
ABSTRACT
Here, we report the characterization of a VHH-derived IgG-like bi- and trispecific antibody platform that essentially relies on the replacement of the VH and VL regions of a conventional antibody by two independently functioning VHH domains. Consequently, a VHH is engrafted onto constant region CH1 while the other VHH-based paratope is engrafted on the constant region of the light chain, Cκ or Cλ, resulting in a tetravalent bispecific IgG-like molecule. Combined with a heavy chain heterodimerization technique, this platform allows facile engineering of bi- and trispecific antibodies with flexible valencies. We demonstrate the general applicability of this generic platform approach and elaborate on the limitations of specific formats.
Introduction
During the past decades, monoclonal antibodies emerged as promising treatment options for various diseases. Today, more than 80 entities have been granted marketing approval and over 570 antibody-based therapeutics are currently being evaluated in clinical trials.Citation1,Citation2 From a structural perspective, conventional antibodies are large and complex hetero-tetrameric proteins composed of two identical heavy chains as well as two identical light chains, and are monospecific by nature. Consequently, they can only target one antigen. However, diseases are typically complex and originate from multiple different factors or mediators.Citation3,Citation4 In order to address diseases more adequately, substantial efforts have been made to engineer antibodies for bispecificity, and a plethora of different formats with different layers of complexity are known today.Citation5,Citation6
In addition to conventional antibodies, sharks and camelids produce antibodies that are composed of heavy chains only. The paratopes of those isotypes consist of a single variable domain that solely facilitate antigen binding and that are referred to as variable domains of New Antigen Receptor (vNARs, in sharks) or variable domains of the heavy chain of a heavy chain only antibody (VHHs, in camelids).Citation7,Citation8 Due to their small size, which might be beneficial for tissue penetrationCitation9 as well as their ease of generationCitation10-Citation12 good stability and simple architecture, which allows multiple reformatting options, single-domain antibodies evolved as promising alternatives to conventional antibodies.Citation13 While the therapeutic engineering of shark-derived single-domain antibodies is still at a preclinical stage, numerous VHH-derived molecules have progressed into clinical development.Citation14 Moreover, one VHH-based therapeutic, caplacizumab was approved for the treatment of patients with acquired thrombotic thrombocytopenic purpura by the US Food and Drug Administration (FDA) in 2019.Citation15
The general modularity of VHHs enables unprecedented possibilities for the construction of bi- and multispecific antibodies. Consequently, a multitude of VHH-based formats with different valencies as well as specificities have been described in the literature, including multispecific tandem arrangements in a ‘beads-on-string’ manner, VHH-Fc fusions and VHH-antibody fusions.Citation16,Citation17 In 2013, Baty and colleagues described a bispecific VHH-based format that uses the constant domains Cκ and CH1 of IgG as natural dimerization domains.Citation18 In this regard, the authors replaced the variable domain of the heavy chain as well as the variable domain of the light chain by two VHHs, targeting carcinoembryonic antigen and FcγRIIIa, respectively. This resulted in an antigen-binding fragment (Fab)-like bispecific molecule eliciting potent tumor lysis by natural killer (NK) cells. More recently, the authors extended the applicability of this format by targeting human epidermal growth factor receptor 2 (HER2)Citation19 and mesothelin.Citation20
In this study, we set out to investigate whether this Fab-derived bispecific building block can be used to generate a generic IgG-like platform that allows for facile reformatting in bi- and trispecific VHH-based antibodies with flexible valencies ()). By genetically fusing this Fab-derived bispecific VHH module on an IgG1 Fc backbone, bivalent (for each antigen) bispecific IgG-like antibodies can be obtained ()). Combining this building block with a heavy chain heterodimerization technique allows facile reformatting into a monovalent bispecific IgG-like entity ()). To this end, the strand-exchanged engineered domain (SEED) technology was applied.Citation22 This method is based on beta-strand exchanges of IgG and IgA CH3 domains, yielding preferentially heterodimers without impairing Fc-mediated effector functions. This also enables the generation of trispecific moieties with different valencies (,e)). By characterizing this platform with respect to biophysical and biochemical properties, we show the benefits as well as limitations of the herein described in different formats. Moreover, when substantial paratope-dependent effects (i.e., steric hindrance) arise, we demonstrate that this can be counteracted by simply placing a 10 amino acid flexible linker between the respective VHH domain and the constant domain region of the light chain. Ultimately, we conclude that most of the different formats of the herein presented platform approach seem to be adequate to expand the ‘playground’ of multispecific VHH-based antibody engineering.
Figure 1. Schematic representation of the overall strategy for the generation of VHH-based IgG-like multispecific antibodies. (a) Model depicting the approach to replace domains VH and VL of a conventional IgG by two independently functioning VHHs for the generation of a bispecific entity. The model is based on pdb:5dk3Citation21 and was generated using PyMOL v0.99. (b) Bivalent bispecific VHH-based IgG-like antibody employing an IgG backbone. (c) Utilization of the strand-exchanged engineered domains (SEED) technology enables the generation of monovalent bispecific moieties. (d) Monovalent trispecific VHH-based SEEDbodies can be generated by grafting a third VHH onto the hinge region of the SEED GA chain. (e) Trispecific VHH-based SEEDbody comprising bivalency of the light chain-engrafted VHH. (f) Schematic depiction of a one-armed VHH-derived control molecule.
Results
Design of different formats
Within this study, we aimed at developing and characterizing a generic platform approach for the facile generation of IgG-like bi- and trispecific VHH-derived antibodies. Essentially, the platform is based on the replacement of the variable domains of a human antibody by two independent VHH-based paratopes ()). Consequently, one VHH is grafted onto domain CH1 of IgG1, whereas the second VHH is grafted either on domain Cκ or Cλ. In order to get a profound understanding of this platform approach, i.e., of the respective formats (,e)), we decided to use four different VHHs. One VHH was directed against HER2 (sequence derived from WO2016016021). Two paratopes were directed against epidermal growth factor receptor (EGFR), wherein clone 9G8 was described by Ferguson and coworkersCitation23 and clone 722C03 was derived from an internal yeast surface display campaign.Citation24 Finally, an NKG2D-targeting VHH sequence was obtained from WO2017081190. Sequences of the respective VHHs can be found in supplementary Figure S1A. Design examples of VHH engraftments on constant antibody regions CH1, Cκ, or Cλ are shown in supplementary Figure S1B.
In regard to the different formats, we wanted to include at least some flexibility regarding specificities (bi- and trispecific versions) as well as valencies for a cognate epitope (mono- and bivalent). When the VHH-engrafted Fab is expressed on an IgG backbone for instance ()), this results in a bispecific IgG-looking molecule, that is tetravalent by design (and bivalent for each antigen). By simply using a heavy chain heterodimerization technique, i.e., SEED technology ()), one can express one-armed versions of the VHH-based bispecific. This enables bivalency and accordingly monovalent targeting of each epitope. Consequently, this also allows for a very thorough characterization of the platform with respect to functional properties, e.g., simultaneous binding on the protein and cellular level as well as investigations on paratope-dependent effects, i.e., steric hindrance. This monovalent bispecific format (i.e., monovalent for the respective target) can easily be extended into a trispecific format that is monovalent for each antigen by simply placing a third VHH-derived paratope onto the hinge region of the second heavy chain ()). Finally, by placing two VHH-engrafted Fabs on each heavy chain, the platform also allows trispecific targeting in which the paratope grafted onto the constant region of the light chain is bivalent ()).
Expression and characterization of monovalent bispecific IgG-like VHH-based antibodies
To gain a thorough understanding of the functionality of each engrafted paratope in this platform approach, we first focused on the generation of monovalent IgG-like VHH-based bispecific antibodies ()). For heavy chain heterodimerization, the SEED technology was applied, which is based on beta-strand exchanges of IgG and IgA CH3 domains, resulting in two non-identical heavy chains referred to as AG chain and GA chain. For the SEED GA chain, it is known that homodimers might form, clearly representing a misassembled side product that might affect the analysis of the respective construct in manifold ways, e.g., kinetic measurements and size exclusion profiles. In order to obviate this, the RF mutation was introduced, preventing binding of GA:GA homodimers to protein A.Citation25 Consequently, only AG-GA heterodimers are purified, since AG-AG species typically do not assemble. In this particular monovalent bispecific format, the VHH-based heavy chain of the Fab was expressed on the AG chain, while the GA chain solely comprised the hinge region (one-armed IgG-like VHH-SEED). To allow for direct comparisons to the parental VHHs, i.e., VHHs not engrafted into this bispecific construct, each camelid-derived variable domain was also expressed as one-armed SEED control ()). To get a more comprehensive view on this format, we produced all four different VHHs in the bispecific monovalent IgG-like scaffold and tried different orientations as well as lambda and kappa backbones for grafting the second paratope onto the constant region of the light chain. The expression of all monovalent bispecific IgG-like VHH-based molecules resulted in fairly equal expression yields compared to one-armed control molecules (). To be more precise, expression yields as determined after protein A purification were in the triple-digit milligram per liter scale, indicating good expression profiles of all the molecules scrutinized in this particular format. Moreover, thermal stabilities of individual bispecific constructs were very similar to the implemented VHH displaying the lower aggregation onset temperature. Aggregation properties were also in general quite similar to the control molecules as well. Except for one molecule (, monovalent bispecific entry MoBi12) size exclusion chromatography (SEC) profiles were above 90% target peak. Hence, the results clearly indicate the favorable biophysical properties of the herein described monovalent bispecific IgG-like VHH-based antibody format.
Table 1. Biophysical and biochemical properties of one-armed control molecules and monovalent bispecific series of engineered molecules. Format designs are indicated as well as expression yields post protein A purification, Tonsets and size exclusion chromatography target monomer peaks. Affinities as determined by biolayer interferometry are also given.
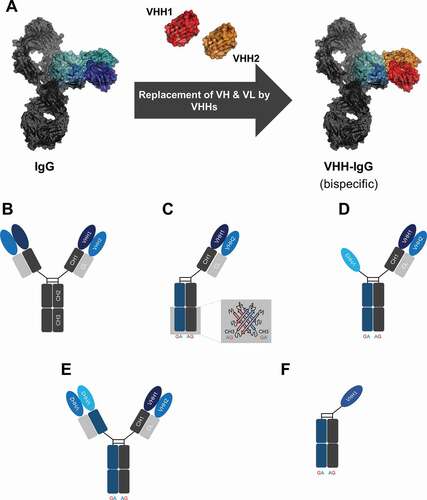
Next, we aimed at characterizing the biochemical properties. To this end, affinities were determined via biolayer interferometry (). For all different VHH-derived paratopes and all the different orientations as well as kappa and lambda backbones, affinities of the bispecific molecules were similar to the parental molecules. However, as shown in Fig. S2, the total interference pattern shift in the association step of all analyzed monovalent bispecifics was slightly diminished compared to the parental clones. Still, association and dissociation rates were nearly identical, indicating no substantial loss of affinities (Tab. S1). To analyze this in more detail in a cellular context, we focused on molecule MoBi8 (), which consists of EGFR-specific VHH 9G8 engrafted on the AG chain as well as a HER2-specific VHH fused to the constant region of the lambda chain. As shown in , both paratopes can independently engage in the cellular binding of target-positive cell lines in a concentration-dependent manner similar to the respective monospecific control molecules (Ctrl1 and Ctrl3), supporting the notion that the functionality of each VHH is not compromised significantly in this bispecific architecture. Importantly, no binding was observed against the antigen-negative control cell line.
Figure 2. Cellular binding of monovalent bispecific antibody MoBi8 (black) and monospecific control molecules Ctrl1 (green) and Ctrl3 (orange) to EGFR-positive MDA-MB-468 (a) and HER2-positive SK-BR-3 cells (b) as well as to EGFR/HER2-negative CHO cells (c). 1 × 105 target cells were incubated with the respective antibody in varying concentrations.
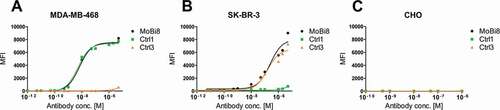
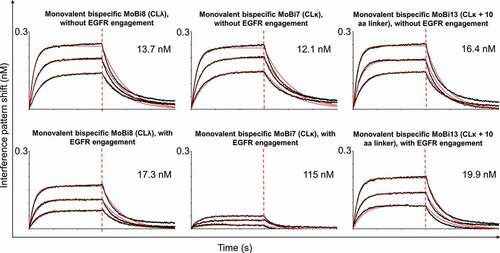
In addition, all bispecific one-armed IgG-like VHH-SEEDs showed simultaneous binding to their antigens on the protein level (Fig. S3). Interestingly, as depicted in ), when grafted onto the kappa constant region, the HER2-specific VHH (, entry MoBi7) showed compromised HER2-binding compared to the lambda counterpart (, entry MoBi8), when EGFR-specific VHH clone 9G8 that was fused to the AG heavy chain already bound its antigen, clearly demonstrating paratope-dependent positioning effects in the kappa context. Surprisingly, this phenomenon was neither observed to a significant extent when EGFR-specific clone 722C03 was grafted onto the AG chain nor when the VHH directed against NKG2D was used for the AG chain (,c)). NKG2D (extra-cellular domain (ECD)) with a molecular weight of 18.4 kDa is a much smaller protein compared to EGFR (ECD) with 71.4 kDa. Consequently, it is tempting to speculate that steric hindrance might be the cause for impeded simultaneous binding. In terms of the strikingly different binding behavior of the HER2 paratope in the EGFR context, it is worth noting that both EGFR-specific VHHs address different non-overlapping epitopes (Fig. S4). Therefore, we assume that 9G8 binds an epitope of EGFR in a way that EGFR negatively impacts the accessibility of HER2 to the kappa-engrafted entity. In order to address this more meticulously, we simply placed a 10 amino acid linker between the HER2-specific paratope and the kappa constant region (Fig. S1B, , entry MoBi13). This restored the binding capacity of the HER2-VHH, clearly corroborating that steric hindrance is the main reason for the positioning effects ()). We also determined the affinities of all three monovalent bispecifics against HER2, when the inner (AG chain) EGFR-directed VHH 9G8 bound its antigen in saturation (). When grafted onto the lambda chain, the overall affinities for HER2 were quite similar, irrespective of the EGFR-specific paratope engaged in antigen-binding or not. Still, even in the lambda context, the total interference pattern shift during association decreased somewhat, indicating also a moderate decline in binding capacity. However, when grafted onto the kappa light chain, not only the total interference pattern shift dropped substantially but also the overall affinities were compromised by a factor of approximately 10 for HER2-binding. This effect could be counteracted in both regards by placing a 10 amino acid linker between the HER2-paratope and the kappa constant region. Interestingly, when the VHH orientation in this format was inverted, i.e., when the HER2-specific paratope was grafted onto the AG chain and EGFR-specific VHH 9G8 was placed onto the light chain, this effect was also observed (Fig. S5), corroborating this paratope-specific positioning effect.
Figure 3. Biolayer interferometry analysis to assess simultaneous binding of one-armed monovalent bispecific molecules. The association of the respective antigen (50 nM) was measured, followed by a second association step using HER2 (50 nM). (a) Association of one armed monovalent bispecific entities MoBi7 (CLκ, black), MoBi8 (CLλ, dark gray) and MoBi13 (CLκ +10 aa linker, gray) against EGFR and HER2 (or kinetics buffer (KB) as control, light gray). (b) Simultaneous binding of one armed monovalent bispecific molecules MoBi9 (CLκ, black), MoBi10 (CLλ, dark gray) and MoBi14 (CLκ +10 aa linker, gray) against EGFR and HER2 (or KB, light gray). (c) Binding of one armed monovalent bispecific moieties MoBi11 (CLκ, black) and MoBi12 (CLλ, dark gray) against NKG2D and HER2 (or KB, light gray).
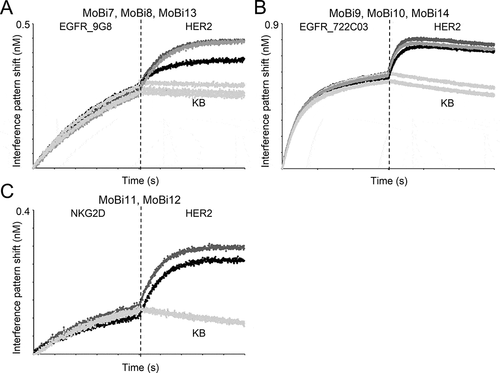
Figure 4. Kinetic measurements of monovalent bispecific VHH-derived IgG-like SEEDbodies MoBi7, MoBi8 and MoBi13 against HER2 in the presence or absence of EGFR binding of the heavy chain-engrafted VHH 9G8 in saturation. After loading and sensor rinsing, a first association step was performed using EGFR in saturating conditions or kinetics buffer (KB). Afterward, kinetics where analyzed against HER2 in the presence or absence of EGFR.
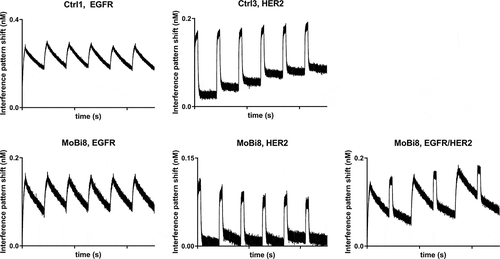
In certain disease settings, a therapeutic (bispecific) antibody must be able to bind to its antigen multiple times throughout its half-life. To investigate whether the architecture of this format permits periodic binding, consecutive association and dissociation cycles were performed for one exemplarily chosen molecule (MoBi8) on the Octet Red system (). The bispecific entity showed periodic binding to EGFR as well as HER2 comparable to the mono-specific controls. Moreover, consecutive association and dissociation against both antigens in an alternating manner was also observed, clearly demonstrating the ability to bind multiple times in a bispecific mode of action.
Figure 5. Periodic binding of mono-specific controls Ctrl1 (EGFR) and Ctrl3 (HER2) as well as bispecific MoBi8 (EGFR/HER2) antibody to their corresponding antigens at a concentration of 50 nM. Consecutive association (300 s) and dissociation (30 min) cycles were performed six times in total.
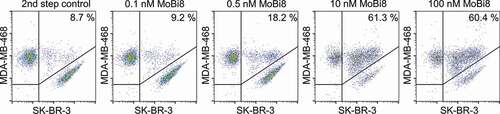
Furthermore, we set out to investigate if this format also enables simultaneous binding on a cellular level. To this end, the EGFR-positive cell line MDA-MB-468 was labeled with CellTracker™ Deep Red Dye whereas HER2-positive SK-BR-3 cells were labeled with CellTrace™ CFSE. Of note, HER2-positive cell line SK-BR-3 is also slightly EGFR-positive. However, since only one EGFR-paratope was engrafted into this monovalent bispecific format, the entity can either bind one cell type or the other, not both. If a bispecific molecule is able to bind both antigens simultaneously, this will result in antibody-mediated clustering of both cells. Consequently, this can be analyzed by fluorescence-activated cell sorting since cell clustering appears as a double-positive event. As shown in , EGFR- and HER2-targeting monovalent bispecific IgG-like VHH-based SEED MoBi8 bound simultaneously to both cell types because clustering was observed in a concentration-dependent manner, whereas no significant increase in double-positive events was detected for EGFR- or HER2-specific one-armed monospecific control molecules (Fig. S6). Importantly, monovalent bispecific IgG-like VHH-based SEED molecules either targeting HER2 and NKG2D or EGFR and NKG2D did not enable the clustering of both cell types, giving clear evidence that the architecture of this platform approach affords the benefit of simultaneous binding also in a cellular context.
Figure 6. Simultaneous cellular binding analysis of monovalent bispecific antibody MoBi8. Binding to EGFR-positive cell line MDA-MB-468 and HER2-positive cell line SK-BR-3 was assessed by flow cytometric analysis. Of note, MDA MB 468 was labeled with CellTracker™ Deep Red Dye and HER2-positive SK-BR-3 cells were labeled with CellTrace™ CFSE to allow for double-positive fluorescence gating.
Similar to Baty and coworkers,Citation18-Citation20 we set out to demonstrate whether this format bears the potential to be used for effector cell redirection. To this end, a tumor cell killing assay was performed using all four different EGFR (9G8) and NKG2D-specific variants of the MoBi series using peripheral blood mononuclear cell (PBMC)-isolated NK cells as effector cell population (Fig. S7). All EGFR- and NKG2D-targeting monovalent bispecific entities elicited the killing of EGFR-overexpressing A431 cells in a dose-dependent manner, whereas no significant tumor cell lysis was observed for unrelated control molecules.
Expression and characterization of bivalent bispecific IgG-like VHH-based antibodies
Bivalent targeting of two different disease mediators might be beneficial for specific therapeutic applications such as cytokine trapping. From a conceptional perspective, the herein characterized monovalent bispecific format can be readily extended into a bivalent counterpart by exchanging the SEED technology for an IgG1 backbone ()). To get a glimpse of the biochemical and biophysical properties of this format, we expressed nine different bivalent bispecific IgG-like VHH-based antibodies, essentially using all four different paratopes. Moreover, the kappa as well as the lambda constant regions were taken into consideration for light-chain engraftments. Interestingly, expression yields after protein A chromatography indicated in general good production profiles (). Some of the molecules, however, were prone to aggregation or showed low molecular weight species as determined by analytical SEC (Fig. S8). In direct comparison to the monovalent counterparts, it is noteworthy that tendencies for aggregation or mis-assembly of the molecule (as indicated by small molecular weight species) were significantly aggravated. In this respect, for one molecule (BiBi4), the target peak declined to only 72%, meaning that substantial efforts in downstream processing would be required in order to consider such a molecule for further development. On the other hand, two molecules exhibited quite decent SEC profiles (BiBi1 and BiBi6). Since some of the molecules with exceptionally good as well as bad aggregation properties were grafted onto the kappa and lambda backbone, it is tempting to speculate that this observation is more related to the paratopes themselves and the individual orientation of the VHHs in the overall architecture. In terms of biochemical properties, all analyzed molecules retained their affinities as compared to the monospecific control molecules (, Ctrl1-4; ). Moreover, all bivalent bispecifics were able to engage in simultaneous binding (Fig. S3). As expected, when EGFR-specific VHH 9G8 was placed onto the heavy chain and the HER2-specific VHH was engrafted onto the kappa constant chain (BiBi1), simultaneous binding was again compromised (Fig. S9). Similar to the monovalent counterpart, this decline in binding capacity could be partially compensated by employing a 10 amino acid linker (BiBi9).
Table 2. Biophysical and biochemical properties of bivalent bispecific series of engineered VHH-based IgG-like antibodies. Format designs are indicated as well as expression yields post protein A purification and size exclusion chromatography target monomer peaks. Affinities as determined by biolayer interferometry are also given.
Expression and characterization of trispecific IgG-like VHH-based SEEDbodies
By simply placing a third VHH-derived moiety on top of the hinge region of the GA chain of the IgG-like VHH-based SEEDbody, we also set out to characterize this platform approach with respect to trispecific targeting (), , MoTri series of molecules). In this regard, we wanted to directly compare this monovalent trispecific format with another format enabling trispecificity, in which the light-chain-based VHH is bivalent (), , BiTri entries). To this end, SEED molecules were produced, where on each chain of the SEEDbody (AG and GA) a different VHH was engrafted on domain CH1. Simultaneous expression of a VHH engrafted onto the constant region of the light chain results in the pairing of both heavy chain-derived paratopes with the light chain-embedded paratope.
Table 3. Biophysical and biochemical properties of trispecific VHH-derived SEEDbodies. Format designs are indicated as well as expression yields post protein A purification and size exclusion chromatography target monomer peaks. Affinities as determined by biolayer interferometry are also given.
The strictly monovalent trispecific SEEDbodies displayed rather beneficial biophysical properties. Expression yields were constantly in the triple-digit mg per liter scale, and SEC profiles (Fig. S8) were reasonably acceptable, even when compared to the bivalent bispecific format (, BiBi series of molecules). This was to our surprise, since the sophisticated architecture of this asymmetric trispecific format seems to be fairly complex, at least in direct comparison to the bivalent bispecific approach, where not even a heavy chain heterodimerization technique is needed. However, this is in stark contrast to the bivalent trispecific format (, BiTri series of entries). While expression yields were still reasonable, the SEC profiles, i.e., the target peaks, declined significantly, to only little more than 50% for some of the scrutinized molecules, clearly demonstrating strongly compromised biophysical properties. Noteworthy, these detrimental attributes are not only related to aggregation propensities but also due to significant amounts of low molecular weight species, clearly indicating mis-assembled molecules (Fig. S8).
Similar to the design of the bivalent bispecific format (BiBi series), the expression of entities of the bivalent trispecific series (BiTri) result in a symmetric overall architecture of the respective molecules. On the contrary, molecules of the MoBi and MoTri series that in general display more adequate biophysical properties, i.e., target monomer peaks in SEC, are asymmetric by design. Consequently, this suggests that an asymmetric molecule design of the herein presented ‘plug-and-play’ approach generally results in fairly favorable biophysical attributes.
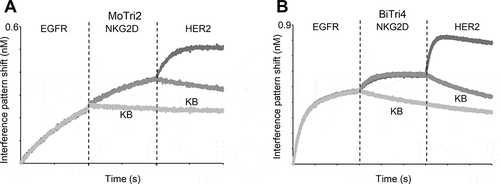
With respect to biochemical properties, as anticipated, no significant decline of affinities was detected for each embedded VHH against the respective antigen. Likewise, all molecules analyzed engaged in trispecific simultaneous binding against each antigen, as exemplarily shown for molecules MoTri 2 and BiTri4 (, Fig. S3).
Figure 7. Simultaneous trispecific antigen binding of (a) monovalent trispecific VHH-derived IgG-like antibody MoTri2 and (b) bivalent trispecific entity BiTri4 against 50 nM EGFR followed by 50 nM NKG2D and 50 nM HER2. For each association step a control in KB is shown.
Discussion
Bi- and multispecific antibodies have recently emerged as promising molecules for disease treatment.Citation5,Citation6 In this respect, bi- and multispecific entities opened up new avenues to address the complex biology of several diseases more appropriately compared to monospecific antibodies.Citation26-Citation30 Accordingly, an unparalleled repertoire of different antibody formats was developed, each with its own characteristics, beneficial attributes, and limitations, as elegantly reviewed elsewhere.Citation5,Citation6 In this context, VHH-derived constructs afford the benefit of multiple reformatting options.Citation16,Citation17 Moreover, antigen-specific single-domain antibodies from camelids can be readily obtained by combining animal immunization with antibody display systems enabling genotype-phenotype coupling.Citation10,Citation11 However, for therapeutic use in patients, it must be noted that the foreign nature of camelid-derived VHHs bears the risk of immunogenicity. In this respect, Vincke et al. described a general strategy for humanization.Citation31 Currently, multiple VHH-derived entities are being investigated in clinical trials, and bivalent single-domain caplacizumab (Cablivi®) has been approved for the treatment of patients with acquired thrombotic thrombocytopenic purpura by the FDA.Citation14,Citation15 To the best of our knowledge, most if not all of these molecules were humanized. This gives clear evidence that the risk of substantial immunogenicity can be circumvented by humanization.
Within this work, we aimed at characterizing a VHH-based platform for facile construction of different IgG-like bi- and multispecific antibody constructs that differ significantly among themselves with respect to their biophysical characteristics as well as biochemical properties. Essentially, the main building block in this platform approach was described some time ago by Baty and coworkers.Citation18 We set out to investigate to what extent this approach can be broadened for the generation of bi- and trispecific moieties comprising flexible valencies without fundamentally compromising essential intrinsic attributes, for instance aggregation behavior or binding capacities.
In regard to biophysical properties, it is remarkable that expression yields, as determined after protein A purification of most of the bi- and trispecific molecules, were fairly high compared to other technologies reported.Citation32-Citation34 Most importantly, yields were quite similar to the parental molecules, i.e., controls. This was also true for aggregation onset temperatures, indicating in general adequate stabilities of VHHs when engrafted into the Fab-based architecture. With respect to aggregate formation and potential mis-assembly of molecules, substantial differences were determined for the different herein described formats. In general, asymmetric formats, i.e., monovalent IgG-like VHH-based bispecific antibodies as well as strictly monovalent trispecific SEEDbodies, displayed rather favorable aggregation propensities while symmetric formats tended to be more prone to malfunction. In this context, SEC profiles for bivalent bispecific VHH-based IgG-like antibodies indicated still acceptable target monomer formation. For the bivalent (for the light chain) trispecific format (BiTri series of molecules), however, aggregate formation and mis-assembled molecules (low molecular weight species) were fundamentally worsened clearly disqualifying this format for further consideration.
It is important to note that for all the different VHHs that were embedded into the Fab scaffold, overall affinities were quite similar to the control molecules. The interference pattern shift, however, was slightly decreased in every instance, indeed indicating impeded binding capacities to a minor extent for engrafted paratopes. Yet, this effect appears to be almost neglectable, since cellular binding did not seem to be impaired substantially. From a functional aspect, we were able to demonstrate that both Fab-engrafted single-domain paratopes can engage in simultaneous binding in a cellular context. This is especially vital for several therapeutic concepts that involve targeting of different cell types at the same time, such as effector cell redirection.Citation35-Citation37 Similar to Baty and coworkers,Citation18 we were able to show that the overall architecture of this platform clearly allows for effector cell recruitment enabling tumor cell killing.
Furthermore, we report that for one VHH analyzed (HER2) in this platform approach, engraftment onto the kappa chain causes a substantial loss of affinity when the other embedded VHH (EGFR, clone 9G8) already engages in antigen binding. Positioning effects have also been described for other bispecific platform technologies, for instance, DVD-Igs or TriFabs and others,Citation34,Citation38-Citation40 typically resulting in compromised binding affinities. Surprisingly, the individual binding affinity of the engrafted paratope against the respective antigen was not impaired substantially. Accordingly, this positioning effect does not diminish the binding capacity of the individual paratope per se, but only in the context of simultaneous binding. We were also able to show that a 10 amino acid linker between the impacted VHH and the kappa chain reversed this effect. In addition, this affinity sink in simultaneous binding was not observed in the lambda context. Stanfield and colleagues were able to demonstrate that Fabs comprising lambda chains tend to have hypervariable elbow angles, i.e., angles between the variable regions and the constant domains.Citation41 On average, these elbow bends are significantly wider than those of kappa chain Fabs. Presumably, this is due to a glycine insertion at the switch region between the variable domain of the light chain and the constant region that appears to increase the flexibility of the Fab. This increased flexibility might be the main reason why this positioning effect was not detected for VHH engrafted onto the lambda light chain. Thus, this gives clear evidence that positioning effects can be circumvented either by simple linker design or by replacing the kappa chain for a lambda chain.
Ultimately, the herein presented VHH-based IgG-like bi- and trispecific antibody platform appears to have the versatility to combine up to three preexisting VHHs in one multispecific molecule. We demonstrated that the different described formats, except for molecules of the BiTri series seem to comprise acceptable properties and that early-stage ‘developability’ is feasible. Essentially, this ‘plug-and-play’ platform affords the benefit to tailor multispecific entities with respect to their biophysical and biochemical properties.
Materials and methods
Antibody expression and purification
All constructs were designed in-house and synthesized by GeneArt (Thermo Fisher Scientific). In general, all antibody-like constructs and SEED molecules were cloned into pTT5 plasmid backbone (GeneArt/Thermo Fisher Scientific). For SEEDbody expression, the RF mutation was introduced to prevent the formation of undesired GA:GA chain homodimers.Citation25 Expi293 cells were transiently transfected with expression vectors according to the manufacturer’s recommendations (Thermo Fisher Scientific). Five days post transfection, the antibody-containing supernatants were harvested by centrifugation and purified via MabSelect antibody purification chromatography resin (GE Healthcare). A final dialysis step using Pur-A-LyzerTM Maxi 3500 Dialysis Kit (Sigma Aldrich/Merck KGaA) was performed at 4°C for 24 h to ensure sample formulation in phosphate-buffered saline (PBS) pH 6.8, followed by an additional protein concentration step via Amicon Ultra-4 Centrifugal Filters (EMD Millipore), if necessary. UV-Vis spectrophotometric measurement (Nanodrop ND-1000, Peqlab) was used to determine protein concentrations. Additionally, the expressed antibodies were analyzed in duplicates by differential scanning fluorometry on Prometheus NT.48 (Nanotemper Technologies) to evaluate the stability of expressed molecules. When measuring the thermal stability of proteins, multiple values like Tonset, Tm1 and Tagg can be reported. The lowest temperature at which the protein starts to unfold is defined as the Tonset while Tm1 is the melting point at which 50% of the thermally least stable domain is unfolded. Multiple unfolding events of protein domains can overlap, and therefore Tm1 can vary more between similar constructs than Tonset. Thus, we chose Tonset as a discriminator to compare the thermal stability of multiple protein variants. Furthermore, aggregate formation was analyzed by analytical SEC of 10 µg protein per sample, using a TSKgel SuperSW3000 column (4.6 × 300 mm, Tosoh Bioscience LLC) in an Agilent HPLC system with a flow rate of 0.35 ml/min.
Biolayer Interferometry
Simultaneous binding, periodic binding, epitope binning, as well as kinetic measurements were performed on the Octet RED96 system (ForteBio, Pall Life Science) at 25°C and 1000 rpm agitation. For kinetic analysis, the antibody-like molecules to be analyzed were loaded on anti-human-FC (AHC) Biosensors at 5 μg/mL in PBS for 3 min. Afterward, tips were transferred to kinetics buffer (KB; PBS, 0.1% Tween-20 and 1% bovine serum albumin, BSA) for 60 s for sensor rinsing. Association to the corresponding antigen human EGFR (in-house), human NKG2D (Sino Biological, Cat: 10575-H07B) and human HER2 (Sino Biological, Cat: 10004-H08H) in varying concentrations ranging from 12.5 nM to 50 nM in KB was measured for 300 s followed by dissociation in KB for 300 s. In each experiment, one negative control was included where the association was monitored against an unrelated antigen (50 nM in KB). Moreover, one reference value was included with the association in KB instead of the antigen. Resulting data were fitted and analyzed with ForteBio data analysis software 8.0 using a 1:1 binding model after Savitzky-Golay filtering.
To demonstrate simultaneous antigen binding, respective antibody-like molecules were captured to the surface of anti-human IgG Fc (AHC) biosensors for 3 min at a concentration of 5 µg/ml and sensor rinsing was conducted in KB for 60 s. Afterward, sensors were transferred to first antigen solution (50 nM in KB) and incubated for 100 s to 300 s, followed by an association in second antigen solution for 100 s to 300 s (50 nM in KB) and consecutively, when applicable, in antigen three for 100 s to 300 s (50 nM in KB).
Furthermore, for the kinetic measurements with EGFR bound in saturation or without prior EGFR binding the antibody was loaded on anti-Penta His biosensors for 180 s, followed by 60 s of sensor rinsing in KB. Afterward, 500 nM EGFR was used to saturate the binding capacity of the antibody for 300 s. Accordingly, KB was used instead of EGFR for the measurements without EGFR binding in saturation. Afterward, the association was determined against HER2 (300 s) using varying concentrations from 0 to 50 nM. To avoid a dissociation of EGFR in this step, 50 nM EGFR was used for every HER2 concentration tested. For the following dissociation step (300 s), EGFR was also used at 50 nM to assess dissociation against HER2 only.
For the assessment of periodic antigen-binding antibodies were loaded on Anti-human-Fc (AHC) biosensors at 1 μg/mL for 3 min. After 60 s of sensor rinsing in KB association was analyzed at a concentration of 50 nM for 300 s followed by a dissociation step in KB for 30 min. The association and dissociation steps were consecutively repeated for six times in total.
Epitope binning analysis was performed by loading EGFR (ECD) via its histidine-tag on anti-Penta His (HIS1K) biosensors at 3 μg/mL in PBS for 3 min. After a step of sensor rinsing in KB for 60 s association to the respective EGFR-specific molecule (at 100 nM) was performed (, Ctrl1 or Ctrl4), followed by either dissociation in KB or by another association step with the second EGFR-derived VHH (at 100 nM). One negative control was measured in parallel, where the sensor was not loaded with antigen to check for unspecific association of the antibodies to the biosensor. Moreover, one reference value using buffer instead of antibody association was measured as ‘association baseline.’
Flow cytometry
Flow cytometric analyses were conducted on the Guava® easyCyte 12HT device (Merck Millipore) using guavaSoft 3.2 Software. For each experiment, 5000 events/well were measured.
To determine cellular binding properties of the expressed antibodies, EGFR-positive MDA-MB-468, HER2-positive SK-BR-3, and EGFR/HER2 negative CHO cells were used. For staining assays, 1 × 105 cells per well were seeded. After two consecutive washing steps with PBS+1% BSA, cells were incubated for 1 h on ice with antibodies in varying concentrations. Afterward, two additional washing steps with PBS+1% BSA were performed, followed by staining with a detection antibody at 4°C for another 1 h. Cell binding was identified with Alexa Fluor® 488 AffiniPure F(ab’)₂ Fragment Goat Anti-Human IgG, Fcγ fragment specific antibody (Jackson ImmunoResearch, Cat: 109–546-008) for EGFR- and HER2-positive cells lines, respectively, as well as Alexa Fluor® 488 AffiniPure Fab Fragment Goat Anti-Human IgG (H + L) antibody (Jackson ImmunoResearch, Cat: 109–547-003) for the negative CHO cells. After two washing steps with PBS+1% BSA, dead cells were stained via 20 µg/ml propidium iodide (Invitrogen) addition in a total volume of 200 µl/well. Additionally, a negative control without antibody treatment as well as a control using the detection antibody only and a control using in-house produced cetuximab (Erbitux®) or trastuzumab (Herceptin®) as positive reaction, was conducted in each experiment for each cell line (not shown).
For the investigation of simultaneous cellular binding, EGFR-positive MDA-MB-468 cells were stained prior to the assay with CellTracker™ Deep Red Dye (ThermoFisher). To this end, the cells were harvested and washed once in PBS before staining 1 × 106 cells/ml with the reagent (1:1000 in PBS) for 15 min at 37°C in the dark. Following incubation, the cells were washed once with respective complete medium and finally washed once with PBS+1% BSA. This staining procedure was performed accordingly for HER2-positive SK-BR-3 cells, except using CellTrace™ CFSE Cell Proliferation Kit as dye (1:100ʹ000 in PBS). For both cell lines, 1 × 105 cells each were combined and inserted per well. As controls, wells containing just one cell line were used, allowing for gating the single-cell populations. After a washing step with PBS+1% BSA, cells were incubated for 1 h on ice with the antibodies in varying concentrations ranging from 0.1 nM to 1 µM. Finally, two more washing steps with PBS+1% BSA were conducted and the cells were resuspended in 200 µl volume for flow cytometric measurements. By applying a rectangular gate, the single-cell populations were discriminated from the simultaneously bound cells forming duplets and showing a positive signal for both fluorescence dyes.
Tumor cell killing assay
PBMCs cells were isolated from whole blood samples of healthy human donors by density gradient centrifugation with consecutive NK cell isolation using EasySep™ Human NK Cell Isolation Kit (Stemcell Technologies). Effector cells were adjusted to 0.625 × 106 vc/ml after overnight incubation in complete medium containing 100 U/ml recombinant human interleukin-2 (R&D systems). A431 target cells were prepared in parallel by staining with CellTracker™ Deep Red Dye (ThermoFisher) according to the manufacturer’s instructions. For the assay, 20 µl target cells (resulting in 2ʹ500 c/well) were seeded in 384-well clear bottom microtiter plate (Greiner Bio-One) and incubated for 3 h. Afterward, 5 µl of the bispecific antibodies were added with varying concentrations ranging from 10−7 M to 10−12 M followed by the addition of 20 µl NK cell suspension, resulting in an effector cell to target cell (E:T) ratio of 5:1. Finally, SYTOX™ Green Dead Cell Stain (Invitrogen) was added to the assay.
NK cells cultivated with A431 cells in the absence of antibodies as well as A431 cultivated with antibodies without adding NK cells as well as A431 cells with the highest antibody concentration without NK cells were used as controls. To assess maximum tumor cell lysis, staurosporine was dispensed to A431 cells as another control with a final concentration of 30 µM. For the incubation (24 h) and on-line imaging of the assay an Incucyte® Live Cell Analysis System (Sartorius) was used. Overlay (green and red fluorescence) signals per well allowed for analysis of dead target cells only.
Disclosure of potential conflicts of interest
BV, MB, LT, SK and SZ are employees of Merck Healthcare KGaA. Besides, the authors declare no conflict of interest.
Abbreviations
| CH1 | = | constant domain 1 of the heavy chain; |
| Cκ | = | constant kappa region of the light chain |
| Cλ | = | constant lambda region of the light chain |
| EGFR | = | epidermal growth factor receptor |
| Fab | = | antigen-binding fragment |
| HER2 | = | human epidermal growth factor receptor 2 |
| NK cell | = | natural killer cell |
| NKG2D | = | natural killer group 2 member D |
| PBMC | = | peripheral blood mononuclear cell |
| PBS | = | phosphate-buffered saline |
| SEC | = | size exclusion chromatography |
| SEED | = | strand-exchanged engineered domain |
| VH | = | variable domain of the heavy chain |
| VHH | = | variable domain of the heavy chain of a heavy chain only antibody |
| VL | = | variable domain of the light chain |
| vNAR | = | variable domain of New Antigen Receptor |
Supplemental Material
Download Zip (29.9 MB)Acknowledgments
The authors kindly thank Laura Unmuth, Dirk Müller-Pompalla, Alexander Müller, Stephan Keller, Sigrid Auth, Marion Wetter, Pia Stroh, Stefan Becker, Janina Klemm and Kerstin Hallstein for experimental support.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- Kaplon H, Reichert JM. Antibodies to watch in 2019. mAbs. 2019 Feb;11(2):219–12. doi:https://doi.org/10.1080/19420862.2018.1556465.
- Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to watch in 2020. mAbs. 2020 Jan;12(1):1703531. doi:https://doi.org/10.1080/19420862.2019.1703531.
- Krah S, Kolmar H, Becker S, Zielonka S. Engineering IgG-like bispecific antibodies—an overview. Antibodies. 2018 Aug;7(3):28. doi:https://doi.org/10.3390/antib7030028.
- Wu C, Ying H, Grinnell C, Bryant S, Miller R, Clabbers A, Bose S, McCarthy D, Zhu -R-R, Santora L, et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotechnol. 2007 Nov;25(11):1290–97. doi:https://doi.org/10.1038/nbt1345.
- Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019 Aug;18(8):585–608.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs. 2017 Feb;9(2):182–212. doi:https://doi.org/10.1080/19420862.2016.1268307.
- Könning D, Zielonka S, Grzeschik J, Empting M, Valldorf B, Krah S, Schröter C, Sellmann C, Hock B, Kolmar H, et al. Camelid and shark single domain antibodies: structural features and therapeutic potential. Curr Opin Struct Biol. 2017 Aug;45:10–16. doi:https://doi.org/10.1016/j.sbi.2016.10.019.
- Zielonka S, Empting M, Grzeschik J, Könning D, Barelle CJ, Kolmar H. Structural insights and biomedical potential of IgNAR scaffolds from sharks. mAbs. 2015 Jan;7(1):15–25. doi:https://doi.org/10.4161/19420862.2015.989032.
- Li Z, Krippendorff B-F, Sharma S, Walz AC, Lavé T, Shah DK. Influence of molecular size on tissue distribution of antibody fragments. MAbs. 2016;8(1):113–19. doi:https://doi.org/10.1080/19420862.2015.1111497.
- Sellmann C, Pekar L, Bauer C, Ciesielski E, Krah S, Becker S, Toleikis L, Kügler J, Frenzel A, Valldorf B, et al. A one-step process for the construction of phage display scFv and VHH libraries. Mol Biotechnol. 2020 Jan;62(4):228–39. doi:https://doi.org/10.1007/s12033-020-00236-0.
- Uchański T, Zögg T, Yin J, Yuan D, Wohlkönig A, Fischer B, Rosenbaum DM, Kobilka BK, Pardon E, Steyaert J, et al. An improved yeast surface display platform for the screening of nanobody immune libraries. Sci Rep. 2019 Dec;9(1). doi:https://doi.org/10.1038/s41598-018-37212-3.
- Pardon E, Laeremans T, Triest S, Rasmussen SGF, Wohlkönig A, Ruf A, Muyldermans S, Hol WGJ, Kobilka BK, Steyaert J, et al. A general protocol for the generation of Nanobodies for structural biology. Nat Protoc. 2014 Mar;9(3):674–93. doi:https://doi.org/10.1038/nprot.2014.039.
- Krah S, Schröter C, Zielonka S, Empting M, Valldorf B, Kolmar H. Single-domain antibodies for biomedical applications. Immunopharmacol Immunotoxicol. 2016;38(1):21–28. doi:https://doi.org/10.3109/08923973.2015.1102934.
- Jovčevska I, Muyldermans S. The therapeutic potential of nanobodies. BioDrugs. 2020 Feb;34(1):11-26. doi: https://doi.org/10.1007/s40259-019-00392-z
- Duggan S. Caplacizumab: first Global Approval. Drugs. 2018 Oct;78(15):1639–42. doi:https://doi.org/10.1007/s40265-018-0989-0.
- Bannas P, Hambach J, Koch-Nolte F. Nanobodies and nanobody-based human heavy chain antibodies as antitumor therapeutics. Front Immunol. 2017 Nov;8. doi:https://doi.org/10.3389/fimmu.2017.01603.
- Chanier T, Chames P. Nanobody engineering: toward next generation immunotherapies and immunoimaging of cancer. Antibodies. 2019 Jan;8(1):13. doi:https://doi.org/10.3390/antib8010013.
- Rozan C, Cornillon A, Petiard C, Chartier M, Behar G, Boix C, Kerfelec B, Robert B, Pelegrin A, Chames P, et al. Single-domain antibody-based and linker-free bispecific antibodies targeting Fc RIII induce potent antitumor activity without recruiting regulatory T cells. Mol Cancer Ther. 2013 Aug;12(8):1481–91. doi:https://doi.org/10.1158/1535-7163.MCT-12-1012.
- Turini M, Chames P, Bruhns P, Baty D, Kerfelec B. A FcγRIII-engaging bispecific antibody expands the range of HER2-expressing breast tumors eligible to antibody therapy. Oncotarget. 2014 Jul;5(14):5304–19. doi:https://doi.org/10.18632/oncotarget.2093.
- Del Bano J, Florès-Florès R, Josselin E, Goubard A, Ganier L, Castellano R, Chames P, Baty D, Kerfelec B. A bispecific antibody-based approach for targeting mesothelin in triple negative breast cancer. Front Immunol. 2019 Jul;10. doi:https://doi.org/10.3389/fimmu.2019.01593.
- Scapin G, Yang X, Prosise WW, McCoy M, Reichert P, Johnston JM, Kashi RS, Strickland C. Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat Struct Mol Biol. 2015 Dec;22(12):953–58. doi:https://doi.org/10.1038/nsmb.3129.
- Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo K-M, Huston JS. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies†. Protein Eng Des Sel. 2010 Apr;23(4):195–202. doi:https://doi.org/10.1093/protein/gzp094.
- Schmitz KR, Bagchi A, Roovers RC, van Bergen En Henegouwen PMP, Ferguson KM. Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure. 2013 Jul;21(7):1214–24. doi:https://doi.org/10.1016/j.str.2013.05.008.
- Roth L, Krah S, Klemm J, Günther R, Toleikis L, Busch M, Becker S, Zielonka S. Isolation of antigen-specific VHH single-domain antibodies by combining animal immunization with yeast surface display. Methods Mol Biol. 2020;2070:173–89.
- Tustian AD, Endicott C, Adams B, Mattila J, Bak H. Development of purification processes for fully human bispecific antibodies based upon modification of protein A binding avidity. mAbs. 2016 May;8(4):828–38. doi:https://doi.org/10.1080/19420862.2016.1160192.
- Dheilly E, Moine V, Broyer L, Salgado-Pires S, Johnson Z, Papaioannou A, Cons L, Calloud S, Majocchi S, Nelson R. Selective Blockade of the Ubiquitous Checkpoint Receptor CD47 Is Enabled by Dual-Targeting Bispecific Antibodies. Mole Ther. 2017 Feb;25(2):523–33. doi:https://doi.org/10.1016/j.ymthe.2016.11.006.
- Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, Dietlein M, Kuhnert G, Kessler J, Buerkle C, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2015 Jun;125(26):4024–31. doi:https://doi.org/10.1182/blood-2014-12-614636.
- Sun LL, Ellerman D, Mathieu M, Hristopoulos M, Chen X, Li Y, Yan X, Clark R, Reyes A, Stefanich E, et al. Anti-CD20/CD3 T cell–dependent bispecific antibody for the treatment of B cell malignancies. Sci Transl Med. 2015 May;7(287):287ra70–287ra70. doi:https://doi.org/10.1126/scitranslmed.aaa4802.
- Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C, Bien-Ly N, Hersom M, Maloney JA, Meilandt WJ, et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med. 2014 Nov;6(261):261ra154–261ra154. doi:https://doi.org/10.1126/scitranslmed.3009835.
- Khan SN, Sok D, Tran K, Movsesyan A, Dubrovskaya V, Burton DR, Wyatt RT. Targeting the HIV-1 spike and coreceptor with Bi- and trispecific antibodies for single-component broad inhibition of entry. J Virol. 2018 Jul;92(18). doi:https://doi.org/10.1128/JVI.00384-18
- Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. 2009 Jan;284(5):3273–84. doi:https://doi.org/10.1074/jbc.M806889200.
- Scheuer W, Thomas M, Hanke P, Sam J, Osl F, Weininger D, Baehner M, Seeber S, Kettenberger H, Schanzer J. Anti-tumoral, anti-angiogenic and anti-metastatic efficacy of a tetravalent bispecific antibody (TAvi6) targeting VEGF-A and angiopoietin-2. mAbs. 2016 Apr;8(3):562–73. doi:https://doi.org/10.1080/19420862.2016.1147640.
- Croasdale R, Wartha K, Schanzer JM, Kuenkele K-P, Ries C, Mayer K, Gassner C, Wagner M, Dimoudis N, Herter S, et al. Development of tetravalent IgG1 dual targeting IGF-1R–EGFR antibodies with potent tumor inhibition. Arch Biochem Biophys. 2012 Oct;526(2):206–18. doi:https://doi.org/10.1016/j.abb.2012.03.016.
- Wu C, Ying H, Bose S, Miller R, Medina L, Santora L, Ghayur T. Molecular construction and optimization of anti-human IL-1α/β dual variable domain immunoglobulin (DVD-Ig TM) molecules. mAbs. 2009 Aug;1(4):339–47. doi:https://doi.org/10.4161/mabs.1.4.8755.
- Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, Trichard S, Cesari C, Sapet M, Bosco F. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell. 2019 Jun;177(7):1701–1713.e16. doi:https://doi.org/10.1016/j.cell.2019.04.041.
- Peipp M, Derer S, Lohse S, Staudinger M, Klausz K, Valerius T, Gramatzki M, Kellner C. HER2-specific immunoligands engaging NKp30 or NKp80 trigger NK-cell-mediated lysis of tumor cells and enhance antibody-dependent cell-mediated cytotoxicity. Oncotarget. 2015 Oct;6(31):32075–88. doi:https://doi.org/10.18632/oncotarget.5135.
- Satta A, Mezzanzanica D, Turatti F, Canevari S, Figini M. Redirection of T-cell effector functions for cancer therapy: bispecific antibodies and chimeric antigen receptors. Future Oncol. 2013 Apr;9(4):527–39. doi:https://doi.org/10.2217/fon.12.203.
- DiGiammarino EL, Harlan JE, Walter KA, Ladror US, Edalji RP, Hutchins CW, Lake MR, Greischar AJ, Liu J, Ghayur T. Ligand association rates to the inner-variable-domain of a dual-variable-domain immunoglobulin are significantly impacted by linker design. mAbs. 2011 Sep;3(5):487–94. doi:https://doi.org/10.4161/mabs.3.5.16326.
- Mayer K, Baumann A-L, Grote M, Seeber S, Kettenberger H, Breuer S, Killian T, Schäfer W, Brinkmann U. TriFabs—trivalent IgG-shaped bispecific antibody derivatives: design, generation, characterization and application for targeted payload delivery. Int J Mol Sci. 2015 Nov;16(11):27497–507. doi:https://doi.org/10.3390/ijms161126037.
- Metz S, Panke C, Haas AK, Schanzer J, Lau W, Croasdale R, Hoffmann E, Schneider B, Auer J, Gassner C. Bispecific antibody derivatives with restricted binding functionalities that are activated by proteolytic processing. Protein Eng Des Sel. 2012 Oct;25(10):571–80. doi:https://doi.org/10.1093/protein/gzs064.
- Stanfield RL, Zemla A, Wilson IA, Rupp B. Antibody elbow angles are influenced by their light chain class. J Mol Biol. 2006 Apr;357(5):1566–74. doi:https://doi.org/10.1016/j.jmb.2006.01.023.