
ABSTRACT
Antibodies mediate effector functions through Fcγ receptor (FcγR) interactions and complement activation, causing cytokine release, degranulation, phagocytosis, and cell death. They are often undesired for development of therapeutic antibodies where only antigen binding or neutralization would be ideal. Effector elimination has been successful with extensive mutagenesis, but these approaches can potentially lead to manufacturability and immunogenicity issues. By switching the native glycosylation site from position 297 to 298, we created alternative antibody glycosylation variants in the receptor interaction interface as a novel strategy to eliminate the effector functions. The engineered glycosylation site at Asn298 was confirmed by SDS-PAGE, mass spectrometry, and X-ray crystallography (PDB code 6X3I). The lead NNAS mutant (S298N/T299A/Y300S) shows no detectable binding to mouse or human FcγRs by surface plasmon resonance analyses. The effector functions of the mutant are completely eliminated when measured in antibody-dependent cell-meditated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) assays. In vivo, the NNAS mutant made on an antibody against a human lymphocyte antigen does not deplete T cells or B cells in transgenic mice, in contrast to wild-type antibody. Structural study confirms the successful glycosylation switch to the engineered Asn298 site. The engineered glycosylation would clash with approaching FcγRs based on reported Fc-FcγR co-crystal structures. In addition, the NNAS mutants of multiple antibodies retain binding to antigens and neonatal Fc receptor, exhibit comparable purification yields and thermal stability, and display normal circulation half-life in mice and non-human primate. Our work provides a novel approach for generating therapeutic antibodies devoid of any ADCC and CDC activities with potentially lower immunogenicity.
Introduction
Antibody therapeutics have been extensively developed for treating cancer and autoimmune diseases.Citation1 Antibodies elicit immune responses through effector functions via the Fc domain by interaction with Fcγ receptors (FcγRs) on immune cells, and activation of complement proteins in circulation.Citation2 This causes cytokine release and cell death by mechanisms including phagocytosis, antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). While often critical to the biological activitiy of therapeutic antibodies for cancer, effector functions may increase toxicity and side effects, and are often undesired for the development of antibody antagonists where neutralization or engagement without effector involvement would be ideal.Citation3,Citation4 Fcs with silenced effector function can also be incorporated into other antibody formats, such as chimeric antigen receptor (CAR) T-cell therapy, antibody-drug conjugates (ADCs), bispecific antibodies, and Fc-fusion proteins, for high therapeutic index toward many disease targets.Citation5–Citation8
Extensive studies have been carried out to map the amino acids and contact surface in the antibody Fc domain important for binding to FcγRs and complements.Citation9,Citation10 The oligosaccharides at the single N-linked glycosylation site at Asn297 were found to be critical for maintaining the structure to present the amino acids for the interaction, while providing direct contacts with the other residues.Citation11 Many engineered antibody variants have been generated based on these studies to either enhance or reduce antibody effector functions, depending on therapeutic applications. Approaches to eliminate effector function include isoforms that have lower intrinsic effector function (IgG4 or hybrid IgG2/IgG4 antibodies), aglycosylated antibodies, or variants with extensive mutations introduced at the Fc-FcγR interaction interface.Citation8,Citation12-Citation22 While those approaches have been proved successful in effector function reduction, there are potential issues associated with each approach. For example, IgG4 antibodies have been shown to form half-antibody species, which can promote formation of hybrid antibodies in circulation through Fab-arm exchanges, and to have reduced stability at low pH.Citation7,Citation23 Reduced stability was also observed with aglycosylated antibodies.Citation20 There are some residual effector functions associated with certain Fc mutations.Citation17,Citation18 Other concerns include immunogenicity from extensive mutagenesis and incomplete elimination of the effector function by these variants.Citation19 We took a completely different approach for the task, by engineering an alternative glycosylation site near Asn297 and the receptor interaction interface to block FcγR binding, and thus the effector function. Since the size of an oligosaccharide (about 2,000–3,000 Da) on a N-linked glycosylation site is much larger than the side chains of natural amino acids (less than 200 Da), we hypothesize that an engineered glycosylation site in the interaction interface would block IgG from binding to FcγRs and complements, thus eliminating the antibody effector function. Specifically, we performed site-directed mutagenesis to switch the glycosylation from inward-pointing Asn297 to an engineered Asn298 site on the antibody surface in several antibody backbones and characterized the variants in vitro and in vivo in animal models. We found that our novel mutations result in glycosylation switch with completely silenced ADCC and CDC.
Results
Glycosylation mutant design and preparation
We performed site-directed mutagenesis to introduce the consensus glycosylation sequence Asn-X-Ser/Thr near the native Asn297 site of IgG1 molecules, aiming to switch the glycosylation from Asn297 to engineered Asn298. IgG has a single N-linked glycosylation site at Asn297, followed by Ser298 and Thr299. Based on published crystal structures of IgG Fc and its complex with FcγRs (1e4k),Citation11 the oligosaccharides attached to Asn297 are located inside the CH2 domain and are partially buried. However, Ser298 is found to sit on a loop on the surface of the CH2 domain in the FcγR binding interface. The mutants with introduced glycosylation sites at Asn298, including NN298A299S3°° (NNAS, or S298N/T299A/Y300S), Q297N298TS3°° (QNTS, or N297Q/S298N/Y300S), and NN298TS3°° (NNTS, or S298N/Y300S), were prepared on a monoclonal antibody IgG1 (mAb1) molecule. Two aglycosylation mutants, N297Q and T299A, were generated as controls. All mutants were purified from media of transiently transfected Chinese hamster ovary (CHO) cells with yields similar to wild-type antibody (~100% for each mutant as compared to wild-type). They were analyzed by SDS-PAGE () and mass spectrometry (). Both tests confirm the successful creation of the alternative glycosylation site at Asn298 with the NNAS or QNTS mutant. As controls, substitution of either Asn297 or Thr299 resulted in an antibody with increased migration on gel electrophoresis, likely due to aglycosylation as expected. On the other hand, the NNAS and QNTS mutants migrated at the same size as wild-type IgG1, suggesting it is glycosylated at the engineered Asn298 site.
Table 1. Site-directed mutagenesis to introduce the consensus N-linked glycosylation sequence NXS/T around the native Asn297 site and LC-MS characterization of mAb1 Fc mutant antibodies.
Figure 1. Protein characterization of mAb1 wild-type and various glycosylation mutants. (a) SDS-PAGE analysis of the mutants under reducing condition (lane 1: protein molecular weight , or MW, standards; lane 2: wild-type mAb1; lane 3: N297Q; lane 4: N299A; lane 5: QNTS; lane 6: NNAS; lane 7: NNTS). Asterisk shows migration of the heavy chains of glycovariants under reducing condition. (b) Deconvoluted spectra from intact protein LC-MS analysis of NNAS. The antibody heavy chains with different N-glycans are shown.
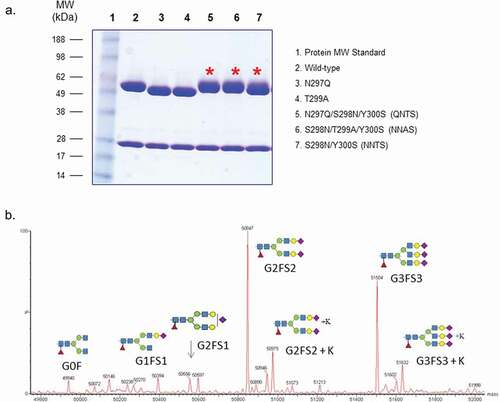
Detailed intact protein LC-MS characterization revealed that the oligosaccharides on the engineered Asn298 site are different from those on the native Asn297 site (). Instead of the typical biantennary G0F (GlcNAc2Man3GlcNAc2Fuc1) and G1F (Gal1GlcNAc2Man3GlcNAc2Fuc1) found on the Asn297 site in recombinant IgG1 molecules, the NNAS and QNTS mutants have both biantennary and triantennary oligosaccharides with terminal sialic acids. In contrast, the NNTS mutant contains both non-sialylated and sialylated biantennary structure, suggesting potential attachments of oligosaccharides at either Asn297 or Asn298 site. Aglycosylated species in these glycosylation mutants, including NNAS, QNTS, and NNTS, were not observed at any significant level. All of the antibodies, including wild-type and mutants, have low aggregation (<3.5% aggregates) and maintain similar antigen and neonatal Fc receptor (FcRn) binding (Figure S1). As expected with other aglycosylation mutants, N297Q shows 6.6°C decreased thermal transition temperature (Tm), while the glycosylation mutants display variably reduced thermal stability with a range from 0.7°C to 5.7°C lower than the wild-type antibody ().
Table 2. Characterization of various Fc mutants of mAb1.
In vitro characterization of FcγR binding by the mutants
Surface plasmon resonance (SPR) was used to test the binding of the engineered mutants to human and mouse FcγRs, including hFcγRII, hFcγRIII and mFcγRIV, which are important for triggering effector function. Human FcγRIIIa is expressed on innate immune cells such as macrophages and nature killer (NK) cells, and it is the most relevant receptors for ADCC of therapeutic antibodies. Both polymorphic forms of the receptor, hFcγRIIIa V158 and hFcγRIIIa F158, were used for characterization. The high-affinity hFcγRI was also tested to better differentiate the effect on binding by the mutants. Mouse FcγRs were included to correlate the binding results with lymphocyte depletion in mouse models.
The two glycosylation mutants NNAS and QNTS show no binding to any of the human or mouse FcγRs being tested, including the high-affinity hFcγRI (Figure S2). The two aglycosylation mutants, N297Q and T299A, exhibit some intermediate binding to hFcγRI and hFcγRII (T299A), but no detectable interaction with hFcγRIII. Moreover, there is little or no binding of glycosylation mutants to mouse FcγRs, including mFcγRI, mFcγRIIb, mFcγRIII, and mFcγRIV (Figure S3).
Based on results from FcγR bindings and other characterizations as described above, NNAS mutant was selected for further studies.
The NNAS mutant was also prepared in another recombinant human IgG1, mAb2, by transient transfection of both CHO cells and HEK293 cells. The N-linked oligosaccharides from these antibodies were analyzed (Figure S4). The mutant was produced together with a few other mutants with reduced effector function as reported in literature, including IgG4 PE (S228P/L235E), IgG1 LALA (L234A/L235A), and IgG1 N297A, another aglycosylation mutant.Citation16,Citation17 The FcγR binding of the NNAS and other mutants was determined using SPR ().
Figure 2. Comparison of FcγR binding of mAb2 wild-type, NNAS and other Fc mutants. (a) The interactions of wild-type IgG1, IgG4 PE, IgG1 LALA, IgG1 N297A, and IgG1 NNAS with human and mouse FcγRs were investigated using SPR. The sensorgrams show the binding profiles of IgG antibodies at various concentrations to different FcγRs. (b) The sensorgrams reveal the interactions of different subclasses of wild-type and NNAS mutants with human and mouse FcγRs. FcγRIIa (R131) and FcγRIIIa (V158) allotypes were used in the characterization.
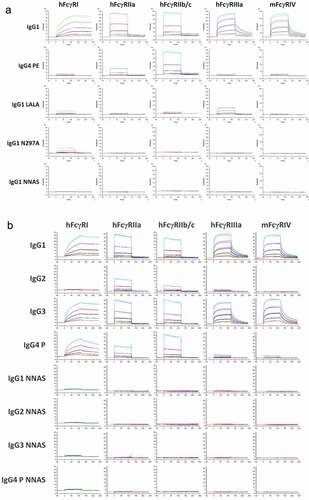
In contrast to wild-type antibody, which has strong binding, IgG1 NNAS shows no detectable binding to hFcγRI, although residual receptor interactions were observed with other mutants including IgG4 PE, IgG1 LALA, and IgG1 N297A. As for hFcγRII, no binding was detected with IgG1 NNAS or N297A, while IgG1 LALA demonstrates residual receptor interaction. Interestingly, there is significant binding of IgG4 PE to hFcγRII, especially to hFcγRIIb/c. Since hFcγRIIIa and mFcγRIV are responsible for ADCC activity in human and mouse, respectively, the receptor binding of wild-type and mutants was investigated. No interaction was detected with IgG1 NNAS or IgG1 N297A for these two receptors. Some residual bindings were observed with both IgG1 LALA and IgG4 PE, although the levels are minimal with the latter. The effects of NNAS mutation on FcγR interaction were also investigated with different IgG subclasses (). Both wild-type IgG1 and IgG3 show strong bindings to FcγRs, including hFcγRI, hFcγRIIa, hFcγRIIb/c, hFcγRIIIa, and mFcγRIV. However, as expected, there is no or very low binding of wild-type IgG2 and IgG4 P (S228P) to hFcγRIIIa and mFcγRIV, and wild-type IgG2 to hFcγRI. Significant residual bindings of wild-type IgG2 and IgG4 P were observed with hFcγRII, including FcγRIIa and FcγRIIb/c, while the latter antibody also has significant interaction with hFcγRI. The parent antibody IgG4, from which the S228P mutation was generated for reduced Fab-arm exchange, also shows similar interactions compared to the mutant. In contrast, there is no binding of NNAS mutant in any of IgG subclass to any of the human and mouse FcγRs tested.
In vitro ADCC and CDC activity
The IgG1 NNAS mutant mAb2 was tested for ADCC and CDC activities, two of the most important mechanisms for antibody effector functions, and compared to wild-type IgG1 and IgG1 LALA, a mutant with reduced effector function.Citation17
In the ADCC assay, antigen-expressing target cells were incubated with various amounts of antibodies and effector cells before the specific lysis was determined using radioactive chromium-release assay. As shown in , there is no or very low specific target cell lysis for either IgG1 NNAS or IgG1 LALA, although significant target cell lysis is present with the wild-type antibody. The result of no ADCC activity obtained with the NNAS mutant is consistent with its lack of FcγRIIIa binding as described above.
Figure 3. Comparison of the in vitro effector functions of mAb2 wild-type, NNAS and LALA mutants. (a) ADCC activities represent percent specific target cell lysis by effector cells in the presence of various concentrations of antibody mAb2. Monoclonal human cytotoxic T lymphocytes transduced with human FcγRIIIa (V158) was used as effector cells. (b) CDC activities show the percentage of specific target cell lysis by complement in the presence of various amounts of antibody mAb2. Data are presented as mean and standard deviation from triplicates.
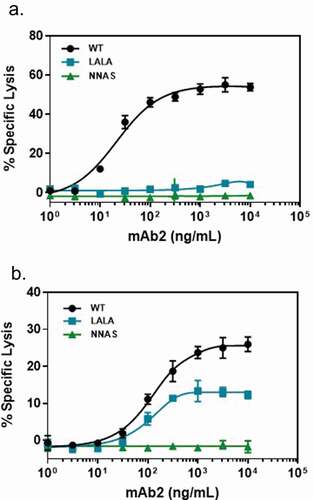
Specific target cell killing via complement activation was monitored after the cells were incubated with various amounts of wild-type or mutant antibodies as well as complements. In contrast to the wild-type antibody, which showed strong CDC activity, there is no specific target cell lysis with the IgG1 NNAS mutant at concentration up to 10 µg/ml after incubation with complement (). Significant amounts of CDC activity were observed with the IgG1 LALA mutant. In addition, strong interaction of the wild-type mAb2 with C1q was observed (Figure S5). In contrast, the NNAS mutant does not bind to this complement, suggesting that lack of CDC of the mutant is due to loss of C1q interaction.
The ADCC and CDC activities of mAb1 wild-type and NNAS mutant were also determined. Human NK cells and CD3-positive T cells expressing mAb1-reactive antigen purified from peripheral blood mononuclear cells (PBMCs) were used as effector cells and target cells, respectively. The result from radioactive chromium-release assay shows no ADCC of NNAS mutant, although the wild-type antibody shows significant activity (Figure S6a). In CDC assay, the wild-type antibody induces significant lysis of human Pfeiffer B cells as target cells, which express mAb1-reactive antigen, in an antibody dose-dependent manner (Figure S6b). In contrast, there is no target cell lysis by mAb1 NNAS mutant when human complements were used.
Lymphocyte depletion in transgenic mice
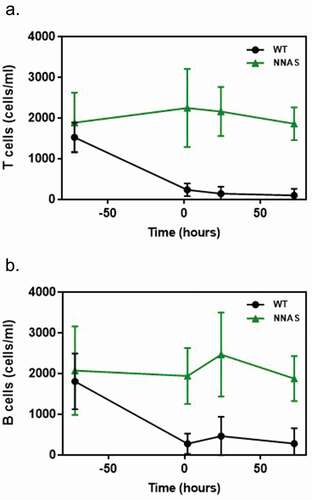
In order to examine the impact of the NNAS mutation on effector function in vivo, we tested lymphocyte depletion in the wild-type and the NNAS mutant mAb1 in transgenic mice expressing a mAb1-reactive human antigen, a classification determinant (CD; also called cluster of differentiation) on leukocytes.Citation24 A single dose of 1 mg/kg was intravenously (IV) administered to the transgenic animals, and whole blood samples were collected 2, 24, and 72 hours post-dose. Flow cytometry was used to determine the number of murine T and B cells in blood from the transgenic mice, monitoring lymphocyte depletion across time points. The wild-type antibody significantly depletes T and B cells () as early as 2 hours after injection, resulting in 94% and 85% of depletions, respectively, at 72 hours post-dose. In contrast, there is no depletion of these cells in animals treated with the NNAS mutant at any time points. The results indicate that the NNAS mutant has no in vivo effector function, as demonstrated by a lack of lymphocyte depletion in transgenic mice.
Figure 4. Comparison of the in vivo lymphocyte depletion of wild-type and NNAS mutant. (a) T cell depletion was determined in human antigen-specific transgenic mice (n = 10 per group) at different timepoints after injected with mAb1 wild-type and NNAS mutant. (b) B cell depletion was measured in human antigen-specific transgenic mice (n = 10 per group) at various timepoints after injected with mAb1 wild-type and NNAS mutant.
Crystal structure of the engineered Fc mutant
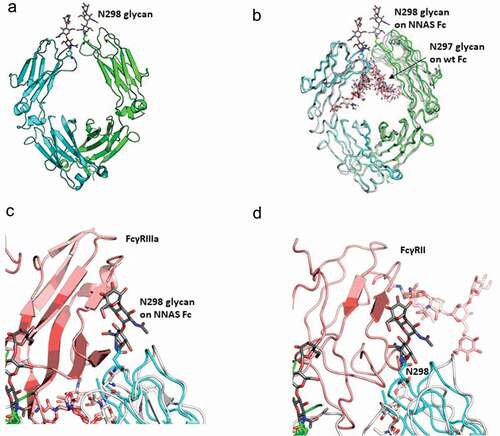
Recombinant antibody IgG1 Fc NNAS mutant was expressed in HEK293 cells in media containing kifunensine, a potent α-mannosidase I inhibitor, to produce an Fc with more homogeneous oligomannose-type glycans, which is better suited for crystallization. The NNAS mutant Fc was purified to homogeneity by a combination of protein A and size-exclusion chromatography. Crystal structures of the oligomannose containing NNAS mutant Fc were solved to a resolution of 2.3 angstroms (, Table S1). Electron densities for up to three sugars can be clearly observed on the engineered Asn298, but not on Asn297 in the NNAS mutant, confirming the successful switching of glycosylation from the inward-pointing Asn297 in the wild-type antibody to the solvent-exposed Asn298 in the NNAS mutant. The NNAS mutant Fc was crystallized with a single Fc chain in each asymmetric unit. Symmetry operation produced the Fc dimer in the classic “horseshoe” shape. We overlaid the PDBs of NNAS Fc with wild-type Fc (PDB: 1FC1, 3AVE), and afucosylated wild-type Fc (PDB: 2DTS) ().Citation25,Citation26 In general, both CH3 domains and one CH2 domain in the Fc dimer superpose very well. The second CH2 domain shows a shift as large as 3Ǻ between the corresponding β strands. The largest local conformational change occurs at the C′E loop where the NNAS mutation sits. The overall root-mean-square deviation (r.m.s.d) of the NNAS Fc dimer with the first-ever solved IgG1 Fc (PDB: 1FC1) was 1.4Ǻ, and 0.7Ǻ with more recently solved Fc structures of similar resolutions (PDB:3AVE and 2DTS).Citation25,Citation26 We superposed the NNAS Fc structure onto the co-crystal structures of the previously published Fc-FcγRIII (PDB: 1E4K, 3AY4) and Fc-FcγRII (PDB:3RY6) to examine how the glycosylation switch affects the binding.Citation11,Citation27,Citation28 The engineered glycosylation on NNAS sits right in the middle of the Fc-FcγR binding interface, with the glycans clashing into FcγRIII and FcγRII, respectively ( and ). Therefore, the NNAS Fc mutant can no longer interact with its receptors, and thus eliminating the downstream signaling cascade.
Figure 5. Crystal structure of NNAS Fc mutant. (a) Overall structure of NNAS Fc. The two Fc chains are colored in cyan and green. Visible glycans at N298 are shown in stick representation, with carbon atoms in gray. (b) Superposition of NNAS Fc with wildtype Fc (PDB 1FC1, 3AVE, and 2DTS.Citation25,Citation26 The carbon atoms in 1FC1, 3AVE, and 2DTS Fc are colored in different shades of gray. NNAS Fc is colored as in A. (c) Superposition of NNAS Fc with Fc-FcγRIII. For simplification, only PDB 3AY4Citation27 was shown, since PDB 1E4KCitation11 gave near identical results. NNAS is colored as in A. Wild-type Fc was colored in white while FcγRIIIα was colored in light pink. (d) Superposition of NNAS Fc with Fc-FcγRII (PDB:3RY6).Citation28 The color scheme is same as C. The glycans on N298 of the NNAS Fc all clash into the respective receptors. All oxygen atoms are colored in red and nitrogen atoms colored in dark blue. All figures are generated in Pymol.Citation29
Pharmacokinetics study in mice and non-human primates
The elimination half-lives of the NNAS mutants were evaluated in vivo for potential pharmacokinetic (PK) impact in circulation due to the mutations or oligosaccharides derived from the introduced glycosylation site. In a mouse PK study, the human mAb1 concentrations in serum were measured using an enzyme-linked immunosorbent assay (ELISA). After a single IV dose of 1 mg/kg, blood was collected from CD-1 mice at the following sampling times: 0.25, 4, 25, 48, 96, 168, and 336 hours. Results show that the NNAS mutant has a long circulation half-life in mice comparable to the wild-type antibody (). In addition, the clearances of the wild-type antibody and NNAS mutant were also compared in transgenic mice expressing the human antigen that is reactive to mAb1. The study was performed similarly as described above with CD-1 mice. There is no significant difference in elimination half-life between mAb1 wild-type and the NNAS mutant (Figure S7).
Figure 6. Comparison of pharmacokinetic profiles of wild-type and NNAS mutant. (a) PK profile in mouse (n = 10 per group) injected with 1 mg/kg of mAb1 wild-type and NNAS mutant. (b) PK profiles in cynomolgus monkeys (n = 3 per group) following administration of mAb2 wild-type and NNAS mutant. The wild-type and NNAS mutant of mAb1 and mAb2 were produced from CHO cells.
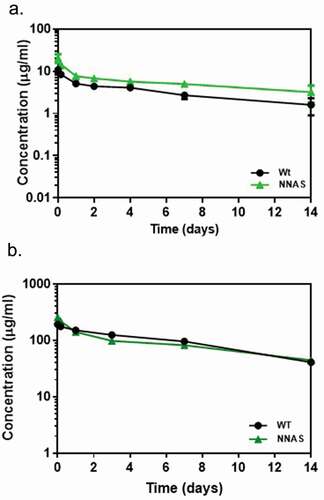
The PK profiles of mAb2 wild-type antibody and NNAS mutant were also compared in non-human primates (NHP). In the study, after a single IV dose of 10 mg/kg, blood was collected from cynomolgus monkeys at 0.08, 4, 24, 72, 168, and 336 hours post injection for quantification by bottom-up liquid chromatography-mass spectrometry (LC-MS/MS) assay method. The NNAS mutant shows comparable PK profiles to the wild-type antibody in NHP (). It has a similar half-life and clearance (Table S2). Our results suggest that the engineered glycosylation site at Asn298 does not have any effect on the PK of mAbs.
Discussion
Antibody effector functions trigger inflammatory signaling and cell death, which in turn can lead to side effects and toxicities for therapeutic antibodies. We report here design, preparation, and characterization of novel glycoengineered IgG1 variants to completely eliminate the effector functions. The lead NNAS mutant shows lack of binding to any human or mouse FcγRs being tested, including high-affinity hFcγRI, resulting in an absence of cell lysis in ADCC and CDC in vitro, and loss of lymphocyte depletion in vivo. The variant has a similar binding affinity to antigen and FcRn, as well as similar elimination half-life compared to wild-type antibodies in mouse and NHP. We have further established that the glycosylation switch can be applied to different IgG1 antibodies and IgG subclasses. There are multiple existing Fc variants and platforms that have been applied to therapeutic antibodies and Fc fusions for reducing effector functions. They include aglycosylated antibodies,Citation30,Citation31 modified IgG1, IgG2 and IgG4,Citation14,Citation17,Citation18,Citation21,Citation22 and IgGs with extensive mutagenesis.Citation14,Citation19 We believe that our glycosylation switched variants reported here are superior to those approaches by completely blocking the FcγR bindings, and thus silencing the effector functions.
Although several structures of Fc variants have been reported, the mechanisms of their reduced effector functions are not very clear except for some changes near the hinge region and the orientation of Pro329 in an IgG2 variant for FcγRs.Citation14,Citation32 As for aglycosylation mutants, early structural studies suggest the relevance of “closed” Fc conformation to reduced FcγR interaction. However, later work indicates the significant disorder in the C′E loop, which contains Asn297, has a strong correlation with reduced FcγRIIIa affinity.Citation33,Citation34 The high-resolution crystal structure of our NNAS mutant reveals unique mechanisms for completely silenced effector functions. First, the removal of glycosylation at Asn297 creates a structural disorder in the loop, as in the aforementioned aglycosylated antibodies. Second, the addition of an alternative glycosylation site to the neighboring engineered Asn298 residue results in changed orientation of oligosaccharides from facing inward between the two Fc chains to facing outwards, directly into the FcγR binding interface ().Citation11,Citation27,Citation28 The combination of the two mechanisms, especially the introduction of a bulky carbohydrate group in the binding interface, likely leads to the observed elimination of effector functions. The oligosaccharides at the Asn298 site are, on average, about 2000 to 3000 Da in mass as measured by mass spectrometry, at least 10 times greater than the average mass of an amino acid (less than 200 Da). This would completely block the interaction of the glycosylation switched variant with FcγRs and complements compared to that from a single or combination of amino acid mutations. Indeed, our results show that the NNAS mutant compared favorably in lowering binding to FcγRs than aglycosylation mutant N297Q and T299A.Citation35 While some of the existing Fc formats are presumed to have silenced effector functions, the commonly used aglycosylated IgG1 and the IgG4 with PFALA backbone still bind FcγRI and engage in phagocytosis of opsonized cells.Citation14 Indeed, we also observed some interactions of both aglycosylated IgG1 and IgG1 with LALA mutation to the FcγRI receptor, although with reduced affinity as expected.Citation30 Our glycosylation switched NNAS mutant shows no detectable binding under the same conditions, and thus is likely to be more effective in eliminating phagocytosis. Moreover, although significant CDC activity was observed with IgG1 LALA, there is no C1q binding and complement-mediated cytotoxicity with the NNAS mutant ().
Due to the fact that the NNAS mutant has the same antigen binding, FcRn interaction, and PK profile as the wild-type antibody, it is likely that undetectable FcγR binding as well as silenced ADCC and CDC activities of the glycosylation variant contribute to the lack of in vivo effector function or lymphocyte depletion in transgenic mice.
Removal of glycosylation from Asn297 has been a concern for reduced thermal stability and increased propensity to aggregation when it was tested at high temperatures in vitro.Citation36,Citation37 Our differential scanning calorimetry (DSC) study did show a 4.6°C decrease in melting temperature of the NNAS mutant, slightly less than 6.6°C decrease of aglycosylated N297Q. However, the structures of oligosaccharides at the engineered Asn298 site are different from those on the native Asn297 of IgG antibody. Although glycans derived from Asn297 are mainly biantennary G0F and G1F, the oligosaccharides at Asn298 are found to be predominantly biantennary and triantennary structures with terminal sialic acids. The oligosaccharides containing sialic acid residues with negative charge on the protein surface could also potentially reduce the surface hydrophobicity.
Our data also effectively addresses the question of whether carbohydrate receptors can significantly affect the clearance of this engineered glycosylation variant. Similar to the wild-type antibody, the engineered NNAS mutants have long circulation half-life in both mouse and NHP, likely due to the fact that the glycosylation variants maintain the same FcRn binding. Our results are consistent with the reports by others on antibodies with reduced effector function.Citation12,Citation20,Citation22 Bas et al. reported that Fc-sialylation also displays synergistically prolonged PK profile of antibody when it was combined with mutation for enhanced FcRn binding.Citation38 Therefore, our NNAS Fc could be a very useful fusion partner for half-life extension of wild-type antibodies and other Fc mutants, especially if combined with such Fc mutations.
The potential immunogenicity of many engineered Fc, including our NNAS mutant, is not completely clear. It is a valid concern, especially when extensive mutagenesis is involved to reduce the effector functions. The NNAS mutation could generate two new epitopes, the one exposed by removal of glycosylation at Asn297, and the other introduced with the new glycosylation site at Asn298. However, no adverse immunogenicity effects have been reported in the ongoing clinical trials of several aglycosylated antibodies, indicating that the unmasking of the region around N297 does not seem to result in the formation of a neoepitope.Citation39 The second potential new epitope could be from the new Asn298 glycosylation site with three amino acid substitutions at residues 298 (Ser to Asn), 299 (Thr to Ala), and 300 (Tyr to Ser). This is unlikely an immunogenicity concern for the following reasons. First, carbohydrates have been shown to be able to shield the underlying protein sequences from the immune system.Citation40 As a matter of fact, it is a common protein engineering practice to introduce a glycosylation site to hot spots of an immunogenic protein to reduce its immunogenicity.Citation41 Secondly, the terminal sialic acids on the oligosaccharides of the engineered Asn298 residue could be particularly beneficial for masking any possible new epitopes due to the fact that Fc sialylation is inversely correlated with autoimmunity.Citation42–Citation44 The sialylation of pathogenic antibodies in vivo has been reported to attenuate autoimmune disease in mouse models.Citation45 Thus, increased sialylation in the NNAS mutant would potentially benefit in immunosuppression.
We have previously reported site-specific antibody conjugation through glycans attached to Asn297 site.Citation46 The same strategy can be applied to the engineered Asn298 glycosylation as reported here. The potential advantage of the NNAS mutant over the wild-type IgGs could be its multi-antennary structures and open access to solvent and conjugation chemistries, leading to a higher drug-to-antibody ratio and more efficient conjugation than those with wild-type antibody.
In summary, the glycosylation switched NNAS mutant completely ablates antibody effector functions through a novel mechanism. This work provides a new platform that can be applied to therapeutic antibodies and many antibody formats, including Fc fusion proteins, ADCs, and bispecific antibodies, to improve safety profiles or to increase therapeutics index.
Materials and methods
Materials
The recombinant proteins, except for those made in-house, were obtained from R&D systems: recombinant human FcγRI (1257-Fc), recombinant human FcγRIIa (1330-CD), recombinant human FcγRIIb/c (1875-CD), recombinant human FcγRIIIa (4325-Fc), recombinant mouse FcγRI (2074-Fc), recombinant mouse FcγRIIb (1460-CD), recombinant mouse FcγRIII (1960-Fc), and recombinant mouse FcγRIV (1974-CD). All chemicals were purchased from Sigma-Aldrich unless stated otherwise.
Site-directed mutagenesis
Mutations were introduced by QuikChange II site-directed mutagenesis kit (Agilent) in pENTR vector encoding antibody constant domains CH1-3. Full-length antibody was created by LIC cloning in pENTR, which was subcloned into the expression vector pCEP4 (-E + I)Dest or pTT5. Mutations were confirmed by DNA sequencing.
Expression and purification of antibody mutants
HEK293 cells or CHO cells were transiently transfected with the plasmids containing antibody cDNAs. mAb1 wild-type and mutants were expressed from NRC CHO cells, while mAb2 wild-type and mutants were generated from both CHO and HEK293 cells. Conditioned media was harvested and filtered through a 0.22 µm filter. Antibodies were purified using Protein A columns (GE Healthcare) and were buffer-exchanged into phosphate-buffered saline (PBS) pH 7.2 using Amicon ultra centrifugal filters (EMD Millipore) before they were concentrated.
Fc NNAS mutant was also expressed and purified for crystallization. Its N-terminus starting from TCPPCP in the hinge region, was purified using Protein A and gel-filtration columns. Supernatant from Expi HEK293 cells treated with kifunensine was centrifuged and filtered through 0.22 µm filter before loading onto a 5 mL HiTrap Protein A column (GE Healthcare). The Protein A column was pre-equilibrated and run in PBS, and the protein was eluted with 0.1 M glycine pH 3.0. The eluate was fractionated and immediately mixed with an equal volume of 1 M HEPES pH 7.5 to neutralize. The fractions corresponding to the Fc mutant were pooled, concentrated, and loaded onto a 24 mL Superdex 200 10/300 GL column (GE Healthcare) in 20 mM HEPES pH 7.4 containing 50 mM sodium chloride. On gel-filtration, the Fc mutant gave a single symmetric UV absorbance peak.
SDS-PAGE
Antibody samples were heated for 10 min at 70°C in reducing sample buffer before they were applied to a 4–20% Tris-Glycine gel. After the run, the gel was stained with Coomassie blue. The gel image was acquired using a scanner.
Size exclusion chromatography
The size exclusion chromatography was performed on an Agilent 1100 HPLC system with a ChemStation software. Antibody samples (25 μg) were diluted in mobile phase (40 mM NaPO4 pH 6.0; 500 mM NaCl) and loaded onto a TSK Gel G3000SWXL column. The HPLC was performed under isocratic conditions in the mobile phase as described above at 0.5 mL/min for 35 min.
Differential scanning calorimetry
The samples were diluted to 0.5 mg/mL in PBS pH 7.2, centrifuged at 1,000 x g for 15 min to remove large particles and microscopic bubbles, and analyzed using a Microcal VP-capillary DSC system equipped with an autosampler. Sample and reference capillaries (containing matching buffer) were equilibrated at 10°C for 15 min and then heated to 110°C at a rate of 1°C/min. Data were acquired using passive feedback and an 8 sec filter. Raw scans were corrected for buffer/buffer scan signal and protein concentration, adjusted with a linear chemical baseline set visually from the pre- and post-transition baselines, and then qualitatively deconvoluted using the non-two state model contained in the analysis software package provided by the instrument manufacturer.
LC-MS intact protein analysis
Antibodies were partially reduced with 20 mM dithiothreitol for 30 min at 37°C at 0.5 mg/mL. The reaction was quenched with 0.1% trifluoroacetic acid. Samples were run on an Acquity/XevoG2 QToF UPLC/MS system using a Waters BEH C4 UPLC column (2.1 mm x 100 mm, 1.7 µm) held at 55°C with mobile phases A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile) within m/z range 500–3600 Da. Data were processed using Masslynx 4.1 software. The five most intense charge states in each spectrum were used for deconvolution using MaxEnt 1 algorithm (resolution:1.0 Da; model: uniform Gaussian 0.5 Da; minimum intensity ratios: 33%, iterate to convergence) in the ranges LC at 20–25 kDa and HC at 40–60 kDa.
FcγR, FcRn, and antigen-binding using surface plasmon resonance
Analysis of antibody binding to FcγRs was performed on a Biacore T200 using anti-His capture or Biacore 3000 using protein A/G capture. Anti-tetra His (Qiagen) was immobilized to a series S CM5 chip to a surface density of 10,000 RU using the amine coupling kit provided by GE Healthcare. Recombinant human and mouse FcγRs were diluted to 0.5 μg/mL in HBS-EP+ (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM ethylenediaminetetraacetic acid or EDTA, 0.05% surfactant P20) and injected for 30 sec at 10 μL/min flowrate. Antibodies were serially diluted threefold from 3000 to 37 nM and injected over the captured receptors for 2 min in duplicate. For binding to hFcγRI, serial dilutions were made from 300 nM to 1.2 nM. The surface was regenerated with 10 mM glycine pH 1.5 for 30 sec.
For results in supplemental figures, each antibody was diluted to 5 μg/mL and injected for 30 sec onto a CM5 chip immobilized with 3500 RU of protein A/G (Sigma). Recombinant human and mouse FcγRs in HBS-EP (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% Tween 20) were injected over the captured antibodies. Receptor binding responses were normalized to antibody capture level by dividing the binding response by a capture level report point and multiplied by 100.
For FcRn binding, a CM5 chip was immobilized with 715 RU mouse FcRn (prepared in-house) using amine chemistry. Each antibody was diluted to 200 nM with PBS-P pH 6.0 (50 mM sodium phosphate pH 6.0, 150 mM NaCl, 0.05% Tween 20) and injected over the immobilized FcRn at 30 μL/min flowrate. The surface was regenerated with 10 mM sodium tetraborate, 1 M NaCl pH 8.5 for 30 sec at 50 μL/min flowrate.
To measure antigen binding, a low level of human antigen was immobilized on a CM5 chip. Antibodies were diluted in HBS-EP running buffer to 90 nM for relative binding comparison. The diluted antibodies were injected for 3 min over the surface to monitor binding at 30 μL/min flowrate.
C1q binding
The C1q binding was determined using ELISA according to a report by Schlothauer et al. with minor modification.Citation18 Briefly, antibodies in eight dilutions (0 to 50ug/mL) in triplicates were immobilized in a 96-well plate overnight before the plate was blocked with SuperBlock blocking buffer in PBS (Thermo Fisher Scientific) containing 0.05% Tween-20. Human C1q (Sigma, C1740) was added to each well and incubated for 1.5 hours at room temperature. After washing, the wells were then incubated with mouse anti-human C1q (Thermo Fisher Scientific, MA183963) for 1 hours. Finally, goat anti-mouse IgG conjugated with horseradish peroxidase (HRP) (Jackson ImmunoResearch,115–035-164) was incubated for 1 hoursbefore TMB substrate was added. The absorbance at 405 nm, which is proportional to the amount of bound C1q, was determined using a plate reader with BD-SpectraMax software.
ADCC assay
ADCC potency of mAb2 wild-type and mutants was evaluated using chromium (51Cr) release assay with the ratio of effector cells to target cells at 10 to 1. The target cells were generated by transfecting CHO cells with the gene encoding a membrane-bound antigen.Citation47 The cells were grown in Ham’s F12 medium containing 10% heat-inactivated fetal bovine serum and 1 mg/mL G418. The effector cells were monoclonal human cytotoxic T lymphocytes transduced with human FcγRIIIa (V158) as described by Clemenceau et al.Citation48 The cytotoxic T cell clone was cultured in RPMI medium containing 5% fetal calf serum. In the assay, 51Cr labeled target cells expressing specific antigen were mixed with both antibody (mAb2) and effector cells. After being incubated for 4 hoursat 37°C, the supernatants were transferred into LumaPlates. The radioactivity, corrected count per minute (CCPM), was measured for gamma counting in triplicates using MicroBeta Jet (Perkin Elmer). The percent specific lysis was calculated using the formula (experimental CCPM − spontaneous CCPM)/(maximum CCPM − spontaneous CCPM) × 100; where maximum CCPM was the radioactivity from the labeled target cells treated with 0.75% Triton X-100 and spontaneous CCPM represents the one from labeled target cells alone without antibody and effector cells.
The ADCC of mAb1 wild-type antibody and NNAS mutant was performed similarly as described for mAb2 except that NK cells purified from human PBMCs, which were isolated using Ficoll-Hypaque-gradient centrifugation from whole blood (Red Cross), were used as effector cells and CD3-positive T cells expressing mAb1-reactive antigen as target cells. After incubation for 5 hours, radioactivity released from 51Cr labeled target cells was determined.
CDC assay
The CDC assay of mAb2 wild-type and mutants was performed similarly except that a fixed quantity of complements was used instead of effector cells. In the assay, 51Cr labeled target cells were incubated with various amount of antibody and 1 CH50 guinea pig complement (Tebubio) for 4 hoursat 37°C before the radioactivity was determined as described above. Percentage of specific lysis was calculated by the formula: (experimental CCPM – spontaneous CCPM)/(maximum CCPM – spontaneous CCPM) x 100. Data given are the means of triplicate determinations. The spontaneous CCPM represents the one from labeled target cells alone without antibody and complement.
The CDC activity of mAb1 wild-type and NNAS mutant was determined using protocol exactly as described by Qiu et al.Citation49 The normal human serum complement (Quidel Corporation, A112) was used in the assay, while Pfeiffer B cells expressing mAb1-reactive antigen (ATCC, CRL-2632) was used as target cells.
Animal experiments
All in vivo studies were conducted in compliance with the Sanofi institutional animal care policy and in accordance with the Helsinki Declaration of 1975. The monkey studies were approved by the French “Ministère de l’Enseignement Superieur et de la Recherche.” The mouse studies were reviewed and approved by the Institutional Animal Use and Care (IACUC) committee at Sanofi. Receipt, acclimation, room temperature, relative humidity, and light/dark cycle were in accordance with Sanofi’s Comparative Medicine standards. The mice were group-housed in rodent cages. Water and rodent diet (PicoLab® Rodent Diet 20) were provided ad libitum throughout the course of the study.
In vivo lymphocyte depletion
The study utilized mAb1 reactive human antigen-expressing transgenic mice. The mice were obtained from Charles River Labs. Heterozygous human antigen expressing transgenic mice were generated by breeding homozygous (+/+) transgenic female mice to wild type (CD-1) male mice. Mice were 8–14 weeks of age at study initiation. Animals (10 per group) were administered a single dose of vehicle (PBS, Thermo Fisher Sci) as well as mAb1 wild-type antibody and NNAS mutant at 1 mg/kg via the lateral tail vein. Doses were based on the animal’s most recent bodyweight.
After injection, serial blood samples in the volume of 100 µL were collected from the retro-orbital plexus of anesthetized transgenic mice at baseline (−3 days), 2, 24, and 72 hours post dose. All samples were collected into tubes containing EDTA and processed the same day.
The lymphocyte depletion was measured using flow cytometric analysis. Cells were incubated with anti-CD16/32 (Fc-block 2.4G2, BD Biosciences) followed by staining with a combination of anti-CD45R/B220 PE Cy7 (RA3-6B2, BD Bioscience) and anti-CD3ε APC (145–2C11, Biolegend) for 10 min at room temperature. Erythrocytes were lysed with BD FACS Lysing Solution (BD Biosciences) and fixed with 0.5% formaldehyde. Prior to acquisition, CountBright™ Absolute Counting Beads (Life Technologies) were added to the samples for cell concentration calculations. Data were collected on a Becton Dickinson FACSCanto II with Diva 6.1.3 and equipped with 488, 405, and 633 nm lasers, and analyzed with FlowJo software (TreeStar).
Pharmacokinetics studies
For the PK study using mouse, animals (ten per group) were administered a single dose of antibody mAb1 at 1 mg/kg via the lateral tail vein. Serial blood samples in the volume of 60 µL were collected from each human antigen-specific transgenic mouse or CD-1 mouse at 0.25, 4, 24, 48, 96, 168, and 336 hours post dose. All samples were collected into micro-hematocrit capillary tubes and spun in a centrifuge at 10,000 rpm for 5 min. The serum was then separated into an eppendorf tube, transported on dry ice, and stored at ≤ −60°C until processed. At the end of the study, all surviving animals were euthanized by CO2 asphyxiation.
The amounts of antibody in mouse serum were determined using ELISA. Ninety-six-well plates were coated with goat anti-human IgG primary antibody (Jackson ImmunoResearch, 109–005-003) in 50 mM carbonate/bicarbonate buffer pH 9.6 and incubated for at least 12 hours at 2–8°C. The plates were washed and blocked with 1% bovine serum albumin (BSA) in PBS for at least 1 hourat 37°C while shaking. Primary antibody incubations were done with either samples (diluted 1:25 to 1:1000) or serial dilutions of the appropriate standard (in PBS containing 0.05% Tween 20 and 0.1% BSA). Detection was carried out with the relevant secondary antibody, goat anti-human IgG biotin conjugated (Sigma-Aldrich, AP112B) after incubation for 1 hourat 37°C with shaking, followed by incubation with HRP-conjugated Streptavidin (KPL, 5950–0004) for 30 minutes, and then with tetramethylbenzidine substrate (KPL, Sure-Blue, 5120–0075). Optical density was measured at 450 nm using a plate reader (Molecular Devices).
For PK studies conducted in cynomolgus monkeys, all animals obtained from Noveprim were male monkeys aged between 1 and 3 years of age at study start, weighing between 3 kg and 5 kg. All antibodies were prepared in 10 mM histidine pH 6.0 and 150 mM NaCl, and dosed at 10 mg/kg as a single intravenous administration with a dose at 1.5 mL/kg, to saturate target binding. A total of three animal replicates were evaluated for each antibody utilizing a serial sampling approach across the study duration of 14 days. Blood samples (0.5 mL) were collected into the tubes containing potassium EDTA as anticoagulant by venipuncture of the saphenous vein at six sampling times post dosing. Once collected, blood samples were centrifuged at 4°C for 10 minutes at 1500 x g and stored at −80°C.
Plasma concentrations of antibodies were determined using a generic LC-MS/MS approach. The concentration of each antibody at each time point was determined by a bottom-up LC-MS/MS assay. After precipitation of a plasma aliquot, the plasma pellet was subjected to protein denaturation, reduction, alkylation, trypsin digestion, and solid-phase extraction prior to analysis of surrogate peptides. Calibration standards were prepared by spiking the antibody into the plasma at 1, 2, 5, 10, 20, 50, 100, 200, and 400 µg/mL. Peptide separation was performed on a Waters Acquity UPLC system with a reverse phase XBridge BEH C18 column (2.1x150 mm, 3.5 µM, 300 Å, Waters) at a flow rate of 300 µL/min in a stepwise gradient of 0.1% formic acid in water and 0.1% formic acid in acetonitrile. For detection, a SCIEX API5500 mass spectrometer was used in positive ion mode, with the source temperature at 700°C, the ionspray voltage at 5500 V, curtain, and nebulizer gases at 40, and the collision gas at mid. Dwell times were 20 ms and the entrance potential was 10 V for each transition. The multiple reaction-monitoring transitions for two unique surrogate peptides of the antibody were used for concentration determination relative to the standards and controls, using the peak area from the MQIII integration algorithm of the Analyst software.
PK parameters were determined from individual animal data using non-compartmental analysis in Phoenix WinNonlin version 6.4 (Certara).
NNAS Fc mutant crystallization, data collection, and structure determination
Purified NNAS Fc was concentrated to 12 mg/mL and set up for sparse matrix crystal screening at 21°C. Crystal clusters were observed from well condition 0.1 M HEPES pH 7.4% and 16% polyethylene glycol 20,000. After microseeding and optimization, single crystals were obtained suitable for diffraction data collection under 30% polyethylene glycol 1,500. Crystals were cryo frozen in corresponding well conditions plus 20% ethylene glycol, and diffraction data sets were collected at CMCF-08ID at Canadian light source. Crystals diffracted to 2.27 angstrom, and data were processed with HKL2000.Citation50 Crystals belonged to space group C2221, with cell dimensions a = 52.1 Å, b = 142.5 Å, c = 75.2 Å, and α = β = γ = 90.0°.
The NNAS Fc structure was determined by molecular replacement with PHASERCitation51 as implemented in the Phenix software packageCitation52 using chain A of PDB 3AVECitation26 as initial search model. The model was refined in Phenix.refine program using default protocols.Citation52 Manual modification to the model between refinement cycles was made with the program Coot.Citation53 Statistics of diffraction data and refinement are summarized in Table S1. The structure factors and coordinates have been deposited into RCSB with PDB code 6X3I.
Abbreviations
| ADCC | = | Antibody-dependent cell-meditated cytotoxicity |
| CDC | = | Complement-dependent cytotoxicity |
| SPR | = | Surface plasmon resonance |
| NHP | = | Non-human primate |
| DSC | = | Differential scanning calorimetry |
| SEC | = | Size exclusion chromatography |
| HPLC | = | High-performance liquid chromatography |
| r.m.s.d | = | Root-mean-square deviation |
| CCPM | = | Corrected count per minute |
| PBMCs | = | Peripheral blood mononuclear cells |
| NK | = | Natural killer |
| HRP | = | Horseradish peroxidase |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download Zip (290.6 KB)Acknowledgments
The authors would like to thank Maria Wendt, William Brondyk, Xiaoying Jin, Tracey McSherry, Mostafa Kabiri, Pascale Vicat, Robert Cost, Jose Sancho, Laura Geagan, Paula Boutin, Ben Greene, Kimberly Bishop, Helene Simonds-Mannes, Brian Mackness for support, suggestions and help in preparing the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to watch in 2020. MAbs. 2020;12:1703531.
- Bournazos S, Ravetch JV. Diversification of IgG effector functions. Int Immunol. 2017;29:303–13. doi:https://doi.org/10.1093/intimm/dxx025.
- Goulet DR, Atkins WM. Considerations for the design of antibody-based therapeutics. J Pharm Sci. 2020;109:74–103. doi:https://doi.org/10.1016/j.xphs.2019.05.031.
- Stein MM, Hrusch CL, Sperling AI, Ober C. Effects of an FcgammaRIIA polymorphism on leukocyte gene expression and cytokine responses to anti-CD3 and anti-CD28 antibodies. Genes Immun. 2019;20:462–72. doi:https://doi.org/10.1038/s41435-018-0038-8.
- Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, Chang W-C, Bretzlaff W, Starr R, Priceman S, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. 2015;23:757–68. doi:https://doi.org/10.1038/mt.2014.208.
- Gillies SD, Lan Y, Lo KM, Super M, Wesolowski J. Improving the efficacy of antibody-interleukin 2 fusion proteins by reducing their interaction with Fc receptors. Cancer Res. 1999;59:2159–66.
- Herbener P, Schönfeld K, König M, Germer M, Przyborski JM, Bernöster K, Schüttrumpf J. Functional relevance of in vivo half antibody exchange of an IgG4 therapeutic antibody-drug conjugate. PLoS One. 2018;13:e0195823. doi:https://doi.org/10.1371/journal.pone.0195823.
- Escobar-Cabrera E, Lario P, Baardsnes J, Schrag J, Durocher Y, Dixit S. Asymmetric Fc engineering for bispecific antibodies with reduced effector function. Antibodies. 2017;6:7.
- Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front Immunol. 2019;10:1296. doi:https://doi.org/10.3389/fimmu.2019.01296.
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276:6591–604. doi:https://doi.org/10.1074/jbc.M009483200.
- Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature. 2000;406:267–73. doi:https://doi.org/10.1038/35018508.
- An Z, Forrest G, Moore R, Cukan M, Haytko P, Huang L, Vitelli S, Zhao JZ, Lu P, Hua J, et al. IgG2m4, an engineered antibody isotype with reduced Fc function. mAbs. 2009;1:572–79. doi:https://doi.org/10.4161/mabs.1.6.10185.
- Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol. 1989;143:2595–601.
- Vafa O, Gilliland GL, Brezski RJ, Strake B, Wilkinson T, Lacy ER, Scallon B, Teplyakov A, Malia TJ, Strohl WR, et al. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods. 2014;65:114–26. doi:https://doi.org/10.1016/j.ymeth.2013.06.035.
- Borrok MJ, Mody N, Lu X, Kuhn ML, Wu H, Dall’Acqua WF, Tsui P. An “Fc-silenced” IgG1 format with extended half-life designed for improved stability. J Pharm Sci. 2017;106:1008–17. doi:https://doi.org/10.1016/j.xphs.2016.12.023.
- Newman R, Hariharan K, Reff M, Anderson DR, Braslawsky G, Santoro D, Hanna N, Bugelski PJ, Brigham-Burke M, Crysler C, et al. Modification of the Fc region of a primatized IgG antibody to human CD4 retains its ability to modulate CD4 receptors but does not deplete CD4(+) T cells in chimpanzees. Clin Immunol. 2001;98:164–74. doi:https://doi.org/10.1006/clim.2000.4975.
- Xu D, Alegre M-L, Varga SS, Rothermel AL, Collins AM, Pulito VL, Hanna LS, Dolan KP, Parren PWHI, Bluestone JA, et al. In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell Immunol. 2000;200:16–26. doi:https://doi.org/10.1006/cimm.2000.1617.
- Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E, et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. PEDS. 2016;29:457–66. doi:https://doi.org/10.1093/protein/gzw040.
- Armour KL, Clark MR, Hadley AG, Williamson LM. Recombinant human IgG molecules lacking Fcgamma receptor I binding and monocyte triggering activities. Eur J Immunol. 1999;29:2613–24. doi:https://doi.org/10.1002/(SICI)1521-4141(199908)29:08<2613::AID-IMMU2613>3.0.CO;2-J.
- Hristodorov D, Fischer R, Joerissen H, Müller-Tiemann B, Apeler H, Linden L. Generation and comparative characterization of glycosylated and aglycosylated human IgG1 antibodies. Mol Biotechnol. 2013;53:326–35. doi:https://doi.org/10.1007/s12033-012-9531-x.
- Reddy MP, Kinney CAS, Chaikin MA, Payne A, Fishman-Lobell J, Tsui P, Dal Monte PR, Doyle ML, Brigham-Burke MR, Anderson D, et al. Elimination of Fc receptor-dependent effector functions of a modified IgG4 monoclonal antibody to human CD4. J Immunol. 2000;164:1925–33. doi:https://doi.org/10.4049/jimmunol.164.4.1925.
- Tam SH,McCarthy SG, Armstrong AA, Somani S, Wu S-J, Liu X, Gervais A, Ernst R, Saro D, Decker R et al. Functional, biophysical, and structural characterization of human IgG1 and IgG4 Fc variants with ablated immune functionality. Antibodies. 2017;6:12.
- Dumet C, Pottier J, Gouilleux-Gruart V, Watier H. Insights into the IgG heavy chain engineering patent landscape as applied to IgG4 antibody development. mAbs. 2019;11:1341–50. doi:https://doi.org/10.1080/19420862.2019.1664365.
- Zalevsky J, Leung IWL, Karki S, Chu SY, Zhukovsky EA, Desjarlais JR, Carmichael DF, Lawrence CE. The impact of Fc engineering on an anti-CD19 antibody: increased Fcgamma receptor affinity enhances B-cell clearing in nonhuman primates. Blood. 2009;113:3735–43. doi:https://doi.org/10.1182/blood-2008-10-182048.
- Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry. 1981;20:2361–70. doi:https://doi.org/10.1021/bi00512a001.
- Matsumiya S, Yamaguchi Y, Saito J-I, Nagano M, Sasakawa H, Otaki S, Satoh M, Shitara K, Kato K. Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J Mol Biol. 2007;368:767–79. doi:https://doi.org/10.1016/j.jmb.2007.02.034.
- Mizushima T, Yagi H, Takemoto E, Shibata-Koyama M, Isoda Y, Iida S, Masuda K, Satoh M, Kato K. Structural basis for improved efficacy of therapeutic antibodies on defucosylation of their Fc glycans. Genes Cells. 2011;16:1071–80. doi:https://doi.org/10.1111/j.1365-2443.2011.01552.x.
- Ramsland PA, Farrugia W, Bradford TM, Sardjono CT, Esparon S, Trist HM, Powell MS, Tan PS, Cendron AC, Wines BD, et al. Structural basis for Fc gammaRIIa recognition of human IgG and formation of inflammatory signaling complexes. J Immunol. 2011;187:3208–17. doi:https://doi.org/10.4049/jimmunol.1101467.
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.3r1; 2010.
- Jung ST, Kang TH, Kelton W, Georgiou G. Bypassing glycosylation: engineering aglycosylated full-length IgG antibodies for human therapy. Curr Opin Biotechnol. 2011;22:858–67. doi:https://doi.org/10.1016/j.copbio.2011.03.002.
- Jacobsen FW, Stevenson R, Li C, Salimi-Moosavi H, Liu L, Wen J, Luo Q, Daris K, Buck L, Miller S, et al. Engineering an IgG Scaffold lacking effector function with optimized developability. J Biol Chem. 2017;292:1865–75. doi:https://doi.org/10.1074/jbc.M116.748525.
- Oganesyan V, Gao C, Shirinian L, Wu H, Dall’Acqua WF. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallogr D Biol Crystallogr. 2008;64:700–04. doi:https://doi.org/10.1107/S0907444908007877.
- Borrok MJ, Jung ST, Kang TH, Monzingo AF, Georgiou G. Revisiting the role of glycosylation in the structure of human IgG Fc. ACS Chem Biol. 2012;7:1596–602. doi:https://doi.org/10.1021/cb300130k.
- Subedi GP, Barb AW. The structural role of antibody N-glycosylation in receptor interactions. Structure. 2015;23:1573–83. doi:https://doi.org/10.1016/j.str.2015.06.015.
- Sazinsky SL, Ott RG, Silver NW, Tidor B, Ravetch JV, Wittrup KD. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc Natl Acad Sci USA. 2008;105:20167–72. doi:https://doi.org/10.1073/pnas.0809257105.
- Kayser V, Chennamsetty N, Voynov V, Forrer K, Helk B, Trout BL. Glycosylation influences on the aggregation propensity of therapeutic monoclonal antibodies. Biotechnol J. 2011;6:38–44. doi:https://doi.org/10.1002/biot.201000091.
- Zhou Q, Qiu H. The mechanistic impact of N-glycosylation on stability, pharmacokinetics, and immunogenicity of therapeutic proteins. J Pharm Sci. 2019;108:1366–77. doi:https://doi.org/10.1016/j.xphs.2018.11.029.
- Bas M, Terrier A, Jacque E, Dehenne A, Pochet-Béghin V, Beghin C, Dezetter A-S, Dupont G, Engrand A, Beaufils B, et al. Fc Sialylation prolongs serum half-life of therapeutic antibodies. J Immunol. 2019;202:1582–94. doi:https://doi.org/10.4049/jimmunol.1800896.
- Ju MS, Jung ST. Aglycosylated full-length IgG antibodies: steps toward next-generation immunotherapeutics. Curr Opin Biotechnol. 2014;30:128–39. doi:https://doi.org/10.1016/j.copbio.2014.06.013.
- Sinclair AM, Elliott S. Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. J Pharm Sci. 2005;94:1626–35. doi:https://doi.org/10.1002/jps.20319.
- Tatsumi Y, Sasahara Y, Kohyama N, Ayano S, Endo M, Yoshida T, Yamada K, Totsuka M, Hattori M. Introducing site-specific glycosylation using protein engineering techniques reduces the immunogenicity of beta-lactoglobulin. Biosci Biotechnol Biochem. 2012;76:478–85. doi:https://doi.org/10.1271/bbb.110753.
- Ohmi Y, Ise W, Harazono A, Takakura D, Fukuyama H, Baba Y, Narazaki M, Shoda H, Takahashi N, Ohkawa Y, et al. Sialylation converts arthritogenic IgG into inhibitors of collagen-induced arthritis. Nat Commun. 2016;7:11205. doi:https://doi.org/10.1038/ncomms11205.
- Mimura Y, Kelly RM, Unwin L, Albrecht S, Jefferis R, Goodall M, Mizukami Y, Mimura-Kimura Y, Matsumoto T, Ueoka H, et al. Enhanced sialylation of a human chimeric IgG1 variant produced in human and rodent cell lines. J Immunol Methods. 2016;428:30–36. doi:https://doi.org/10.1016/j.jim.2015.11.009.
- Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science. 2008;320:373–76. doi:https://doi.org/10.1126/science.1154315.
- Pagan JD, Kitaoka M, Anthony RM. Engineered Sialylation of pathogenic antibodies in vivo attenuates autoimmune disease. Cell. 2018;172(564–577):e513. doi:https://doi.org/10.1016/j.cell.2017.11.041.
- Zhou Q, Stefano JE, Manning C, Kyazike J, Chen B, Gianolio DA, Park A, Busch M, Bird J, Zheng X, et al. Site-specific antibody-drug conjugation through glycoengineering. Bioconjug Chem. 2014;25:510–20. doi:https://doi.org/10.1021/bc400505q.
- Lallemand C, Liang F, Staub F, Simansour M, Vallette B, Huang L, Ferrando-Miguel R, Tovey MG. A novel system for the quantification of the ADCC activity of therapeutic antibodies. J Immunol Res. 2017;2017:3908289. doi:https://doi.org/10.1155/2017/3908289.
- Clémenceau B, Congy-Jolivet N, Gallot G, Vivien R, Gaschet J, Thibault G, Vié H. Antibody-dependent cellular cytotoxicity (ADCC) is mediated by genetically modified antigen-specific human T lymphocytes. Blood. 2006;107:4669–77. doi:https://doi.org/10.1182/blood-2005-09-3775.
- Qiu H, Wei R, Jaworski J, Boudanova E, Hughes H, VanPatten S, Lund A, Day J, Zhou Y, McSherry T, et al. Engineering an anti-CD52 antibody for enhanced deamidation stability. mAbs. 2019;11:1266–75. doi:https://doi.org/10.1080/19420862.2019.1631117.
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26.
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–74. doi:https://doi.org/10.1107/S0021889807021206.
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi:https://doi.org/10.1107/S0907444909052925.
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi:https://doi.org/10.1107/S0907444904019158.