), 2 significantly inhibited food intake only for the first 2 days with a decrease in body weight of 11.5%. At 4 nmol/kg, 2 reduced food intake for the duration of the study and reduced weight by 21.9%. To determine the food intake-independent effects of 2 on body weight loss, DIO mice were pair-fed to the average daily food intake amounts observed with the three doses of 2. Significant differences in weight loss between treated groups and their respective pair-fed groups became evident by day 4. The 2 and 4 nmol/kg dosed groups lost more body weight (11.5 ± 0.9% and 21.9 ± 1.6%, respectively) than their pair-fed groups (6.9 ± 0.4% and 13.3 ± 0.3% respectively). At 2 or 4 nmol/kg 2 significantly decreased fasting blood glucose and plasma insulin. At the higher dose 2 also significantly increased FGF21 and decreased plasma triglycerides and total cholesterol (Table S1).
Figure 5. Repeat dosing effects of 2 on (a) food intake and (b) weight change in DIO mice over 9 days. Weight change was measured in pair-fed animals where the food intake between the dosed and non-dosed arms is matched. For (a), groups were compared using Linear mixed model, followed by Tukey’s multiple comparison post-hoc test (*p < .05, Vehicle versus 4 nmol/kg; ^p < .05, Vehicle versus 1 & 2 nmol/kg; #p < .05, 4 nmol/kg versus 1 & 2 nmol/kg). For (b), groups were compared using Two-way ANOVA repeated measures, followed by Tukey’s multiple comparison post-hoc test (*p < .05, Vehicle versus all groups; ^p < .05, between all groups treated with 2; #p < .05, between all groups treated with 2 and their respective PF [pair-fed] groups).
![Figure 5. Repeat dosing effects of 2 on (a) food intake and (b) weight change in DIO mice over 9 days. Weight change was measured in pair-fed animals where the food intake between the dosed and non-dosed arms is matched. For (a), groups were compared using Linear mixed model, followed by Tukey’s multiple comparison post-hoc test (*p < .05, Vehicle versus 4 nmol/kg; ^p < .05, Vehicle versus 1 & 2 nmol/kg; #p < .05, 4 nmol/kg versus 1 & 2 nmol/kg). For (b), groups were compared using Two-way ANOVA repeated measures, followed by Tukey’s multiple comparison post-hoc test (*p < .05, Vehicle versus all groups; ^p < .05, between all groups treated with 2; #p < .05, between all groups treated with 2 and their respective PF [pair-fed] groups).](/cms/asset/1dabc326-2dfd-4c0a-a9f4-bece75ab89f6/kmab_a_1794687_f0005_oc.jpg)
Translation of in vitro avidity to in vivo pharmacology in DIO mice – single dose study of food intake and body weight in DIO mice with 1 and 2
To investigate whether the in vitro differences in binding and potency between 1 and 2 translated in vivo, DIO mice were given a single SC dose at 3 or 10 nmol/kg (). After a single dose, both 1 and 2 significantly inhibited food intake (data not shown) and body weight for 3 days. The dimer 2 at 3 nmol/kg had 80% of the efficacy of the monomer 1 at 10 nmol/kg despite having ~3.2-fold lower levels of active OXM peptide. The larger in vitro avidity effect at GCGR (28.5-fold) versus GLP-1R (1.5-fold) suggests that the greater than additive weight loss induced by 2 is driven predominantly by increased agonism of the glucagon receptor. This was confirmed by measuring FGF21 levels. FGF21 is known to be upregulated in response to GCGR activation.Citation30,Citation31 Monomer, 1, at 3 nmol/kg did not induce any significant increase in FGF21, while 2 at the same dose increased FGF21 some 9-fold. This difference was not significant at the higher dose of 10 nmol/kg, possibly due to saturation of the GCGR.
Figure 6. (a) Single dose terminal exposures of 1 and 2 in DIO mice. Exposures were measured by LCMS of the intact conjugates. (b) Effects of a single dose of 1 and 2 on body weight. (c) Effects of a single dose of 1 and 2 on plasma FGF21. For (a), groups were compared using One-way ANOVA, followed by Tukey’s multiple comparison post-hoc test (*p < .05, 2 [10 nmol/kg] versus 1 & 2 [3 nmol/kg]; ^p < .05, 1 [10 nmol/kg] versus 2 [10 nmol/kg]). For (b), groups were compared using Two-way ANOVA repeated measures, followed by Tukey’s multiple comparison post-hoc test (*p < .05, Vehicle versus all groups; ^p < .05, 2 [3 nmol/kg] versus 1 [3 nmol/kg]; #p < .05, 1 & 2 [3 nmol/kg] versus 1 & 2 [10 nmol/kg]; $p < .05, Vehicle versus all groups except 1 [3 nmol/kg]; @p < .05, 1 [10 nmol/kg] versus 2 [3 & 10 nmol/kg]). For (c), groups were compared using One-way ANOVA, followed by Tukey’s multiple comparison post-hoc test (*p < .05, Vehicle versus all groups except 1 [3 nmol/kg]; ^p < .05, 2 [3 nmol/kg] versus 2 [10 nmol/kg] & 1 [3 nmol/kg]; #p < .05, 1 [10 nmol/kg] versus 1 [3 nmol/kg]).
![Figure 6. (a) Single dose terminal exposures of 1 and 2 in DIO mice. Exposures were measured by LCMS of the intact conjugates. (b) Effects of a single dose of 1 and 2 on body weight. (c) Effects of a single dose of 1 and 2 on plasma FGF21. For (a), groups were compared using One-way ANOVA, followed by Tukey’s multiple comparison post-hoc test (*p < .05, 2 [10 nmol/kg] versus 1 & 2 [3 nmol/kg]; ^p < .05, 1 [10 nmol/kg] versus 2 [10 nmol/kg]). For (b), groups were compared using Two-way ANOVA repeated measures, followed by Tukey’s multiple comparison post-hoc test (*p < .05, Vehicle versus all groups; ^p < .05, 2 [3 nmol/kg] versus 1 [3 nmol/kg]; #p < .05, 1 & 2 [3 nmol/kg] versus 1 & 2 [10 nmol/kg]; $p < .05, Vehicle versus all groups except 1 [3 nmol/kg]; @p < .05, 1 [10 nmol/kg] versus 2 [3 & 10 nmol/kg]). For (c), groups were compared using One-way ANOVA, followed by Tukey’s multiple comparison post-hoc test (*p < .05, Vehicle versus all groups except 1 [3 nmol/kg]; ^p < .05, 2 [3 nmol/kg] versus 2 [10 nmol/kg] & 1 [3 nmol/kg]; #p < .05, 1 [10 nmol/kg] versus 1 [3 nmol/kg]).](/cms/asset/b7c71c15-fb77-446d-9474-06cc512bdf7d/kmab_a_1794687_f0006_oc.jpg)
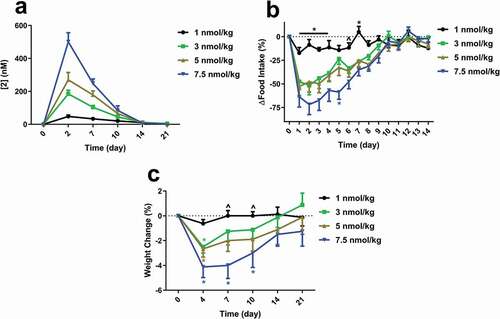
Food intake and body weight in overweight cynomolgus monkeys – single dose study with 2
Aged overweight male cynomolgus monkeys were dosed once with 2 at 1, 3, 5, and 7.5 nmol/kg (). All but the lowest dose significantly inhibited food intake for 7–8 days in a dose-dependent manner. Weekly food intake was reduced by 38, 41, and 57% for the 3, 5, and 7.5 nmol/kg groups, respectively, resulting in a 2.5–4.1% weight loss by day 4. Food intake and body weight returned to baseline levels within 2 or 3 weeks, respectively, corresponding to decreases in plasma compound levels.
Figure 7. (a) Single dose exposures of 2 in overweight cynomolgus monkeys. Exposures were measured by LCMS of the intact conjugate. Effects of a single dose of 2 on (b) food intake and (c) weight change. Animals were dosed subcutaneously at day 0. For (b), groups were compared using Two-way ANOVA repeated measures, followed by Tukey’s multiple comparison post-hoc test (*p < .05, 1 nmol/kg versus all other doses; ^p < .05, 1 nmol/kg versus 5 & 7.5 nmol/kg). For (c), groups were compared using Two-way ANOVA repeated measures, followed by Tukey’s multiple comparison post-hoc test (*p < .05, versus respective baseline).
Discussion
Presentation of pharmacologically active peptides on proteins with long circulating half-lives is well established for Fc and HSA platforms. mAb-derived molecules such as Fc fusions and conjugates are intrinsically homodimeric, and thus provide bivalent display of their payloads with marked increases in potency due to target binding avidity in many cases. However, in cases where the payload tends to self-associate (as is true for glucagon-derived peptides), there is potential for loss of potency unless care is taken to ensure that the payloads can be independently posed for optimal target binding. In such cases the antigen-binding arms of the mAb provide excellent spatial separation (of our hydrophobic/amphiphilic glucagon-derived peptides) compared to the N- or C-termini of an Fc. Where mAbs have previously been used to deliver conjugated peptides, the key features of the platform have tended to center on the target binding offered by the CDR (targeted delivery of payloads conjugated remotely from the CDR), and the cytotoxic effector functions offered by FcγIIIR binding (targeted cell killing). The full-length mAb has remained essentially unexplored as a platform purely for half-life extension, although such an embodiment is implicit in a recent report from Biswas et al.Citation32 Here, we have described such a construct, wherein the HC CDR3 loop has been engineered for efficient conjugation to a synthetic peptide payload.
Potential sites for conjugation were chosen based on their solvent accessibility in a crystal structure. Surprisingly, the reactivity of cysteine residues introduced at these sites varied widely both in terms of their susceptibility to TCEP reduction, and to the facility with which they underwent subsequent reaction with an electrophilic peptide. The mAb selected for this work, a VH I102 C point mutant, can be rapidly and selectively reduced at C102, and this partially reduced form is stable at 4°C for several days at pH<6. The free thiol reacts rapidly, selectively, and efficiently with maleimide and haloacetamide electrophiles. To realize a viable pharmaceutical for the treatment of T2DM in humans, we targeted OXM potencies at GCGR similar to that of native oxyntomodulin, and at GLP-1R some five to six times higher than native oxyntomodulin. The latter (EC50 at humanGLP-1R ~ 0.4 nM) was accomplished by substituting the native serine residue at position 2 of OXM with aminoisobutyrate (a change that also mitigates dipeptidyl peptidase-4 (DPPIV)-mediated proteolysis at this site), and by stabilizing the helical region of the peptide with arginine-glutamate salt bridges. Achieving the former (EC50 at humanGCGR ~2 nM) was enabled by a profound avidity effect – the potency of the analogous monomeric conjugate at humanGCGR is almost twenty times lower than the homodimer. The translation of this avidity to in vivo potency was clearly demonstrated by the fact that 2 elicited similar pharmacodynamic effects to its monomer 1 at a half-log lower dose, despite having lower total active OXM peptide levels. That this avidity affected potency by GCGR agonism rather than GLP-1R agonism (the avidity effect at GLP-1R is modest) was confirmed in vivo by the large difference in respective changes in plasma FGF21 induced by the homodimer 2 relative to the monomer 1. That this effect was not observed at the GLP-1R in this work suggests that the effect may be exploitable to different degrees with different receptor ligand pairs and that positioning of the conjugation site may need to be explored more systematically to optimize the effect for a specific receptor ligand pair.
mAb conjugate 2 demonstrated dramatically increased half-life in DIO mice, rats and non-human primates compared to native oxyntomodulin. A single dose of 2 improved glucose tolerance in DIO mice and decreased body weight for 3 days in DIO mice, and rats. This prolonged half-life in (62 versus the reported 3–5 hours for lipidated GLP-1R/GCGR dual agonistsCitation16,Citation17) allowed for decreased dosing frequency, as well as lower doses in a sub-chronic DIO mouse study (animals were dosed every 3 days). A dose of 4 nmol/kg administered only 3 times over 9 days resulted in a 22% weight loss. For comparison, lipidated dual agonists only decreased body weight of DIO mice by ~17% when dosed daily at 30 nmol/kgCitation16 or 11.3 nmol/kg twice a dayCitation17 over a span of 9 days.
The crucial role of the GCGR agonism in GLP-1R/GCGR dual agonist-mediated weight loss has been shown in GCGR knockout mice.Citation18,Citation33 A lipidated dual agonist has no effect on food intake on GLP-1R knockout mice, while the GCGR agonist IUB288 has no effect on food intake either.Citation16 IUB288 however, increases energy expenditure via FGF21,Citation31 as do a variety of dual agonists.Citation16,Citation19 Pair-feeding of mice in the control group to match the lower food intake achieved with 2 resulted in significantly less weight loss, indirectly supporting the contention that GCGR agonism increases energy expenditure. This was supported by increases seen in FGF21. While it has been shown that OXM or glucagon increases energy expenditure in humans,Citation9,Citation10,Citation11 glucagon has been shown to both increaseCitation30,Citation31 or decreaseCitation11 FGF21 in humans. The need for a sensitive and robust GCGR-specific biomarker in higher species remains a key obstacle to the development of dual agonists.
The half-life of 2 in cynomolgus monkeys was 52 hours, and a single dose of 2 resulted in sustained food intake inhibition and weight loss, pharmacodynamic effects that corresponded to and can be explained by the improved PK of the molecule. Given the array of additional benefits (e.g., renal and cardiovascularCitation34,Citation35) conferred by GLP-1R agonism and/or sustained weight loss on other co-morbidities such as nonalcoholic fatty liver diseaseCitation36 and nonalcoholic steatohepatitis,Citation37 better pharmacotherapies that are capable of inducing significant (and sustained) weight loss are desperately needed. This novel platform enables chronic, once-weekly dosing of a dual agonist and offers an attractive alternative for developing other peptide-based therapeutics with half-lives suitable for once weekly dosing.
Materials and methods
Preparation and characterization of OXM peptide, mAb, 1 and 2, and CCL2 binding experiments are described in Supplementary Material. Reagent and control antibodies used in this work are as follows:
Mouse monoclonal anti-human Fc (R10Z8E9) produced in Janssen
Rabbit polyclonal anti-glucagon, Proscience 49-161
Sulfo-tagged goat polyclonal anti-rabbit IgG, Meso Scale Diagnostics R32AB-1
Mouse monoclonal anti-CCL2 clone E10052, Thermo M2MCP1I
Human monoclonal anti-RSV-F (B21M) IgG4 PAA isotype control produced in-house
Human monoclonal anti-B7H3 IgG4 PAA isotype control produced in-house
GLP-1R competition binding assay
Radioligand competition binding studies (n = 3 per agonist) were performed with cell membranes containing recombinant human GLP-1R obtained from EMD Millipore. Membranes were mixed with Citation1Citation25I-GLP1(7–36) (Perkin Elmer # NEX308) in binding buffer (50 mM HEPES, pH 7.4, 5 mM MgCl2, 1 mM CaCl2, 0.2% bovine serum albumin (BSA)) in a nonbinding 96-well plate. Each well of the 96 well assay plate contained diluted test compounds, 0.3 nM radioligand and 10 μg/well human GLP-1R membrane suspension in a total volume of 200 μL. After equilibration for 60 minutes at room temperature, bound and free radioligand were separated by collecting membrane-bound fractions onto GF/B filter plates impregnated with PEI 0.5% and pre-wetted with assay buffer (50 mM HEPES, pH 7.4, 500 mM NaCl, 0.1% BSA), and washed 3 times with ice-cold binding buffer using a Harvester Filtermate 96 (Perkin Elmer). Filter plates were dried for 2 hours and 50 µL of Microscint O was added to each well. Retained radioactivity was counted using the Topcount scintillation counter (Packard). Duplicate determinations were made at each concentration. Nonspecific binding was determined in the presence of 300 nM unlabeled GLP1(7–36). Data analysis was performed using the program Prism (GraphPad) with non-linear regression analysis and reported as Ki values.
In vitro GLP-1R/GCGR functional bioassays
Compounds were screened for functional activity and in vitro potency in cell-based assays measuring cAMP in clonal HEK293 cells stably expressing mouse, rat, cynomolgus monkey, or human GLP-1R or GCGR. Cells were resuspended in HBSS, 5 mM HEPES, 0.1% BSA, 1.0 mM IBMX and added to 384-well white Opti-plates with compounds diluted in HBSS, 5 mM HEPES, 0.1% BSA. After incubation, the cells were lysed and cAMP was quantitated in the LANCE competitive cAMP immunoassay (Perkin Elmer). Compounds were also screened for in vitro potency in cell-based assays measuring cAMP in primary frozen hepatocytes. The day after seeding cells into collagen treated 384-well plates (5000 cells/well), the media was replaced with stimulation buffer (HBSS, 5 mM HEPES, 0.1% BSA, 0.5 mM IBMX) followed by the compounds diluted in stimulation buffer. Cells were lysed for cAMP immunoassay after 30 minutes.
The levels of bioactive compounds in plasma samples were quantitated in a cell-based bioassay using clonal HEK293 cells stably expressing the human GLP-1R. Cells were treated with either compound standards or diluted plasma and intracellular cAMP was measured. The concentrations of functional OXM in the plasma samples were determined by interpolation from a standard curve generated with the compound standards. Standards and quality control compounds were prepared in plasma diluted to 50% in assay diluent (HBSS, 5 mM HEPES, 0.1% BSA, 5 mM EDTA, 0.5 mM IBMX, protease inhibitors (Roche Diagnostics). Unknown plasma samples were prepared at various dilutions in normal assay diluent with control plasma added when needed to hold the final concentration of plasma at 50% and assayed in quadruplicate. Compound EC50 values and plasma concentrations were calculated using Prism.
Ex vivo human plasma stability
Compounds were incubated at 20 nM with fresh (never frozen) heparinized plasma at 37°C for up to 168 hours. Bioactivity was assessed as described above. Sample concentrations for each compound were interpolated from its own reference standard, and the percent remaining over time was calculated.
In vivo pharmacokinetics
Twenty-week-old male diet-induced obese (DIO, on 60 kcal% high fat diet [Research Diet D12492] for 15 weeks) C57BL/6NTac mice (Taconic) were dosed subcutaneously (SC) with 2 (10 nmol/kg) and blood was collected 7, 24, 48, 72, 96, and 120 hours later. Mice lacking the mouse FcRn and expressing the human FcRn (Tg32 mice;Citation30 Sage Laboratories) were dosed SC with 2 (10 nmol/kg). Mice were sacrificed at 1, 2, 6, 9, 13, 17, and 21 days after dosing, and blood was collected. Male Sprague-Dawley rats (Charles River Laboratories) were dosed intravenously (IV) or SC with 2 (2.5 nmol/kg). Blood was collected at 1, 2, 3, 6, 9, 13, 17 and 21 days after dosing. Male cynomolgus monkeys (MPI Research, Mattawan, MI) were dosed IV or SC with 2 (6.45 nmol/kg). Blood was collected at 1, 6, 24, 48, 72, 120, 240, 336, 432, and 528 hours after dosing. Blood samples in all studies were collected in K2EDTA coated tubes containing complete protease inhibitor (4% volume, Roche Diagnostics) and DPPIV inhibitor (1% volume, Sigma). Pharmacokinetics (PK) were calculated from bioassay measurements described above, and were consistent with trypsin digestion-based LCMS analysis (data not shown).
LC-MS/MS measurements
Where indicated, PK measurements were analyzed using an immuno-affinity capture, trypsin digestion LC-MS/MS method for concentrations of total mAb and OXM. The method was the same as previously reported,Citation38 except that an N-terminal 12 amino acid fragment (H-Aib-QGTFTSDYSK) was monitored as a surrogate of OXM.
Acute and sub-chronic pharmacodynamic studies in DIO mice and growing rats
All rodents used in these studies were maintained in accordance with the protocols approved by the Institutional Animal Care & Use Committee (IACUC) at Janssen R&D, Spring House, PA. Animals were housed on a 12-hour light/12-hour dark cycle at standard temperature and humidity conditions with ad libitum access to food and water (unless noted otherwise). Individually housed twenty-week-old male DIO (fed a 60 kcal% high fat diet [Research Diet D12492] for 15 weeks) C57BL/6 J mice (Jackson Laboratories) were used. Animals were randomized into groups based on body weight. For IPGTT, mice were dosed SC with vehicle (10 mM sodium-acetate, pH = 5, 5% mannitol) or 2 (n = 8/group). Twenty-four hours later (after a 6 hour fast), mice were dosed IP with dextrose (1 g/kg). Blood glucose was measured at indicated times with a One Touch Ultra glucometer (LifeScan), and plasma insulin was measured at 0 and 10 minutes (Meso Scale Discovery). Terminal FGF21 was measured by ELISA (Millipore). For acute food intake and weight loss studies, individually housed DIO mice or lean growing rats (Charles River Laboratories) were dosed with vehicle, 1, or 2, and food and body weights were measured for 3 days (n = 8/group). Animals were fasted for 6 hours on day 3, prior to the final measurements of food and body weight, for fasted glucose and insulin measurements. For 9-day sub-chronic food intake/weight loss studies, DIO mice were dosed SC daily with either vehicle or every 3 days with 2 (mice were given vehicle injections on non-dosing days). Separate groups of animals were pair-fed to the average food intake amounts of animals dosed with 2. On day 9, body composition was measured by NMR (Bruker). Free glycerol and fatty acids (Zenbio), triglycerides (Wako), and total cholesterol (Wako) were measured from terminal plasma samples.
Pharmacodynamic studies in non-human primates
For nonhuman-primate studies, 33 male cynomolgus monkeys, 7–15 years of age with an average body weight of 8.0 (6.2–11.8) kg, were individually housed, maintained and treated in accordance with the guidelines approved by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Water was available ad libitum. All monkeys were fed twice daily with a nutritionally balanced diet rich with seasonal fruits and vegetables. Animals were randomized to groups based on 14-day average/baseline food intake, body weight, and fasting blood glucose. To determine food intake, the weight of the balanced diet consumed was monitored daily. After monitoring food intake for 14 days to ensure a stable baseline food intake, animals were dosed SC with 2 (n = 8–9 monkeys/dose). Monkeys were monitored at least twice daily for signs of nausea (gaping and yawning), malaise (refusal of enrichment foods, hunched posture and decreased activity) and emesis. No malaise or emesis was observed, but some animals exhibited yawning, in a non-dose-dependent manner.
Plasma compound concentrations were measured by incubating samples in Streptavidin Gold Multi-Array 96-well plates (Meso Scale Diagnostics) coated with a biotinylated anti-human Fc antibody. Plates were incubated for 1 hour with a Sulfo Tag anti-human glucagon antibody and read on a Sector S 6000 plate reader (Meso Scale Diagnostics). Concentrations of compound were back calculated based on standard curves included on each plate.
Data analysis
Graphs and statistical analyses were made with GraphPad Prism software. Results are expressed as mean ± standard error of the mean (SEM). Data was analyzed with either one-way or two-way repeated measures ANOVA (where appropriate), followed by Tukey’s multiple comparison post-hoc test. The sub-chronic mouse food intake data was analyzed by a linear mixed model to correct for missing values (due to pieces of food too small to accurately measure) followed by Tukey’s multiple comparison post-hoc test. Differences with a P value <.05 were considered significant
Supplemental Material
Download Zip (6.7 MB)Supplemental Material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Kreymann B, Ghatei MA, Williams G, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet. 1987;2(8571):1300–11. doi:https://doi.org/10.1016/S0140-6736(87)91194-9.
- Gutniak M, Ørkov C, Holst JJ, Ahrén B, Efendić S. Antidiabetogenic effect of glucagon-like peptide-1 (7-36)amide in normal subjects and patients with diabetes mellitus. N Engl J Med. 1992;326(20):1316–22. doi:https://doi.org/10.1056/NEJM199205143262003.
- Wettergren A, Schjoldager B, Mortensen PE, Myhre J, Christiansen J, Holst JJ. Truncated GLP-1 (proglucagon 78-107-amide) inhibits gastric and pancreatic functions in man. Dig Dis Sci. 1993;38(4):665–73. doi:https://doi.org/10.1007/BF01316798.
- Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. 1998;101(3):515–20. doi:https://doi.org/10.1172/JCI990.
- Schulman JL, Carleton JL, Whitney G, Whitehorn JC. Effect of glucagon on food intake and body weight in man. J Appl Physiol. 1957;11(3):419–21. doi:https://doi.org/10.1152/jappl.1957.11.3.419.
- Cohen MA, Ellis SM, Le Roux CW, Batterham RL, Park A, Patterson M, Frost GS, Ghatei MA, Bloom SR. Oxyntomodulin suppresses appetite and reduces food intake in humans. J Clin Endocrinol Metab. 2003;88(10):4696–701. doi:https://doi.org/10.1210/jc.2003-030421.
- Bagger JI, Holst JJ, Hartmann B, Andersen B, Knop FK, Vilsbøll T. Effect of oxyntomodulin, glucagon, GLP-1, and combined glucagon +GLP-1 infusion on food intake, appetite, and resting energy expenditure. J Clin Endocrinol Metab. 2015;100(12):4541–52. doi:https://doi.org/10.1210/jc.2015-2335.
- Wynne K, Park AJ, Small CJ, Patterson M, Ellis SM, Murphy KG, Wren AM, Frost GS, Meeran K, Ghatei MA. Subcutaneous oxyntomodulin reduces body weight in overweight and obese subjects: a double-blind, randomized, controlled trial. Diabetes. 2005;54(8):2390–95. doi:https://doi.org/10.2337/diabetes.54.8.2390.
- Wynne K, Park AJ, Small CJ, Meeran K, Ghatei MA, Frost GS, Bloom SR. Oxyntomodulin increases energy expenditure in addition to decreasing energy intake in overweight and obese humans: a randomised controlled trial. Int J Obes (Lond). 2006;30(12):1729–36. doi:https://doi.org/10.1038/sj.ijo.0803344.
- Tan TM, Field BCT, McCullough KA, Troke RC, Chambers ES, Salem V, Gonzalez Maffe J, Baynes KCR, De Silva A, Viardot A. Coadministration of glucagon-like peptide-1 during glucagon infusion in humans results in increased energy expenditure and amelioration of hyperglycemia. Diabetes. 2013;62(4):1131–38. doi:https://doi.org/10.2337/db12-0797.
- Chakravarthy M, Parsons S, Lassman ME, Butterfield K, Lee AYH, Chen Y, Previs S, Spond J, Yang S, Bock C. Effects of 13-hour hyperglucagonemia on energy expenditure and hepatic glucose production in humans. Diabetes. 2017;66(1):36–44. doi:https://doi.org/10.2337/db16-0746.
- Shankar SS, Shankar RR, Mixson LA, Miller DL, Pramanik B, O’Dowd AK, Williams DM, Frederick CB, Beals CR, Stoch SA. Native oxyntomodulin has significant glucoregulatory effects independent of weight loss in obese humans with and without type 2 diabetes. Diabetes. 2018;67(6):1105–12. doi:https://doi.org/10.2337/db17-1331.
- Zhu L, Tamvakopoulos C, Xie D, Dragovic J, Shen X, Fenyk-Melody JE, Schmidt K, Bagchi A, Griffin PR, Thornberry NA. The role of dipeptidyl peptidase IV in the cleavage of glucagon family peptides: in vivo metabolism of pituitary adenylate cyclase activating polypeptide-(1-38). J Biol Chem. 2003;278(25):22418–23. doi:https://doi.org/10.1074/jbc.M212355200.
- Liu Y-L, Ford HE, Druce MR, Minnion JS, Field BCT, Shillito JC, Baxter J, Murphy KG, Ghatei MA, Bloom SR. Subcutaneous oxyntomodulin analogue administration reduces body weight in lean and obese rodents. Int J Obes (Lond). 2010;34(12):1715–25. doi:https://doi.org/10.1038/ijo.2010.110.
- Kervran A, Dubrasquet M, Blache P, Martinez J, Bataille D. Metabolic clearance rates of oxyntomodulin and glucagon in the rat: contribution of the kidney. Regul Pept. 1990;31(1):41–52. doi:https://doi.org/10.1016/0167-0115(90)90194-2.
- Henderson SJ, Konkar A, Hornigold DC, Trevaskis JL, Jackson R, Fritsch Fredin M, Jansson‐Löfmark R, Naylor J, Rossi A, Bednarek MA, et al. Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetes Obes Metab. 2016;18(12):1176–90. doi:https://doi.org/10.1111/dom.12735.
- Evers A, Haack T, Lorenz M, Bossart M, Elvert R, Henkel B, Stengelin S, Kurz M, Glien M, Dudda A. Design of novel exendin-based dual glucagon-like peptide 1 (GLP-1)/glucagon receptor agonists. J Med Chem. 2017;60(10):4293–303. doi:https://doi.org/10.1021/acs.jmedchem.7b00174.
- Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L, Du X, Petrov A, Lassman ME, Jiang G. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes. 2009;58(10):2258–66. doi:https://doi.org/10.2337/db09-0278.
- Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, Findeisen H, Bruemmer D, Drucker DJ, Chaudhary N, et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5(10):749–57. doi:https://doi.org/10.1038/nchembio.209.
- Boccara F, Dent R, Ruilope L, Valensi P. Practical considerations for the use of subcutaneous treatment in the management of dyslipidaemia. Adv Ther. 2017;34(8):1876–96. doi:https://doi.org/10.1007/s12325-017-0586-8.
- Doppalapudi VR, Tryder N, Li L, Aja T, Griffith D, Liao -F-F, Roxas G, Ramprasad MP, Bradshaw C, Barbas CF. Chemically programmed antibodies: endothelin receptor targeting CovX-Bodies. Bioorg Med Chem Lett. 2007;17(2):501–06. doi:https://doi.org/10.1016/j.bmcl.2006.10.009.
- Murphy RE, Kinhikar AG, Shields MJ, Del Rosario J, Preston R, Levin N, Ward GH. Combined use of immunoassay and two-dimensional liquid chromatography mass spectrometry for the detection and identification of metabolites from biotherapeutic pharmacokinetic samples. J Pharm Biomed Anal. 2010;53(3):221–27. doi:https://doi.org/10.1016/j.jpba.2010.04.028.
- Wang Y, Du J, Zou H, Liu Y, Zhang Y, Gonzalez J, Chao E, Lu L, Yang P, Parker H. Multifunctional antibody agonists targeting glucagon-like peptide-1, glucagon, and glucose-dependent insulinotropic polypeptide receptors. Angew Chem Int Ed Engl. 2016;55(40):12475–78. doi:https://doi.org/10.1002/anie.201606321.
- Vauquelin G, Charlton SJ. Exploring avidity: understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. Br J Pharmacol. 2013;168(8):1771–85. doi:https://doi.org/10.1111/bph.12106.
- Angal S, King DJ, Bodmer MW, Turner A, Lawson ADG, Roberts G, Pedley B, Adair JR. A single amino acid substitution abolishes the heterogeneity of chimeric mouse/human (IgG4) antibody. Mol Immunol. 1993;30(1):105–08. doi:https://doi.org/10.1016/0161-5890(93)90432-B.
- Alegre M-L, Peterson LJ, Xu D, Sattar HA, Jeyarajah DR, Kowalkowski K, Thistlethwaite JR, Zivin RA, Jolliffe L, Bluestone JA. A non-activating “humanized” anti-CD3 monoclonal antibody retains immunosuppressive properties in vivo. Transplantation. 1994;57(11):1537–43. doi:https://doi.org/10.1097/00007890-199457110-00001.
- Santoprete A, Capitò E, Carrington PE, Pocai A, Finotto M, Langella A, Ingallinella P, Zytko K, Bufali S, Cianetti S. DPP-IV-resistant, long-acting oxyntomodulin derivatives. J Pept Sci. 2011;17(4):270–80. doi:https://doi.org/10.1002/psc.1328.
- Teplyakov A, Obmolova G, Malia TJ, Luo J, Muzammil S, Sweet R, Almagro JC, Gilliland GL. Structural diversity in a human antibody germline library. MAbs. 2016;8(6):1045–63. doi:https://doi.org/10.1080/19420862.2016.1190060.
- Obmolova G, Teplyakov A, Malia TJ, Grygiel TLR, Sweet R, Snyder LA, Gilliland GL. Structural basis for high selectivity of anti-CCL2 neutralizing antibody CNTO 888. Mol Immunol. 2012;51(2):227–33. doi:https://doi.org/10.1016/j.molimm.2012.03.022.
- Arafat AM, Kaczmarek P, Skrzypski M, Pruszyńska-Oszmalek E, Kołodziejski P, Szczepankiewicz D, Sassek M, Wojciechowicz T, Wiedenmann B, Pfeiffer AFH. Glucagon increases circulating fibroblast growth factor 21 independently of endogenous insulin levels: a novel mechanism of glucagon-stimulated lipolysis? Diabetologia. 2013;56(3):588–97. doi:https://doi.org/10.1007/s00125-012-2803-y.
- Habegger KM, Stemmer K, Cheng C, Muller TD, Heppner KM, Ottaway N, Holland J, Hembree JL, Smiley D, Gelfanov V. Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes. 2013;62(5):1453–63. doi:https://doi.org/10.2337/db12-1116.
- Biswas K, Nixey TE, Murray JK, Falsey JR, Yin L, Liu H, Gingras J, Hall BE, Herberich B, Holder JR. Engineering antibody reactivity for efficient derivatization to generate Na V 1.7 inhibitory GpTx-1 Peptide–antibody conjugates. ACS Chem Biol. 2017;12(9):2427–35. doi:https://doi.org/10.1021/acschembio.7b00542.
- Kosinski JR, Hubert J, Carrington PE, Chicchi GG, Mu J, Miller C, Cao J, Bianchi E, Pessi A, SinhaRoy R. The glucagon receptor is involved in mediating the body weight-lowering effects of oxyntomodulin. Obesity (Silver Spring). 2012;20(8):1566–71. doi:https://doi.org/10.1038/oby.2012.67.
- Mann JFE, Ørsted DD, Brown-Frandsen K, Marso SP, Poulter NR, Rasmussen S, Tornøe K, Zinman B, Buse JB. Liraglutide and renal outcomes in type 2 diabetes. N Engl J Med. 2017;377(9):839–48. doi:https://doi.org/10.1056/NEJMoa1616011.
- Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375(4):311–22. doi:https://doi.org/10.1056/NEJMoa1603827.
- Lazo M, Solga SF, Horska A, Bonekamp S, Diehl AM, Brancati FL, Wagenknecht LE, Pi-Sunyer FX, Kahn SE, Clark JM. Effect of a 12-month intensive lifestyle intervention on hepatic steatosis in adults with type 2 diabetes. Diabetes Care. 2010;33(10):2156–63. doi:https://doi.org/10.2337/dc10-0856.
- Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, Friedman SL, Diago M, Romero-Gomez M. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149(2):367–78.e5. quiz e14-5. doi:https://doi.org/10.1053/j.gastro.2015.04.005.
- Kang L, Camacho RC, Li W, D’Aquino K, You S, Chuo V, Weng N, Jian W. Simultaneous catabolite identification and quantitation of large therapeutic protein at the intact level by immunoaffinity capture liquid chromatography–high-resolution mass spectrometry. Anal Chem. 2017;89(11):6065–75. doi:https://doi.org/10.1021/acs.analchem.7b00674.