
ABSTRACT
Antibody-based delivery of bioactive molecules represents a promising strategy for the improvement of cancer immunotherapy. Here, we describe the generation and characterization of R6N, a novel fully human antibody specific to the alternatively spliced domain D of Tenascin C, which is highly expressed in the stroma of primary tumors and metastasis. The R6N antibody recognized its cognate tumor-associated antigen with identical specificity in mouse and human specimens. Moreover, the antibody was able to selectively localize to solid tumors in vivo as evidenced by immunofluorescence-based biodistribution analysis. Encouraged by these results, we developed a novel fusion protein (termed mIL12-R6N) consisting of the murine interleukin 12 fused to the R6N antibody in homodimeric tandem single-chain variable fragment arrangement. mIL12-R6N exhibited potent antitumor activity in immunodeficient mice bearing SKRC52 renal cell carcinoma, as well as in immunocompetent mice bearing SMA-497 glioma. The experiments presented in this work provide a rationale for possible future applications for the R6N antibody for the treatment of cancer patients.
Introduction
Over the past two decades, antibody-based therapy has become an established and successful strategy to treat hematological and solid malignancies.Citation1–3 The targeted delivery of bioactive payloads (e.g., radionuclides, cytotoxic drugs, cytokines, procoagulant factors) to the tumor environment, by means of antibodies specific to tumor-associated antigens, represents an avenue for the development of selective anti-cancer agents.Citation4–7 In addition to antibodies specific to cell surface antigens (e.g., carcinoembryonic antigen, fibroblast activating protein, carbonic anhydrase IX), markers of tumor angiogenesis expressed in the extra-cellular matrix (ECM) of the tumor microenvironment exemplify attractive molecules for the targeted delivery of therapeutics.Citation8 ECM components are well accessible to antibodies due to their low shedding profile and their abundancy and stability.Citation9 Our group has extensively studied ECM-associated antigens, such as the spliced extra domains A (EDA) and B (EDB) of fibronectinCitation10,Citation11 and Tenascin C (TNC).Citation9,Citation12
In this work, we describe the generation and validation of a new antibody specific to the alternatively spliced domain D of Tenascin C (TNC-D). TNC is a highly conserved glycoprotein, which comprises multiple fibronectin type 3 (FNIII) like domains. A total of eight FNIII domains are constitutionally present in the protein, whereas nine repeats (FNIII A-D) located between FNIII5-6 undergo alternative splicing giving rise to small and large isoforms of TNC.Citation13–15 In vitro assay demonstrates that the alternative splicing of normal cells can be controlled by extracellular pH, whereas malignant cells accumulate large TNC isoforms regardless of the pH of the culture media.Citation16 The large isoform of TNC is physiologically expressed during embryogenesis, but it is not detectable in adult healthy tissues. However, it can be rapidly expressed de novo in response to pathological stress conditions such as chronic inflammation and cancer.Citation16–18 Domain D of Tenascin C is highly conserved between mouse and man,Citation12 thus representing an ideal target for the development of novel fully human monoclonal antibodies, which can be used for preclinical experiments in mice and for subsequent clinical application.
Several antibodies have been raised against Tenascin C, in particular against large Tenascin C isoforms.Citation19–21 The murine antibody BC2 in IgG1 format (specific to an epitope of the alternative spliced A4 domain of TNC) was radiolabeled with 131I or111I and used as an imaging agent in patients with glioma and malignant glioblastoma.Citation22,Citation23 Furthermore, a three-step pre-targeted imaging method consisting of the injection of biotinylated BC2 followed by avidin and (99mTc)PnAO-biotin was tested to detect cerebral gliomas.Citation24 The murine 81C6 antibody (specific to TNC-D) was investigated in Phase 2 clinical trials as 131I radio-conjugate for the treatment of glioma patients.Citation25,Citation26 In order to minimize murine Fc-mediated immunogenicity in patients, the murine variable regions of the 81C6 antibody were grafted in a human IgG2 background.Citation27 The monoclonal chimeric antibody was studied in Phase 1 clinical trials for the treatment of non-Hodgkin lymphoma.Citation28 Sigma Tau (Italy) developed murine antibodies specific to the large (named ST2485) and small (named ST2146) isoform of TNC.Citation29,Citation30 ST2146 (later called tenatumomab) was investigated in a Phase 1 clinical trial as a delivery agent for radionuclides to neoplastic lesions (NCT02602067). An antibody against TNC-D (named P12) had previously been reported by our group. P12 displayed excellent in vitro properties, but sub-optimal in vivo targeting performance.Citation12 For this reason, the isolation of novel antibodies against this target is still needed.
Phage display is one of the most versatile and reliable platforms for the discovery and affinity maturation of monoclonal antibodies, peptide and protein therapeutics.Citation31–33 The large number of clinical-grade antibodies generated by phage display technology (e.g., Humira®, Benlysta®, Lucentis®), suggest the value of this in vitro methodology.Citation34 Phage-derived antibodies directed against ECM components are particularly suited for the delivery of bioactive payloads (e.g., drugs, radionuclides, cytokines) to the tumor site, helping spare normal tissues.Citation35–37 Among the cytokine payloads, interleukin-12 (IL12) is one of the most attractive candidates for tumor therapy because of its unique ability to potently activate natural killer (NK) cells, CD4 + T cells and CD8 + T cells.Citation38,Citation39 We and others have previously described antibody-IL12 fusion proteins,Citation40 and two of these products have moved to clinical trials for the treatment of various types of malignancies (NCT00625768, NCT04303117).
Here we describe the generation by phage display technology of R6N, an antibody selective for the TNC-D. The antibody was capable of recognizing the antigen both in human and mouse specimens and showed encouraging targeting properties in the mouse model of cancer. To investigate the therapeutic potential of R6N for the active delivery of payloads to the tumor site, we fused it to IL12. The resulting novel fusion protein, termed mIL12-R6N, consisted of a murine IL12 sequentially fused to the N-terminus of R6N in homodimeric tandem single-chain variable fragment (scFv) arrangement.Citation41 Preclinical studies were performed with the murine analog of IL12 since the human protein is not able to recognize the cognate receptor in mice.Citation42 mIL12-R6N was able to induce cancer regression in SKRC52 and SMA-497 orthotopic syngeneic tumor-bearing mice, without showing signs of toxicity. The fully human product (IL12-R6N) may find clinical applications for cancer immunotherapy.
Results
Isolation and characterization of antibodies specific to the spliced domain D of TNC
Monoclonal antibodies were isolated from the synthetic human scFv ETH-2 Gold libraryCitation43 by phage display technology. The BirA Substrate Peptide (BSP) was fused to human TNC-D () at the N-terminus of the protein, in order to coat magnetic beads used for the selection (Supplementary Figure S1). The addition of BSP sequence to the antigen allowed a site-specific biotinylation of the antigen.Citation44 After two rounds of panning, the clone L7D (Supplementary Figure S2) was chosen based on the enzyme-linked immunosorbent assay (ELISA) signal and used for further studies (Supplementary Figure S3). L7D in scFv format was expressed in Chinese hamster ovary (CHO) cells and characterized by analytical methods, showing homogeneity in size-exclusion chromatography and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (,c)). Monomeric fractions from gel-filtration experiments were used for affinity measurements. Surface plasmon resonance (SPR) analysis on TNC-D coated chip showed a biphasic dissociation profile, with a first fast dissociation phase followed by a slow dissociation phase. The calculated KD value was 41 nM. ()). To confirm the cross-reactivity of L7D for the mouse antigen, BIAcore analyses were performed on a mouse TNC domains BCD-coated chip ()). A microscopic immunofluorescence analysis on U87 glioblastoma section confirmed that the L7D antibody could recognize its cognate antigen in tissues ()).
Figure 1. Characterization of antibodies against the human spliced isoform D of Tenascin C (hTNC-D)
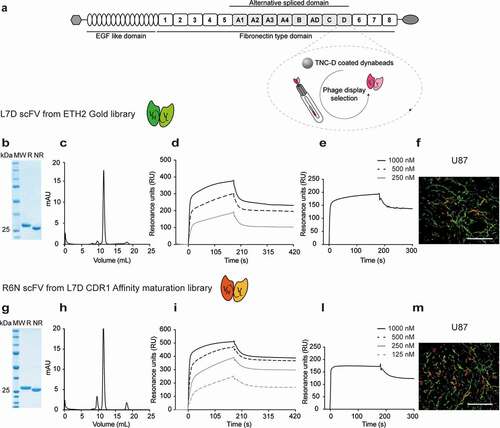
In vitro CDR1 affinity maturation of L7D scFv
The binding properties of the L7D antibody were further improved using a previously described in vitro affinity maturation methodology.Citation11 Sequence variability in the complementarity-determining region 1 (CDR1) loops of the antibody heavy and light chains was introduced. Since residue 88 of the light chain encoded for a potential glycosylation site, the amino acid was mutated into glutamine during the cloning of the library.Citation45 The best clone isolated from affinity maturation library selections was R6N ( and Supplementary Figure S4). BIAcore analysis on TNC-D coated chip showed an improvement in binding properties with an affinity in the two-digit nanomolar range, with a KD value of 24 nM. The cross-reactivity for the mouse antigen was further confirmed by SPR (KD value of 3 nM) ()).
Antigen specificity was evaluated for L7D and R6N scFvs by ELISA. Both antibodies bound to their antigen in a specific manner (TNC-D and TNC domains CD6) and were cross-reactive for the mouse TNC-BCD. As expected, L7D and R6N did not bind to closely-related proteins, including the alternatively-spliced extra-domains A (EDA) and B (EDB) of fibronectin, as well as domain A1 of TNC and recombinant human tumor necrosis factor (Supplementary Figure S5). Furthermore, the antibody specificity for the Domain D of Tenascin C was confirmed by immunofluorescence analysis using R6N IgG2a (Supplementary Figure S6) co-stained with P12 IgG1Citation12 (Supplementary Figure S7) on SKRC52 tissue slides. Both antibodies showed a comparable pattern of staining, thus verifying antigen specificity (Supplementary Figure S8).
Immunofluorescence analysis on xenografts, mouse tissue sections and tissue microarray
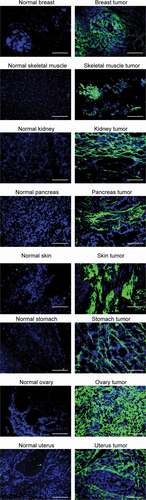
R6N in IgG1 (Supplementary Figure S9) format was studied by immunofluorescence staining on tissue sections. The protein was labeled with fluorescein isothiocyanate (FITC-labeled) and used to stain xenograft tumor models (SKRC52, U87, A431 and A375) and mouse tumor sections (Colon 26, C51, SMA-540 and SMA-497) (). R6N exhibited an intense staining in all sections, confirming the capability of the antibody to recognize both human and mouse antigen in vitro. Moreover, R6N-IgG1-FITC exhibited intense staining in most human solid tumors (particularly ovarian, breast and uterine tumors), but not in healthy adult organs (). Immunofluorescence analysis on brain sections from a glioma patient revealed that the expression of TNC-D and EDB (detected with the L19 antibody) was comparable (Supplementary Figure S10).
Figure 2. Microscopic fluorescence analysis of TNC-D expression on xenografts and tumors of mouse origin section with R6N IgG1-FITC
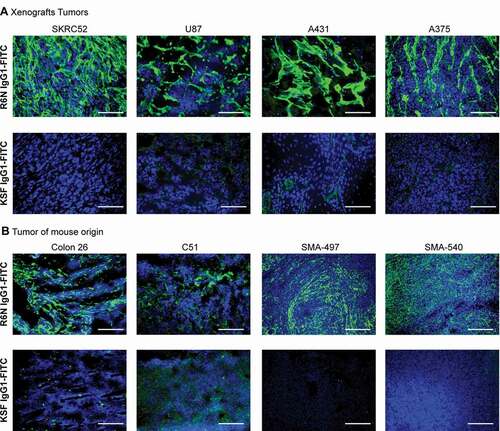
Figure 3. Microscopic fluorescence analysis of TNC-D expression of frozen tumor and normal tissues
Immunofluorescence-based biodistribution analysis
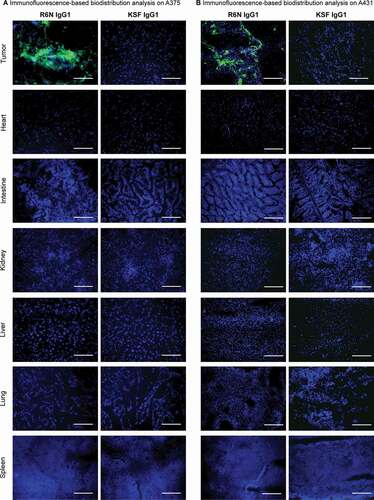
The in vivo tumor-targeting performance of R6N was evaluated through immunofluorescence-based biodistribution in A375 ()) or A431 ()) tumor-bearing mice. Tumor cell lines were inoculated in immunodeficient mice and when the tumor reached a size of 100–250 mm3, R6N in IgG1 (in A375 tumor-bearing mice) or diabody (in A431 tumor-bearing mice) (Supplementary Figure S11) format was injected intravenously (KSF in IgG1 or diabody formats were used as negative control). After 24 hours, mice were sacrificed, organs and tumors were examined by immunofluorescence procedure. R6N in both IgG1 and diabody format localized to tumors, while no detectable antibody was found in the other organs. No preferential accumulation of the KSF antibody was observed in the neoplastic mass, confirming the requirement of TNC-D binding for an active targeting process. Signal quantification validated the expression of TNC-D only in the tumor mass and not in healthy organs (Supplementary Figure 12A). Quantitative biodistribution analysis in immunodeficient mice bearing subcutaneously grafted U87 glioblastoma (Supplementary Figure 13A) or A431 epidermoid carcinoma (Supplementary Figure 13B) confirmed the capability of R6N diabody to localize to solid tumors with a good tumor:blood ratio (11.07 in U87 tumor-bearing mice and 7.15 in A431 tumor-bearing mice).
Figure 4. Immunofluorescence-based biodistribution analysis with R6N IgG1
Generation of mIL12-R6N and tumor therapies
We first evaluated the potential antitumor activity of R6N in IgG2a and diabody formats in A375 tumor-bearing mice. Both formats without the cytokine were not able to induce any tumor growth retardation (Supplementary Figure S14).
depicts the amino acid sequence and a schematic representation of mIL12-R6N in the format previously described.Citation40 The product was well behaved in biochemical assays and mIL12 retained its biological activity as evidenced by an interferon (IFN)-γ release experiment ()). In vivo therapy studies with mIL12-R6N revealed strong anti-cancer activity in immunodeficient mice, bearing SKRC52 renal cell carcinoma ()) and in immunocompetent mice bearing SMA-497 glioma model ()). Both in xenograft and orthotopic syngeneic cancer models, a sustained tumor growth retardation was observed, with a significant difference compared to the untargeted antibody mIL12-KSF ()). No significant change in body weight was detected during the therapy in SKRC52, indicating good tolerability of the fusion protein at the dose used (Supplementary Figure S15).
Figure 5. Therapy in BALB/c nude mice bearing SKRC52 human renal cell carcinoma and in VM/Dk mice bearing SMA-497 glioma

Discussion
In this work, we describe the generation, in vitro characterization and in vivo targeting properties of a novel antibody specific to domain D of Tenascin C. L7D scFv was isolated by phage display technology from ETH-2 Gold libraryCitation43 and affinity matured in vitro by randomization of CDR1 loops of antibody heavy and light chains. The resulting antibody, R6N, featured an increased affinity for the target antigen (with a Kd value in the two digits nanomolar range). Unlike the F16 antibody, specific to the domain A1 of Tenascin C discovered by Brack and colleagues,Citation12 R6N had the advantage of being cross-reactive due to the high sequence homology between the mouse and human antigen,Citation46 as confirmed by SPR analysis (-e)) and immunofluorescence studies (). Cross-reactivity for the mouse antigen facilitates the preclinical evaluation of the antibody’s performance.
R6N was sub-cloned in three different formats, diabody, IgG1 and IgG2a, resulting in bivalent antibodies with improved molecular stability and increased avidity for the cognate antigen. R6N could be expressed in mammalian cells and purified to homogeneity with excellent yield regardless of the format chosen (i.e., scFv, diabody, IgG1 and IgG2a) (Supplementary Figure S6, S9 and S11). Recombinant proteins manufacturing is a critical area in today’s pharmaceutical industry:Citation47 efficient protein expression is essential for the scaling up process, which is required for industrial development.
The in vivo targeting performance of R6N was evaluated by immunofluorescence-based biodistribution analysis ( and ), which showed a positive pattern of staining in the neoplastic region but not in healthy organs, reinforcing the fact that TNC is an attractive target for cancer immunotherapy.
A novel IL12-based immunocytokine was generated and its therapeutic efficacy was evaluated in immunodeficient SKRC52 tumor-bearing mice and in immunocompetent SMA-497 orthotopic syngeneic tumor-bearing mice (). A strong anti-cancer activity was observed in both tumor models, with an improvement of survival in mice treated with R6N antibody.
In an SMA-497 glioma model, the antibody-mediated delivery of mIL12 to the tumor mass clearly enhanced the therapeutic performance of this cytokine, compared to the irrelevant antibody mIL12-KSF ()). Glioblastoma remains one of the most challenging cancer, with still poor prognosis.Citation48 It is thus conceivable, that using a targeted approach with a fully human IL12-based product (Supplementary Figure S16) may provide a benefit to certain groups of patients with malignant gliomas and glioblastomas.
The ability of mIL12-R6N to induce a tumor growth retardation was confirmed also in xenograft model SKRC52 in nude mice ()). In an immunodeficient mice setting, NK cells stimulated by IL12 may play an important role in the tumor rejection process.Citation39,Citation49
In conclusion, we developed an antibody selective for TNC-D with excellent targeting properties confirmed by immunofluorescence-based biodistribution staining. The novel mIL12-R6N fusion protein exhibited potent single-agent activity, leading to tumor growth inhibition in treated mice. The data presented here provide a rationale for the clinical development of this human IL12-R6N fusion protein.
Materials and methods
Cell lines
The human renal cell carcinoma SKRC52 was kindly provided by Professor E. Oosterwijk (Radboud University Nijmegen Medical Center, Nijmegen, the Netherlands). CHO cells, U87 cells, A431 cells, A375 cells, Colon 26 cells and C51 cells were obtained from the ATCC. Cell lines were received between 2018 and 2020, expanded and stored as cryopreserved aliquots in liquid nitrogen. SMA-497 and SMA-540 cells were obtained from Dr. D. Bigner (Duke University Medical Center, Durham, North Carolina, USA) and cultured as previously described.Citation50
Cells were grown according to the supplier’s protocol and kept in culture for no longer than 14 passages. Authentication of the cell lines also including checks of post-freeze viability, growth properties and morphology test for mycoplasma contamination, isoenzyme assay and sterility test was performed by the cell bank before shipment.
Cloning, expression and biotinylation of BSP-hTNC-D
The gene encoding for TNC-D was amplified and cloned into the bacterial expression vector pQE-12 with the BirA target sequence “BirA Substrate Peptide” (BSP), LHHILDAQKMVWNHR, to specifically biotinylate the antigen. Primers were designed to fuse the BSP tag sequence to the N-terminus of human TNC-D. PCR fragments were assembled and cloned into the expression vector by EcoRI and HindIII restriction site as described previously.Citation51 The BSP-hTNC-D protein was expressed in E. coli TG-1 as described before.Citation44,Citation52 The protein biotinylation was carried with BirA enzyme, an E. coli enzyme able to achieve precise biotin modification. The biotinylation was carried in BirA buffer (10 mM Tris pH 7.5, 200 mM NaCl, 5 mM MgCl2) following the protocol described by Fairhead et al.Citation44
In vitro protein characterization
The fusion proteins described here were produced through transient gene expression (TGE) in CHO-S cellsCitation53 and purified from the cell culture medium by protein A Sepharose (Sino Biological) affinity chromatography, dialyzed against phosphate-buffered saline (PBS) and stored in PBS at −80°C. SDS-PAGE was performed with 10% gels under reducing and non-reducing conditions. Purified proteins were analyzed by size-exclusion chromatography using a Superdex 75 increase or 200 increase 10/300 GL column on an ÄKTA FPLC (GE Healthcare, Amersham Biosciences). Affinity measurements were performed by SPR using BIAcoreX100 instrument (BIAcore AB, Uppsala, Sweden) on human TNC-D coated CM5 chip and on mouse TNC domains BCD-coated CM5 chip. Differential scanning fluorimetry was performed on an Applied Biosystem StepOnePlus RT-PCR instrument for R6N diabody and R6N IgG1. Protein Thermal Shift Dye Kit (Thermo Fisher) was used for thermal stability measurements, where the temperature range spanned from 25°C to 95°C with a scan rate of 1°C/min. For electrospray ionization-mass spectrometry (ESI-MS) analysis samples were diluted to 0.1 mg/mL and LC-MS was performed on a Waters Xevo G2XS Qtof instrument (ESI-ToFMS) coupled to a Waters Acquity UPLC H-Class System using a 2.1 Å ~ 50 mm Acquity BEH300 C4 1.7 μm column (Waters).
Selections of antibodies from the ETH-2 Gold library by phage display
Human monoclonal antibody L7D specific to human TNC-D was isolated by two rounds of biopanning from ETH-2 Gold libraryCitation43 following the protocol described by Viti et al.Citation54 Briefly, 120 pmol of biotinylated BSP-hTNC-D was incubated with 60 µL of streptavidin-coated magnetic beads (Invitrogen, M-280); coated dynabeads were subsequently incubated with preblocked 10−12 transforming units of phage antibodies. Beads were washed with 0.1% Tween 20 in PBS and with PBS (100 mmol/L NaCl, 50 mmol/L phosphate, pH 7.4). Bound phage was eluted with triethylamine (Sigma) and amplified in E. coli TG-1 using VCS-M13 Interference-Resistant Helper Phage (Agilent, Santa Clara, CA). Phage particles were precipitated from culture supernatant using a solution of 20% polyethylene glycol/2.5 M NaCl. Two rounds of panning were performed against the target antigen. Induced supernatants of individual clones were screened by ELISA on human TNC-D and on mouse TNC domains BCD as described by Viti et al.Citation54
Affinity maturation of scFv L7D
The scFv(L7D) affinity maturation library was constructed in a phagemid vector by introducing sequence variability in the CDR1 loops of both heavy and light chain using degenerated primers as described by Brack et al.Citation12 A point mutation N-Q was introduced at position 88 of the light chain to remove a potential glycosylation site.Citation45 The ligation was electroporated into E. coli TG-1 cells; phage was rescued by superinfection with helper phage VCS-M13. The library had a theoretical variability of 6.4 × 107; the obtained library had a size of 1.4 × 106. The library was subjected to two rounds of panning following the same protocol used for the parental antibody selection. R6N, together with L7D, were subcloned into the mammalian expression vector pcDNA 3.1 (+) by NheI/HindIII restriction site, produced and characterized as described.
Cloning, expression and purification of R6N in IgG1 and IgG2a format and of P12 in IgG1 format
R6N IgG1, IgG2a and P12 IgG1 cloning started from the cloning of the light chain of the immunoglobulin into a suitable vector by HpaI/SpeI restriction sites. Cloning procedures continued with R6N/P12 IgG1/IgG2a heavy chain cloning using HindIII and XhoI as restriction sites. The same cloning strategy was used to design KSF IgG1. R6N IgG1, IgG2a and P12 IgG1 format were expressed using TGE in CHO-S cells, purified and characterized in vitro as described.
Cloning, expression and purification of R6N in diabody format
R6N was reformatted in diabody format into the mammalian expression vector pcDNA 3.1 (+) using NheI and NotI restriction enzymes. The same cloning strategy was used to design KSF diabody (specific for an irrelevant antigen, here used as negative control). R6N in diabody format was expressed using TGE in CHO-S cells, purified and characterized as described.
Cloning, expression and purification of mIL12-R6N and IL12-R6N
The fusion protein mIL12-R6N contains the R6N antibody in homodimeric tandem scFv arrangement fused to murine IL12 at N-terminus. The gene encoding for R6N in diabody format and the gene encoding for the murine IL12 or human IL12 were PCR amplified, PCR assembled and cloned into pcDNA 3.1 (+) by NheI/HindIII restriction site. mIL12-R6N and IL12-R6N were expressed using TGE in CHO-S cells, purified and characterized in vitro as described. The biological activity of mIL12-R6N was evaluated by an IFN-γ release assay as described by Puca et al.Citation49
Immunofluorescence studies
Antigen expression was confirmed on ice-cold acetone-fixed 10-µm cryostat sections of A431, U87, SKRC52, A375, Colon 26, C51, SMA-540, SMA-497, of frozen tumor and normal tissue specimens in microarray (Amsbio, T2635700) and of patient-derived glioblastoma stained with R6N IgG1-FITC (protein was FITC labeled according to manufacturer protocol; Sigma) (final concentration 10 µg/mL) and detected with rabbit anti-FITC (Bio-Rad, 4510–7804) and anti-rabbit AlexaFluor488 (Invitrogen; A11008). For vascular staining, rat anti-CD31 (BD Biosciences, 550274) and anti-rat AlexaFluor594 (Invitrogen; A21209) antibodies were used. Cell nuclei were stained with DAPI (Invitrogen; D1306). For the immunofluorescence-based biodistribution analysis, mice were injected with 200 µg/mouse of R6N IgG1 or KSF IgG1 and of R6N Diabody or KSF Diabody when tumor size reached 100–250 mm3 and sacrificed 24 hours after injection. Tumors were excised and embedded in cryo-embedding medium (Thermo Fisher) and cryostat sections (10 µm) were stained and detected with Protein A-Alexa 488 conjugate (Thermo Fisher, P11047). Slides were mounted with fluorescent mounting medium (Dako) and analyzed with Axioskop2 mot plus microscope (Zeiss). Quantification of the fluorescent signal, using Image J software, is depicted in Supplementary Figure S12.
Animal study design
The immunofluorescence-based biodistribution analysis was performed with six BALB/c nude mice bearing A431 epidermoid carcinoma or A375 malignant melanoma. One control group (mice injected with KSF IgG or Diabody) was used for the experiment. The quantitative biodistribution analyses were performed with groups of 4-5 BALB/c nude mice bearing U87 glioblastoma or A431 epidermoid carcinoma, respectively, and one control group (mice injected with KSF diabody) was used for each experiment. The therapy with mIL12-R6N was performed with 10 BALB/c nude mice bearing SKRC52 renal cell carcinoma; one control group with mice injected with saline was used during the experiment. The therapy with R6N IgG2a and diabody was performed with 15 BALB/c nude mice bearing A375 malignant melanoma. The therapy with mIL12-R6N and mIL12-KSF was performed with 15 VM/Dk mice bearing SMA-497 glioma. Mice were randomized into groups according to their tumor volume; tumor volume measurements were taken by the same experimenter to minimize any subjective bias.
Experimental animals
A total of 51 female BALB/c nude mice, aged 8 weeks with an average weight of 20 g were used in this work. Mice were purchased from Janvier (Route du Genest, 53940 Le Genest-Saint-Isle, France) and raised in a pathogen-free environment with a relative humidity of 40–60%, at a temperature between 18°C and 26°C and with daily cycles of 12 hours light/darkness according to guidelines (GV-SOLAS; FELASA). The animals were kept in a specific pathogen-free (OHB) animal facility in cages of maximum five mice, left for 1-week acclimatization upon arrival, and subsequently handled under sterile BL2 workbenches. Specialized personnel were responsible for their feeding; food and water were provided ad libitum. Mice were monitored daily (in the morning) in weight, tumor load, appearance (coat, posture, eyes and mouth moisture) and behavior (movements, attentiveness and social behavior). Euthanasia criteria adopted were bodyweight loss >15% and/or ulceration of the subcutaneous tumor and/or tumor diameter >1500 mm and/or mice pain and discomfort. Mice were euthanized in CO2 chambers.
VM/Dk mice were bred in pathogen-free facilities at the University of Zurich. Female and male mice of 6 to 12 weeks of age were used in all experiments.
Ethical statement
Mouse experiments were performed under a project license (license number 04/2018) granted by the Veterinäramt des Kantons Zürich, Switzerland, in compliance with the Swiss Animal Protection Act (TSchG) and the Swiss Animal Protection Ordinance (TSchV).
Tumor models and biodistribution studies
The in vivo targeting performance of the R6N antibody was evaluated by quantitative biodistribution analysis, as previously described.Citation10 Ten micrograms of 125I radio-iodinated R6N diabody protein was injected into the lateral tail vein of U87 and A431 tumor-bearing BALB/c nude mice. Mice were sacrificed 24 hours after injection, organs were excised, weighed, and the radioactivity of organs and tumors was measured using a Cobra gamma counter and expressed as a percentage of injected dose per gram of tissue (%ID/g ± SEM; n = 4 mice/group). An immunoreactivity test on TNC-D coated CNBr sepharose resin (GE Healthcare) was conducted as a quality control analysis of the radiolabeling process of the antibody. Pooled fractions of radiolabeled antibody were loaded on a resin previously coated with TNC-D (according to manufacturer protocol) and the flow through was collected. Afterward, TNC-D resin was washed with 1 column volume of PBS, and fractions were collected. Finally, radiolabeled antibody was eluted with 1 M triethylamine pH 11.0, and fractions were collected. The radioactivity of the single fractions and of the resin was measured and normalized to the initial radioactivity input.
Tumor model and therapy studies
1 × 107 SKRC52 and 5 × 106 cells A375 tumor cells were implanted subcutaneously in the flank of BALB/c nude mice with 0.5 ml 29 G insulin syringes (MicroFine™+, BD medical). Mice were monitored daily; tumor volume was measured with a caliper and volume was calculated using the formula: tumor size = (Length[mm]*Width2[mm])/2. When tumors reached a suitable volume (approx. 100 mm3), mice were injected three times into the lateral tail vein with the pharmacological agents. mIL12-R6N was dissolved in PBS (pH: 6.9) and administered at a dose of 24 µg/mouse every 48 hours. A saline group was included as a control. R6N Diabody and IgG2a were dissolved in PBS (pH: 7.4) and administered at a dose of 100 µg/mouse every 72 hours for three times. Intracranial tumor cell SMA-497 implantation has been performed as previously described.Citation55 SMA-497 tumor-bearing mice were treated intravenously at days 5 and 10 after tumor implantation with 100 µl PBS, 100 µl containing 8 µg mIL12-KSF or 100 µl containing 8 µg mIL12-R6N µg. Survival data are presented as Kaplan–Meier plots.
Statistical analysis
Data were analyzed using Prism 7.0 (GraphPad Software, Inc.). Differences in tumor volume between therapeutic groups (until day 27, when n = 5) were evaluated with the two-way ANOVA followed by Bonferroni as a posttest. P < .05 was considered statistically significant (*P < .05, **P < .01, ***P < .001, ****P < .0001). Kaplan–Meier survival analysis was performed to assess survival differences among the treatment groups and p values were calculated with Gehan-Breslow-Wilcoxon test. Significance was tested at *p < .05 and **p < .01.
Abbreviations
Disclosure of potential conflicts of interest
Dario Neri is a co-founder and shareholder of Philogen (www.philogen.com), a Swiss-Italian Biotech company that operates in the field of ligand-based pharmacodelivery. Lisa Nadal, Riccardo Corbellari, Alessandra Villa and Roberto De Luca are employees of Philochem AG, daughter company of Philogen acting as a discovery unit of the group.
Authors contributions
LN and RD: conception and design, development of methodology, acquisition, analysis and interpretation of data. RC, TW and MW: acquisition and analysis of data. LN, AV, DN and RD: writing, review and revision of the manuscript. RD: study supervision.
Supplemental Material
Download PDF (702.5 KB)Acknowledgments
We would like to thank Dr. Emanuele Puca for their help with experimental procedures.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12(4):278–12. doi:https://doi.org/10.1038/nrc3236.
- Kaplon H, Reichert JM. Antibodies to watch in 2019. mAbs. 2019;11(2):219–38. doi:https://doi.org/10.1080/19420862.2018.1556465.
- Walsh G. Biopharmaceutical benchmarks 2018. Nat Biotechnol. 2018 Nature Publishing Group;36(12):1136–45. doi:https://doi.org/10.1038/nbt.4305.
- Lane DM, Eagle KF, Begent RHJ, Hope-Stone LD, Green AJ, Casey JL, Keep PA, Kelly A, Ledermann JA, Glaser MG, et al. Radioimmunotherapy of metastatic colorectal tumours with iodine-131-labelled antibody to carcinoembryonic antigen: phase I/IL study with comparative biodistribution of intact and F(Ab’) antibodies. Br J Cancer. 1994;7:521–25. doi:https://doi.org/10.1038/bjc.1994.338.
- Dan N, Setua S, Kashyap VK, Khan S, Jaggi M, Yallapu MM, Chauhan SC. Antibody-drug conjugates for cancer therapy: chemistry to clinical implications. Pharmaceuticals. 2018;11:32.
- Nilsson F, Kosmehl H, Zardi L, Neri D. Targeted delivery of tissue factor to the ED-B domain of fibronectin, a marker of angiogenesis, mediates the infarction of solid tumors in mice. Cancer Res. 2001;61:711–16.
- Lustgarten J, Cui Y, Li S. Targeted cancer immune therapy. Springer: Springer New York; 2009.
- Zagzag D, Frdlander DR, Miller DC, Dosik J, Cangiarella J, Kostianovsky M, Cohen H, Grumet M and Greco MA et al. Tenascin expression in astrocytomas correlates with angiogenesis. Cancer Res. 1995;55(4):907–14.
- Silacci M, Brack SS, Späth N, Buck A, Hillinger S, Arni S, Weder W, Zardi L and Neri D. Human monoclonal antibodies to domain C of Tenascin-C selectively target solid tumors in vivo. Protein Eng Design Select. 2006;19:471–78. doi:https://doi.org/10.1093/protein/gzl033.
- Villa A, Trachsel E, Kaspar M, Schliemann C, Sommavilla R, Rybak JN, Rösli C, Borsi L and Neri D. A high-affinity human monoclonal antibody specific to the alternatively spliced EDA domain of fibronectin efficiently targets tumor neo-vasculature in vivo. Int J Cancer. 2008;122:2405–13. doi:https://doi.org/10.1002/ijc.23408.
- Pini A, Viti F, Santucci A, Carnemolla B, Zardi L, Neri P, and Neri. Design and use of a phage display library. J Biol Chem. 1998;34:21769–76. doi:https://doi.org/10.1074/jbc.273.34.21769.
- Brack SS, Silacci M, Birchler M, Neri D. Tumor-targeting properties of novel antibodies specific to the large isoform of Tenascin-C. Clini Cancer Res. 2006;12:3200–08. doi:https://doi.org/10.1158/1078-0432.CCR-05-2804.
- Midwood KS, Hussenet T, Langlois B, Orend G. Advances in Tenascin-C biology. Cell Mol Life Sci. 2011;68:3175–99.
- Tucker RP, Chiquet-Ehrismann R. Tenascin-C: its functions as an integrin ligand. Int J Biochem Cell Biol. 2015;65:165–68. doi:https://doi.org/10.1016/j.biocel.2015.06.003.
- Dobbertin A, Czvitkovich S, Theocharidis U, Garwood J, Andrews MR, Properzi F, Lin R, Fawcett JW, Faer A. Analysis of combinatorial variability reveals selective accumulation of the fibronectin type III domains B and D of Tenascin-C in injured brain. Exp Neurol. 2010;225(1):60–73. doi:https://doi.org/10.1016/j.expneurol.2010.04.019.
- Borsi L, Allemanni G, Gaggero B, Zardi L. Extracellular pH controls pre-mRNA alternative splicing of Tenascin-C in normal, but not in malignantly transformed, cells. Int J Cancer. 1996;66(5):632–35.
- Giblin SP, Midwood KS. Tenascin-C: form versus function. Cell Adh Migr. 2015;9(1–2):48–82. doi:https://doi.org/10.4161/19336918.2014.987587.
- Chiquet-Ehrismann R. Tenascins, a growing family of extracellular matrix proteins. Experientia. 1995;51(9–10):853–62. doi:https://doi.org/10.1007/BF01921736.
- Bourdon MA, Wikstrand CJ, Furthmayr H, Matthews TJ, Bigner DD. Human glioma-mesenchymal extracellular matrix antigen defined by monoclonal antibody. Cancer Res. 1983;43:2796–805.
- Balza E, Siri A, Ponassia M, Caocci F, Linnala A, Virtanen I, Zardi L. Production and characterization of monoclonal antibodies specific for different epitopes of human tenascin. Eur Biochem Soc. 1993;332:39–43. doi:https://doi.org/10.1016/0014-5793(93)80479-E.
- Siri A, Carnemolla B, Saginati M, Leprini A, Casari G, Baralle F, Zardi L. Human tenascin: primary structure, pre-mRNA splicing patterns and localization of the epitopes recognized by two monoclonal antibodies. Nucleic Acids Res. 1991;19(3):3525–3231. doi:https://doi.org/10.1093/nar/19.3.525.
- Riva P, Sturiale AA, Sturiale C, Moscatelli G, Tison V, Mariani M, Seccamani E, Lazzari S, Fagioli L, Franceschi G. Treatment of intracranial human glioblastoma by direct intratumoral administration of 1311-labelled anti-Tenascin monoclonal antibody BC-2. Int J Cancer. 1992;51:7–13. doi:https://doi.org/10.1002/ijc.2910510103.
- Riva P, Arista A, Franceschi G, Frattarelli M, Sturiale C, Riva N, Casi M, Rossitti R. Local treatment of malignant gliomas by direct infusion of specific monoclonal antibodies labeled with 131I: comparison of the results obtained in recurrent and newly diagnosed tumors. Cancer Res. 1995;55:5952–56.
- Paganelli G, Magnani P, Zito F, Lucignan G, Sudati F, Truci G, Motti E, Terreni M, Pollo B, Giovanelli M. Pre-targeted immunodetection in glioma patients: tumour localization and single-photon emission tomography imaging of [99mTc]PnAO-biotin. Eur J Nucl Med. 1994;21(4):314–32. doi:https://doi.org/10.1007/BF00947966.
- Bigner DD, Brown MT, Friedman AH, Coleman RE, Akabani G, Friedman HS, Thorstad WL, McLendon RE, Bigner SH, Zhao XG, et al. Iodine-131-labeled antitenascin monoclonal antibody 81C6 treatment of patients with recurrent malignant gliomas: phase I trial results.. J clin oncol. 1998;16(6):2202–12. doi:https://doi.org/10.1200/JCO.1998.16.6.2202.
- Reardon DA, Zalutsky MR, Akabani G, Coleman RE, Friedman AH, Herndon JE, McLendon RE, Pegram CN, Quinn JA, Rich JN, et al. A pilot study: 131I-AntiTenascin monoclonal antibody 81C6 to deliver a 44-Gy resection cavity boost. Neuro-Oncology. 2008;10(2):182–89. doi:https://doi.org/10.1215/15228517-2007-053.
- He X, Archer GE, Wikstrand CJ, Morrison SL, Zalutsky MR, Bigner DD, Batra SK. Generation and characterization of a mouse/human chimeric antibody directed against extracellular matrix protein tenascin. J Neuroimmunol. 1994;52(2):127–37. doi:https://doi.org/10.1016/0165-5728(94)90106-6.
- Rizzieri DA, Akabani G, Zalutsky MR, Coleman RE, Metzler SD, Bowsher JE, Toaso B, Anderson E, Lagoo A, Clayton S. Phase 1 trial study of 131I-labeled chimeric 81C6 monoclonal antibody for the treatment of patients with non-Hodgkin lymphoma. Blood. 2004;104(3):642–48. doi:https://doi.org/10.1182/blood-2003-12-4264.
- De Santis R, Anastasi AM, D’Alessio V, Pelliccia A, Albertoni C, Rosi A, Leoni B, Lindstedt R, Petronzelli F, Dani M, et al. Novel antiTenascin antibody with increased tumour localisation for Pretargeted Antibody-Guided RadioimmunoTherapy (PAGRITR). Br J Cancer. 2003;88(7):996–1003. doi:https://doi.org/10.1038/sj.bjc.6600818.
- Petronzelli F, Pelliccia A, Anastasi AM, D'Alessio V, Albertoni C, Rosi A, Leoni B, De Angelis C, Paganelli G, Palombo G. et al. Improved tumor targeting by combined use of two antiTenascin antibodies. Clini Cancer Res. 2005;11:7137–45. doi:https://doi.org/10.1158/1078-0432.CCR-1004-0007.
- Rami A, Behdani M, Yardehnavi N, Habibi-Anbouhi M, Kazemi-Lomedasht F. An overview on application of phage display technique in immunological studies. Asian Pac J Trop Biomed. 2017;6:599–602. doi:https://doi.org/10.1016/j.apjtb.2017.06.001.
- Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol. 1994;12(1):433–55. doi:https://doi.org/10.1146/annurev.iy.12.040194.002245.
- Alfaleh MA, Alsaab HO, Mahmoud AB, Alkayyal AA, Jones ML, Mahler SM and Hashem AM. Phage display derived monoclonal antibodies: from bench to bedside. Front Immunol. 2020;11.
- Frenzel A, Schirrmann T, Hust M. Phage display-derived human antibodies in clinical development and therapy. mAbs. 2016;8(7):1177–94. doi:https://doi.org/10.1080/19420862.2016.1212149.
- Neri D, Sondel PM. Immunocytokines for cancer treatment: past, present and future. Curr Opin Immunol. 2016;40:96–102. doi:https://doi.org/10.1016/j.coi.2016.03.006.
- Neri D. Antibody–cytokine fusions: versatile products for the modulation of anticancer immunity. Cancer Immunol Res. 2019;7:348–54. American Association for Cancer Research Inc.
- Pasche N, Neri D. Immunocytokines: A novel class of potent armed antibodies. Drug Discov Today. 2012;17(11–12):583–90. doi:https://doi.org/10.1016/j.drudis.2012.01.007.
- List T, Neri D. Immunocytokines: A review of molecules in clinical development for cancer therapy. Clin Pharm Adv Appl. 2013;5:29–45.
- Fallon J, Tighe R, Kradjian G, Guzman W, Bernhardt A, Neuteboom B, Lan Y, Sabzevari H, Schlom J, Greiner JW, et al. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget. 2014;5(7):1869–84. doi:https://doi.org/10.18632/oncotarget.1853.
- Pasche N, Wulhfard S, Pretto F, Carugati E, Neri D. The antibody-based delivery of interleukin-12 to the tumor neovasculature eradicates murine models of cancer in combination with paclitaxel. Clini Cancer Res. 2012;18(15):4092–103. doi:https://doi.org/10.1158/1078-0432.CCR-12-0282.
- Huston JS, Levinson D, Mudgett-Huntert M, Tai M-S, Novotnt J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R, et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci. 1988;85:5879–83. doi:https://doi.org/10.1073/pnas.85.16.5879.
- Dwyer G, Mccomas W, Familletti PC, Schoenhaut DS, Chua AO, Wolitzky AG, Quinn PM, Gately MK, Gubler U. Cloning and expression of murine IL-12. J Immunol. 1992;148:3433–40.
- Silacci M, Brack S, Schirru G, Mårlind J, Ettorre A, Merlo A, Viti F, Neri D. Design, construction, and characterization of a large synthetic human antibody phage display library. Proteomics. 2005;5(9):2340–50. doi:https://doi.org/10.1002/pmic.200401273.
- Fairhead M, Howarth M. Site-specific biotinylation of purified proteins using BirA. Methods Mol Biol. 2015;1266:171–84.
- Gébleux R, Stringhini M, Casanova R, Soltermann A, Neri D. Non-internalizing antibody–drug conjugates display potent anti-cancer activity upon proteolytic release of monomethyl auristatin E in the subendothelial extracellular matrix. Int J Cancer. 2017;140:1670–79. doi:https://doi.org/10.1002/ijc.30569.
- Saga Y, Tsukamoto T, Jing N, Kusakabe M, Sakakura T. Murine Tenascin: cDNA cloning, structure and temporal expression of isoforms. Gene. 1991;104(2):177–85. doi:https://doi.org/10.1016/0378-1119(91)90248-A.
- Thoring L, Dondapati SK, Stech M, Wüstenhagen DA, Kubick S. High-yield production of “difficult-to-express” proteins in a continuous exchange cell-free system based on CHO cell lysates. Sci Rep. 2017;7(1):11710. doi:https://doi.org/10.1038/s41598-017-12188-8.
- Weller M, van den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-Jonathan-Moyal E, Henriksson R, Rhun EL, Balana C, Chinot O. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017;18(6):315–29. doi:https://doi.org/10.1016/S1470-2045(17)30194-8.
- Puca E, Probst P, Stringhini M, Murer P, Pellegrini G, Cazzamalli S, Hutmacher C, Gouyou B, Wulhfard S, Matasci M, et al. The antibody-based delivery of interleukin-12 to solid tumors boosts NK and CD8+ T cell activity and synergizes with immune checkpoint inhibitors. Int J Cancer. 2020;146(9):2518–30. doi:https://doi.org/10.1002/ijc.32603.
- Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res. 2018;78(4):1031–43. doi:https://doi.org/10.1158/0008-5472.CAN-17-1788.
- Weber M, Bujak E, Putelli A, Villa A, Matasci M, Gualandi L, Hemmerle T, Wulhfard S, Neri D. A highly functional synthetic phage display library containing over 40 billion human antibody clones. PLoS ONE. 2014;9(6):6. doi:https://doi.org/10.1371/journal.pone.0100000.
- Beckett D, Kovaleva E, Schatz PJ. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999;8:921–29. doi:https://doi.org/10.1110/ps.8.4.921.
- Pasche N, Woytschak J, Wulhfard S, Villa A, Frey K, Neri D. Cloning and characterization of novel tumor-targeting immunocytokines based on murine IL7. J Biotechnol. 2011;154:84–92. doi:https://doi.org/10.1016/j.jbiotec.2011.04.003.
- Viti F, Nilsson F, Demartis S, Huber A, Neri D. Design and use of phage display libraries for the selection of antibodies and enzymes. Method Enzymol. 2000;326:480–505.
- Weiss T, Schneider H, Silginer M, Steinle A, Pruschy M, Polić B, Weller M, Roth P. NKG2D-Dependent antitumor effects of chemotherapy and radiotherapy against glioblastoma. Clini Cancer Res. 2018;24(4):882–95.s. doi:https://doi.org/10.1158/1078-0432.CCR-17-1766.