
ABSTRACT
A major asset of many monoclonal antibody (mAb)-based biologics is their persistence in circulation. The MHC class I family Fc receptor, FCGRT, is primarily responsible for this extended pharmacokinetic behavior. Engagement of FCGRT with the crystallizable fragment (Fc) domain protects IgG from catabolic elimination, thereby extending the persistence and bioavailability of IgG and related Fc-based biologics. There is a need for reliable in vivo models to facilitate the preclinical development of novel IgG-based biologics. FcRn-humanized mice have been widely accepted as translationally relevant surrogates for IgG-based biologics evaluations. Although such FCGRT-humanized mice, especially the mouse strain, B6.Cg-Fcgrttm1Dcr Tg(FCGRT)32Dcr (abbreviated Tg32), have been substantially validated for modeling humanized IgG-based biologics, there is a recognized caveat – they lack an endogenous source of human IgG that typifies the human competitive condition. Here, we used CRISPR/Cas9-mediated homology-directed repair to equip the hFCGRT Tg32 strain with a human IGHG1 Fc domain. This replacement now results in mice that produce human IgG1 Fc-mouse IgG Fab2 chimeric antibodies at physiologically relevant levels, which can be further heightened by immunization. This endogenous chimeric IgG1 significantly dampens the serum half-life of administered humanized mAbs in an hFCGRT-dependent manner. Thus, such IgG1-Fc humanized mice may provide a more physiologically relevant competitive hFCGRT-humanized mouse model for the preclinical development of human IgG-based biologics.
Introduction
Monoclonal antibody (mAb)-based biologics are among the fastest growing and most promising treatments for a broad range of human disorders, including cancer, autoimmune and inflammatory diseases, diabetes, respiratory diseases, and ophthalmologic diseases. A major asset of many mAb-based biologics is their persistence in circulation (~21-day serum half-life in humansCitation1). This extended pharmacokinetic (PK) behavior requires the crystallizable fragment (Fc) domain. For this reason, the majority of therapeutic mAbs, as well as many other protein-based biologics, include the human immunoglobulin G (IgG) Fc domain.
The Fc receptor of IgG was originally described as the neonatal Fc receptor (FcRn). This unusual MHC class I family Fc receptor is, however, used throughout life and is now named ‘Fc fragment of IgG receptor and transporter’ (FCGRT). It is primarily expressed in the early endosomes of cells within the circulatory system and is engaged in fluid-phase endocytosis. IgG-Fc binds the Fc of IgG at acidic pH, rescuing it from normal lysosomal catabolism by redirecting it to the plasma membrane, which, upon encountering a neutral pH, is released back into the circulation. This mechanism greatly slows IgG destruction and is the primary mechanism for the extended persistence and bioavailability of IgG (reviewed in Ref.Citation2,Citation3). Appreciation of FCGRT salvage has stimulated the redesign of many IgG-based biologics with Fcs engineered to enhance their persistence.Citation4–Citation6
However, the PK behavior of novel IgG-based biologics is still unpredictable, with modifications to either the Fab or Fc domains requiring extensive in vivo validation. Thus, the ability to relevantly and economically assess their PK in vivo is a critical aspect of the preclinical development of new IgG-based biologics. In vivo testing of mAb-based biologics in standard rodent models falls short in translational relevance owing to species differences between mouse and human FcRn.Citation7–Citation10 Non-human primates, e.g., cynomolgus monkeys, do provide a close evolutionary surrogate, but their use is encumbered by practical and ethical concerns. To overcome these, we have previously described FCGRT-humanized mouse models that serve for preclinical evaluations of IgG-based biologics in vivo. In particular, the mouse strain B6.Cg-Fcgrttm1Dcr Tg(FCGRT)32Dcr/DcrJ (abbreviated here to Tg32), which carries a null mutation for the mouse Fcgrt α-chain gene and a transgene expressing the human FCGRT α-chain transgene under the control of its natural human promoter, has been rigorously validated and is regarded to be the most reliable non-primate surrogate for preclinical testing of IgG-based biologics.Citation9,Citation11–Citation14
Although such FCGRT-humanized mice have been substantially validated for modeling of humanized IgG-based biologics, they lack an endogenous source of human IgG that typifies the human competitive condition. Of particular relevance here is that FCGRT trafficking is saturable. It operates with high efficiency when serum IgG concentrations are low, but declines asymptotically when serum IgG is in excess. This phenomenon is referred to as the concentration–catabolism effect in which the persistence of therapeutic mAbs is inversely correlated with the endogenous levels of competing endogenous IgG.Citation1,Citation15 Preexisting FcRn-humanized models have very low levels of endogenous mouse IgG due to the inability of hFcRn to effectively bind and recycle mouse IgG.Citation9,Citation16 Thus, PK assessments of human IgG-based biologics in such FcRn-humanized mice lack competing human IgG required for evoking the concentration–catabolism effect. While preloading mice by using intravenous immunoglobulin (IVIg) has been employed as a means to provide a bolus of competing human IgG, an endogenous source of humanized IgG will provide a more appropriate physiological context.
To address this need, we used CRISPR/Cas9-mediated homology-directed repair (HDR) to further engineer the hFCGRT Tg32 strain, replacing the mouse hinge and Ighg1 Fc domain with the equivalent human IgG1 domains. The replacement resulted in the mouse strain, B6.Cg-Fcgrttm1Dcr Ighg1em2(IGHG1)Mvw Tg(FCGRT)32Dcr/Mvw (abbreviated to Tg32-hFc) that produce hIgG1 Fc-mouse IgG Fab2 chimeric antibodies in serum concentrations at physiologically relevant levels, which can be further heightened by immunization. We show that the endogenous production of the chimeric IgG1 significantly dampens serum half-life of administered humanized mAbs in an FCGRT-dependent manner consistent with an appreciable concentration-competition effect. Thus, such IgG1-humanized, FCGRT humanized mice provide a more physiologically appropriate mouse model for the preclinical assessment of human IgG-based biologics.
Results
Construction of Tg32-hFc model
To provide a more relevant humanized IgG recycling environment, we genetically modified the Tg32 mouse strain to produce human Fc-mouse Fab2 chimeric IgG1 antibodies. The modification strategy outlined in uses CRISPR/Cas9-mediated HDR leading to a switching out of the mouse Ighg1 locus hinge (Exon 2), CH2 (exon 3) and CH3 (exon 4) regions replacing it with the human equivalent regions while preserving splice junctions (). From 95 micro-injected embryos, six survived to term (3 F, 3 M). Long-range PCRs extending beyond the homology arms identified two females as having transgene replacement. These were backcrossed to the parental Tg32 strain. While both candidates faithfully transmitted the transgenic allele, a single strain was ultimately established: B6.Cg-Fcgrttm1Dcr Ighg1em2(IGHG1)Mvw Tg(FCGRT)32Dcr/Mvw, abbreviated here to Tg32-hFc. This strain was brought to homozygosity and characterized in depth, including sequence on the modified region; see Supplemental .
Figure 1. (a) Targeting strategy to create Tg32-hFc mouse strain. Two sgRNAs 1652 and 1630 were combined with Cas9 in the presence of human donor DNA to mediate HDR replacement of mouse Igh1 exons 2-4 with the human equivalents, whilst maintaining splice accepter and donor sites; see Supplemental Figure 1. Mouse segments in blue, human in green. Correct integration and expression are predicted to give two chimeric mRNAs isoforms. CH1, constant heavy chain 1; CH2, constant heavy chain 2; CH3, constant heavy chain 3; Fc, fragment crystallizable region. (b) Shows predicted mRNAs from the region which were verified by RT-PCR, amplicons generated from the cDNA template used a common forward primer targeting the mouse Ighg1 and a reverse primer specific to the 3’ UTR of each variant; note, both chimeric isoforms were found and their correct splicing and predicted sequence confirmed; see Supplemental . Mouse regions shown in blue, human in green.. (c) Schematic of the predicted chimeric IgG1 antibody, with the human hinge and hFc (CH2 and CH3) domains (in green).
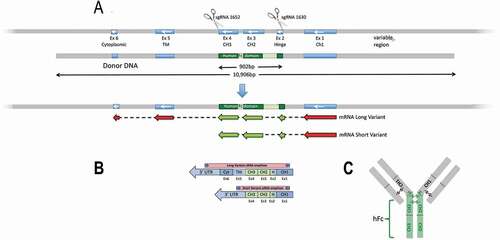
To confirm that the modified gene is appropriately transcribed and spliced, RNA was isolated from Tg32-hFc spleen and subjected to RT-PCR and Sanger sequencing. Analysis of the resulting sequence products demonstrated that the chimeric mouse-human in-frame mRNA spliced as predicted (). Two isoforms were detected, with and without sequences encoding the transmembrane and cytoplasmic tail domains; see Supplemental . These data demonstrate that the Ighg1 locus has undergone a functional modification and now expresses an Ighg1 chimeric mRNA predicted to give rise to intact human Fc and/mouse (Fab)2 IgG1 antibodies.
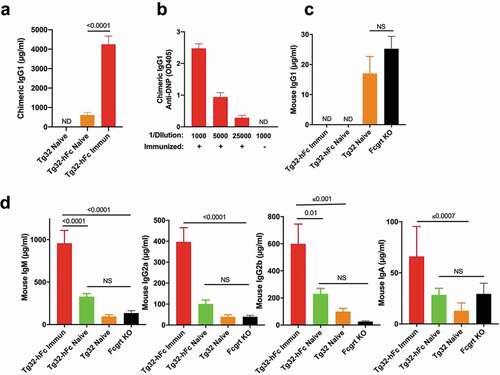
Figure 2. Serological analysis of naive and immunized Tg32 and Tg32-hFc mice. (a) Levels of chimeric IgG1 from plasma of naïve Tg32-hFc, immunized and naive Tg32 mice. ND, not detected above background. (b) Titers of anti-DNP activity from plasma of immunized Tg32-hFc mice. Naive mice yielded no detectable anti-DNP activity upon a plasma dilution of 1/1000 (not shown). (c) Mouse IgG1 quantification from the same plasma. (d) IgM, IgG2a, IgG2b, and IgA quantification from the same plasma. Data from 19 week old, female mice. Values are plotted as mean ± SEM (N=7-8).
Serological analysis of naive and immunized Tg32 and Tg32-hFc mice
To determine if the targeting strategy resulted in chimeric IgG1, we determined the levels of chimeric IgG1 plasma in naïve and immunized Tg32 and Tg32-hFc mice. Naïve Tg32-hFc mice yielded chimeric IgG1 levels that ranged from 280 to 1,320 μg/ml (mean 600 μg/ml) and following immunization of Tg32-hFc mice with 2,4-dinitrophenyl-keyhole limpet hemocyanin (DNP-KLH), chimeric IgG1 was significantly increased (mean value 4,200 μg/ml) (). Consistent with the targeting strategy, chimeric IgG1 was not detected in Tg32 mice (). We additionally assessed the antigen specificity of chimeric IgG developed by the immunized Tg32-hFc mice. Robust activity was evident even at a 1/25,000 dilution, indicating that the human genetic substitution did not impede the ability of the locus to elicit a strong antigen-specific chimeric IgG1 response after immunization ().
We also determined the impact of the modified Ighg1 locus on the production of mouse FCGRT-dependent (IgG1, IgG2a, IgG2b) and mouse FCGRT-independent (IgM and IgA) immunoglobulins from the same mice as well as Fcgrt KO mice. As expected for the targeted genetic modification, we did not detect fully mouse IgG1 in naïve or immunized Tg32-hFc mice, while significant but low levels of mouse IgG1 (averaging 17 μg/ml) were detected in naive Tg32 mice (). Furthermore, immunization of Tg32-hFc mice leads to significant increases in mouse IgM, IgM IgG2a, IgG2b and IgA, indicating that the modified Ighg1 locus did not negatively impact the overall antibody response (). The data indicated that the hFc knock-in strategy resulted in Tg32 mice that undergo class switch, affinity maturation and produce antibodies that include chimeric IgG1. Moreover, the appreciable plasma concentrations of chimeric IgG1 found in naïve and immunized Tg32-hFc mice, ~600 μg/ml, and ~4,200 μg/ml, respectively, provide strong evidence that chimeric IgG1 is actively recycled by human FCGRT.
Pharmacokinetics of humanized mAbs in Tg32 models
Having confirmed that the Tg32-hFc mice have significant levels of chimeric IgG1 antibodies, we addressed if this constitutive endogenous production significantly dampens the half-lives of administered humanized mAbs in a human FCGRT-dependent manner. We tested two different mAbs: 1) HuLys11, a humanized mAb containing the CH1-3 region of human IgG1 that was raised against hen egg-white lysozyme,Citation17 and 2) trastuzumab, a humanized mAb that binds the extracellular domain of the HER2 receptor and is in clinical useCitation18 (). The experimental cohorts included the naïve and immunized Tg32-hFc and naïve Tg32 mice whose plasma were analyzed above, as well as similarly aged and sex-matched Fcgrt knockout (KO) mice. As preloading with human IgG is known to enhance the clearance of administered humanized mAbs,Citation9 the study also included naïve Tg32 mice injected 2 days earlier with purified human IgG (IVIg); their human IgG levels in plasma averaged 2,300 μg/ml on day 0 of the experiment. Each cohort was injected intravenously with an admixture of trastuzumab and HuLys11 (10 mg/kg each) and their blood was monitored serially. As is evident by the plasma concentration-time courses (; see for detailed PK analyses), naïve Tg32-hFc mice cleared HuLys11 significantly faster than naive Tg32 mice, with β-phase T1/2 of 8.7 days and 17.4 days, respectively (p = .01) (). Previously immunized Tg32-hFc mice also increased clearance (T1/2 8.2) but did not differ significantly from naïve Tg32-hFc mice. IVIg-preloaded Tg32 mice showed the most rapid clearance (T1/2 5.4 days); see for details. Dependence on FCGRT was evident by the inability to detect HuLys11 in Fcgrt KO mice.
Table 1. Mouse models with trastuzumab and HuLys11 pharmacokinetic analysis.
Figure 3. Clearance of mAbs in naïve Tg32, naïve Tg32-hFc, immunized Tg32, IVIg-preloaded Tg32, and naïve Fcgrt KO mice. (a) Plasma concentration-time course and (b) β-phase half-lives of HuLys11. (c) Log-linear plasma concentration-time course and (d) β-phase half-lives of trastuzumab. Mean ± SEM of cohorts from (a). ND, Half-life of Fcgrt KO mice too low to be computed.
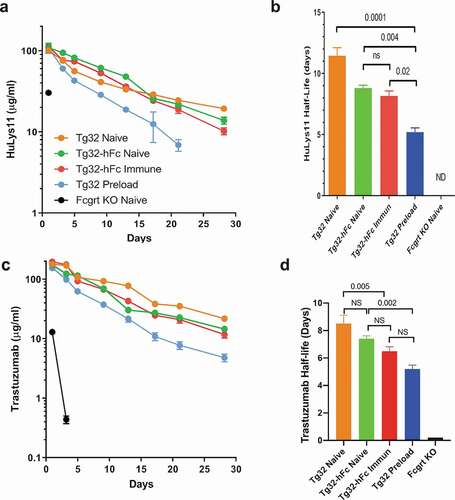
Very similar patterns were found for trastuzumab, despite its lower overall persistence (8.5 days in Tg32 mice; see ,d). Compared with naïve Tg32 counterparts, naive and immunized Tg32-hFc mice increased clearance of trastuzumab to a similar extent (T1/2 7.4 and 6.5 days, respectively), while Tg32 mice preloaded with IVIG exhibited a more appreciable reduction (T1/2 5.3 days). Again, dependence on FCGRT in all cases was evident by the very short half-lives found for mice lacking FCGRT (T1/2 ± 0.02 days); see for details. These combined data strongly support the contention that the endogenous production of chimeric IgG1 in Tg32-hFc mice, both naïve and immunized, significantly dampens the half-lives of administered humanized mAbs in a human FCGRT-dependent manner, but not to the extent found in bolus IVIg-preloaded mice.
Discussion
FCGRT-humanized mice have been substantially validated for modeling of humanized IgG-based biologics, but lack an endogenous source of competing human IgG that typifies the human condition. Here, we used gene editing to seamlessly humanize the mouse IgG1 Fc and hinge domains. Our targeting strategy resulted in Tg32 mice, Tg32-hFc, that develop fully mouse antibodies of the expected classes, subclasses, and abundance, but with the IgG1 gene carrying a humanized Fc domain, leading to its substantial plasma elevation.
Tg32 mice are hypergammaglobulinemic because of the failure of human FCGRT to bind and recycle mouse IgGs. In keeping with this, plasma concentrations of IgG1 in naïve Tg32 mice averaged 17 μg/ml, whereas that of naïve Tg32-hFc mice averaged 600 μg/ml. This 35-fold elevation of circulating IgG1 is almost certainly due to active hFCGRT-specific recycling, extending the IgG half-life and thus its plasma concentrations. The level of chimeric IgG1 now observed is comparable to that of native IgG1 reported in wildtype C57BL/6 J mice (~280 μg/ml mg/ml).Citation19 Furthermore, after immunization with DNP-KLH, we found that the Tg32-hFc mice mount a robust antibody response, with chimeric IgG1 plasma levels increasing substantially. The elevated plasma elevations of chimeric IgG1 in comparison to mouse IgG2a and IgG2b provide further confirmation that the chimeric IgG1 is uniquely protected by hFCGRT in Tg32-hFc mice. Moreover, the detection of all immunoglobulin classes in plasma of naive Tg32-hFc mice and its elevation post immunization indicates that the hFc substitution did not impair the overall capacity and capability of B cells to respond, class switch and differentiate to plasma cells. Combined, these data provide functional evidence that the chimeric IgG1 is appropriately regulated and uniquely protected by hFCGRT in the Tg32-hFc model.
Our key prediction is that the chimeric IgG1 in Tg32-hFc mice would compete for hFCGRT recycling with administered humanized mAbs. In support of this, we evaluated two humanized mAbs, trastuzumab and HuLys11. Our data show reduced β-phase half-lives when these mAbs are administered to Tg32-hFc mice compared with the noncompetitive environment of standard Tg32 mice. This more rapid clearance clearly documents the ability of chimeric IgG, endogenously produced and maintained homeostatically by human FCGRT-mediated recycling, to diminish the persistence of administered therapeutic mAbs. Although these data cannot be directly equated to cynomolgus monkeys or humans, Tg32-hFc mice do provide a more physiologically relevant competitive FCGRT-humanized rodent model for assessing the behavior of Fc-based therapeutics and human FCGRT biology.
IgG recycling by FCGRT is a logarithmic function of serum IgG concentrations. It operates at high efficiency when IgG is limiting and diminishes when IgG is in excess.Citation1,Citation15,Citation20 Thus, the persistence of IgG is predicted to be inversely related to its concentration in circulation. Our findings that the half-lives of the test therapeutic mAbs were longest in Tg32 mice (lacking Fc-humanized IgG1), naïve Tg32-hFc mice (having ~600 μg/ml chimeric IgG1) being intermediate, and with IVIG-preloaded Tg32 mice (having 2,300 μg/ml IgG at Day 0 of the experiment) showing the greatest reduction were consistent with this inverse relationship. However, our finding that immunized Tg32-hFc mice (having ~4,200 μg/ml chimeric IgG1) showed only an intermediate reduction is a conspicuous exception not readily explained by the standard concentration–catabolism model.
It is well established that FCGRT is a saturable Fc receptor. The FCGRT-dependent concentration–catabolism effect incorporates two variables: FCGRT availability and IgG concentrations in circulation. While modeling of this phenomenon usually assumes that FCGRT availability is invariant, it is well established in transgenic models that hIgG concentrations correlate positively with the transgenic hFCGRT copy number.Citation9 Increases in FCGRT thus increase serum concentrations of IgG. The reticuloendothelial system is a major repository of functionally competent FCGRT expression and IgG recycling.Citation21–Citation24 Immunization protocols that include a strong adjuvant, such as the Freund’s adjuvant used in our study, are highly immunostimulatory to this organ system, with a substantial expansion of myeloid derivatives, their activation state, and upregulated levels of FCGRT expression.Citation25–Citation27 We thus posit that the relatively anemic half-life reductions of HuLys11 and trastuzumab in immunized Tg32-hFc mice occurred because immunization increased the overall capacity of hFCGRT to recycle IgG.
Overall, the development of these Tg32-hFc mice provides a relevant and economic model to rapidly evaluate novel engineered Fc-based biologics within a more competitive environment, and could provide a more reliable preclinical PK analyses model. This Tg32-hFc model also provides a convenient vehicle for the rapid generation of Fc-humanized IgG1 monoclonal antibodies.
Materials and methods
Generation of Tg32-hFc mice
Mice used and created in this study are listed with genotype, full and abbreviated names in . An overview of the targeting strategy is shown in . Fertilized oocytes isolated from the Tg32 strain were microinjected with two sgRNAs (#1630, #1652) targeting the sequence encoding the beginning of the Ighg1 hinge domain and the end of the CH3 domain, respectively. The sgRNAs were designed as TRU-guides with 18nt of target-specific sequence.Citation28 sgRNA #1630 required pre-pending with a non-homologous 5ʹ G to facilitate in vitro transcription. Guide target sequences are shown with the PAM in parentheses: #1630: 5ʹ gCACAATCCCTGGGCACTG(tgg) and #1652 5ʹ GAGCCTCTCCCACTCTCC(tgg); see . Each sgRNA was microinjected at 30 ng/µl, with Cas9 mRNA (Trilink) at 60 ng/µl, and plasmid donor at 20 ng/µl. The plasmid donor (partly synthesized by Genscript) consisted of a 902 bp human sequence (hinge (Exon 2), CH2 (exon 3) and CH3 (exon 4)) flanked by ~5kb homology arms. A total of 95 embryos derived from Tg32 were microinjected and transferred into 5 pseudopregnant females. Three transfers failed to deliver any liveborn, the remaining two females delivered 6 mice (3 F/3 M) from 38 embryos implanted, two of which were identified as candidates for the transgene replacement by PCR. Following backcrossing to Tg32 mice, both strains transmitted the transgenic allele. Ultimately, ahomozygous single strain was established and characterized; B6.Cg-Fcgrttm1Dcr Ighg1em2(IGHG1)Mvw Tg(FCGRT)32Dcr/Mvw, JAX # 029686, abbreviated here to Tg32-hFc.
Table 2. Mouse strains used or created for this study with genotype and abbreviated name. All mice are available from The Jackson Laboratory.
Genotyping of the hFc allele used PCR primers flanking the humanized region (1731 F, 5ʹ GAAATGGATCTCAGCCCAGAAG and 1732 R, 5ʹ AGCTAGAGGAAGGTCTGGTAAG), generating a 1,484 bp amplicon. Sanger sequencing of the PCR product was used to determine the zygosity of the chimeric allele. Partial verification of the replacement event used long-range PCR primers 1838 F, 5ʹ CAGCCGGAGAACAACTACAA and 1839 R, 5ʹ AACAGTCCCAGGCCTAAATG generating a 5,825 bp amplicon, one located outside of the homology arm and the other located within the humanized portion of the chimeric allele followed by sequencing. Additionally, Long Read Sequencing (PacBio) was used to verify the complete sequence across both homology arms as base-perfect; see Supplemental . This sequence was generated using genomic DNA isolated from the liver of a mouse homozygous for the humanized Ighg1 allele. Long-Range PCR (LongAmp® Taq, New England Biolabs) was performed with primers (1839 R, 5ʹ AACAGTCCCAGGCCTAAATG; and 1840 F, 5ʹGACAGGTGGAAGTGTGTTGA) to generate an 11,665 bp amplicon for Long Read Sequencing (PacBio). Alignment of the LR Sequence to the predicted allele confirmed that no unintended mutations were incorporated into the chimeric allele.
Verification of the Tg32-hFc humanized immunoglobulin heavy constant gamma 1 transcript and splicing
Total spleen RNA from of a 12-week-old homozygous male Tg32-hFc mouse was isolated and used to make poly-T primed cDNA (Invitrogen 18080–044; First Strand cDNA Synthesis Kit). This cDNA was subject to PCR followed by Sanger Sequencing verifying the predicted splice variants (Ighg1-v1 and Ighg1-v2), and that splicing of the human sequence occurred as predicted. Amplicons generated from the cDNA template used a common forward primer targeting the mouse Ighg1 Exon 1 (F1, 5ʹ AGTGACCTGGAACTCTGGAT), and a reverse primer specific to the 3ʹ UTR of each variant: either Rv1 (5ʹ CAGTGCTGGGTGCTTTATTTAC) or Rv2 (5ʹ CTGTGACCAGAAGGAGGATCTA); see . The resulting product was sequenced; see Supplemental .
Immunoglobulin quantification
Chimeric IgG1 was quantified from the plasma of Tg32-hFc mice by capture with mouse anti-human IgG1 (clone G17-1, BD Biosciences) and detection with mouse anti-human kappa-HRP (clone SB81a, Southern Biotech). Human IgG1 antibodies purified from healthy donors were used as a standard (Enzo Life Sciences). Some mice were primed with 100 µg of DNP-KLH in complete Freund’s adjuvant, challenged with DNP-KLH in incomplete Freund’s adjuvant, and bled 2 weeks later. Anti-DNP quantification of chimeric IgG1 was determined by capture with DNP-albumin conjugate (Sigma-Aldrich) coated at 5 µg/ml phosphate-buffered saline (PBS), and detection with mouse anti-human IgG1-HRP (clone G17-1, BD Biosciences). Mouse Ig isotypes were quantified using the Meso Scale Discovery mouse isotyping panel per manufacturer’s instructions with the exception of mouse IgG1. Mouse IgG1 was quantified by capture with goat anti-mouse IgG1 (Southern Biotech) at 5 µg/ml PBS, and detection with goat anti-mouse kappa-AP (Southern Biotech). A mouse IgG1 isotype control (Southern Biotech) was used as a standard. Statistical analysis of antibody concentrations was determined with PRISM software by a 2-sided T-test with Welch’s correction or ANOVA using the Turkey’s multiple sample comparison test.
PK analysis
We essentially followed the approach of Petkova et al.Citation9 In brief, groups of naïve Tg32, naïve Tg32-hFc, Tg32-hFc mice previously immunized with DNP-KLH, naïve Tg32, preloaded with 250 mg/kg human IVIg, and Fcgrt KO mice were pre-bled at day 0, and intravenously co-injected with the humanized mAbs trastuzumabCitation18 and HuLys11Citation17 (10 mg/kg each). The mice were then bled serially (25 µl samples) into K3EDTA at 1, 3, 5, 7, 13, 17, 21, and 28 days, and the blood processed to plasma. The plasma was diluted 1 to 10 in 50% glycerol in PBS and stored at −20°C until analysis. HuLys11 was quantified by capturing with lysozyme purified from chicken egg white (Sigma-Aldrich) coated at 5 µg/ml in carbonate buffer (pH 9.6) and detected with mouse anti-human IgG1-HRP (clone G17-1, BD Biosciences). Trastuzumab was quantified in the plasma samples by capturing activity on ELISA plates with anti-idiotype antibody AbD18018 (Bio-Rad) coated at 5 µg/ml PBS, detecting with mouse anti-human IgG Fc CH2 domain with HRP conjugation (Bio-Rad) at 2 µg/ml. HuLys11 was quantified by capturing with lysozyme purified from chicken egg white (Sigma-Aldrich) coated at 5 µg/ml in carbonate buffer (pH 9.6) and detected with mouse anti-human IgG1-HRP (clone G17-1, BD Biosciences). Trastuzumab and HuLys11 plasma concentrations were analyzed using PK Solutions Software (Summit Research Services). Human IgG1 was quantified from the plasma of Tg32-hFc mice by capture with mouse anti-human IgG1 (clone G17-1, BD Biosciences), detection with mouse anti-human kappa-HRP (clone SB81a, Southern Biotech), with human IgG1 isotype control purified from healthy donors (Enzo Life Sciences) used as a standard. Anti-DNP activity resulting from the DNP-KLH immunization was quantified by capture with DNP-albumin conjugate (Sigma-Aldrich) coated at 5 µg/ml PBS, and detection with mouse anti-human IgG1-HRP (clone G17-1, BD Biosciences). Mouse Ig isotypes were quantified using the Meso Scale Discovery mouse isotyping panel per manufacturer’s instructions with the exception of mouse IgG1. Mouse IgG1 was quantified by capture with goat anti-mouse IgG1 (Southern Biotech) at 5 µg/ml PBS, detection with goat anti-mouse kappa-AP (Southern Biotech), with mouse IgG1 isotype control (Southern Biotech) used as a standard. PK analyses of HuLys11 and trastuzumab were determined using PK Solutions Software (Summit Research Services). Statistical analysis of half-life determinations was evaluated with PRISM software by ANOVA using the Turkey’s multiple sample comparison test.
Abbreviations
| A | = | Adenosine |
| B6 | = | C57BL/6J |
| bp | = | Base pairs |
| C | = | Cytosine |
| Cas9 | = | CRISPR associated protein 9 |
| CH1 | = | Constant heavy domain 1 |
| CH2 | = | Constant heavy domain 2 |
| CH3 | = | Constant heavy domain 3 |
| CRISPR/Cas9 | = | Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 |
| DNA | = | Deoxyribonucleic acid |
| DNP-KLH | = | 2,4-dinitrophenyl-keyhole limpet hemocyanin |
| Fab | = | Fragment antigen-binding |
| FCGRT | = | Fc fragment of IgG, receptor, transporter, alpha - human |
| Fcgrt | = | Fc fragment of IgG, receptor, transporter, alpha - mouse |
| FcRn | = | Fragment crystallizable receptor neonate (old name for Fcgrt) |
| G | = | Guanine |
| HDR | = | Homology directed repair |
| IgA | = | Immunoglobulin A |
| IgG | = | Immunoglobulin G |
| IGHG1 | = | Immunoglobulin heavy constant gamma 1 |
| IgM | = | Immunoglobulin M |
| IV | = | Intravenous |
| K3EDTA | = | Tripotassium ethylenediaminetetraacetic acid |
| mAbs | = | Monoclonal antibody |
| mg/kg | = | Milligram per kilogram |
| MHC | = | Major histocompatibility complex |
| mRNA | = | Massager ribonucleic acid |
| PBS | = | Phosphate-buffered saline |
| PCR | = | Polymerase chain reaction |
| pH | = | Potential of hydrogen |
| PK | = | Pharmacokinetic |
| RNA | = | Ribonucleic acid |
| T | = | Thymine |
| T | = | Time |
| UTR | = | Untranslated region |
| µg/ml | = | Microgram per milliliter |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Ethics statement
All animals experiments are done under The Jackson Laboratory Animal Care and Use Committee (IACUC), those specific to MVW used IACUC summary # 11006. Animals were euthanized by cervical dislocation.
Supplemental Material
Download Zip (120.8 KB)Acknowledgments
The authors would like to thank Cindy Avery and John Wilson for their excellent technical assistance in this work. Also, Pete Kutny and the microinjection core for their expertise and dedication as well as Simon Lesbirel and the Genome Technology core at The Jackson Laboratory. We also gratefully acknowledge the contribution of Melissa Berry, Derry Roopenian for suggested experimental design and writing and also the Genome Technologies Service and Genetic Engineering Technologies at The Jackson Laboratory for expert assistance with the work described in this publication.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Morell A, Terry WD, Waldmann TA. Metabolic properties of IgG subclasses in man. J Clin Invest. 1970;49:673–8. doi:https://doi.org/10.1172/JCI106279.
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–25.
- Pyzik M, Sand KMK, Hubbard JJ, Andersen JT, Sandlie I, Blumberg RS. The neonatal Fc receptor (FcRn): a misnomer? Frontiers in Immunology. Published online 2019 Jul 10.
- Huang C. Receptor-Fc fusion therapeutics, traps, and MIMETIBODY™ technology. Current Opinion in Biotechnology. 2009;20:692–99. doi:https://doi.org/10.1016/j.copbio.2009.10.010.
- Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Current Opinion in Biotechnology. 2011;22:868–76. doi:https://doi.org/10.1016/j.copbio.2011.06.012.
- Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. mAbs. 2011;3:422–30. doi:https://doi.org/10.4161/mabs.3.5.16983.
- Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. International Immunology. 2001;13:1551–59. doi:https://doi.org/10.1093/intimm/13.12.1551.
- Zhou J, Mateos F, Ober RJ, Ward ES. Conferring the binding properties of the mouse MHC class I-related receptor, FcRn, onto the human ortholog by sequential rounds of site-directed mutagenesis. J Mol Biol. 2005;345:1071–81. doi:https://doi.org/10.1016/j.jmb.2004.11.014.
- Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, Presta LG, Meng YG, Roopenian DC. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18:1759–69. doi:https://doi.org/10.1093/intimm/dxl110.
- Dostalek M, Prueksaritanont T, Kelley RF. Pharmacokinetic de-risking tools for selection of monoclonal antibody lead candidates. mAbs. 2017;9:756–66. doi:https://doi.org/10.1080/19420862.2017.1323160.
- Tam SH, McCarthy SG, Brosnan K, Goldberg KM, Scallon BJ. Correlations between pharmacokinetics of IgG antibodies in primates vs. FcRn-transgenic mice reveal a rodent model with predictive capabilities. mAbs. 2013;5:397–405. doi:https://doi.org/10.4161/mabs.23836.
- Haraya K, Tachibana T, Nanami M, Ishigai M. Application of human FcRn transgenic mice as a pharmacokinetic screening tool of monoclonal antibody. Xenobiotica. 2014;44:1127–34. doi:https://doi.org/10.3109/00498254.2014.941963.
- Avery LB, Wang M, Kavosi MS, Joyce A, Kurz JC, Fan YY, Dowty ME, Zhang M, Zhang Y, Cheng A, et al. Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. mAbs. 2016;8:1064–78. doi:https://doi.org/10.1080/19420862.2016.1193660.
- Betts A, Keunecke A, van Steeg TJ, van der Graaf PH, Avery LB, Jones H, Berkhout J. Linear pharmacokinetic parameters for monoclonal antibodies are similar within a species and across different pharmacological targets: a comparison between human, cynomolgus monkey and hFcRn Tg32 transgenic mouse using a population-modeling approach. mAbs. 2018;10:751–64. doi:https://doi.org/10.1080/19420862.2018.1462429.
- Brambell FWR, Hemmings WA, Morris IG. A theoretical model of γ-globulin catabolism. Nature. 1964;203:1352–55. doi:https://doi.org/10.1038/2031352a0.
- Stein C, Kling L, Proetzel G, Roopenian DC, de Angelis MH, Wolf E, Rathkolb B. Clinical chemistry of human FcRn transgenic mice. Mamm Genome. 2012;23:259–69. doi:https://doi.org/10.1007/s00335-011-9379-6.
- Foote J, Winter G. Antibody framework residues affecting the conformation of the hypervariable loops. Journal of Molecular Biology. 1992;224:487–99. doi:https://doi.org/10.1016/0022-2836(92)91010-M.
- Goldenberg MM. Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clinical Therapeutics. 1999;21:309–18. doi:https://doi.org/10.1016/S0149-2918(00)88288-0.
- Klein-Schneegans AS, Kuntz L, Fonteneau P, Loor F. Serum concentrations of IgM, IgG1, IgG2b, IgG3 and IgA in C57BL 6 mice and their congenics at the lpr (lymphoproliferation) locus. Journal of Autoimmunity. 1989;2:869–75. doi:https://doi.org/10.1016/0896-8411(89)90013-9.
- Roopenian DC, Sun VZ. Clinical ramifications of the MHC family Fc receptor FcRn. J Clin Immunol. 2010;30:790–97. doi:https://doi.org/10.1007/s10875-010-9458-6.
- Zhu X, Meng G, Dickinson BL, Li X, Mizoguchi E, Miao L, Wang Y, Robert C, Wu B, Smith PD, et al. MHC class I-related neonatal Fc receptor for IgG is functionally expressed in monocytes, intestinal macrophages, and dendritic cells. Journal of Immunology. 2001;166:3266–76. doi:https://doi.org/10.4049/jimmunol.166.5.3266.
- Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol. 2007;179:4580–88. doi:https://doi.org/10.4049/jimmunol.179.7.4580.
- Kobayashi K, Qiao S, Yoshida M, Baker K, Lencer WI, Blumberg RS. An FcRn-dependent role for anti-flagellin immunoglobulin G in pathogenesis of colitis in mice. Gastroenterology. 2009;137(1746–56.e1). doi:https://doi.org/10.1053/j.gastro.2009.07.059.
- Richter WF, Christianson GJ, Frances N, Grimm HP, Proetzel G, Roopenian DC. Hematopoietic cells as site of first-pass catabolism after subcutaneous dosing and contributors to systemic clearance of a monoclonal antibody in mice. mAbs. 2018;10:803–13. doi:https://doi.org/10.1080/19420862.2018.1458808.
- Liu X, Ye L, Christianson GJ, Yang J-Q, Roopenian DC, Zhu X. NF-kappaB signaling regulates functional expression of the MHC class I-related neonatal Fc receptor for IgG via intronic binding sequences. Journal of Immunology (Baltimore, Md: 1950). 2007;179:2999–3011. doi:https://doi.org/10.4049/jimmunol.179.5.2999.
- Guo J, Li F, Qian S, Bi D, He Q, Jin H, Luo R, Li S, Meng X, Li Z, et al. TGEV infection up-regulates FcRn expression via activation of NF-kappaB signaling. Sci Rep. 2016;6:32154. doi:https://doi.org/10.1038/srep32154.
- Cervenak J, Doleschall M, Bender B, Mayer B, Schneider Z, Doleschall Z, Zhao Y, Bősze Z, Hammarström L, Oster W, et al. NFkappaB induces overexpression of bovine FcRn: a novel mechanism that further contributes to the enhanced immune response in genetically modified animals carrying extra copies of FcRn. mAbs. 2013;5:860–71. doi:https://doi.org/10.4161/mabs.26507.
- Wang Y, Zhang WY, Hu S, Lan F, Lee AS, Huber B, Lisowski L, Liang P, Huang M, de Almeida PE, et al. Genome editing of human embryonic stem cells and induced pluripotent stem cells with zinc finger nucleases for cellular imaging. Circ Res. 2012;111:1494–503. doi:https://doi.org/10.1161/CIRCRESAHA.112.274969.