
ABSTRACT
Activation of T cells specific for insulin B chain amino acids 9 to 23 (B:9–23) is essential for the initiation of type 1 diabetes (T1D) in non-obese diabetic mice. We previously reported that peptide/MHC complexes containing optimized B:9–23 mimotopes can activate most insulin-reactive pathogenic T cells. A monoclonal antibody (mAb287) targeting these complexes prevented disease in 30–50% of treated animals (compared to 10% of animals given an isotype control). The incomplete protection is likely due to the relatively low affinity of the antibody for its ligand and limited specificity. Here, we report an enhanced reagent, mAb757, with improved specificity, affinity, and efficacy in modulating T1D. Importantly, mAb757 bound with nanomolar affinity to agonists of both “type A” and “type B” cells and suppressed “type B” cells more efficiently than mAb287. When given weekly starting at 4 weeks of age, mAb757 protected ~70% of treated mice from developing T1D for at least 35 weeks, while mAb287 only delayed disease in 25% of animals under the same conditions. Consistent with its higher affinity, mAb757 was also able to stain antigen-presenting cells loaded with B:9–23 mimotopes in vivo. We conclude that monoclonal antibodies that can block the presentation of pathogenic T cell receptor epitopes are viable candidates for antigen-specific immunotherapy for T1D.
Introduction
Accumulating evidence strongly suggests that epitopes within amino acids 9 to 23 of the insulin B chain (B:9–23) are critical targets of islet autoimmunity in the non-obese diabetic (NOD) mouse,Citation1–6 and likely in humans as well.Citation7–9 The NOD mouse only expresses a single MHC class II glycoprotein (I-AgCitation7). Like HLA-DQ8, the molecule conferring the greatest genetic risk for T1D in humans,Citation10 I-AgCitation7 lacks an Asp at position 57 of the beta chain. This residue normally forms a salt bridge with a conserved Arg at position 76 of the α chain to “close” the P9 peptide-binding pocket. Substitution of AspβCitation5,Citation7 with Ser (I-Ag) or Ala (DQ8) “loosens” the pocket and creates a preference for peptides with acidic residues at P9.Citation11,Citation12 A large number of CD4+ T cell clones and hybridomas reactive with B:9–23/I-Ag7 have been generated from insulitic lesions from NOD mice,Citation2,Citation13 and many cause disease when adoptively transferred to immunodeficient animals.Citation1,Citation13 Broadly speaking, these T cells can all be categorized as either “type A” or “type B,” depending upon their relative ability to be stimulated by antigen-presenting cells (APCs) loaded with exogenous insulin.Citation13,Citation14 However, the precise nature of their endogenous ligands has proved controversial. One hypothesis is that the “type A” cells recognize an energetically favorable complex in which Glu occupies the P9 pocket, while “type B” cells recognize a lower affinity register-shifted complex in which Gly occupies P9 instead.Citation14,Citation15 However, to our surprise, our initial epitope mapping studies using anchored peptide/MHC complexes rather than free peptides were not consistent with this conclusion.Citation16 Instead, our data strongly suggested that representative members of both sub-types recognize the native peptide bound in the same, energetically unfavorable alternative register termed “register 3” in which Arg occupies P9, and that the difference in reactivity in fact reflects their relative tolerance for a Glu residue at the T cell receptor-contacting p8 position.Citation16,Citation17 Recent structural analyses of ligand-receptor complexes from canonical “type A” and “type B” cells support this conclusion.Citation8
A major goal of T1D research is to develop novel methods of antigen-specific immunotherapy (ASI) to prevent or arrest T1D progression efficiently. The seminal study by Nakayama and Eisenbarth in 2005 demonstrated that selective disruption of the critical B:9–23 epitope(s) by creating NOD mice that only express a B16Tyr to Ala variant completely prevented disease.Citation6 Accordingly, we postulated that monoclonal antibodies (mAbs) able to selectively block presentation of these epitopes should be similarly effective. Our initial efforts to test this hypothesis resulted in the generation of “mAb287.”Citation18 When given weekly after weaning, this antibody showed weak protection in delaying T1D progression. A likely explanation for the weak protection by mAb287 is that it has a relatively low affinity for its ligands, especially for the ligands to “type B” T cells.Citation18 We hypothesized that antibodies with broad specificity for the ligands of insulin B:9–23 reactive T cells should exert stronger disease protection efficacy. Therefore, we generated the new antibody mAb757, which showed broader specificity and higher affinity, particularly to the ligands of “type B” cells, than the original mAb287. As predicted from its enhanced binding profile, mAb757 was significantly more effective in protecting young mice from developing T1D than mAb287. Importantly, mAb757 was effective at late pre-diabetic stages. We conclude that mAbs targeting pathogenic peptide/MHC complexes represent a viable approach for selective immunomodulation to prevent T1D.
Results
MAb757 blocks activation of both “type A” and “type B” T cells
Our previous studies identified two mimotopes derived from B:9–23 that are potent agonists for a panel of previously defined “type A” and “type B” T cells.Citation16,Citation17 Each mimotope has optimal P1 and P9 anchor residues for binding in “register 3,” with one (p8E) having the native Glu at P8, and the other (p8G) having a Gly at P8 instead. “Type A” cells preferentially respond to p8E, while “Type B” cells preferentially respond to p8G,Citation8,Citation16,Citation17,Citation19 and a combination of I-AgCitation7 tetramers loaded with p8E and p8G can stain the vast majority of B:9–23 reactive T cells in islet infiltrates from unmanipulated NOD mice.Citation17 Consistent with its limited efficacy, while mAb287 effectively blocked activation of “Type A” T cells in vitro, it was much less potent against “Type B” T cells.Citation18 To generate a more potent antibody we immunized a new cohort of mice using a mixture of p8E and p8G containing complexes rather than p8E and p8A as we had done previously.Citation18 From 665 initial wells, hybridomas grew in 528 wells, with 500 producing antibodies that bound to at least one of the immunogens (Fig S1). Forty-six lines produced antibodies that bound both p8E and p8G, albeit to differing extents, while the majority bound only p8E with only a few lines specific for the p8G complex. Six “dual specificity” lines were cloned by limiting dilution and their antibodies purified for assessments of binding affinity and specificity. Clone 757, which secretes antibodies that bind both p8E and p8G complexes but has negligible binding to I-AgCitation7-HEL ()) was selected for further studies.
Figure 1. Binding of mAb757 to immobilized ligands and cells. A-C: mAb757 was bound to protein G-coated sensor chips and binding of I-Ag7/p8E (a), I-Ag7/p8G (b), or I-Ag7-HEL (c), measured by bio-layer interferometry. Results shown are representative of 3 independent experiments. D, E: Immortalized “type A” or “type B” T cells were incubated with cognate tetramers in the presence or absence of mAb757 and analyzed by flow cytometry. Plots show profiles in the absence of tetramer (red), tetramer alone (blue), or tetramer + 2 μg (orange), 5 μg (light green), or 10 μg (dark green) mAb757. One of 3 independent replicates is shown. D. PCR1-10 + p8G; E. BDC12-4.4 + p8G
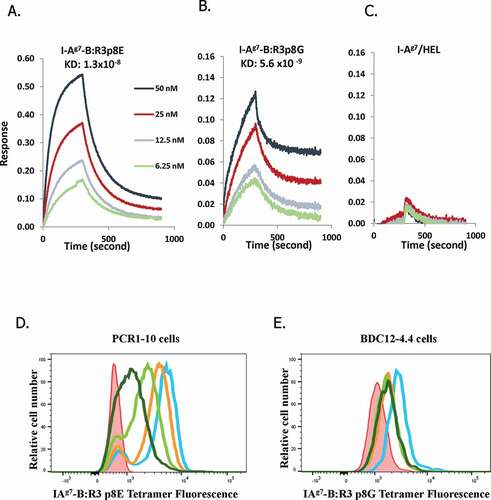
The affinity of mAb757 for the test and control complexes was measured by biolayer interferometry (BLI) (, Table S2). MAb757 bound with nanomolar affinity to I-Ag7/p8G complexes (Kd 5.6 × 10−9 M; )), and similarly to I-Ag7/p8E complexes (Kd 1.3 × 10−8 M; )). This behavior is in marked contrast to that of mAb287, which binds p8G with ~36-fold lower affinity than p8E (referenceCitation18 and Fig S2). As expected, binding of mAb757 to I-Ag7/HEL complexes was undetectable ()). I-Ag7/p8G and I-Ag7/p8E tetramers have been used to identify B:9–23 reactive CD4+ T cells.Citation17 Accordingly, we next asked whether mAb757 was able to selectively block these interactions. As expected, mAb757 showed a dose-dependent inhibition of both I-Ag7/p8E staining of PCR1-10 cells ()) and I-Ag7/p8G staining of BDC12-4.4 T cells ()), but had no effect on I-Ag7/HEL staining of 5F2 cells (data not shown).
MAb757 does not bind to free insulin peptides or I-AgCitation7/B:9-23 complexes in other registers
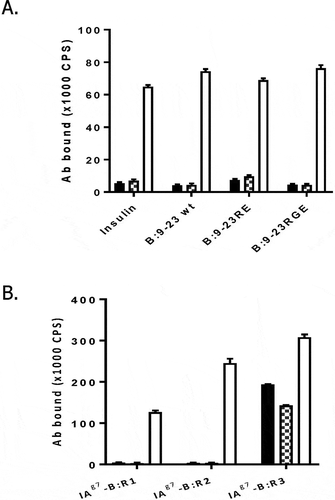
Antibodies that bind the insulin protein with high affinity would likely interfere with the metabolic function of the hormone, rendering them unsafe for in vivo use. Thus, we next used an enzyme-linked immunosorbent assay (ELISA) to test whether mAb757 recognizes free insulin, the unbound B:9–23 peptide, or the various unbound mimotopes. As expected, only background binding was observed ()), suggesting that mAb757 is unlikely to disturb insulin metabolic action in vivo. As a further assessment of specificity, we also examined binding to I-Ag7/B:9–23 complexes having the peptide trapped in “register 1” (R1) or “register 2” (R2), which place the key B16Tyr at P5 and P4, respectively. Like mAb287 (checkered bars), mAb757 only bound significantly to “register 3” complexes (where B16Tyr is at P3), and not those with the peptide trapped in R1 or R2 (), filled bars). In contrast, the I-Af,k,r,s restricted mAb 10–3.6,Citation20 which cross-reacts with I-Ag7,Citation21 recognized all of the complexes regardless of the position of the peptide (), open bars).
Figure 2. Binding characteristics of mAb757 A. ELISA plates were coated with 2 µg/ml human insulin, or the B:9–23 wt, R3:p8E, R3:p8G, or TT peptides (Table S1). After washing and blocking steps, specific binding of test and control antibodies was determined.Citation18 Binding to mAb757 (black bars), mAb287 (checkered bars; negative control), or AIP-46.12 (open bars; positive control) is shown. B: ELISA plates were coated with 1 µg/ml mAb757 (filled bars), mAb287 (checkered bars) or 10–3.6 (positive control; open bars). After washing and blocking steps, specific binding of biotinylated I-Ag7-B:R1, I-Ag7-B:R2, or I- Ag7-B:R3 complexes was measured by ELISA.Citation18 Data is the mean ±SD of 3 replicates
MAb757 blocks in vitro activation of “Type B” T cells more efficiently than mAb287
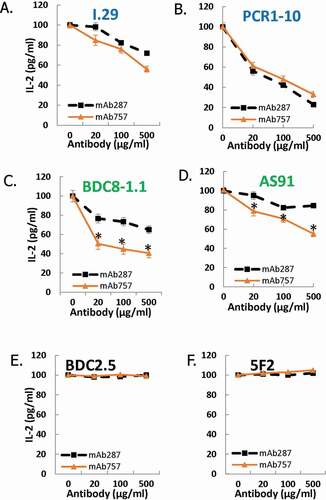
We next compared the relative abilities of mAb287 and mAb757 to block antigen-specific activation of a panel of 6 immortalized T cell lines. Both antibodies were equally effective in blocking IL-2 secretion by the two “type A” lines I.29 and PCR1-1 in response to p8E stimulation (). In contrast mAb757 was significantly more effective than mAb287 in blocking p8G-mediated activation of the 2 representative “type B” lines BDC8-1.1 and AS91 (). As expected, neither antibody had any effect on antigen-specific activation of the two I-Ag7-restricted control lines BDC2.5 and 5F2Citation22,Citation23 ().
Figure 3. Effect of mAb757 on T cell activation. Fixed M12(C3):I-Ag7 APCs were incubated with agonist peptides as described in Methods, then co-cultured overnight with T cell lines and mAb287 (black squares) or mAb757 (orange triangles). Secreted IL-2 was measured by ELISA. Graphs show the percent of the response in the absence of antibody and are the mean of 3 independent experiments. A: I-29 + R3:p8E (100 µg/ml); B: PCR 1–10 + R3:p8E (10 µg/ml); C: BDC8-1.1 + R3:p8G (10 µg/ml); D: AS91 + R3:p8G (10 µg/ml); E: BDC-2.5 + HRPI (1 µg/ml); F: 5F2 + HEL (1 µg/ml). *p < .05 analyzed by student-t test
MAb757 can detect primed APCs ex vivo
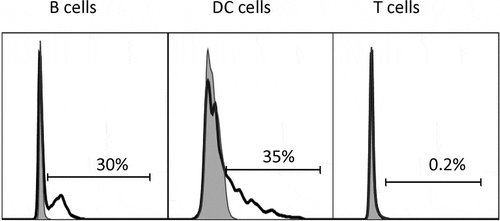
To confirm that mAb757 is able to selectively recognize primary APCs that are expressing I-Ag7/p8E complexes, we immunized mice subcutaneously with mimotope or control HEL peptides and analyzed cells harvested 90 min later from the draining lymph nodes by flow cytometry.Citation24 As shown in (filled gray profiles), mAb757 stained a significant fraction of CD19+ B cells (A) and CD11c+ dendritic cells (DCs) (B), but not T cells, that in mice do not express of MHC class II (C). In contrast, only background staining of cells from animals immunized with the HEL peptide was observed (, open profiles).
Figure 4. Binding of mAb757 to APCs ex vivo. Draining popliteal lymph node cells were harvested after peptide immunization and analyzed by flow cytometry as described in Methods. Profiles show binding of mAb757 (open profiles) or control 15G1a (filled gray profiles) to A: B lymphocytes, B: Dendritic cells, C: T cells. Cells were gated on CD19+ cells, CD11c+ cells and CD4+ cells, respectively
MAb757 is a more effective therapeutic than mAb287
As mAb757 has superior binding properties to those of mAb287, we next examined if it would also be a more effective therapeutic. The sub-class of mAb287 is IgG1Citation18 whereas mAb757 is an IgG2c. Thus, to minimize any potential confounding effects of the different constant domains, we engineered an IgG1 version of mAb757 by replacing its native IgG2c constant domains with those from mAb287 (Fig S3 A), and expressed it in transfected HEK 293 cells. BLI measurements suggested that the binding properties of the IgG1 and IgG2c variants to p8E and p8G containing complexes were not significantly different (Table S2, Fig S3 B-D).
Beginning at 4–5 weeks, female NOD mice were treated with test or control antibodies (100 µg/animal; ~6.25 mg/kg at baseline), or saline buffer, for three consecutive days, followed by weekly injections until they either developed T1D or reached 35 weeks of age when the experiment was terminated. Consistent with the behavior of unmanipulated mice in our colony, mock, or isotype control-treated mice started to develop T1D at 12 weeks of age and only 10–20% survived to the end of the experiment (); red and black symbols). In contrast, no animal given the IgG1 variant of mAb757 developed T1D before 16 weeks of age, and almost 70% remained euglycemic at 35 weeks of age ; blue symbols; p = .003 vs.IgG1 isotype control). As expected, mAb287 was much less effective, with only 25% of these mice remaining diabetes-free at 35 weeks ; green symbols; p = ns vs.IgG1 isotype control; p = .011 vs. mAb757).
Figure 5. Effect of mAb757 on T1D incidence after early intervention. Groups of 4–5-week-old female NOD mice were treated with antibodies or saline as described in Methods and monitored for up to 35 weeks of age. A: Effect of low (100 µg) dose therapy with IgG1 variants. Graphs show treatment with mAb757 (blue circles; n = 19), mAb287 (green triangles; n = 18), isotype control (red squares; n = 32), or saline control (black open circles; n = 22). B: Comparison of low (100 µg) and high (500 µg) doses of mAb757 isotype variants. Graphs show treatment with mAb757-IgG1-low (blue circles; n = 19); mAb757-IgG2c-low (purple triangles; n = 18), IgG2c isotype control-low (purple triangles; n = 13), mAb757-IgG2c-high (purple stars; n = 20), IgG2c isotype control-high (black filled circles; n = 13), IgG1 isotype control-low (n = 32; red squares), or saline control (black open circles; n = 22). Statistically significant variation between groups was determined using a Mantel-Cox test. C: The percentage of insulin positive islets from surviving mAb757-treated animals at 35 weeks or diabetic control-treated mice after diagnosis was measured by immunohistochemistry. Data show average results from all islets from each individual mouse. Asterisks indicate p < .001 relative to diabetic animals
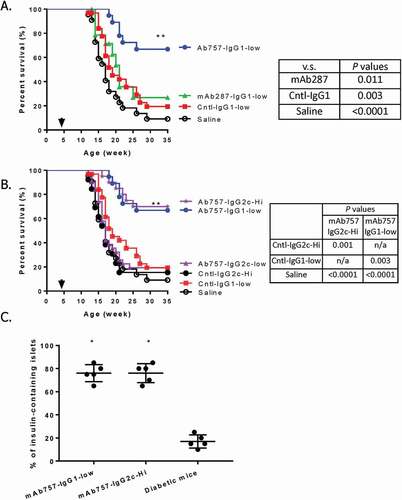
IgG1 isotype of mAb757 provides stronger T1D protection than IgG2c isotype
To gain further insight into the properties of mAb757, we also compared the efficacy of the two isotypes. Surprisingly, 100 µg doses of the IgG2c variant, which was highly effective in the IgG1 format, did not provide significant protection (); purple triangles; p = ns vs. isotype control), with only 20% of the treated mice remaining euglycemic at the end of the experiment. However, at a dose of 500 µg/injection (~31 mg/kg at baseline; high dose arm), the IgG2c variant showed significant efficacy () purple stars), with only 30% of the mice given this higher dose developing T1D by 35 weeks (p = .001 vs. IgG2c isotype).
MAb757 reduces insulitis in treated mice
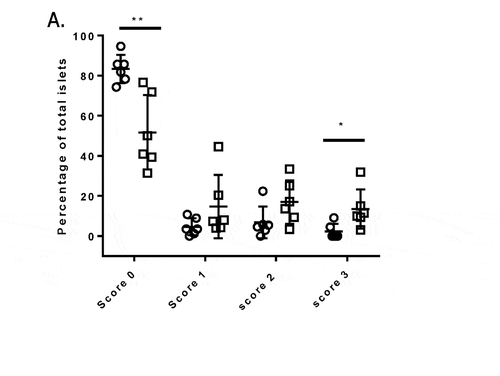
To gain insight into the mechanism of action of mAb757, we first investigated the severity of insulitis of the treated animals. Consistent with their euglycemia, the pancreata of surviving mAb757-treated mice had significantly more insulin-positive islets than control mice that had developed T1D ()). The most likely explanation for the observed preservation of ß cells is that mAb757 selectively delays or prevents islet lymphocytic infiltration. To directly test this possibility, we sacrificed a small cohort of test and control animals at 8 weeks of age, a time point at which insulitis should have been established, but all animals remain normoglycemic. As shown in , mAb757-treated mice had significantly more intact islets (score 0) than the control mice. At 8 weeks of age, at least 75% of the islets in all six mAb757-treated mice remained pristine (range 75–95%). In contrast, only 1/6 control animals had such a low level of infiltration at this stage of disease, with 4/6 having at least 50% of their islets infiltrated by this time (range 50–70%; p = .003). Similarly, only 2/6 test mice, but 6/6 control mice, had any islets with >75% infiltration (Grade 3 insulitis) at this age (; p = .027). Consistent with the insulitis score, more B cells, DCs and T cells were detected in the islets of control mice (representative images shown in Fig S6).
Figure 6. Histological analysis of islets from pre-diabetic mice. Groups of mice (n = 6) from the early prevention trial were sacrificed 4 weeks after the initiation of therapy. Pancreata were harvested and serial sections stained with hematoxylin & eosin. Insulitis was scored based on established criteria (Fig. S5) by a blinded pathologist. Mean scores for mice in the mAb757-IgG1-low (circles) and IgG1 isotype control-low (squares) groups are shown. Statistically significant differences were identified using an unpaired student t test. ** p = .003; *p = .026
Irrespective of T1D status, NOD mice also develop spontaneous autoimmune sialitis.Citation25,Citation26 To evaluate possible off-target effects of mAb757, we also measured the development of sialadenitis histologically. As expected, irrespective of treatment group and the degree of insulitis, by 35 weeks of age all of the mice examined exhibited autoimmune sialadenitis (data not shown). Similarly, there were no significant differences in the percentage of B cells, DCs, CD4, and CD8 T cells of spleens between test and control animals analyzed by flow cytometry (data not shown), suggesting that mAb757 acts selectively to inhibit insulitis rather than as a global immunosuppressant.
Like humans, the earliest noninvasive indicator of islet autoimmunity in NOD mice is often the appearance of insulin autoantibodies (IAA) in the serum.Citation27 Therefore, we also investigated whether mAb757 treatment influenced the development of IAAs. However, we did not observe significant difference of IAA development between any test and control group (data not shown). This suggests that mAb757 does not deplete insulin-specific B cells, although it may still alter their APC functionality.
MAb757 remains effective after epitope spreading has occurred
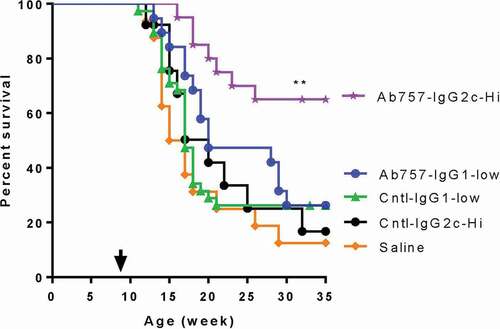
As expected, female NOD mice in our colony typically start to develop insulitis at ~5–6 weeks of age, with the earliest cases of overt diabetes appearing ~6–7 weeks later. Thus, by the time they are 9 weeks old most unmanipulated animals have already developed a severe insulitis and show evidence of epitope spreading.Citation28 A critical question, therefore, is whether mAb757, which targets an initiating epitope, remains effective once spreading has occurred. To answer this key question, we treated a second cohort of test and control animals, initiating therapy when the animals were 9 weeks old and following them until they were 35 weeks of age or diabetes was diagnosed. At baseline, all animals were normoglycemic and there was no significant difference in blood glucose levels between groups (data not shown). As shown in (purple stars), the higher dose of the mAb757 IgG2c variant remained effective at this later pre-diabetic stage, with 12/19 (63.2%) of the mice in this group remaining normoglycemic when the experiment was terminated compared with 2/13 (15.4%) of the matched isotype control animals (p = .005). In contrast the lower dose of the mAb757 IgG1 variant, which was highly effective when therapy was initiated at 4–5 weeks, did not provide any significant protection at this later stage of disease. It should be noted that we did not adjust the antibody dose to compensate for changes in body weight (16 ± 1.2 g at 5 weeks, 22 ± 1.5 g at 12 weeks), which may have contributed to the failure of the IgG1variant to prevent disease progression in the older animals with this protocol.
Figure 7. Effect of mAb757 on T1D incidence after intervention late in pre-diabetes. Groups of 9-week-old female NOD mice were treated with antibodies or saline as described in Methods and monitored for up to 35 weeks of age. Graphs show treatment with mAb757-IgG2c-high (purple stars; n = 19), mAb757-IgG1-low (blue circles; n = 19), IgG1 isotype control-low (green triangles; n = 38), IgG2c isotype control-high (black filled circles; n = 13), or saline (orange diamonds; n = 16). The proportions of animals remaining diabetes-free in each group are shown. Statistically significant variation between groups was determined using a Mantel-Cox test. Double asterisk shows p = .005 vs. IgG2c isotype control-high
Using the prediabetic protocol mAb757 cannot induce durable remission in newly diagnosed diabetic mice
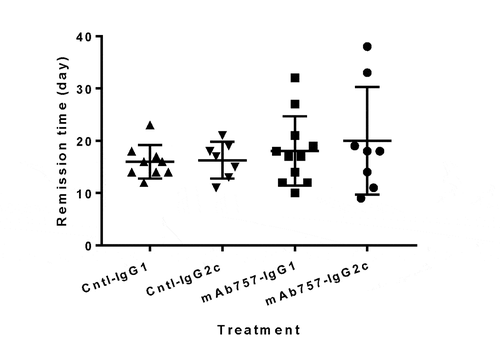
Finally, we asked whether mAb757 treatment could induce durable remission in newly diabetic mice. To provide sufficient time for the antibodies to reach therapeutic levels, all animals were implanted subcutaneously with a single LinBit sustained-release insulin pellet to render them normoglycemic.Citation29,Citation30 As expected, after insulin implantation blood glucose levels fell below 250 mg/dL in all the treated mice. This level of glucose control was maintained for a median period of 16–18 days in animals given isotype control antibodies (IgG1 median 16d, range 12–23d; IgG2c median 18d, range 11–21d; , triangles). Similarly, the median period for the mAb757-treated animals before blood glucose levels exceeded 250 mg/dL was 17–18 days (IgG1 median 17d, range 10–32d; IgG2c median 18d, range 9–38d; p = ns; squares and circles). There was no significant difference in the age of onset of the test and control groups (Fig S7) suggesting that this potential confounder is unlikely to be responsible for the intriguing fact that ~20% of the treated animals had remission times that extended beyond those of any control animals.
Figure 8. Effect of mAb757 on recurrence of hyperglycemia after intervention post-onset. Newly diabetic female NOD mice with blood glucose levels between 250–400 mg/dL at baseline were randomly assigned to a treatment group, implanted with a sustained-release insulin pellet to restore normoglycemia, then treated with antibodies as described in Methods, and monitored for up to 25 additional weeks. Animals in the two IgG1 groups received 200 µg/injection, animals in the two IgG2c groups received 1 mg/injection. The number of days before overt hyperglycemia recurred is shown for each group: IgG1 isotype control (upright triangles, n = 9), IgG2c isotype control (inverted triangles, n = 7), mAb757-IgG1 (squares, n = 11), mAb757-IgG2c (circles, n = 8)
Discussion
An effective antigen-specific immune intervention represents a “Holy Grail” for current T1D research.Citation31 Most previous studies in this area have relied upon immunization with the free antigen in either protein, peptide, or cDNA formats, with several showing promise in early phase human trials.Citation31 However, the inherent promiscuity of the immune system means that the potential for off-target effects of this modality cannot be entirely eliminated. Herein, we describe an alternative approach, namely the use of a “T cell receptor mimetic” mAb that targets defined pathogenic peptide-MHC complexes.
Insulin, the signature product of pancreatic ß cells, comprises ~10% of the total protein in the cell,Citation32 and is generally regarded as a primary target of islet autoimmunity in both animal models and human.Citation33 Indeed, polymorphisms in the INS gene that decrease thymic expression are the second highest genetic risk factor for developing T1D.Citation34 Thus, (pro)insulin is a logical target of efforts to develop an effective ASI for T1D.Citation31 Although most research has focused on the native protein, there is growing awareness that post-translational modifications that create neo-antigens within the target tissue may be of particular importance in breaking immune tolerance in T1D.Citation35 One such mechanism is protein-splicing, which we believe is critical to the generation of the highly pathogenic epitopes from the insulin B chainCitation8 that are recognized by mAb757. As discussed above, B:9–23 reactive CD4+ T cells isolated from the islets of NOD mice can be segregated into two types termed “type A” and “type B.” The fact that mAb757 is able to readily block activation of both specificities, but has minimal affinity for I-Ag7 loaded with irrelevant peptides, appears inconsistent with the hypothesis that the two T cell types recognize the same peptide bound in different registers as originally proposed.Citation14 Rather it strongly supports our alternate proposal that “type A” and “type B” cells each recognize complexes in which the peptide is bound in the same register, but has different post-translational modifications to the carboxy-terminus.Citation8 Given that the relative importance of the two specificities remains uncertain, and may vary at different stages of disease, we reasoned that a broad specificity antibody capable of blocking activation of both T cell types would have greater therapeutic potential than one that shows a significant preference for either “type A” or “type B,” such as mAb287. The results described above support this concept.
Consistent with our previous study using mAb287,Citation18 treatment with mAb757 suppressed disease when administration began either early or late in pre-diabetes. However, the same treatment protocol that was effective at these time points was unable to reverse persistent hyperglycemia post-onset. This implies that, as a monotherapy, agents such as mAb757 are probably most suited for disease prevention, rather than intervention. At present, the reason for the apparent loss of efficacy at advanced stages of disease remains uncertain. However, it should be noted that, although mAb757 treatment was able to protect the majority of pre-diabetic animals from developing overt disease, under the experimental conditions used about one-third of the animals still progressed. It is known that in NOD mice islet inflammation triggers an amplification loop that leads to a progressive accumulation of mature APCs within islets and pancreatic draining lymph nodes that are capable of stimulating autoreactive T cells.Citation36 Thus, one potential explanation is that as disease progresses the number of cells that our antibodies must target increases to a point where too many escape, rendering our experimental doses, which were selected mainly to allow direct comparison with our prior studies with mAb287,Citation18 ineffective. Many variables dictate the pharmacokinetics of mAbs,Citation37 some of which (such as biodistribution and antigen density) can vary with disease state due to changes in vascular permeability and APC function, and others (such as isotype and diversity of glycoforms) are specific to the antibody and/or producer cell. Such complex behavior cannot entirely be predicted a priori, but our results suggest that our current therapeutic protocol could be modified in future studies, for example by increasing the total dose, using more advanced formulations designed to increase bioavailability such as those based on nanotechnology,Citation38 or the use of combination therapies.
A key finding in our study was that the IgG1 variant provided stronger protection than IgG2c mAb757. Specifically, while a low dose of the IgG1variant of mAb757 was highly protective when given at early stages of disease, the same amount of the native IgG2c antibody was not. Indeed, 4–5 times as much was required to achieve the same level of protection as the low dose of the IgG1 variant. The key residues governing binding to mouse FcRn are entirely conserved between mouse IgG1, IgG2a, and IgG2c. Although the clearance rate of IgG2c has not been reported, the half-lives of IgG1 and IgG2a are indistinguishable,Citation39 and so we expect both variants of mAb757 will behave similarly in this regard. We suspect that the significant difference in potency we observed most likely stems from their relative affinity for mouse FcγRIIb,Citation40 which has been implicated previously as a key regulator of therapeutic antibody function,Citation41 and the heterogeneity of the disease-related T cells. Broadly speaking, therapeutic mAbs targeting cell surface receptors can act in one of the three ways: 1) by depleting their target cell, 2) acting as a direct agonist or antagonist for their target receptor to induce a desired cellular response, or 3) blocking the interaction between the receptor and its cognate ligand.Citation41 Numerous previous studies have shown that binding of mAbs to the inhibitory FcγRIIb receptor is generally detrimental to depleting mAbs, but beneficial to those that act as agonists, and is largely irrelevant to mAbs that act mainly by receptor blockade.Citation41 Consequently, our current hypothesis is that mAb757 (and presumably mAb287 as well) likely acts either by inducing a “tolerogenic” phenotype in APCs expressing pathogenic B:9–23/I-Ag7 complexes following cross-linking of these molecules in the absence of the other receptor-ligand interactions that would normally occur with binding of a cognate T cell. This “infectious tolerance” mechanism is consistent with the robust protection provided by mAb757 at late pre-diabetic time points when epitope spreading has occurred, and its ability to suppress islet infiltration of T cells recognizing epitopes from ß cell proteins other than insulin. Compared to IgG2c, Ab757-IgG1 has a similar affinity to p8G, but slightly lower affinity to p8E epitope. The p8E and p8G epitopes activate two groups of T cells.Citation14–17 Our result is consistent with previous studies that the p8G-reacting T cells may play a more important role in causing disease. We believe additional studies will be required to test the hypothesis and explore the mechanisms in-depth. In addition, it will be important to determine if therapeutic efficacy requires continuous administration of the antibody, or if treatment with mAb757 is capable of inducing a population of regulatory cells that could potentially sustain a durable tolerance if therapy were withdrawn.
In summary, our data provide convincing evidence that in this setting a broad specificity antibody that can recognize multiple variants of a pathogenic epitope has better therapeutic efficacy than a more focused one. Indeed, our study suggests that a bispecific antibody that combines mAb757 with an antibody targeting a second pathogenic peptide-MHC complex, such as the BDC-2.5 ligand,Citation22 may provide even stronger disease protection, a hypothesis we are actively exploring.
Materials and methods
Animals and Reagents
Female NOD/LtJ mice were purchased from Jackson Laboratories (Bar Harbor, ME) and maintained under specific pathogen-free conditions, with 12-h light/dark cycles and food and water ad libitum. All animal experiments were performed in accordance with protocols approved by the organization’s animal care and use committee.
Peptides were synthesized at greater than 95% purity. Their sequences and those covalently linked to I-Ag7 are listed in Table S1. The I.29, AS91,Citation42 BDC8-1.1, PCR1-10,Citation43,Citation44 5F2, and BDC2.5Citation45 T cell hybridomas/transfectomas, and M12(C3):I-AgCitation7 B lymphoma were maintained as previously described.Citation43,Citation46
Generation of mAb757
Recombinant proteins were generated as previously described.Citation16,Citation17 Five female NOD mice were immunized with a 1:1 mixture of I-Ag7/p8E and I-Ag7/p8G complexes in the complete Freund’s adjuvant, then boosted 4 further times at 3-week intervals with proteins emulsified in the incomplete Freund’s adjuvant.Citation18 All boosts used the 1:1 mixture except the second that only used I-Ag7/p8G. After three boosts, serum antibodies were measured by ELISA with plate-bound I-Ag7-HEL as a negative control. The two animals with the highest titers were given a final boost, and 3 days later splenocytes and draining lymph node cells were harvested, pooled, and hybridomas generated by fusion with the Sp2/0-Ag14 myeloma.Citation18,Citation47 Cells secreting antibodies that bound to both I-Ag7/B:9–23 complexes, but not to the control complexes, were selected for re-cloning (Fig S1). After secondary screening, clone 757 was chosen for all subsequent experiments.
Sequencing of cDNA encoding mAb757 revealed that it has a κ light chain and IgG2c heavy chain. To allow direct comparison with mAb287 (IgG1/κ), a chimeric heavy chain construct containing the mAb757 variable region fused to the mAb287 constant domains was created by splice-overlap PCR (Fig S3A). The IgG1 variant of mAb757 was expressed recombinantly in HEK 293 cells by co-transfection with two constructs containing the kappa light chain and mouse IgG1 heavy chain, respectively. The antibody IgG1 was purified using protein G resin (Bio-Rad, CA) according to manufacturer’s protocol. Briefly, HEK293 cell culture supernatants were loaded on protein G agarose column to capture expressed IgG in the medium, then washed with phosphate-buffered saline (PBS), pH7.4, before elution with glycine-HCl, pH 3.0. The purified IgG was further equilibrated in PBS and concentrated using Centricon centrifugal concentrator (30KD MW cutoff, Fisher Scientific) at 4°C. IgG2c of mAb757 was purified with nProtein A Sepharose beads (GE Healthcare 17–5280-04) from the supernatants of the B cell hybridoma serum-free culture. Clone 15G1a, an IgG1/κ antibody raised to the ecdysone receptorCitation48 (deposited to the DSHB by Thummel, C./Hogness, D. (DSHB Hybridoma Product 15G1a)) was used as isotype controls. The mouse native IgG1 was isolated from the medium of hybridoma cells. An IgG2c variant was constructed using the domain switch strategy described above, and expressed in HEK293 cells. Levels of endotoxin in each batch of purified antibody were determined to be <1 EU/mg of antibody measured by LAL chromogenic quantitation kit (Thermo Scientific).
Characterization and binding affinity of mAb757
The binding of purified mAb757 to insulin, B:9–23 peptides or I-Ag7/peptide complexes were measured by ELISA as previously described.Citation18 The binding affinities of the IgG1 and IgG2c isotypes of mAb757 were determined using label-free BLI.Citation49 Briefly, antibodies (20 µg/ml) were bound to protein G-coated biosensors for 180 seconds, then incubated with antigens or controls at concentrations ranging from 100 to 0.325 nM with constant shaking at 1,000 rpm. Incubation of the loaded sensors in buffer alone for 1 minute was used for background correction, 5 minutes for association, and 10 minutes for dissociation analysis. The optical interference patterns were monitored continuously and the on and off-rates and equilibrium binding affinities determined using the Octet data analysis software (Pall ForteBio Corp.CA). Kinetic binding affinities were measured three times and analyzed using 1:1 fitting model with the software Octet 96Red. Kinetic constants (kon, koff, and KD) are listed in Table S2. The purity and molecular size of antibodies were confirmed by SDS-PAGE (Fig S3E) and size exclusion chromatography (SEC) (Fig S3 F&G). Antibodies (1 μg per lane) were mixed with 2× SDS/PAGE sample buffer in the presence or absence of 2-mercaptoethanol (reducing agent), heated for 5 min before loading on 10% gels. The gels were stained with Coomassie blue and de-stained for imaging (Supplemental Figure 3E). SEC analysis showed both IgG1 and IgG2c mAb757 have purity above >98%, and no aggregation was observed in either isotypes.
In vitro T cell activation assays
The ability of mAb757 to inhibit activation of selected T cell hybridoma and transfectomas was tested essentially as previously reported.Citation17 Briefly, fixed M12(C3):I-Ag7 APCs (105) were incubated with control or test peptides at concentrations previously shown to elicit a response from the T cell line under investigation of 4 ~ 50 pg/ml/h IL-2, in a final volume of 100 µl for 2 h at 37°C. T cell hybridomas or transfectomas (105) were then added in the presence of increasing concentrations of mAb287 or mAb757, and culture supernatants harvested after overnight culture. Secreted IL-2 was measured by ELISA (BD Biosciences).
Staining of APCs after in vivo peptide loading
Six NOD mice were injected with 50 µg B:9–23p8E or HEL peptides in 50 µl saline in hind limb footpads. Ninety minutes later the popliteal lymph nodes were collected, minced, and digested in buffer containing 2% bovine serum albumin, collagenase D (40 U/ml), and DNase I (250 mg/ml) for 30 min at 4°C. Cells were then washed and stained with 1 µg Alexa Fluor 647 labeled mAb757 or 15G1a (Alexa Fluor™ 647 antibody labeling kit; Invitrogen), and fluorescent antibodies against CD11c, CD19, and CD3 (Biolegend).
Diabetes prevention and reversal studies in NOD mice
The protocols used at the three intervention time points are summarized in Fig S4. At each stage (4–5 w, 9 w, post-onset), female NOD mice were randomly assigned to one of the test or control groups. In disease prevention studies, a low dose arm (100 µg/injection) and a high dose arm (500 µg/injection) were used. For the disease reversal experiments, only animals having blood glucose levels between 250 mg/dL and 400 mg/dL in two consecutive tests were included. These animals were implanted subcutaneously with a LinBit sustained-release insulin pellet (LinShin Canada, Inc., Toronto)Citation29 immediately before antibody therapy was begun. Initially, all mice were given intraperitoneal injections of 100 µl saline or antibody solution (high dose) for 3 consecutive days, followed by weekly injections until the animals either became diabetic or reached the end of the study. Blood glucose levels were monitored weekly from 9 weeks of age with a OneTouch Ultra2 Blood Glucose Meter, or daily post-onset. Animals with blood glucose values of ≥250 mg/dl were re-tested the following day and considered diabetic after two consecutive positive values. In prevention experiments, bodyweights were measured weekly from 4 − 20 weeks of age. Similarly, in the early prevention study serum, IAA were monitored by fluid-phased radioimmunoassay every 2 weeks from 4 to 10 weeks.Citation27 All mice reaching a study endpoint (remaining diabetes-free at 35 weeks, or either developing T1D or experiencing disease recurrence) were euthanized and pancreata harvested for analysis of insulitis.Citation50 In some animals, autoimmune sialitis was also assessed histologically.
Islet immunohistochemistry
To assess insulin content and the composition of islet infiltrates, a subset of animals from the early prevention trial were sacrificed at 8 weeks. Pancreata were collected and either fixed in formaldehyde or flash frozen for analysis by immunohistochemistry.Citation51 Primary antibodies used for fixed tissue were guinea pig anti-insulin (ThermoFisher, PA1-26938), rat anti-mouse B220 (BD Biosciences, 550286), and mouse anti CD3 (Abcam, ab17143). DCs were stained using frozen sections and hamster anti-mouse CD11c (Biolegend, 117301). Secondary antibodies used were donkey anti-guinea pig IgG(H + L) FITC (706–095-148), donkey anti-rat Cy3 (712–165-150), donkey anti-mouse Cy 5 (715–175-150), and goat anti-Armenian hamster Cy3 (127–165-160) (all from Jackson ImmunoResearch Inc.).
Statistics
Survival curves were analyzed using a log-rank (Mantel-Cox) test. Other parameters were analyzed with an unpaired student’s t-test. p values < .05 were considered statistically significant. Analysis was performed using PRISM7.03 software (Graphpad, San Diego, CA).
Abbreviations
| ASI | = | Antigen-specific immunotherapy |
| APC | = | antigen-presenting cell |
| BLI | = | biolayer interferometry |
| DC | = | dendritic cell |
| ELISA | = | enzyme-linked immunosorbent assay |
| HLA | = | human leukocyte antigen |
| MHC | = | major histocompatibility complex |
| mAb | = | monoclonal antibody |
| NOD mice | = | non-obese diabetic mice |
| PBS | = | phosphate-buffered saline |
| SEC | = | size exclusion chromatography |
| T1D | = | type 1 Diabetes |
| TCR | = | T cell receptor |
Disclosure of Potential Conflicts of Interest
LZ and JWK are holders of a US patent in which mAb757 is a listed product. Other authors have no potential conflicts of interest to disclose.
Supplemental Material
Download MS Word (70.8 MB)Acknowledgments
The authors would like to thank Dr. Matthew Bettini and Dr. Alexandre F Carisey for providing flow cytometry and imaging equipment. This work was supported by the Juvenile Diabetes Research Foundation (JDRF 2-SRA-2016-238-S-B and JDRF2-SRA-2018-648-S-B to LZ), the National Institutes of Health (1R03AI139811-01A1 to LZ), the Cancer Prevention and Research Institute of Texas (PR150551 and RP190561 to ZA) and the Welch Foundation (AU-0042-20030616 to ZA). We also thank Dr. Massimo Pietropaolo for critical reading of the manuscript, this work was partly supported by the McNair Medical Institute at The Robert and Janice McNair Foundation (to Dr. Pietropaolo). L.Z. conceived the study, designed experiments, interpreted data, wrote and edited the manuscript. N.Z. and Z.A. were involved in antibody generation and manuscript editing. J.R.C., J.H., N.S., W.X., L.Y., and S.D. performed experiments. J.W.K. and H.W.D. contributed to discussions, critical reading, and editing of the manuscript. L.Z. is the guarantor of the data.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Daniel D, Gill RG, Schloot N, Wegmann D. Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur J Immunol. 1995;25:1056–12. doi:https://doi.org/10.1002/eji.1830250430.
- Wegmann DR, Norbury-Glaser M, Insulin-specific DD. T cells are a predominant component of islet infiltrates in pre-diabetic NOD mice. Eur J Immunol. 1994;24:1853–57. doi:https://doi.org/10.1002/eji.1830240820.
- Jasinski JM, Yu L, Nakayama M, Li MM, Lipes MA, Eisenbarth GS, Liu E. Transgenic insulin (B:9-23) T-cell receptor mice develop autoimmune diabetes dependent upon RAG genotype, H-2g7 homozygosity, and insulin 2 gene knockout. Diabetes. 2006;55:1978–84. doi:https://doi.org/10.2337/db06-0058.
- Stadinski B, Kappler J, Eisenbarth GS. Molecular targeting of islet autoantigens. Immunity. 2010;32:446–56. doi:https://doi.org/10.1016/j.immuni.2010.04.008.
- Yi W, Seth NP, Martillotti T, Wucherpfennig KW, Sant’Angelo DB, Denzin LK. Targeted regulation of self-peptide presentation prevents type I diabetes in mice without disrupting general immunocompetence. J C linInvest. 2010;120:1324–36. doi:https://doi.org/10.1172/JCI40220.
- Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 2005;435:220–23. doi:https://doi.org/10.1038/nature03523.
- Yang J, Chow IT, Sosinowski T, Torres-Chinn N, Greenbaum CJ, James EA, Kappler JW, Davidson HW, Kwok WW. Autoreactive T cells specific for insulin B:11-23 recognize a low-affinity peptide register in human subjects with autoimmune diabetes. Proc Natl Acad Sci U S A. 2014;111:14840–45.
- Wang Y, Sosinowski T, Novikov A, Crawford F, White J, Jin N, Liu Z, Zou J, Neau D, Davidson HW, et al. How C-terminal additions to insulin B-chain fragments create superagonists for T cells in mouse and human type 1 diabetes. Sci Immunol. 2019;4(34) :eaav7517. doi: https://doi.org/10.1126/sciimmunol.aav7517.
- Michels AW, Landry LG, McDaniel KA, Yu L, Campbell-Thompson M, Kwok WW, Jones KL, Gottlieb PA, Kappler JW, Tang Q, et al. Islet-derived CD4 T cells targeting proinsulin in human autoimmune diabetes. Diabetes. 2017;66(3):722–34. doi:https://doi.org/10.2337/db16-1025.
- Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes. 2008;57:1084–92. doi:https://doi.org/10.2337/db07-1331.
- Corper AL, Stratmann T, Apostolopoulos V, Scott CA, Garcia KC, Kang AS, Wilson IA, Teyton L. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science. 2000;288:505–11. doi:https://doi.org/10.1126/science.288.5465.505.
- Suri A, Vidavsky I, van der DK, Kanagawa O, Gross ML, Unanue ER. APCs, the autologous peptides selected by the diabetogenic I-Ag7 molecule are unique and determined by the amino acid changes in the P9 pocket. J Immunol. 2002;168:1235–43. doi:https://doi.org/10.4049/jimmunol.168.3.1235.
- Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol. 2010;11:350–54.
- Mohan JF, Petzold SJ, Unanue ER. Register shifting of an insulin peptide-MHC complex allows diabetogenic T cells to escape thymic deletion. J Exp Med. 2011;208:2375–83. doi:https://doi.org/10.1084/jem.20111502.
- Gioia L, Holt M, Costanzo A, Sharma S, Abe B, Kain L, Nakayama M, Wan X, Su A, Mathews C, et al. Position beta57 of I-A(g7) controls early anti-insulin responses in NOD mice, linking an MHC susceptibility allele to type 1 diabetes onset. Sci Immunol. 2019;4(38):eaaw6329. doi:https://doi.org/10.1126/sciimmunol.aaw6329.
- Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci U S A. 2010;107:10978–83. doi:https://doi.org/10.1073/pnas.1006545107.
- Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, Marrack P, Eisenbarth G, Kappler JW. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci U S A. 2011;108:16729–34. doi:https://doi.org/10.1073/pnas.1113954108.
- Zhang L, Crawford F, Yu L, Michels A, Nakayama M, Davidson HW, Kappler JW, Eisenbarth GS. Monoclonal antibody blocking the recognition of an insulin peptide-MHC complex modulates type 1 diabetes. Proc Natl Acad Sci U S A. 2014;111:2656–61. doi:https://doi.org/10.1073/pnas.1323436111.
- Wang Y, Sosinowski T, Novikov A, Crawford F, Neau DB, Yang J, Kwok WW, Marrack P, Kappler JW, Dai S, et al. C-terminal modification of the insulin B:11–23 peptide creates superagonists in mouse and human type 1 diabetes. Proc Natl Acad Sci U S A. 2018;115(1):162–67. doi:https://doi.org/10.1073/pnas.1716527115.
- Oi VT, Jones PP, Goding JW, Herzenberg LA, Herzenberg LA. Properties of monoclonal antibodies to mouse Ig allotypes, H-2, and Ia antigens. Curr Top Microbiol Immunol. 1978;81:115–20.
- Boitard C, Bendelac A, Richard MF, Carnaud C, Bach JF. Prevention of diabetes in nonobese diabetic mice by anti-I-A monoclonal antibodies: transfer of protection by splenic T cells. Proc Natl Acad Sci U S A. 1988;85:9719–23. doi:https://doi.org/10.1073/pnas.85.24.9719.
- Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K. Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes. 2012;61:3239–46. doi:https://doi.org/10.2337/db12-0112.
- Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Piganelli JD, Barbour G, Bradley B, Crawford F, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;11:225–31. doi:https://doi.org/10.1038/ni.1844.
- Spanier JA, Frederick DR, Taylor JJ, Heffernan JR, Kotov DI, Martinov T, Osum KC, Ruggiero JL, Rust BJ, Landry SJ, et al. Efficient generation of monoclonal antibodies against peptide in the context of MHCII using magnetic enrichment. Nat Commun. 2016;7(1):11804. doi:https://doi.org/10.1038/ncomms11804.
- Yamano S, Atkinson JC, Baum BJ, Fox PC. Salivary gland cytokine expression in NOD and normal BALB/c mice. Clin Immunol. 1999;92:265–75. doi:https://doi.org/10.1006/clim.1999.4759.
- Jonsson MV, Delaleu N, Brokstad KA, Berggreen E, Skarstein K. Impaired salivary gland function in NOD mice: association with changes in cytokine profile but not with histopathologic changes in the salivary gland. Arthritis Rheum. 2006;54:2300–05. doi:https://doi.org/10.1002/art.21945.
- Yu L, Robles DT, Abiru N, Kaur P, Rewers M, Kelemen K, Eisenbarth GS. Early expression of anti-insulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc Natl Acad Sci U S A. 2000;97:1701–06. doi:https://doi.org/10.1073/pnas.040556697.
- Pearson JA, Wong FS, Wen L. The importance of the non-obese diabetic (NOD) mouse model in autoimmune diabetes. J Autoimmun. 2016;66:76–88.
- Gill RG, Pagni PP, Kupfer T, Wasserfall CH, Deng S, Posgai A, Manenkova Y, Bel Hani A, Straub L, Bernstein P, et al. A preclinical consortium approach for assessing the efficacy of combined anti-CD3 Plus IL-1 blockade in reversing new-onset autoimmune diabetes in NOD Mice. Diabetes. 2016;65:1310–16. doi:https://doi.org/10.2337/db15-0492.
- Atkinson MA, Maclaren NK, Luchetta R. Insulitis and diabetes in NOD mice reduced by prophylactic insulin therapy. Diabetes. 1990;39:933–37. doi:https://doi.org/10.2337/diab.39.8.933.
- Roep BO, Wheeler DCS, Peakman M. Antigen-based immune modulation therapy for type 1 diabetes: the era of precision medicine. Lancet Diabetes Endocrinol. 2019;7:65–74. doi:https://doi.org/10.1016/S2213-8587(18)30109-8.
- Howell SL. The mechanism of insulin secretion. Diabetologia. 1984;26:319–27. doi:https://doi.org/10.1007/BF00266030.
- Zhang L, Nakayama M, Eisenbarth GS. Insulin as an autoantigen in NOD/human diabetes. Curr Opin Immunol. 2008;20:111–18. doi:https://doi.org/10.1016/j.coi.2007.11.005.
- Wicker LS, Clark J, Fraser HI, Garner VE, Gonzalez-Munoz A, Healy B, Howlett S, Hunter K, Rainbow D, Rosa RL, et al. Type 1 diabetes genes and pathways shared by humans and NOD mice. J Autoimmun. 2005;25(Suppl):29–33.
- Mannering SI, Di Carluccio AR, Elso CM. Neoepitopes: a new take on beta cell autoimmunity in type 1 diabetes. Diabetologia. 2019;62:351–56. doi:https://doi.org/10.1007/s00125-018-4760-6.
- Melli K, Friedman RS, Martin AE, Finger EB, Miao G, Szot GL, Krummel MF, Tang Q. Amplification of autoimmune response through induction of dendritic cell maturation in inflamed tissues. J Immunol. 2009;182:2590–600. doi:https://doi.org/10.4049/jimmunol.0803543.
- Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6:576–88.
- Davidson HW, Zhang L. Immune therapies for autoimmune diabetes targeting pathogenic peptideMHC complexes. J Mol Cell Biol. 2020. doi:https://doi.org/10.1093/jmcb/mjaa037.
- Vieira P, Rajewsky K. The half-lives of serum immunoglobulins in adult mice. Eur J Immunol. 1988;18:313–16. doi:https://doi.org/10.1002/eji.1830180221.
- Dekkers G, Bentlage AEH, Stegmann TC, Howie HL, Lissenberg-Thunnissen S, Zimring J, Rispens T, Vidarsson G. Affinity of human IgG subclasses to mouse Fc gamma receptors. MAbs. 2017;9:767–73. doi:https://doi.org/10.1080/19420862.2017.1323159.
- Beers SA, Glennie MJ, White AL. Influence of immunoglobulin isotype on therapeutic antibody function. Blood. 2016;127:1097–101. doi:https://doi.org/10.1182/blood-2015-09-625343.
- Levisetti MG, Suri A, Petzold SJ, Unanue ER. The insulin-specific T cells of nonobese diabetic mice recognize a weak MHC-binding segment in more than one form. J Immunol. 2007;178:6051–57. doi:https://doi.org/10.4049/jimmunol.178.10.6051.
- Nakayama M, Castoe T, Sosinowski T, He X, Johnson K, Haskins K, Vignali DAA, Gapin L, Pollock D, Eisenbarth GS, et al. Germline TRAV5D-4 T-cell receptor sequence targets a primary insulin peptide of NOD mice. Diabetes. 2012;61:857–65. doi:https://doi.org/10.2337/db11-1113.
- Michels AW, Ostrov DA, Zhang L, Nakayama M, Fuse M, McDaniel K, Roep BO, Gottlieb PA, Atkinson MA, Eisenbarth GS, et al. Structure-based selection of small molecules to alter allele-specific MHC class II antigen presentation. J Immunol. 2011;187(11):5921–30. doi:https://doi.org/10.4049/jimmunol.1100746.
- Haskins K, Portas M, Bergman B, Lafferty K, Bradley B. Pancreatic islet-specific T cell clones from nonobese diabetic mice. Proc Natl Acad Sci USA. 1989;86:8000–04. doi:https://doi.org/10.1073/pnas.86.20.8000.
- Zhang L, Jasinski JM, Kobayashi M, Davenport B, Johnson K, Davidson H, Nakayama M, Haskins K, Eisenbarth GS. Analysis of T cell receptor beta chains that combine with dominant conserved TRAV5D-4*04 anti-insulin B:9-23 alpha chains. J Autoimmun. 2009;33:42–49. doi:https://doi.org/10.1016/j.jaut.2009.02.003.
- Shulman M, Wilde CD, Kohler G. A better cell line for making hybridomas secreting specific antibodies. Nature. 1978;276:269–70. doi:https://doi.org/10.1038/276269a0.
- Talbot WS, Swyryd EA, Hogness DS. Drosophila tissues with different metamorphic responses to ecdysone express different ecdysone receptor isoforms. Cell. 1993;73:1323–37. doi:https://doi.org/10.1016/0092-8674(93)90359-X.
- Noh K, Mangala LS, Han HD, Zhang N, Pradeep S, Wu SY, Ma S, Mora E, Rupaimoole R, Jiang D, et al. Differential effects of EGFL6 on tumor versus wound angiogenesis. Cell Rep. 2017;21:2785–95. doi:https://doi.org/10.1016/j.celrep.2017.11.020.
- Zhang L, Stadinski BD, Michels A, Kappler JW, Eisenbarth GS. Immunization with an insulin peptide-MHC complex to prevent type 1 diabetes of NOD mice. Diabetes Metab Res Rev. 2011;27:784–89. doi:https://doi.org/10.1002/dmrr.1252.
- Leiter EH. The NOD mouse: a model for insulin-dependent diabetes mellitus. Curr Protoc Immunol. 2001;Chapter 15:Unit 15 9.