
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Monoclonal antibodies (mAbs) are among the fastest growing and most effective therapies for myriad diseases. Multispecific antibodies are an emerging class of novel therapeutics that can target more than one tumor- or immune-associated modulators per molecule. The combination of different binding affinities and target classes, such as soluble or membrane-bound antigens, within multispecific antibodies confers unique pharmacokinetic (PK) properties. Numerous factors affect an antibody’s PK, with affinity to the neonatal Fc receptor (FcRn) a key determinant of half-life. Recent work has demonstrated the potential for humanized FcRn transgenic mice to predict the PK of mAbs in humans. However, such work has not been extended to multispecific antibodies. We engineered mAbs and multispecific antibodies with various Fc modifications to enhance antibody performance. PK analyses in humanized FcRn transgenic mouse (homozygous Tg32 and Tg276) and non-human primate (NHP) models showed that FcRn-binding mutations improved the plasma half-lives of the engineered mAbs and multispecific antibodies, while glycan engineering to eliminate effector function did not affect the PK compared with wild-type controls. Furthermore, results suggest that the homozygous Tg32 mouse model can replace NHP models to differentiate PK of variants during lead optimization, not only for wild-type mAbs but also for Fc-engineered mAbs and multispecific antibodies. This Tg32-mouse model would enable prediction of half-life and linear clearance of mAbs and multispecific antibodies in NHPs to guide the design of further pharmacology/safety studies in this species. The allometric exponent for clearance scaling from Tg32 mice to NHPs was estimated to be 0.91 for all antibodies.
Introduction
Monoclonal antibodies (mAbs) are a burgeoning class of therapeutics indicated for a wide range of neurologic, oncologic, respiratory, ophthalmologic, rheumatologic, cardiovascular, and bone disorders. Twelve new mAbs entered the market in 2018 alone, and marketing approval is granted at twice the rate for mAbs as for small-molecule drugs.Citation1 Currently, numerous companies are developing more than 570 mAbs, with approximately 90% of these in Phase 1 or 2 studies.Citation1 Multispecific antibodies are an emerging class of therapeutic antibodies that have 2 or more different antigen-binding sites.Citation2 Over 100 bispecific antibody (bi-Ab) formats currently exist, with over 85 in various stages of clinical development and 2 having achieved regulatory approval.Citation2 Bispecific antibodies offer several advantages over combination therapy with 2 or more mAbs, including possible increased avidity and a lower overall dose burden and consequent reduced risk of immunogenicity.Citation3 Although multispecific antibodies may have certain functional features in common with canonical mAbs, their structure and binding targets also entail a unique set of pharmacokinetic (PK) characteristics.Citation4 For example, bi-Abs have been shown to have shorter in vivo half-lives than their parental mAb components, and the reasons for this remain to be fully elucidated.Citation5 Design and engineering of multispecific antibodies thus must be responsive to numerous challenges, including those related to PK characterization.Citation6 Bispecific antibodies, because of high pharmacologic potency, may require low starting doses in first-in-human studies, necessitating highly sensitive PK assays.Citation6 These and other considerations – for both multispecific antibodies and mAbs – foster an acute need to screen candidate antibodies to exclude in the early discovery stages those antibodies with less promising PK characteristics.
Numerous approaches to engineering mAbs and multispecific antibodies exist to improve their function. These include altering the Fc domain to increase or decrease binding to Fc gamma receptors (FcγRs), with consequent increased or decreased effector function, respectively,Citation7 as well as several approaches to increase an antibody’s half-life and optimize its clearance (CL). The unique PK behavior of many mAbs results from various factors, including target-mediated drug disposition (TMDD), off-target binding, isoelectric point (pI), glycosylation patterns, neonatal Fc receptor (FcRn) binding patterns, and interaction with anti-drug antibodies (ADAs).Citation8,Citation9 TMDD, for example, for some antibodies exhibits nonlinear PK, which affects half-life and creates a challenging situation for interspecies comparisons and dose selection.Citation9 A key determinant of half-life is the affinity of immunoglobulin (IgG) to FcRn, which has become a guiding principle in engineering therapeutic antibodies to have longer serum half-lives.Citation10,Citation11 A crucial characteristic of IgG-FcRn binding is a pH dependency that is strongest at slightly acidic pH and negligible at neutral or slightly basic pH.Citation11,Citation12 The long circulation half-lives of IgGs largely result from the enhanced binding affinity to FcRn, pH-dependent FcRn-mediated protection from lysosomal proteolytic degradation, and subsequent recycling from the endosome to the cell surface.Citation13–15
Direct modification of specific amino acids in the Fc region, such as YTE (M252Y/S254T/T256E,Citation11 including in humansCitation16) or LS (M428L/N434SCitation17), has been shown to enhance the efficacy and to prolong half-life in some marketed mAbs by a factor of 2–4. Recently, our group engineered a panel of novel Fc variants, namely, YD (M252Y/T256D), DQ (T256D/T307Q), and DW (T256D/T307W), that showed improved binding to FcRn and prolonged half-lives versus their wild-type (WT) counterparts, in both Tg32 mouse and cynomolgus monkey models.Citation18 Long circulating half-life is paramount to the clinical success of mAbs,Citation19 enabling lower dosing and less frequent administration, which may result in better compliance and reduced costs.Citation20
The most frequently used species for preclinical testing of mAbs and other therapeutic proteins are non-human primates (NHPs), in particular the cynomolgus monkey (Macaca fascicularis). The high degree of genetic similarity between cynomolgus monkeys and humans has made them useful in predicting human PK profiles to support first-in-human studies.Citation21 However, high costs, large amounts of investigational product needed for studies, time and resource constraints, impracticability of screening large panels of antibodies,Citation21 and, importantly, ethical concernsCitation22 limit NHP studies to high-potential preclinical candidates. Human and mouse FcRn are distinct in their binding specificities; therefore, WT mice are not a reliable in vivo model for PK profiling of human mAbs.Citation23 Moreover, ADA formation can hamper reliable determinations of PK parameters, even if those ADAs are mainly due to heterogeneity of species not predictive of the situation in humans. Therefore, a substantial demand exists for the development of easy-to-use and less expensive in vivo mammalian models to accurately predict PK behavior in humans. Murine models are an obvious choice, but the use of WT mice is limited in PK studies because of differences in species-specific binding affinities between human IgG1 and the different rodent and human Fc receptors, including FcRn.Citation24,Citation25 Murine FcRn shows an enhanced binding affinity to human IgGs compared with human FcRn (hFcRn),Citation26 and thus would not provide a suitable PK prediction for human IgGs. Nonetheless, the design of humanized mouse models has been achieved to fill this critical gap for an in vivo predictive PK model. Humanized FcRn transgenic mouse strains with an appropriate gene expression profile have been generatedCitation27 and introduced as potential surrogates for the in vivo PK evaluation of Fc-engineered human antibodies or other Fc-based biologics. These mice are transgenic for the hFcRn α-chain under the control of the native and tissue-specific promoter (Tg32 strain) or the chicken β-actin promoter (Tg276 strain).Citation27–29 The relevance of the Tg32 mouse model to demonstrate in vivo half-life prolongation of engineered or native mAbs and to predict NHP or human PK has been recently reported.Citation18,Citation30–32
Despite the broad range of reports on the PK of mAbs in these mouse models, there is less information on the Fc modification and applicability of the hFcRn transgenic mouse models for the CL and half-life determination of multispecific antibodies. Accordingly, we performed a comprehensive comparative study in both mouse and NHP models using a panel of Fc-silent and FcRn-enhancing mutations that were introduced into mAbs and bi- and trispecific IgG1 antibodies (tri-Abs). These antibodies were then examined in cynomolgus monkeys and the hFcRn mouse strains Tg32 and Tg276. The results strongly support the Tg32 mouse model as a robust model for investigating the PK profile of Fc-engineered mAbs and multispecific antibodies.
Results
Antibody dataset
We evaluated 16 human antibodies and selected 7 different backbones of which mAb1, bi-Ab4, bi-Ab5, and mAb7 are humanized antibodies from rat or mouse, while mAb2, mAb3 and tri-Ab6 are fully human antibodies. For the mAbs, 3 were WT and 9 were engineered through the Fc region (). Two mAbs are commercially available (bevacizumab and adalimumab). Among the Fc-mutated mAbs, 2 were engineered to abolish immune effector functions (antibody-dependent cell-mediated cytotoxicity [ADCC]) and 7 were engineered to prolong elimination half-life. Variants with the LS, DQ, DW, and YD mutations were previously developed in-house.Citation18 Four multispecific antibodies were included in our study: 2 bi-Abs and 2 tri-Abs. Of the multispecific antibodies, 2 were WT and 2 were engineered through the Fc region. The underlying format for the bi-Ab and tri-Ab we tested was cross-over dual variable (CODV) Ig-like proteins.Citation33
Table 1. Panel of antibodies used in the study. All tested antibodies were of the IgG1 backbone type. The listed mAbs are not parental to the bi- or trispecific molecules
Fc modifications for altered Fc-receptor binding
To examine the relationship between the incorporated Fc mutations with the antibodies’ PK, we measured concentration-dependent binding to FcRn and FcγRIIIa using a Biacore-based assay. The LS, DQ, DW, and YD mutations for enhanced FcRn binding showed a clear 1.3- to 5.8-fold enhancement in binding affinity at pH 6.0 to human and cynomolgus monkey compared with the WT antibody. In addition, the LS mutations increase FcRn binding several fold regardless of the target antigen (mAb1 vs. mAb2) or antibody format (mAbs vs. Tri-Ab6; ). Low FcRn binding at neutral pH (pH 7.4) was maintained for all the studied FcRn-binding variants. Some FcRn-binding variants, like YD, simultaneously reduced the binding to FcγRIIIa by 2-fold (), despite the mutations being located far from this interface.
Table 2. In vitro characterization of antibodies carrying Fc-backbone modifications. Binding affinities to the FcRn receptor of humans and cynomolgus monkeys; binding to FcγRIIIa, and pI
With regard to NNAS, the carbohydrate switch mutations completely eliminated binding to FcγRIIIa and the other FcγRs, as expected (data not shown), but maintained a WT-like FcRn-binding profile in multiple antibody backgrounds (). The DQ, DW, and YD mutations resulted in a 0.2-pH-unit lower pI profile, whereas NNAS resulted in a highly complex pI profile, compared with respective WT antibodies (Supplementary Figure 1, ).
Pharmacokinetics and selection of the best transgenic mouse model
To assess the PK-predictive capacities of Tg32 and Tg276 homozygous mice as relevant murine models for WT antibodies, Fc-engineered mAbs, and multispecific antibodies (bi-/trispecific; whether mutated or not), we performed PK studies in these strains and, in parallel, in cynomolgus monkeys for comparison. A 2-step process was used for ethical and operational reasons. In a first round of experiments, 10 antibodies were selected and profiled in both transgenic mouse models and cynomolgus monkeys, followed by an interim analysis, before the remaining antibodies were tested in the most reliable mouse strain based on results of the first dataset. As described in the Materials and Methods section, all the doses were chosen to be appropriate to determine linear PK parameters, sufficiently high to saturate TMDD for mouse or monkey cross-reactive antibodies (i.e., adalimumab).
shows the obtained PK parameters of the antibodies. In Tg276 mice, several of the antibodies tested (mAb1-LS/NNAS, mAb2-LS) entailed a sharp drop in concentrations early in the kinetics due to the presumed presence of ADAs. This precluded accurate calculation of the PK parameters, which are therefore not reported. Overall, the comparisons between Tg276 mice and cynomolgus monkeys were performed on 7 antibodies. Among them, 2 corresponded to WT mAbs, 3 were mAbs engineered through the Fc region, and 1 was a tri-Ab (WT and Fc-mutated). As shown in , the concentration-time profiles in Tg276 mice were not parallel to those observed in Tg32 mice and in cynomolgus monkeys. The terminal elimination half-life in Tg276 mice was shorter in comparison with that found in Tg32 mice (1.7- to 4-fold faster elimination) and cynomolgus monkeys (1.5- to 4.5-fold faster elimination). Likewise, a higher CL was observed in Tg276 mice compared with Tg32 mice (1.2- to 3.1-fold higher) and cynomolgus monkeys (1.6- to 4.4-fold higher). The trend was exactly the same independent of the antibody, Fc-mutation status, and whether it was a mAb or tri-Ab.
Table 3. Summary of PK parameters of the different antibodies tested in cynomolgus monkeys and Tg32 and Tg276 homozygous mice
Figure 1. Comparison of pharmacokinetic profiles in cynomolgus monkeys and homozygous Tg32 and Tg276 mice following administration of (a) mAb1-WT or (b) with the LS mutation; (c) tri-Ab6-WT or (d) with the LS mutation. For mouse studies, pooled plasma samples were plotted per sampling time. Abbreviations: ADA = anti-drug antibody, mAb = monoclonal antibody, tri-Ab = trispecific antibody, WT = wild type
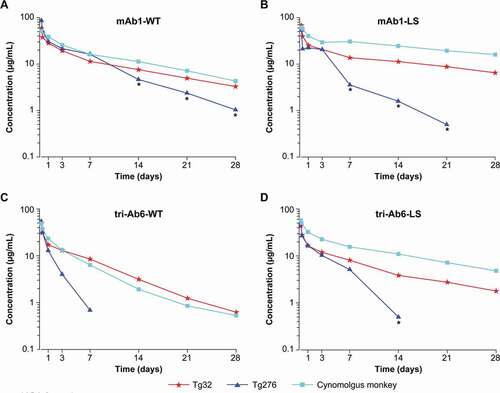
In contrast to the data from Tg276 mice, in Tg32 mice no sharp drop in concentrations was observed for all of the 10 antibodies tested in the first experimental setup. Therefore, the second set of molecules was evaluated only in the Tg32 strain. The antibody selection listed in was used for the comparison between Tg32 mice and cynomolgus monkeys.
In cynomolgus monkeys, ADAs developed in a mean of 30% of treated animals, which affected the PK profiles of some individuals. This ADA impact could be observed as soon as between 7 and 14 days after a single dosing, showing a characteristic sharp drop in concentrations in corresponding animals. Depending on the time of ADA impact on the PK profile, we either excluded the monkey or omitted the concentration data affected by ADAs when appearing after 14 days.
Elimination half-lives of antibodies tested in Tg32 mice and cynomolgus monkeys were in the same range of magnitude; the cynomolgus monkey/Tg32 mouse half-life ratio ranged from 0.8 to 1.3 for WT mAbs and bi-Abs/tri-Abs and from 0.8 to 1.4 for Fc-engineered mAbs. shows an example of a parallel elimination profile observed for mAbs (WT or mutated) and tri-Ab (WT or mutated) in Tg32 mice and in cynomolgus monkeys.
Comparison of PK parameters between monoclonal and multispecific antibodies (WT or Fc-mutated)
In this experiment, 4 different antibodies were mutated on the Fc region to increase the binding to FcRn. Three were monospecific mAbs (mAb1, mAb2, and mAb3) and 1 was a tri-Ab. For mAb1, mAb3, and tri-Ab6, the enhancement of the elimination half-life due to the mutation was observed in both Tg32 mice and in cynomolgus monkeys. ,b) show an example of the plasma PK profile in Tg32 mice and cynomolgus monkeys for WT and mutated mAb1. For the 3 constructs, the increase in elimination half-life due to the mutation was apparent in both species. Furthermore, the amount of increase was in the same range, with on average an improvement in the elimination half-life by a factor of 2.2 in the cynomolgus monkeys and 1.6 in the Tg32 mice for mAb1 and mAb3, and for tri-Ab6, by a factor of 1.9 and 1.7, respectively. These results establish that the Tg32 homozygous mouse model can distinguish PK differences in mAbs containing Fc-backbone mutations and in tri-Abs in the same range of order as in cynomolgus monkeys.
Figure 2. Comparison of pharmacokinetic profiles in (A/C) Tg32 mice and (B/D) cynomolgus monkeys. Administration of (A/B) FcRn-modulating LS and YD mutations; (C/D) mAb1 or mAb3, WT and mutated Fc backbones, Fc-silencing FcγRIIIa (NNAS). Abbreviations: mAb = monoclonal antibody, WT = wild type
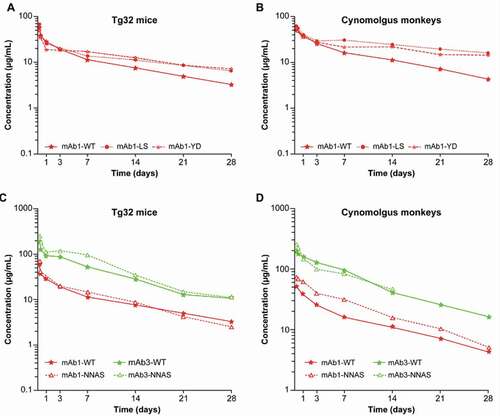
However, for some mAbs the effect of the LS mutation of the Fc region to increase plasma elimination half-life compared with their WT counterpart is not obvious, mainly when the elimination half-life of the WT mAb is already long. For mAb2, either in Tg32 mice or in cynomolgus monkeys, the increase in the elimination half-life due to the LS mutation was not significant, with an increase lower than 1.2-fold despite a 5-fold higher binding affinity to FcRn ().
We also assessed the influence of the NNAS mutation on the PK profile of mAb1 and mAb3. As illustrated in ,), for the 2 backbones the PK profiles of the WT mAb and NNAS-mutated corresponding mAb were parallel and superimposed in Tg32 mice and cynomolgus monkeys. No influence of the NNAS mutation on PK behavior was seen.
Correlation of linear clearance of antibodies and in vitro data
Clearance values depicted in ) represent the linear CL observed for each antibody, independently of any TMDD. For WT mAbs, the linear CL values ranged from 3.2 to 8.2 mL/day/kg in Tg32 mice and from 3.7 to 6.6 mL/day/kg in cynomolgus monkeys. For Fc-mutated mAbs and bi-Abs/tri-Abs, the linear CL values ranged from 2.0 to 16.2 mL/day/kg in Tg32 mice and from 3.2 to 16.2 mL/day/kg in cynomolgus monkeys. The highest CL was observed for bi-Abs/tri-Abs, with a 2- to 5-fold higher CL compared with the comparison mAbs. The very high CL of the bi-Ab4 in cynomolgus monkeys was removed from the comparisons because of the strong additional target-mediated CL demonstrated in this species, without any possibility to increase the dose for target saturation because of safety issues.
Figure 3. (a) Interspecies clearance comparison of the panel of mAbs and bi-Abs/tri-Abs tested. (b) Relationship between antibody clearance in cynomolgus monkeys/Tg32 mice and binding affinity (KD pH6) for human FcRn. Abbreviations: bi-Ab = bispecific antibody, FcRn = neonatal Fc receptor, KD = dissociation constant, mAb = monoclonal antibody, tri-Ab = trispecific antibody, WT = wild type
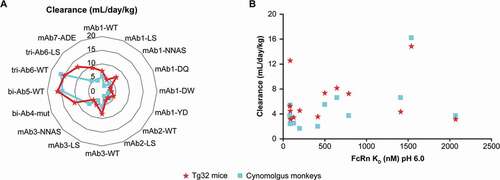
We evaluated the relationship between antibody CL in cynomolgus monkeys/Tg32 mice and binding affinity (KD pH6) for hFcRn ()). Our results show that the highest CL observed for some antibodies was not due to an altered low pH binding affinity to hFcRn. This was true in both Tg32 mice and cynomolgus monkeys.
Correlation of Tg32 mice and cynomolgus monkey half-lives
A significant correlation of the elimination half-life between Tg32 mice and the cynomolgus monkeys was shown for the panel of 16 antibodies tested in (r = 0.91; p < .001; )), suggesting excellent potential for the Tg32 mice to be used instead of NHPs for research drug testing, not only for WT mAbs but also for Fc-engineered mAbs, bi-Abs, and tri-Abs.
Figure 4. Correlation of antibodies’ (a) elimination half-lives and (b) clearance between homozygous Tg32 mice and cynomolgus monkeys
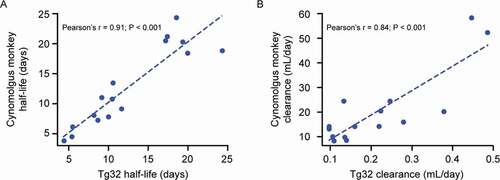
A slightly higher CL (on average 1.5-fold higher) was observed in Tg32 mice compared with cynomolgus monkeys, ranging from 0.7- to 2.3-fold. Nevertheless, the Pearson correlation coefficient still showed that the relationship was significant (r = 0.84; p < .001; )).
Allometric scaling from Tg32 mice to cynomolgus monkeys
We estimated the allometric exponents to scale CL and volume of distribution at steady state (Vss) from Tg32 mice to cynomolgus monkeys for WT mAbs and for Fc-mutated antibodies and bi-Abs/tri-Abs. All the CL and Vss data described in were used for the estimation. As shown in , the allometric exponent for CL, estimated to be 0.97 with WT mAbs, was estimated to be 0.91 for all WT or Fc-mutated antibodies. For Vss, this exponent was estimated to be 0.96 for WT mAbs and 0.93 for all other antibodies. As shown in , all of the predicted Vss or CL values in cynomolgus monkeys using those exponents were within 2-fold of the observed values.
Figure 5. Estimation of α and β allometric exponents for CL and Vss, respectively, to scale from Tg32 mice to cynomolgus monkeys (NHP) using the equation YNHP = YTg32 * (body weightNHP/body weightTg32)α, β, γ. Abbreviations: bi-Ab = bispecific antibody, CL = clearance, NHP = non-human primate, tri-Ab = trispecific antibody, Vss = volume of distribution at steady state, WT = wild type, Y = variable of interest
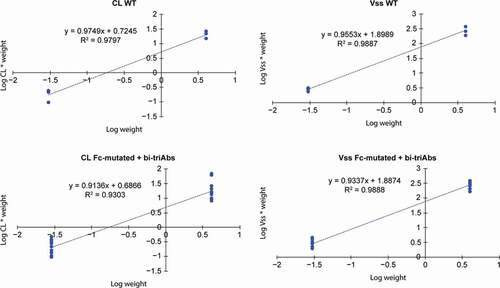
Figure 6. Ratios of predicted versus observed cynomolgus monkey Vss and CL values for the antibodies shown in Table 1. Only antibodies administered with a saturating dose to assess linear pharmacokinetics are presented; therefore, the bi-Ab4 antibody was excluded from the analysis. The predicted values were determined using the allometric equation with the α (for CL) or β (for Vss) allometric exponent estimated in Figure 5. Abbreviations: bi-Ab = bispecific antibody, CL = clearance, mAb = monoclonal antibody, tri-Ab = trispecific antibody, Vss = volume of distribution at steady state, WT = wild type
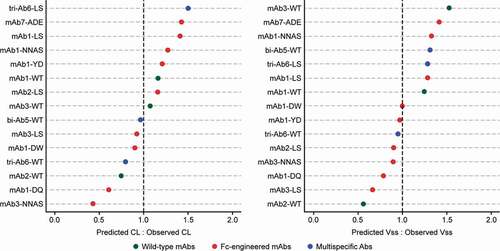
The accuracy and precision of the overall prediction of CL and Vss were also examined in our study, and the results suggest that both these parameters are well predicted, with an average fold-error (afe) of 0.97 and an average absolute fold-error (aafe) of 1.8 for CL, and afe of 1.08 and aafe of 1.08 for Vss.
Discussion
Ideally, development of an Fc mutant for potential clinical testing should balance optimal FcRn binding with intact or attenuated effector functions, depending on the clinical goal.Citation10,Citation20 Other mutations of the Fc region of antibodies besides FcRn are therefore of interest; indeed, the engineering of the Fc part of IgG to decrease the affinity to FcγRs on immune cells has been reported to reduce effector functions such as ADCC.Citation20 We showed that while mutations enhancing FcRn binding changed the PK behavior by increasing the elimination half-life by around 2-fold, the Fc-silent mutation NNAS (which decreases binding to FcγRIIIa as determined by in vitro assays []) did not exert any effect on the PK behavior of the tested antibody format. The lack of impact on plasma PK of the NNAS mutation in Tg32 mice and cynomolgus monkeys is a novel finding. For NNAS molecules, no binding to the FcγRIIIa receptor (and other FcγR) was detectable (silent backbone) and the pI value was heterogeneous. Based on their FcRn binding properties alone, NNAS would be expected to maintain a WT-like PK profile in the in vivo models, while DQ, DW, YD, LS, and the other FcRn-binding variants would contribute to a prolonged plasma PK.Citation18 In summary, the Tg32 hFcRn mouse model is well suited to screen the absence of impact of the NNAS mutation on the PK profile of mAbs as also seen in cynomolgus monkeys.
Although immunogenicity of a humanized antibody in NHPs often does not reflect the situation in humans,Citation34 however and importantly, the high incidence (in around 1 of 3 animals) of ADA formation, which we suspect from the characteristic sharp drop in concentration in cynomolgus monkeys – even after a single dose – does affect the reliability of antibody’s PK analysis in this species.Citation22,Citation34 The absence of ADAs in Tg32 mice in contrast with cynomolgus monkeys could therefore help predict PK profiles in humans in the absence of reliable NHP data, and represents another benefit of the transgenic mouse model. Although some strategies to mitigate the impact of ADA formation in NHPs have been advanced in the literature, these involve either dosing more monkeys than needed and only using data from the ADA-negative monkeys, or selective prescreening,Citation22 two options that are not always successful and that each undermine one of the rationales for using NHPs in the first place.
With respect to TMDD, the prediction of the PK profile from Tg32 mice in cynomolgus monkeys at a dose that saturates the target (linear CL), possible with the scaling factor determined in our experiment, can enable determination of a potential TMDD at a lower dose if observed data show a much higher CL than the one predicted (i.e., bi-Ab4). Information about both linear and nonlinear components (TMDD) of the CLs in NHPs enables further development of mechanistic PK/TMDD models in these species, which allow more reliable PK extrapolation in humans. These PK models can be further used to propose doses and regimens for a greater probability of success in the clinic. Although we did confirm an increased half-life for the LS mutation for mAb1, mAb3, and tri-Ab6, this was not confirmed for mAb2. It is possible that WT antibodies that already have long elimination half-lives, such as mAb2, make it difficult to further increase the half-life due to certain Fc mutations.
One of our findings, which has been confirmed in other studies,Citation35 is that in vitro data alone cannot be relied upon to select the highest elimination half-life of an antibody. The poor relationship is because, even though KD, for example, is a key parameter, affinity to FcRn is not the only factor that affects the linear catabolic CL and elimination half-life of antibodies. Changes in CL are multifactorial, and the quality attributes, diverse receptor binding, or physicochemical features of the mAb molecule (e.g., hydrophobicity, charge) can contribute to nonspecific off-target CL mechanisms possibly influencing PK.Citation32,Citation36 For example, the charges of mAbs can have a substantial impact on their PK and disposition,Citation37 and it has been demonstrated that lowering the pI of an antibody can result in significantly increased half-life and reduced CL.Citation38 A large positive cluster in the variable region might increase the nonspecific binding to the extracellular matrix and subsequently increase the CL. In our experiment, if, for instance, we rank the WT mAbs tested based on in vitro affinity for FcRn binding from the highest KD to the lowest, the ranking is mAb1> mAb2> mAb3. If we consider the pI, the ranking becomes mAb3> mAb2> mAb1 (). Yet in cynomolgus monkeys and Tg32 mice, the half-lives were mAb2> mAb1> mAb3, with mAb2 having the longest half-life, close to 2-fold higher than the others (). Thus, a single in vitro parameter in isolation cannot be considered when predicting PK behavior.
The usefulness of transgenic mouse models for PK profiling of mAbs has been shown in a series of different studies.Citation18,Citation30–32,Citation39 We report here for the first time that hFcRn transgenic mouse models – in particular, Tg32 mice – are highly sensitive for differentiating PK of mAbs and multispecific antibodies compared with a NHP model. Our results confirm literature reports of a differential impact in hFcRn transgenic mice of various antibodies on PK behavior depending on the antibodies’ antigen-binding fragments (Fabs).Citation18,Citation30–32,Citation39,Citation39 Overall, our data suggest Tg32 mice are a better model than NHPs for differentiating the PK of variants during the lead optimization of Fc-engineered mAbs and multispecific antibodies. Ethical concerns, the large quantity of compounds required at the lead optimization stage, and the high probability of development in NHPs of ADAs against humanized antibodies contribute to this view.
Based on data available when the study commenced, we analyzed 2 strains of mice containing the hFcRn transgene: Tg32 and Tg276. IgGs in homozygous Tg276 mice have been shown to have shorter half-lives than in homozygous Tg32 mice.Citation27 Although other studies have found Tg276 mice to be useful in distinguishing the PK of mAbs,Citation39 we found that our model of homozygous Tg276 mice was not appropriate for accurately estimating the elimination half-life and CL of antibodies in NHPs (). Given that NHPs are good predictors of PK parameters in humans, and the large differences we observed between Tg276 mice and cynomolgus monkeys, the Tg276 mouse model is not considered appropriate for predicting human PK profiles. In contrast to the Tg32 strain that uses the endogenous human promoter, in the Tg276 strain expression of the human FcRn is driven by a CAG promoter inducing high levels of receptor expression. The high and unspecific expression of FcRn in Tg276 might explain some of the results observed in this strain.
Our results add to a growing body of evidence attesting to the utility of the Tg32 mouse model for accurate PK prediction and scaling in NHPs or humans. Previously, we engineered several of the novel Fc variants used in this study, showing them to have improved PK properties in both Tg32 mice and cynomolgus monkeys.Citation18 Tam et al.Citation32 found that hemizygous Tg32 mice could serve as a useful model for PK predictions of human antibodies in NHPs, while Avery et al.Citation30 reported homozygous Tg32 to more accurately predict human CL for a wide range of WT mAbs than did hemizygous Tg32 mice, NHPs, or WT mice. Betts et al.Citation31 found good results with Tg32 mice in predicting linear human PK of mAbs, leading the authors to suggest replacing use of cynomolgus monkeys with Tg32 mice for human PK predictions of mAbs. In contrast with the Avery et al. and Betts et al. studies, however, by comparing Tg32 mice with cynomolgus monkeys at the same time we mitigated any concerns regarding the use of historical data.
We found that CL and Vss in cynomolgus monkeys might be accurately predicted from homozygous Tg32 mouse data, using simplified allometric scaling with the recommended exponent of 0.91 for CL and 0.93 for Vss, which is useful, among other things, for guiding the design of further pharmacology and safety studies in NHPs. These findings are consistent with previous conclusions that, for some therapeutic proteins, CL values follow well-defined, size-related physiologic relationships.Citation40 The same approach with Tg32 mice also gives a good prediction of human PK parameters; the allometric exponents to scale from preclinical species to humans for CL were 0.81 for NHP and 0.90 for Tg32 mice, and the V1/V2 values were 1.04/1.07 for NHP and 0.97/0.93 for Tg32 mice.Citation31 The lack of human data on engineered antibodies (notwithstanding some promising new clinical results for multispecific antibodies [data not shown]) did not allow us to make this comparison to scale from Tg32 mice or NHPs to humans, as has been done in the literature for WT mAbs. Moreover, because our results showed that, regardless of the type of construct, the interspecies allometric exponent (from Tg32 mice to NHPs) is in the same range (estimated at 0.91 for CL), this indicates the potential of the allometric exponent described in the literature to predict human CL of mAbs for Fc-engineered antibodies and bi-Abs/tri-Abs.
Although we do not yet have clinical PK data for our investigational antibodies, and therefore cannot perform an interspecies scaling for all investigated antibodies, the comparison of the half-lives of bevacizumab and adalimumab in humans with that in mice and monkeys shows that they are comparable (Supplementary Table 1). The mean elimination half-lives of bevacizumab and adalimumab in humans are slightly higher than their half-lives in monkeys and Tg32 mice, but considering the variability of half-lives in human subjects, the animal data are at an acceptable level and provide a good estimate of the human PK profile. This finding is in agreement with previous reportsCitation31,Citation39 and supports the suggestion of the Tg32 mouse model being a potential replacement for cynomolgus monkey for studies of human PK predictions of mAbs.Citation39
Overall, our analysis includes a limited number of multispecific antibodies tested, mainly because of limited availability. However, platforms for creating higher-order bi-Abs/tri-Abs are entering drug discovery research, and hence will allow future work to expand the panel and correlate results with clinical data to confirm the relevance of the Tg32 model to directly predict human PK profiles.
In conclusion, this study provides the first PK profiling of multispecific antibodies with various backbones and Fc mutations in hFcRn transgenic mice compared with NHPs. The PK evaluation of engineered mAbs and multispecific antibodies in the transgenic rodent and NHP models showed that FcRn-binding mutations improved the plasma half-lives of the engineered mAbs and multispecific antibodies, while glycan engineering to eliminate effector function did not affect the PK compared with WT controls. Further, we showed that the Tg32 homozygous mouse model can distinguish PK differences in mAbs and multispecific antibodies containing Fc-backbone mutations in the same range of order as in cynomolgus monkeys. Tg32 mice homozygous for the FcRn transgene may serve as convenient, cost-effective surrogates for in vivo prediction of humanized IgG-antibody half-life and CL in NHPs. These results could facilitate a screening strategy to select antibodies in Tg32 mice with the most favorable PK characteristics, and could be helpful to characterize the PK behavior of the most promising candidates in NHPs. By extension, a first estimation of half-life and linear CL in humans can be directly deduced from Tg32 mice, not only for mAbs but for all antibodies. Our data could enable substantial acceleration of the delivery of novel protein-based therapeutics.
Our research, along with other recent studies, clearly shows the benefit of Tg32 mice for PK profiling of mAbs. However, efforts to further improve the existing models are underway, aiming at incorporation of further Fcγ receptors or expression of the human IgG repertoire to overcome some of the drawbacks.Citation20 But methodologically sound research and further studies are needed to evaluate and show the benefit of such improved models for PK analysis of mono- and particularly multispecific antibodies.
Materials and methods
Animal experiments
All in vivo studies were conducted in compliance with the Sanofi institutional animal care policy and in accordance with the Helsinki Declaration of 1975. The monkey and mouse studies were approved by the French “Ministère de l’Enseignement Superieur et de la Recherche” and the German “Regierungspraesidium Darmstadt.”
PK studies conducted in hFcRn transgenic mice
The mice experiments were performed in transgenic Tg32 (B6.Cg-FcgrttmCitation1Dcr Tg(FCGRT)32Dcr/DcrJ) and Tg276 (B6.Cg-FcgrttmCitation1Dcr Tg(FCGRT)276Dcr/DcrJ) mice derived from C57BL/6 mice and purchased from The Jackson Laboratory (Bar Harbor, Maine). FcRn-/- hFcRn (line 32) Tg mice carry a null mutation for the mouse gene and a transgene expressing the hFcRn α-chain transgene under the control of its natural human promoter. FcRn-/- hFcRn (line 276) Tg mice carry a null mutation for the mouse gene and a transgene expressing the hFcRn α-chain under the control of the ubiquitous CAG promoter.
All mice were treatment-naive females between the ages of 8 and 12 weeks at study start. For dosing, the antibodies were prepared in a 10 mM histidine, 150 mM NaCl, pH 6.0 formulation buffer and administered as single intravenous doses of 2.5 mg/kg into the tail vein with a dose volume of 5 mL/kg, with the exception of adalimumab, which was dosed at 10 mg/kg to saturate target binding, thus mitigating any TMDD. Bi-Ab5 and mAb7 were dosed at 3 and 5 mg/kg, respectively, for specific internal project support needs. A total of 6 animal replicates were evaluated for each antibody utilizing a serial sampling approach (0.08, 4, 24, 72, 168, 336, 504 and 672 hours) across the study duration of 28 days. Blood samples (20 µL) were collected by sampling time. At each time points, plasma samples were pooled to obtain enough plasma volume for quantification by LC MS/MS. Once collected, blood samples were centrifuged at 4°C for 10 minutes at 1500 g and stored at −80°C until analysis.
PK studies conducted in cynomolgus monkeys
Pharmacokinetic studies were conducted in cynomolgus monkeys obtained from Noveprim Group (Mahebourg, Mauritius). All animals were males aged between 1 and 3 years at study start, weighing between 3 and 5 kg and housed in groups on a litter of corn cob and pine chips mixture. Just before administration, animals were separated and single-housed to the end of the sampling time. The provided food was 107 C (approximately 150 g/day/animal) from SAFE plus one piece of fresh fruit or vegetable, and delicacies every day. The animals had ad libitum access to filtered mains water. For dosing, all antibodies were prepared in a 10 mM histidine, 150 mM NaCl, pH 6.0 formulation buffer, and dosed at 2.5 mg/kg as a single intravenous administration with a dose volume of 1.5 mL/kg, with the exception of adalimumab, which was dosed at 10 mg/kg to saturate target binding, thus mitigating any TMDD. Bi-Ab4 was administered at a lower dose, that is, 0.003 mg/kg, for safety reasons. Bi-Ab5 and mAb7 were dosed at 3 and 5 mg/kg, respectively, for specific internal project support needs. A total of 3 animal replicates were evaluated for each antibody utilizing a serial sampling approach across the study duration of 28 days. The sampling time was 0.003, 0.17, 1, 3, 7, 14, 21, and 28 days post-dosing.
Blood samples (0.5 mL) were collected by venipuncture of the saphenous vein at 8 sampling times post-dosing. Once collected, blood samples were centrifuged at 4°C for 10 minutes at 1500 g and stored at −80°C until analysis.
PK analysis
Plasma concentrations of antibodies were determined using a generic liquid chromatography-mass spectrometry (LC-MS)/MS approach described below. PK parameters were determined from individual animal data in monkeys and from a pooled concentration per sampling time for Tg32 or Tg276 mice, using non-compartmental analysis in Phoenix WinNonlin version 6.4 (Certara L.P.).
PK profiles from an animal that showed a sharp drop in concentration, typical of ADA interference, were excluded from PK calculations. The criterion for exclusion was tlast shorter than 14 days with a sharp drop in concentration between day 7 and day 14, indicating an aberrant change in the drug concentration-time profile with a sudden change in the elimination slope. This is commonly used in preclinical PK analyses when the drug target is not expressed in the animal model under investigation. Therefore, 1 animal has been excluded for mAb1 NNAS, 1 for mAb1 DQ, 1 for mAb2LS, 1 for triAb6 WT, 1 for triAb6LS and 1 for mAb3 NNAS. Globally, 6, over 48 NHPs used in total, were excluded from the PK analysis.
For evaluable animals, terminal data points (D21 and/or D28) in PK profiles, that were presumed to be affected by ADAs, showing a clear change in the slope of elimination, avoiding the use of 3 sampling time for t1/2 estimation, were excluded from PK analysis
Plasma quantitation of antibodies by LC-MS/MS and LBA
The concentration of each antibody at each time point was determined by a bottom-up LC-MS/MS assay, except for bi-Ab4 which was determined by an exploratory ligand-binding assay (LBA), because of the low dose administered in monkeys to avoid toxicity.
LC-MS/MS method: After precipitation of a plasma aliquot, the plasma pellet was subjected to protein denaturation, reduction, alkylation, trypsin digestion, and solid-phase extraction prior to analysis of surrogate peptides. The surrogate peptides, belonging to Fab or Fc regions, were selected for each antibody for quantification, depending on their selectivity and response factor. Calibration standards were prepared by spiking the antibody into the plasma at 1.00, 2.00, 5.00, 10.0, 20.0, 50.0, 100, 200, and 400 µg/mL. Peptide separation was performed on a Waters Acquity UPLC system with a reverse phase XBridge BEH C18 column (2.1x150 mm, 3.5 µM, 300 Å, Waters) at a flow rate of 300 µL/min in a step-wise gradient of 0.1% formic acid in water and 0.1% formic acid in acetonitrile. For detection, a Sciex API5500 mass spectrometer was used in positive ion mode, with the source temperature at 700°C, the ionspray voltage at 5500 V, curtain and nebulizer gases at 40, and the collision gas at mid. Dwell times were 20 ms and the entrance potential was 10 V for each transition. The multiple reaction-monitoring transitions for 2 unique surrogate peptides of the antibody were used for concentration determination relative to the standards and controls, using the peak area from the MQIII integration algorithm of the Analyst software.
LBA method: The assay format used was a sandwich immunoassay running on a SMCxPro platform.
Samples wre incubated in solution phase with magnetic streptavidin-beads pre-coated with biotin rabbit purified polyclonal antibodies against one of the binding sites (targeted antigen A) of the bi-Ab4 molecule. Next unbound materials were washed away during the subsequent exchange and wash steps (using an automated liquid handling device [Bravo Agilent]). Then fluor-labeled target B tracer was added to each well, which binds to the molecule captured on beads. During the following wash step, the beads were transferred to a clean plate (using an automated liquid handling device [Bravo Agilent]). Elution buffer was then added and the bound antibody sandwich chemically dissociated from the beads surface. Finally, these eluted antibodies were transferred to a 384-well microplate (using an automated liquid handling device [Bravo Agilent]) and loaded into the SMCxPro platform where the labeled molecules were detected and counted. The number of fluor-labeled detection antibodies counted was directly proportional to the amount of bi-Ab4 molecule in the sample when captured. Fluorescence data measured by the SMCxPro platform were plotted against nominal standard concentrations to construct the standard calibration curves. Concentration values were interpolated from these curves using a 1/Y2 (1/squared response) weighted 4PL parameters regression fitting model.
Allometric scaling
Interspecies scaling is often used for the prediction of human PK (linear component) from preclinical data for mAbs. The use of cynomolgus monkeys as a single-species approach with a fixed exponent results in good predictions.Citation41–47
Allometric exponents α, β, and γ, estimated for CL, Vss, and elimination half-life, respectively, to scale from Tg32 mice (using a mean weight of 0.03 kg for scaling) to cynomolgus monkeys (NHP, using a mean weight of 4 kg for scaling), were determined using the following equation (BW = body weight, Y = variable of interest):
By plotting the log weight (species) versus the log CL (or Vss)/species weight for WT mAbs (n = 3 each for Tg32 mice and for cynomolgus monkeys), we estimated the allometric exponent for WT mAbs as reference and did a similar plot with Fc-mutated antibodies and bi-Abs/tri-Abs to estimate the exponent for those modalities.
Accuracy and precision assessment
Assessment of the accuracy and the precision of the overall prediction of CL and Vss from Tg32 mice to cynomolgus monkeys using the estimated allometric exponent was done by the calculation of the afe and the aafe using the following equations:
FcRn proteins, antibody expression and purification
The following proteins were expressed and isolated in-house: rat FcRn (rFcRn, UniProt: P1359, p51 subunit: residues 23–298; UniProt: P07151, β2-m: residues 21–119); biotinylated cynomolgus FcRn (UniProt: Q8SPV9, p51 subunit: residues 24–297 with a C-terminal Avi-tag; UniProt: Q8SPW0, β2-m: residues 21–119); biotinylated hFcRn (hFcRn, UniProt: P55899, p51 subunit: residues 24–297 with a C-terminal Avi-tag; UniProt: P61769, β2-m: residues 21–119). NNAS is a Sanofi proprietary mutation. The ADE mutation (G239A/S242D/I335E) is an ADCC-enhanced backbone.
From the first set of 10 molecules, 2 corresponded to WT mAbs, 6 were mAbs engineered through the Fc region (mAb1 [bevacizumab] and mAb2), 1 was a WT tri-Ab (tri-Ab6-WT), and 1 was a Fc-mutated tri-Ab (tri-Ab6-LS). The molecules were administered under controlled and highly standardized experimental procedures to allow data comparison and differentiation. All antibodies tested in our study are IGg1 type.
Details are as previously described, as are those of FcRn binding kinetics.Citation18
Abbreviations
| aafe | = | average absolute fold-error |
| ADA | = | anti-drug antibody |
| ADCC | = | antibody-dependent cell-mediated cytotoxicity |
| afe | = | average fold-error |
| AUC | = | area under the concentration-time curve, from time zero to infinity |
| bi-Ab | = | bispecific antibody |
| CL | = | clearance |
| Cmax | = | maximum plasma drug concentration |
| CODV | = | cross-over dual variable |
| ELISA | = | enzyme-linked immunosorbent assay |
| Fc | = | crystallizable fragment of antibody |
| FcγR | = | Fc gamma receptor |
| FcRn | = | neonatal Fc receptor |
| FL | = | fluorescence |
| hFcRn | = | human/humanized neonatal Fc receptor |
| IgG | = | immunoglobulin G |
| LBA | = | ligand-binding assay |
| LOQ | = | limit of quantification |
| mAb | = | monoclonal antibody |
| NHP | = | non-human primate |
| pI | = | isoelectric point |
| PK | = | pharmacokinetics |
| TMDD | = | target-mediated drug disposition |
| tri-Ab | = | trispecific antibody |
| Vss | = | volume of distribution at steady state |
| WT | = | wild type |
Disclosure of interest
DM, CM, TS, IF, JB, BM, PV, AK, SR, JD, and MK are employees of Sanofi. H Q is an employee of Xilio Therapeutics. KR is an employee of Lonza Group LTD.
Supplemental Material
Download MS Word (175.4 KB)Acknowledgments
The authors are grateful to the following individuals for their key contributions to this work: Olivier Pasquier, Norbert Zombori, Marie Hélène Pascual, and Hervé Laplace. Medical writing, funded by Sanofi, was provided by Steven Tresker of Cactus Life Sciences (part of Cactus Communications).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Kaplon H, Reichert JM. Antibodies to watch in 2019. MAbs. 2019;11(2):219–13. doi:https://doi.org/10.1080/19420862.2018.1556465.
- Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18:585–608.
- Kareva I, Zutshi A, Kabilan S. Guiding principles for mechanistic modeling of bispecific antibodies. Prog Biophys Mol Biol. 2018;139:59–72. doi:https://doi.org/10.1016/j.pbiomolbio.2018.08.011.
- Chen Y, Xu Y. Pharmacokinetics of bispecific antibody. Curr Pharmacol Rep. 2017;3:126–37. doi:https://doi.org/10.1007/s40495-017-0090-5.
- Datta-Mannan A, Brown RM, Fitchett J, Heng AR, Balasubramaniam D, Pereira J, Croy JE. Modulation of the biophysical properties of bifunctional antibodies as a strategy for mitigating poor pharmacokinetics. Biochemistry. 2019;58(28):3116–32. doi:https://doi.org/10.1021/acs.biochem.9b00074.
- Ma M, Colletti K, Yang TY, Leung S, Pederson S, Hottenstein CS, Xu X, Warrino D, Wakshull E. Bioanalytical challenges and unique considerations to support pharmacokinetic characterization of bispecific biotherapeutics. Bioanalysis. 2019;11(5):427–35. doi:https://doi.org/10.4155/bio-2018-0146.
- Shah DK. Pharmacokinetic and pharmacodynamic considerations for the next generation protein therapeutics. J Pharmacokinet Pharmacodyn. 2015;42(5):553–71. doi:https://doi.org/10.1007/s10928-015-9447-8.
- Avery LB, Wade J, Wang M, Tam A, King A, Piche-Nicholas N, Kavosi MS, Penn S, Cirelli D, Kurz JC, et al. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. MAbs. 2018;10(2):244–55. doi:https://doi.org/10.1080/19420862.2017.1417718.
- Liu L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell. 2018;9(1):15–32. doi:https://doi.org/10.1007/s13238-017-0408-4.
- Borrok MJ, Wu Y, Beyaz N, Yu XQ, Oganesyan V, Dall’Acqua WF, Tsui P. pH-dependent binding engineering reveals an FcRn affinity threshold that governs IgG recycling. J Biol Chem. 2015;290(7):4282–90. doi:https://doi.org/10.1074/jbc.M114.603712.
- Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. 2006;281(33):23514–24. doi:https://doi.org/10.1074/jbc.M604292200.
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7(9):715–25. doi:https://doi.org/10.1038/nri2155.
- Datta-Mannan A, Witcher DR, Lu J, Wroblewski VJ. Influence of improved FcRn binding on the subcutaneous bioavailability of monoclonal antibodies in cynomolgus monkeys. MAbs. 2012;4(2):267–73. doi:https://doi.org/10.4161/mabs.4.2.19364.
- Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):576–88. doi:https://doi.org/10.1002/psp4.12224.
- Ward ES, Zhou J, Ghetie V, Ober RJ. Evidence to support the cellular mechanism involved in serum IgG homeostasis in humans. Int Immunol. 2003;15(2):187–95. doi:https://doi.org/10.1093/intimm/dxg018.
- Robbie GJ, Criste R, Dall’acqua WF, Jensen K, Patel NK, Losonsky GA, Griffin MP. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother. 2013;57(12):6147–53. doi:https://doi.org/10.1128/AAC.01285-13.
- Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IWL, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28(2):157–59. doi:https://doi.org/10.1038/nbt.1601.
- Mackness BC, Jaworski JA, Boudanova E, Park A, Valente D, Mauriac C, Pasquier O, Schmidt T, Kabiri M, Kandira A, et al. Antibody Fc engineering for enhanced neonatal Fc receptor binding and prolonged circulation half-life. MAbs. 2019;11(7):1276–88. doi:https://doi.org/10.1080/19420862.2019.1633883.
- Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. 2018;9(1):63–73. doi:https://doi.org/10.1007/s13238-017-0473-8.
- Lee CH, Kang TH, Godon O, Watanabe M, Delidakis G, Gillis CM, Sterlin D, Hardy D, Cogné M, Macdonald LE, et al. An engineered human Fc domain that behaves like a pH-toggle switch for ultra-long circulation persistence. Nat Commun. 2019;10(1):5031. doi:https://doi.org/10.1038/s41467-019-13108-2.
- Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov. 2018;17:197–223.
- Han C, Gunn GR, Marini JC, Shankar G, Han Hsu H, Davis HM. Pharmacokinetics and immunogenicity investigation of a human anti-interleukin-17 monoclonal antibody in non-naïve cynomolgus monkeys. Drug Metab Dispos. 2015;43(5):762–70. doi:https://doi.org/10.1124/dmd.114.062679.
- Vaccaro C, Bawdon R, Wanjie S, Ober RJ, Ward ES. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc Natl Acad Sci U S A. 2006;103(49):18709–14. doi:https://doi.org/10.1073/pnas.0606304103.
- Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Jiang W, Wroblewski VJ. Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: relationship to pharmacokinetics in mice and primates. Drug Metab Dispos. 2007;35(1):86–94. doi:https://doi.org/10.1124/dmd.106.011734.
- Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol. 2001;13(12):1551–59. doi:https://doi.org/10.1093/intimm/13.12.1551.
- Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol. 2002;169(9):5171–80. doi:https://doi.org/10.4049/jimmunol.169.9.5171.
- Proetzel G, Roopenian DC. Humanized FcRn mouse models for evaluating pharmacokinetics of human IgG antibodies. Methods. 2014;65(1):148–53. doi:https://doi.org/10.1016/j.ymeth.2013.07.005.
- Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, Presta LG, Meng YG, Roopenian DC. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18(12):1759–69. doi:https://doi.org/10.1093/intimm/dxl110.
- Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, Petkova S, Avanessian L, Choi EY, Shaffer DJ, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J Immunol. 2003;170(7):3528–33. doi:https://doi.org/10.4049/jimmunol.170.7.3528.
- Avery LB, Wang M, Kavosi MS, Joyce A, Kurz JC, Fan YY, Dowty ME, Zhang M, Zhang Y, Cheng A, et al. Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. MAbs. 2016;8(6):1064–78. doi:https://doi.org/10.1080/19420862.2016.1193660.
- Betts A, Keunecke A, van Steeg TJ, van der Graaf PH, Avery LB, Jones H, Berkhout J. Linear pharmacokinetic parameters for monoclonal antibodies are similar within a species and across different pharmacological targets: A comparison between human, cynomolgus monkey and hFcRn Tg32 transgenic mouse using a population-modeling approach. MAbs. 2018;10(5):751–64. doi:https://doi.org/10.1080/19420862.2018.1462429.
- Tam SH, McCarthy SG, Brosnan K, Goldberg KM, Scallon BJ. Correlations between pharmacokinetics of IgG antibodies in primates vs FcRn-transgenic mice reveal a rodent model with predictive capabilities. MAbs. 2013;5:397–405.
- Steinmetz A, Vallée F, Beil C, Lange C, Baurin N, Beninga J, Capdevila C, Corvey C, Dupuy A, Ferrari P, et al. CODV-Ig, a universal bispecific tetravalent and multifunctional immunoglobulin format for medical applications. MAbs. 2016;8(5):867–78. doi:https://doi.org/10.1080/19420862.2016.1162932.
- van Meer PJK, Kooijman M, Brinks V, Gispen-de Wied CC, Silva-Lima B, Moors EHM, Schellekens H. Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. MAbs. 2013;5(5):810–16. doi:https://doi.org/10.4161/mabs.25234.
- Datta-Mannan A, Chow CK, Dickinson C, Driver D, Lu J, Witcher DR, Wroblewski VJ. FcRn affinity-pharmacokinetic relationship of five human IgG4 antibodies engineered for improved in vitro FcRn binding properties in cynomolgus monkeys. Drug Metab Dispos. 2012;40(8):1545–55. doi:https://doi.org/10.1124/dmd.112.045864.
- Hötzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. A strategy for risk mitigation of antibodies with fast clearance. MAbs. 2012;4(6):753–60. doi:https://doi.org/10.4161/mabs.22189.
- Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem. 2010;21(12):2153–63. doi:https://doi.org/10.1021/bc100261d.
- Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel. 2010;23(5):385–92. doi:https://doi.org/10.1093/protein/gzq009.
- Haraya K, Tachibana T, Nanami M, Ishigai M. Application of human FcRn transgenic mice as a pharmacokinetic screening tool of monoclonal antibody. Xenobiotica. 2014;44(12):1127–34. doi:https://doi.org/10.3109/00498254.2014.941963.
- Mahmood I. Interspecies scaling of protein drugs: prediction of clearance from animals to humans. J Pharm Sci. 2004;93(1):177–85. doi:https://doi.org/10.1002/jps.10531.
- Wang W, Prueksaritanont T. Prediction of human clearance of therapeutic proteins: simple allometric scaling method revisited. Biopharm Drug Dispos. 2010;31:253–63.
- Kazazi-Hyseni F, Beijnen JH, Schellens JHM. Bevacizumab. Oncologist. 2010;15(8):819–25. doi:https://doi.org/10.1634/theoncologist.2009-0317.
- Lu JF, Bruno R, Eppler S, Novotny W, Lum B, Gaudreault J. Clinical pharmacokinetics of bevacizumab in patients with solid tumors. Cancer Chemother Pharmacol. 2008;62(5):779–86. doi:https://doi.org/10.1007/s00280-007-0664-8.
- Food and Drug Administration. Avastin® (bevacizumab). Highlights of prescribing information. 2018 [accessed 2020 Aug 27]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125085s323lbl.pdf.
- Ternant D, Ducourau E, Fuzibet P, Vignault C, Watier H, Lequerré T, Le Loët X, Vittecoq O, Goupille P, Mulleman D, et al. Pharmacokinetics and concentration-effect relationship of adalimumab in rheumatoid arthritis. Br J Clin Pharmacol. 2015;79(2):286–97. doi:https://doi.org/10.1111/bcp.12509.
- Weisman MH, Moreland LW, Furst DE, Weinblatt ME, Keystone EC, Paulus HE, Teoh LS, Velagapudi RB, Noertersheuser PA, Granneman GR, et al. Efficacy, pharmacokinetic, and safety assessment of adalimumab, a fully human anti-tumor necrosis factor-alpha monoclonal antibody, in adults with rheumatoid ar thritis receiving concomitant methotrexate: a pilot study. Clin Ther. 2003;25(6):1700–21. doi:https://doi.org/10.1016/S0149-2918(03)80164-9.
- Food and Drug Administration. Humira® (adalimumab). Highlights of prescribing information. 2020 [accessed 2020 Aug 27]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125057s415lbl.pdf.