
ABSTRACT
T-cell bispecific antibodies (TCBs) are a novel class of engineered immunoglobulins that unite monovalent binding to the T-cell receptor (TCR) CD3e chain and bivalent binding to tumor-associated antigens in order to recruit and activate T-cells for tumor cell killing. In vivo, T-cell activation is usually initiated via the interaction of the TCR with the peptide-HLA complex formed by the human leukocyte antigen (HLA) and peptides derived from intracellular proteins. TCR-like antibodies (TCRLs) that recognize pHLA-epitopes extend the target space of TCBs to peptides derived from intracellular proteins, such as those overexpressed during oncogenesis or created via mutations found in cancer. One challenge during lead identification of TCRL-TCBs is to identify TCRLs that specifically, and ideally exclusively, recognize the desired pHLA, but not unrelated pHLAs. In order to identify TCRLs suitable for TCRL-TCBs, large numbers of TCRLs have to be tested in the TCB format. Here, we propose a novel approach using chimeric antigen receptors (CARs) to facilitate the identification of highly selective TCRLs. In this new so-called TCRL-CAR-J approach, TCRL-candidates are transduced as CARs into Jurkat reporter-cells, and subsequently assessed for their specificity profile. This work demonstrates that the CAR-J reporter-cell assay can be applied to predict the profile of TCRL-TCBs without the need to produce each candidate in the final TCB format. It is therefore useful in streamlining the identification of TCRL-TCBs.
Introduction
The concept of T-cell recruitment is a growing field of cancer immunotherapy, and a multitude of different molecular formats have been conceived for that purpose during recent years.Citation1–3 We have described 2 + 1 T-cell bispecific antibodies (TCBs) using an effector function silent Fc partCitation4–10 ()). Two antigen-binding fragments (Fabs) harbor paratopes toward a selected tumor-associated antigen (TAA) on the surface of cancer cells, while the additional third Fab is able to bind to CD3 on T cells, bringing the two cell types into close proximity ()). This leads to T-cell recruitment, activation, and subsequent killing of the bound cancer cell. The potency with which a particular TCB can mediate killing, in the following referred to as “TCB killing capability”, is mainly governed by the affinity, epitope, and geometry of binding with which the TAA is recognized. To improve therapeutic options and broaden the target space for TCBs, access to novel tumor-specific targets is desired.Citation9 Most proteins produced by tumor cells reside intracellularly, such as overexpressed oncogenic proteins or neoantigens, including mutated tumor suppressor proteins and translocated genes. Intracellular proteins are processed by proteasomes into small peptides and transported by the transporter associated with antigen processing (TAP) to the endoplasmic reticulum (ER). Here the peptides can be loaded onto nascent major biocompatibility complex (MHC) class I molecules, also referred to as human leukocyte antigen (HLA) class I in the human system, and eventually transported to the cell surface as peptide-MHC complex (pMHC/pHLA). T-cell receptors (TCRs) on effector T cells recognize the composite epitopes of pMHC.Citation11,Citation12 An emerging approach that builds on this concept is the development of TCR-like antibodies (TCRLs) that recognize epitopes similar to those recognized by a TCR.Citation13–15 These composite epitopes are composed of linear peptide sequences bound to and presented by the HLA. For the treatment of cancer, peptides derived from overexpressed self-antigens and transcription factors, oncofetal antigens, cancer-testis antigens, translocation-derived amino acid sequences, viral neoantigens, or mutated neoantigens can be imagined,Citation16 representing ample opportunities for targeted therapy approaches using TCRLs.Citation16–19 Wilms tumor protein (WT1) was ranked by the National Cancer Institute as the Number 1 target for cancer immunotherapy in the year 2009.Citation20 WT1 gene is overexpressed in hematological malignancies, e.g., acute myeloid leukemia (AML).Citation21 The nonameric peptide 126–134RMFPNAPYL (RMF) presented by HLA-A*0201, a WT1-derived CD8+ T-cell human leukocyte antigen (HLA)–A0201 epitope, is a validated target for T-cell–based immunotherapy such as TCRs or TCRLs.Citation22
Figure 1. Schematic showing the structure of a TCRL-TCB. (a). The two TCRL-Fab domains confer bivalent binding to pMHC. One of these Fabs is fused “head-to-tail” via a flexible linker to the CD3ε-binding Fab, which enables the TCB to bridge T-cells to tumor cells. Fc heterodimerization is assured by “knobs-in-holes” (KiH) mutations in the CH3 domain, and the Fc-region furthermore carries the P329G LALA mutation that prevents activation of innate immune effector cells, while still extending serum half-life via binding to the neonatal Fc receptor (FcRn). (b). Illustration of the mode of action of a TCB, including bispecific target engagement leading to immune-synapse formation and T-cell killing of the target cell. (c). Overall scheme of the Fab-CAR-constructs used for transduction into Jurkat cells. The Fab coding sequences were assembled by Gibson Assembly from building blocks coding for the light chain, IRES and heavy chain. (d). Representation of the CAR-J assay set up. Co-incubation of CAR-J cells and target cells leads to immune synapse formation and T-cell activation, which can be read out and quantified as luciferase signal
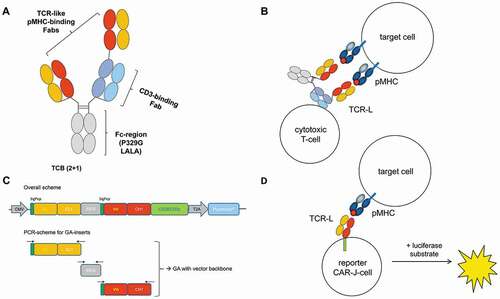
A major challenge in generating pMHC-specific TCBs is to identify TCRLs that specifically, and ideally exclusively, recognize the desired pMHC (of, for example, HLA-A2 allotype), but do not exhibit off-target binding to unrelated peptides in context of the MHC.Citation23 Experience from lead identification in different TCRL-projects showed that, while the generation of such TCRLs with reasonably high binding affinities (e.g., determined by surface plasmon resonance (SPR) or fluorescence-activated cell sorting (FACS)) is feasible by, for example, phage display or immunization, many TCRL antibodies ranked as “clean” in simple binding assays induce killing of cells with unrelated pMHC when converted into TCBs. This is due to the fact that TCBs only require very low-affinity interactions with tumor antigens in the micromolar range in order to mediate significant killing or T cell activation. At the same time, the high affinity of a TCRL does not automatically translate into potent cytotoxic activity and specificity; rather, the optimal TCRL affinity has an upper threshold that differs from case to case.Citation24 Therefore, it is crucial to enable the lead identification process of TCRL candidates with a reliable preselection process that is able to sort out candidates harboring affinity toward MHC-displayed off-target-peptides. In order to identify highly specific TCRLs with minimal off-target reactivity (e.g., recognition and killing of unrelated pMHC complex bearing cells) that are suitable for use in TCB formats, a large number of TCRLs has to be converted into the TCB format to test their biological activity. This is of particular importance when aiming for high affinity TCRLs with KD values in the single-digit nM or pM range.
Here, we propose a novel approach for the identification and screening of unique, highly selective TCRLs suitable for use in the TCBs, representative for any T-cell recruiting format. To date, TCRL-Fab candidates had to be converted into the final TCB format to allow functional screening. Even though the characteristics in TCB-killing assays can be anticipated from TCB activation assays, i.e., reporter Jurkat cells that are co-incubated with target cells in the presence of the TCB-candidates in question,Citation25 these assays still require the labor-intense production and purification of TCB molecules.
Our aim was to establish a screening approach that would be independent from production of TCBs. We present here a cell-based screening approach that builds on chimeric antigen receptors (CARs), into which the TCRL-candidates have to be embedded. CAR molecules are composed of an extracellular binding moiety, typically an IgG-derived single-chain variable fragment (scFv), fused to the intracellular zeta chain of the T-cell receptor (TCR) as signaling domain. In order to screen TCRL-Fabs, pre-selected by phage display, regarding their capability as TCRL-TCB drug leads, the candidates are converted into TCRL-Fab-CAR molecules ()) and expressed in Jurkat reporter-cells. The resulting pools of transduced cells, herein referred to as “CAR-Jurkat” (CAR-J) cells harbor an NFAT-luciferase reporter system, yielding a cellular screening tool that we use in so-called “Fab-CAR-J” assays ()). To establish the assay, we compared various TCRL-candidates, initially selected by phage display against RMF-HLA, regarding their biological activity in CAR-J assays and final TCB-activation and -killing assays, respectively. The pools of transduced CAR-J cells were polyclonal in the sense that they traced back to more than one single-transduced Jurkat cell, but monoclonal in terms of TCRL-Fab-CAR candidate gene that had been used for transduction. That is to say that each given TCRL-Fab-CAR-J pool does express one particular TCRL-Fab-CAR candidate. Comparing the performance of different TCRL-Fab candidates in Fab-CAR- vs. TCB-format, i.e., biological activity in the Fab-CAR-J assay with killing activity in the final TCB-activation and -killing assays, illustrates the predictive strength of the CAR-J assay to assess TCRL-candidates for their potential killing capability, without the need of screening the final TCB format. This shortens the lead identification process, and can thereby accelerate and facilitate the identification of highly specific TCRLs for TCB development, representative for any other biologics format that pairs the concepts of T-cell recruiting and TCR-like targeting.
Results
T-cell activation involves a cascade of intracellular signaling events that are integrated in the well-characterized NFkB (Nuclear factor kappa B), AP-1 (Activator Protein-1), and NFAT (Nuclear factor of activated T-cells) pathways.Citation26–29 The members of both classes of transcription factors localize to the nucleus, and thereby drive productive immune responses. This is why response elements from each have been used to engineer reporter-cell lines that measure T-cell activation.Citation30 We selected Jurkat cells, a well-studied, immortalized human T-cell line engineered to express luciferase under the control of a cassette of NFAT response elements. These NFAT-Luc-Jurkat cells show robust luciferase expression, which can be measured already at 4 hours upon activation.
We hypothesized that the activation of the NFAT-Luc-Jurkat cells via CAR-derived signaling would correlate to the activation-strength derived from activation via respective TCBs (with CAR and TCB having the same TAA-binder), and that the CAR-derived signal strength could therefore serve as a predictive assay for TCB potency.
Many TCRLs previously “clean” as IgG show allo-reactivity as TCBs
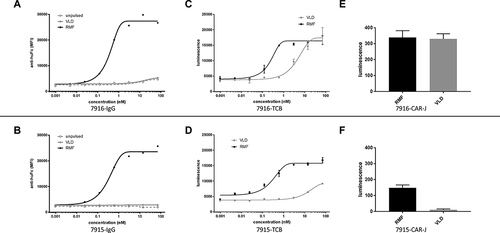
In TCBs that harbor TCRL-Fabs as anti-tumor moiety (TCRL-TCBs), a very transient interaction between TCRL-Fab and pHLA might already be sufficient to elicit T-cell activation. This is in line with affinities of TCRs toward their cognate antigens that are known to be in a rather broad, micromolar range.Citation31–33 In terms of safety, this represents a hurdle regarding the required specificity toward the TCRL-Fab. After selecting TCRL-Fabs binding to different pHLA-targets by means of phage display, we faced the issue that screening for binding by classical assays like ELISA or flow-cytometry was not sufficient to assess the specificity governing activity in the final TCRL-TCB format: Fab-candidates from a pool of binders that had been selected against pHLA harboring the RMF peptide (RMF-HLA) were assessed regarding their specificity. When comparing the binding of selected candidates to T2 cells pulsed with two different WT1-derived peptides (RMF and VLD (VLDFAPPGA, WT1 p37–45), respectively), we observed stronger binding on RMF-pulsed cells. T2 cells were used here because of their defect in peptide loading. A favorable signal-to-background ratio. i.e., specificity window in flow cytometry, using the IgG format (,b)) in many cases, however, did not fully translate to the TCB-activity of the respective binders: In TCB-killing assays performed with the same candidates upon conversion into TCBs, candidates with high RMF/VLD signal in FACS proved active on VLD as well (,d)). To better sample the transient but critical interaction between TCRL-Fab and pHLA, we aimed to assess the biological activity, i.e., the TCB killing capability toward different pHLA complexes, in a cell-based assay that would mimic the TCB-induced interaction of T-cell and target cell. For this, we established a CAR-based cellular screening tool. Here, preselected Fab-candidates are embedded into a CAR-framework and transduced into NFAT-luciferase Jurkat reporter-cells.
Figure 2. Assessment of TCRL-candidates selected against RMF-HLA by three different cell-based assays involving T2 cells pulsed with two different WT1-derived peptides (RMF and VLD). (a, b) binding of candidates 7916 and 7915 in IgG-format assessed by FACS. (c, d) TCB-activity of the respective binders in TCB-killing assays performed with the same candidates upon conversion into TCBs. (e, f) Luciferase signal from a CAR-J assay performed with both candidates in CAR-J format indicates that binder 7915 gives a favorable signal window between SRMF and SVLD when compared to binder 7916
CAR-embedded Fab binders can serve as reporter in a cellular assay
Using Gibson assembly,Citation34 we conveniently embedded the coding sequences of different TCRL-Fab candidates, preselected for binding to the respective pHLA into the backbone of a lentiviral transfer plasmid.Citation35 The extracellular domain of the CAR is composed of the different Fab candidates. In contrast to the classical CAR-build up, which uses the immunoglobulin binding moiety in scFv format, we aimed to display our binders in Fab format to enable straightforward conversion, e.g., from Fab-based phage display libraries. For this purpose, we subcloned light and heavy chain of the respective Fab with N-terminal leader peptides, connected by an internal ribosomal entry site (IRES). The overall constructs are encoded by ssLC-IRES-ssHC_CAR_T2A_PuroR () and can be easily embedded into the lentiviral transfer plasmid backbone in a one-pot one-step Gibson assembly. For each candidate in question, we performed transduction into reporter Jurkat cells that express luciferase under a 3× NFAT response element.Citation30 Transduced Jurkat cells were allowed to grow in pools upon spinfection for 5 d, enriching positive transductants by puromycin selection starting on d 3 after spinfection. The expression levels of the CAR-embedded Fabs, displayed on the cell surface of the Jurkat cells, were comparable, as assessed by anti-Fab flow cytometry (data not shown). The different CAR-J pools were co-incubated with T2 cells that had previously been pulsed with different peptide-epitopes as indicated. After 4 hours of co-incubation, we measured luciferase expression, indicative for T-cell activation derived from the interaction of TCRL and pHLA. The comparison of CAR-J pools of binders 7915 and 7916, which had performed differently in FACS vs. killing assays, showed that the CAR-J assay read-out might serve as a predictive assessment of later TCB potency. The two binders performed differently regarding their ratio of SRMF/SVLD. While CAR-J pool 7916 gave strong luciferase signal on both peptides ()), pool 7915 showed a signal window between SRMF and SVLD that was similar to the window seen in the TCB killing assay ()).
Fab-CAR-J for specificity screening on panels of potential off-target peptides
Next, we set out to compare the signal strength of further individual TCRL-Fab candidates in the CAR-J reporter assay with the characteristics of the respective candidates assessed in TCB-activation-assays (upon conversion into the TCB format). In addition to expanding the number of candidates assessed with both assays, we aimed to broaden our screening toward a bigger panel of potential off-target peptides (POTPs) as they occur theoretically in the human peptidome and could be presented on HLA-A02.
To identify POTPs in silico to any given target peptide-HLA, we screened the relevant proteomes (human, including splice variants and single nucleotide polymorphisms, human pathogenic viruses, and intra-cellular bacteria) for different pre-defined peptide patterns (with one or multiple amino acids per position resulting from structural considerations and modeling approaches) homologous to the respective target peptide. These patterns might have two or multiple wildcards (at least for expected anchor positions). The proteome searches were performed with allowance of none to multiple mismatches. According to the rationale of the very proteome search, the resulting hits were filtered and prioritized for their fit to the target peptide and to the search pattern. In a second filtering step, the hits were run through several different HLA binding prediction algorithms and categorized according to and filtered for their predicted affinity to the HLA. In parallel, all hits were looked up in the IEDB database (https://www.iedb.org/) and any experimental confirmations were prioritized either way. All hits passing these filters were defined as POTPs.
Signal output from TCRL-Fab-CAR-J reporter-cells is predictive for TCB characteristics
To assess the predictive potential of the CAR-J assay regarding the specificity of our TCRL-Fab-candidates, and thereby regarding their valuation for conversion into the TCB format, we scaled up the comparison of the different assay systems and compared respective TCRL-Fab candidates in the CAR-J- and the TCB format via correlation plots. Since we were especially interested to assess the CAR-J assay’s predictive potential regarding the off-target risk related to the different POTPs, we screened preselected TCRL-candidate for their cross-reactivity on 22 different POTPs. The correlation plot between CAR-J assay and TCB-activation assay demonstrates that the two methods give very similar rankings of specificity toward POTPs (). The POTP sequences analyzed are provided in supplementary Figure 1.
Figure 3. Correlation analysis of TCB-activation to the CAR-J assay. TCRL-Fab candidate 7915 is compared in TCB- vs. Fab-CAR-J-format. Target cells being pulsed with 10 µM of each peptide. Values represent the strength of activation, read out as luciferase signal and normalized to RMF-control, by each format upon co-incubation with T2-cells, pulsed with indicated peptides. Cross reactivity was tested on 22 POTPs and obtained signals (luminescence) were normalized to positive control (RMF). The graph of TCB-activation vs. CAR-J activation demonstrates the strong correlation between the two methods
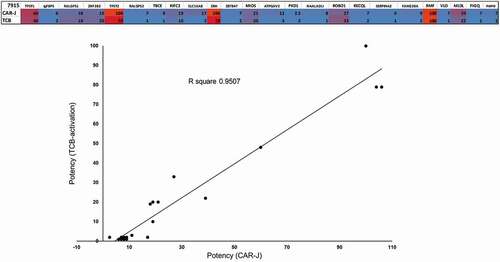
Taken together, these results show that TCRL-Fab-CARs not only induce signaling upon successful and specific TCRL-Fab-pHLA interaction, but they also predict TCB activation and potency.
CAR-J assay on T2 cells pulsed with peptide dilutions
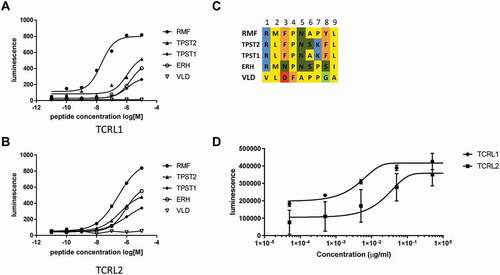
One limitation of the CAR-J assay in comparison to TCB-activation assays is the inability to easily vary the binder concentration. Whereas TCB-killing experiments can be feasibly set up as dilution series, the concentration of binder candidate in a CAR-J assay is fixed, since the binder is expressed as CAR by the transduced reporter cells. In order to assess the TCRL-candidates not only regarding a favorable specificity-profile, but also with regard to affinity, i.e., high biological activity on target pHLA, we modified the CAR-J assay by assessing different densities of pHLA on the cell surface of the target cells. While in the initial screenings the peptide concentrations during pulsing had been kept constant, we now pulsed the target T2 cells with dilution series of the respective peptides. For comparison, we picked the Fab-CAR-J pools of two different TCRL-candidates from another initial phage display selection on RMF-HLA, “TCRL1” and “TCRL2”, that had shown quite similar specificity-profile patterns when assessed on the POTP-panel. The CAR-J-signals of the respective pools on T2 cells pulsed with dilution series of five different peptides are depicted in (a,b), with an alignment of the respective peptide sequences given in ). Importantly, the different EC50 of the two TCRL-candidates in this peptide dilution series is in line with a killing assay with the respective TCRL-TCBs on T2 cells pulsed with the target peptide (RMF) ()). In both assays, CAR-J with peptide dilution and TCB-killing, candidate TCRL1 was active at lower peptide concentration, or showed killing at lower TCB concentration, respectively. The peptide dilution series is at the same time a good indication that the peptide concentration of 10 µM, which was used in all standard CAR-J screenings described above, is appropriate for screening: The sigmoidal curves of all peptide dilutions in (a,b) are close to or in saturation. Hence, using a peptide concentration of 10 µM for screening should assure that a TCRL-Fab candidate´s activity on a particular POTP does not remain unnoticed, which is an important requirement for a screening tool.
Figure 4. Modified CAR-J assay with different densities of pHLA on T2 cells via pulsing with dilution series of the respective peptides. Comparison of CAR-J pools (a, b) of two different TCRL-candidates (TCRL1 and TCRL2) and a killing assay (c) with the respective TCRL-TCBs on T2 cells pulsed with the target peptide (RMF) both indicate higher activity for candidate TCRL1
pHLA-CD3 fusion proteins can serve to report interaction of TCRL and pHLA
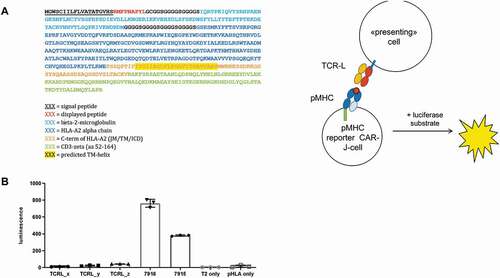
To further test the versatility of such cellular co-incubation assays, we flipped the components of the CAR-J system: switching the TCRL-Fab-CD3 for a pHLA-CD3 allows for screening different peptides instead of different TCRL-Fabs using NFAT-luciferase Jurkat reporter-cells. The extracellular part of the new reporter constructs is built-up from a single-chain fusion of peptide-epitope, beta-2 microglobulin, and HLA heavy chain (), described earlier.Citation36 To test this flipped system for its predictive potential, we expressed the RMF-peptide as pHLA-CD3 fusion on JNL cells, co-incubated those cells with T2 cells expressing different TCRL-Fab molecules as membrane-bound fusions (linked via G4S-linkers to the transmembrane domain of CD28) and assessed the luciferase signal after 4 hours of co-incubation. In line with expectations, the two binders 7915 and 7916, previously compared in the original CAR-J format ()), showed again a strong signal on RMF-pHLA, this time derived from the pHLA-CD3 fusion protein ()). Importantly, unrelated TCRL-candidates (TCRL_x, TCRL_y, and TCRL_z), selected by phage display against different pHLA-targets, did not give luciferase signal upon co-incubation with the JNL-cell-displayed RMF-pHLA-CD3 fusion. Of note, the fusions described here differ in two ways from similar constructs that have been described in two recent reports for the determination of TCR specificity:Citation37,Citation38 (1) they contain a stabilizing disulfide bridge between the first linker and position 227 of the H chain; and (2) the transmembrane domain is not derived from CD28 (like in CARs), but the extracellular single-chain peptide-HLA fusion is fused to the TM of wildtype H-chain instead.
Figure 5. pHLA-CD3 fusion proteins can serve to report interaction of TCRL and pHLA. (a) Schematic showing the design of the peptide-MHC NFAT-luciferase reporter system. (b) Primary sequence of ECD and TM-part of a single chain pHLA fusion protein that is fused to the intracellular domain of human CD3ζ (Uniprot Entry P20963, aa 52–164, not shown). The signal peptide, needed for translocation into the plasma membrane is cleaved off during translation. (c) Luciferase signal following co-incubation of JNL cells expressing the RMF- HLA-CD3 fusion with T2 cells expressing different TCRL-Fab molecules as membrane-bound fusions
Discussion
TCBs are highly potent antibody derivatives, able to mediate T-cell activation and killing. The TAA-targeting Fab moieties of a TCB can be selected by in vitro selection methods like phage display or immunization, and typically show affinities in the single-digit nanomolar range. T-cell-mediated killing can, however unintendedly, be mediated through binding interactions between targeting domain and putative off-target with KD values in the micromolar range. Fabs selected as target antigen-binding moieties for incorporation into the TCB format therefore need to be highly selective to avoid unfavorable reactivity, e.g., killing of off-target cells or alloreactivity. In early assessment and development of TCBs, it is a drawback that the conventional classical binding assays used in lead identification of novel binders (e.g., SPR, flow cytometry) measure affinity, which does not always reflect the specificity in functional terms, i.e. highly potent biological activity occurring at low receptor occupancy, in the TCB format. Here, we present a method that can be used to assess and predict the final specificity profile of TCRL-Fab moieties selected for TCBs, representative for any T-cell recruiting format. It is predictive for the final Fab characteristics upon conversion into the TCB format, without the need of converting the binders in question into the final TCB format, and can therefore be used to facilitate and accelerate the screening of new binders. The described CAR-J reporter assay is based on the interaction of CAR-embedded TCRL-Fabs (displayed on reporter Jurkat cells) with peptide-pulsed endogenous HLA molecules (pHLA) on T2 cells. This transient interaction leads to cell-cell proximity and subsequent activation of the reporter T-cell according to the kinetic-segregation model,Citation39,Citation40 which can in turn be read out as luciferase activity.
Our aim was to substitute the link between the TCB’s anti-CD3 moiety and the T-cell’s TCR by using the concept of CARs that constitute this link in the form of a chimeric receptor, which is stably expressed by the T cells. Incorporation of the TCRL-Fab binders in question as extracellular binding moiety, linked within the CAR to the CD3 chain that acts as signaling domain, allows binding and cell–cell interaction to be linked to a direct functional read out. For the latter, the CARs are transduced into JNL cells that express the luciferase reporter proportionally to the strength of the transient cell-cell contact governed by the characteristics of the interaction between TCRL-candidate and respective pHLA in question. A high level of expression of the luciferase reporter upon co-cultivation of CAR-J and target cells is indicative of cell-cell interaction, mediated by the TCRL-Fab, and allows prediction of the respective TCRL-candidates’ performance in the TCB format. Comparison of different candidates in FACS, CAR-J, and TCB activation assays indicates the predictive potential of the TCRL-Fab-CAR-J screening assay. Binders that appeared specific in FACS, showing strong binding to RMF-pulsed T2 cells and low binding to VLD-pulsed cells, but that proved rather unspecific in TCB-activation, revealed this characteristic already in the CAR-J assay.
The method is particularly suitable to assess and select novel TCRL-Fab moieties for TCBs from pre-selected pools of binders because it measures T-cell activation using a comparable mechanism of action like the intended final TCB format, thereby adding another variation of such cellular assays putting the selection into the context of the immunological synapse.Citation41 We could show that, out of a pool of TCRL candidates incorporated into either TCB- or CAR-format, T-cell activation patterns in both systems were comparable. In other words, a particular TCRL unspecific in the TCB format is expected to mediate unspecific activation in the CAR-format, and vice versa. This predictivity of the CAR-J for the TCB format could now also be used to move from screening to selection, i.e. by converting a library of binders – previously pre-selected and narrowed by a few rounds of phage display – into TCRL-CAR-J cells and screen those subsequently either in an individual or arrayed fashion or in a CAR-J pool. The latter would ideally connect TCR signaling activation with a reporter that can be sorted by FACS, similar to a recent report using the output of fluorescent protein reporters for the accelerated selection of lead CAR candidates for clinical translation.Citation42 Especially in the framework of TCRL binder selection, where the off-target space is huge due to the above mentioned reasons, a second reporter signal within the CAR-J cells could be implemented as survival signal, such as an NFAT-inducible thymidine kinase (TK). Following library transduction, one would then firstly expose the pool to target cells displaying off-target pHLA complexes, followed by negative selection via the TK suicide substrate ganciclovir.
Similar to another CAR-J assay we recently describedCitation43 that also uses an anti-IgG modCAR format,Citation44 the assay we described here is robust, suitable for use in high-throughput, and efficient in terms of hands-on time needed to accomplish the assay. Furthermore, it renders conversion into and production of a huge number of TCBs unnecessary, since the candidate proteins in question are directly expressed by the reporter cells. Thereby, TCB-production within the process of lead identification can be reduced to a final set of candidates that appears to be most potent according to the CAR-J assay, and can then be screened in the final TCB format.
In comparison to conventional cell-killing assays, the CAR-J assay requires shorter cell-co-incubation times, and a facile assay procedure, since it tolerates the presence of dead cells in the cell pools used for co-incubation. This is another advantage in contrast to TCB-killing assays, wherein the functionality of an antibody is determined by measuring cell death. One further advantage of the described assay is that no washing steps are required. This approach could also be applied to other platform used for the selection of TCR-like binders, like nanobodies, DARPins, and anticalins, that can be expressed on Jurkat cells in a CAR-T like fashion.Citation45
Taken together, in the context of lead identification, the reporter-assay presented here has several advantages over any classical cell-killing assay: (1) the number of candidates to be produced in the final TCB format can be largely reduced; (2) assay incubation times are faster (several hours as opposed to overnight for killing assays), allowing the assay to be performed in a single day; and (3) there is no need to distinguish target cells from effector cells by labeling.
To indicate the versatility of the CAR-J assay, we could both establish an assay-variant of the TCRL-Fab-CAR-J assay in which series of peptide dilutions on the T2-cells help to predict the TCRL-candidate’s potency, and another variation in which the cellular signal underlying the assay is derived from a pHLA-b2m-CD3 fusion protein, leading to a pHLA-CAR-J assay. This concept of pHLA-CD3 fusions allows flipping of the interacting components TCRL-Fab and pHLA in terms of the cells by which they are presented. The pHLA-CAR-J assay allows peptides presented by the HLA to be screened for their interaction with a given TCRL-Fab, similar to approaches used for the identification of pMHC-specific TCRs described recently by Baltimore et al.Citation37 and Kopf et al.Citation38 This might be useful, for example, to be able to screen TCRL-Fab candidates displayed by cells that are easier to transduce than Jurkat cells, which is especially interesting if one aims to screen whole libraries of candidates in a high throughput fashion, and is therefore dependent on high transduction efficiency. Of note, the original CAR-J assay and the pHLA-CAR-J assay differ significantly in that, upon pulsing onto T2 cells, the peptides have defined, and varying half-lives as free peptides, whereas in the flipped CAR-J assay, the peptides are covalently fixed and hence have no decay. Therefore, the affinity between HLA and peptide, which might clearly be rate-limiting for the overall interaction between TCRL and pHLA, becomes irrelevant for the assay read-out. This may dramatically change the sensitivity on some off-targets. However, since this will result in false-positives rather than in false-negatives, this aspect has to be kept in mind, but is not worrisome per se.
Under some circumstances, this strengthening of the pHLA might increase the range of application. Jurkat reporter cells expressing a TCR-like CAR of a defined peptide-MHC specificity could, for instance, be applied for diagnostic purposes for the detection of the presentation of the respective peptide in a peptide-MHC complex on tumor/target cells derived, for example, from patient specimens like blood or tumors. Last but not least, TCR-like binders identified through this methodology could also be used in TCR-like CAR-T cell therapy,Citation46,Citation47 as it has been previously shown for TCR-like CAR-T cells specific for WT1 RMF pMHC,Citation48 NY-ESO-1,Citation49 Alpha-Fetoprotein,Citation50,Citation51 or the melanoma-related gp100/HLA-A2-targeting.Citation52
Materials and methods
Cell lines
T2 cells were obtained from ATCC and maintained as per instructions provided. JNL (Jurkat cells engineered with an NFAT-dependent Luciferase gene) cells were obtained from Signosis (Signosis, Inc. Catalog v#SL-0032-FP) and maintained in Advanced RPMI-1640 medium supplemented with 2% fetal bovine serum. Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2 and passaged every 3–4 d.
Peptide pulsing of T2 cells
T2 cells were pulsed with indicated concentrations of chemically synthesized peptide by simple coincubation for 30 minutes at 37 °C, followed by consecutive washes in (phosphate-buffered saline (PBS).
Flow cytometry
Cells were incubated with respective antibodies at indicated concentrations in 100 µl of FACS-buffer (PBS containing 0.1% (v/v) Tween20, 0.2% (w/v) bovine serum albumin, and 0.05% (w/v) NaN3) per condition. After two washes with buffer, the cells were incubated with secondary antibody (anti-huFc) (polyclonal anti-human IgG F (ab`)2 fragment-specific and R-phycoerythrin (PE)-conjugated AffiniPure F (ab`)2 goat fragment, Jackson ImmunoResearch #109–116-097) for 45 minutes on ice, followed by three washes as above. Finally, the cells were resuspended in 200 µl FACS buffer and analyzed using a Fortessa flow cytometer. FACS data were analyzed using FlowJo software (FlowJo_V10).
Construction of lentiviral-based chimeric antigen receptors
Fab-CARs were constructed by amplifying the individual Fab-chains (including leader peptides) by PCR, and assembling them together with an IRES by means of Gibson Assembly into the lentiviral transfer plasmid pCDH, kindly provided by Patrick Salmon, University of Geneva, Geneva, Switzerland. The resulting Fab-CARs were build up from TCRL-Fab, fused via a G4S-linker extending the CH1-domain to the CD28-transmembrane and -intracellular domain (Uniprot Entry P10747, aa153-2020) and the intracellular domain of human CD3-zeta (Uniprot Entry P20963, aa 52–164). To allow for feasible monitoring of transduction efficiency and for enrichment of transductants within transduced pools, the CD3-zeta was followed by a T2A-linked eGFP as fluorescent reporter and a T2A-linked puromycin resistance gene for selection.
Preparation of VLPs and lentiviral transduction of JNL cells
Lentiviral virus-like particles (VLPs) were produced following Lipofectamine LTX™ based transfection of HEK293T cells (ATCC CRL3216) grown to 60–70% confluency with a mixture of Fab-CAR encoding transfer plasmids as well as packing plasmids psPAX2 and pCAG-VSVG-MD2G at 1:1:0.55 weight ratio. 66 hours post-transfection supernatants were collected, centrifuge for 5 minutes at 250 g to remove cell debris, and filtered through a 0.45 µm polyethersulfone filter. VLP-containing supernatants were used directly to transduce JNL cells via spinfection for 90 minutes (32 °C, 1000 × g). Transduced cells were enriched as pools under puromycin selection (1 µg/ml) applying selection pressure on the third day post spinfection.
CAR-J assay and luminescence detection
CAR-expressing JNL cells (CAR-J cells) and peptide-pulsed T2 cells were seeded each with 104 cells in a total volume of 30 µl medium per well for co-incubation in white flat clear-bottom 384-well plates. After 4–6 hours of incubation at 37 °C, 6 µl of ONE-GloTM luficerase assay (E6120, Promega) was added and luminescence was read immediately using a Tecan microplate reader.
TCB activation assay
TCB-mediated activation of JNL cells was performed by mixing 8000 JNL cells with the same number of target cells in presence of indicated TCB concentrations in white flat clear-bottom 384-well plates. After 12–16 hours of incubation (37 °C, 5% CO2) luciferase substrate (ONE-GloTM, Promega) was added and relative luminescence units (RLUs) were read out after 15 minutes incubation at room temperature using a TECAN Spark10M plate reader.
TCB killing assay
Peripheral blood mononuclear cells (PBMCs) were obtained from the blood donation center Zurich in accordance with the WMA Declaration of Helsinki. Donors signed a written informed consent before sample collection. Buffy coats were diluted 2:1 with PBS. PBMCs were then isolated by density gradient centrifugation (450 × g, 30 minutes at room temperature) over Histopaque-1077 (Sigma-Aldrich). PBMCs were harvested from the interphase, washed three times with PBS, and directly used for killing assays. TCB-mediated cell death was assessed using the CytoTox-Glo cytotoxicity assay (Promega) according to the vendor’s instructions. In brief, 3 × 104 target cell and 6 × 104 PBMCs were co-incubated per 96-well round-bottom microtiter plate in 150 µl medium in the presence of the respective TCB at indicated concentrations for 24 up to 48 hours (37 °C, 5% CO2). Afterward, 75 µl of supernatant were transferred into wells of a white 96-well plate and mixed with 25 µl of CytoTox-Glow reagent. Luminescence was read on a TECAN plate reader after 15 minutes of incubation at room temperature.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software version 7. In CAR-J assays, differences with a P < .05 were considered statistically significant. For dose-response curves, EC50 values were determined using nonlinear regression curve fitting. All experiments were repeated a minimum of three times. Data are shown as mean with SD (n = 3) or individual curves (n < 3).
Abbreviations
AML Acute myeloid leukemia
CAR Chimeric antigen receptor
CAR-J Acute myeloid leukemia
CAR-T Chimeric antigen receptor T cell
Fab Antigen binding fragment
FACS Fluorescence-activated cell sorting
Fc Constant fragment
IRES Internal ribosomal entry sequence
NFAT Nuclear factor of activated T-cells
MHC Major histocompatibility complex
pMHC Peptide MHC complex
POTP Potential off-target peptide
PBMC Peripheral blood mononuclear cells
RMF RMF peptide
scFv Single chain variable fragment
SPR RMF peptide
TAA Tumor-associated antigen
TAP Transporter associated with antigen processing
TCB T cell bispecific
TCR T cell receptor
TCRL TCR-like
TK Thymidine kinase
VLD VLD peptide
VLP Virus-like particle
WT1 Wilms tumor protein
Disclosure of Potential Conflict of Interest
CJ, CK, DD, EM, LJH, PU, VP, WX are named as co-inventors on a patent applications relating to the described technology. All authors are employees of Roche except REK. CK, and PU hold Roche stock. RK served/serves as a consultant to Roche.
Supplemental Material
Download Zip (1.2 MB)Acknowledgments
CJ performed this work as a fellow of the Roche Postdoctoral Fellowship (RPF) program. The authors would like to thank Camille Delon, Desirée Leisibach and Dr. Tina Weinzierl and Dr. Tanja Fauti for their helpful advice on various technical issues examined in this paper.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- Clynes RA, Desjarlais JR. Redirected T cell cytotoxicity in cancer therapy. Annu Rev Med. 2019;70:437–10. doi:https://doi.org/10.1146/annurev-med-062617-035821.
- Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18:585–608.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9(2):182–212. doi:https://doi.org/10.1080/19420862.2016.1268307.
- Bacac M, Colombetti S, Herter S, Sam J, Perro M, Chen S, Bianchi R, Richard M, Schoenle A, Nicolini V, et al. CD20-TCB with obinutuzumab pretreatment as next-generation treatment of hematologic malignancies. Clin Cancer Res. 2018;24:4785–97. doi:https://doi.org/10.1158/1078-0432.CCR-18-0455.
- Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, Bodmer W, Lehmann S, Hofer T, Hosse RJ, et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res. 2016;22:3286–97. doi:https://doi.org/10.1158/1078-0432.CCR-15-1696.
- Seckinger A, Delgado JA, Moser S, Moreno L, Neuber B, Grab A, Lipp S, Merino J, Prosper F, Emde M, et al. Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell. 2017;31:396–410. doi:https://doi.org/10.1016/j.ccell.2017.02.002.
- Bacac M, Klein C, Umana P. CEA TCB: a novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology. 2016;5:e1203498. doi:https://doi.org/10.1080/2162402X.2016.1203498.
- Klein C, Schaefer W, Regula JT, Dumontet C, Brinkmann U, Bacac M, Umaña P. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods. 2019;154:21–31. doi:https://doi.org/10.1016/j.ymeth.2018.11.008.
- Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E, et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29:457–66. doi:https://doi.org/10.1093/protein/gzw040.
- Rius Ruiz I, Vicario R, Morancho B, Morales CB, Arenas EJ, Herter S, Freimoser-Grundschober A, Somandin J, Sam J, Ast O, et al. p95HER2-T cell bispecific antibody for breast cancer treatment. Sci Transl Med. 2018;10:eaat1445. doi:https://doi.org/10.1126/scitranslmed.aat1445.
- Petrova G, Ferrante A, Gorski J. Cross-reactivity of T cells and its role in the immune system. Crit Rev Immunol. 2012;32:349–72. doi:https://doi.org/10.1615/CritRevImmunol.v32.i4.50.
- Hebeisen M, Allard M, Gannon PO, Schmidt J, Speiser DE, Rufer N. Identifying individual T cell receptors of optimal avidity for tumor antigens. Front Immunol. 2015;6:582. doi:https://doi.org/10.3389/fimmu.2015.00582.
- He Q, Liu Z, Liu Z, Lai Y, Zhou X, Weng J. TCR-like antibodies in cancer immunotherapy. J Hematol Oncol. 2019;12:99. doi:https://doi.org/10.1186/s13045-019-0788-4.
- Hoydahl LS, Frick R, Sandlie I, Loset GA. Targeting the MHC ligandome by use of TCR-like antibodies. Antibodies (Basel). 2019;8(2):32. doi:https://doi.org/10.3390/antib8020032.
- Yang X, Xie S, Yang X, Cueva JC, Hou X, Tang Z, Yao H, Mo F, Yin S, Liu A, et al. Opportunities and challenges for antibodies against intracellular antigens. Theranostics. 2019;9(25):7792–806. doi:https://doi.org/10.7150/thno.35486.
- Chang AY, Gejman RS, Brea EJ, Oh CY, Mathias MD, Pankov D, Casey E, Dao T, Scheinberg DA. Opportunities and challenges for TCR mimic antibodies in cancer therapy. Expert Opin Biol Ther. 2016;16:979–87. doi:https://doi.org/10.1080/14712598.2016.1176138.
- Dubrovsky L, Dao T, Gejman RS, Brea EJ, Chang AY, Oh CY, Casey E, Pankov D, Scheinberg DA. T cell receptor mimic antibodies for cancer therapy. Oncoimmunology. 2016;5:e1049803. doi:https://doi.org/10.1080/2162402X.2015.1049803.
- Dao T, Pankov D, Scott A, Korontsvit T, Zakhaleva V, Xu Y, Xiang J, Yan S, de Morais Guerreiro MD, Veomett N, et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol. 2015;33:1079–86. doi:https://doi.org/10.1038/nbt.3349.
- Dao T, Liu C, Scheinberg DA. Approaching untargetable tumor-associated antigens with antibodies. Oncoimmunology. 2013;2(e24678):e24678. doi:https://doi.org/10.4161/onci.24678.
- Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–37. doi:https://doi.org/10.1158/1078-0432.CCR-09-0737.
- Rossi G, Minervini MM, Carella AM, Melillo L, Cascavilla N. Wilms’ tumor gene (WT1) expression and minimal residual disease in acute myeloid leukemia. In: Tumor W, van den Heuvel-Eibrink MM, editor. Wilms tumor. Brisbane (AU): Codon Publications; 2016. p. 273–285.
- Doubrovina E, Carpenter T, Pankov D, Selvakumar A, Hasan A, O’Reilly RJ. Mapping of novel peptides of WT-1 and presenting HLA alleles that induce epitope-specific HLA-restricted T cells with cytotoxic activity against WT-1(+) leukemias. Blood. 2012;120:1633–46. doi:https://doi.org/10.1182/blood-2011-11-394619.
- Gejman RS, Jones HF, Klatt MG, Chang AY, Oh CY, Chandran SS, Korontsvit T, Zakahleva V, Dao T, Klebanoff CA, Scheinberg DA. Identification of the targets of T cell receptor therapeutic agents and cells by use of a high throughput genetic platform. Cancer Immunol Res. 2020;8(5):672–684. doi:https://doi.org/10.1158/2326-6066.CIR-19-0745.
- Oren R, Hod-Marco M, Haus-Cohen M, Sharyn T, Blat D, Duvshani N, Denkberg G, Elbaz Y, Benchetrit F, Eshhar Z, et al. Functional comparison of engineered T cells carrying a native TCR versus TCR-like antibody-based chimeric antigen receptors indicates affinity/avidity thresholds. J Immunol. 2014;193(11):5733–43. doi:https://doi.org/10.4049/jimmunol.1301769.
- Lee HY, Register A, Shim J, Contreras E, Wu Q, Jiang G. Characterization of a single reporter-gene potency assay for T-cell-dependent bispecific molecules. MAbs. 2019;11:1245–53. doi:https://doi.org/10.1080/19420862.2019.1640548.
- Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Identification of a putative regulator of early T cell activation genes. Science. 1988;241:202–05. doi:https://doi.org/10.1126/science.3260404.
- Rincon M, Flavell RA. Regulation of AP-1 and NFAT transcription factors during thymic selection of T cells. Mol Cell Biol. 1996;16:1074–84. doi:https://doi.org/10.1128/MCB.16.3.1074.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–54. doi:https://doi.org/10.4049/jimmunol.173.2.945.
- Rosskopf S, Leitner J, Paster W, Morton LT, Hagedoorn RS, Steinberger P, Heemskerk MHM. A Jurkat 76 based triple parameter reporter system to evaluate TCR functions and adoptive T cell strategies. Oncotarget. 2018;9:17608–19. doi:https://doi.org/10.18632/oncotarget.24807.
- Fiering S, Northrop JP, Nolan GP, Mattila PS, Crabtree GR, Herzenberg LA. Single cell assay of a transcription factor reveals a threshold in transcription activated by signals emanating from the T-cell antigen receptor. Genes Dev. 1990;4:1823–34. doi:https://doi.org/10.1101/gad.4.10.1823.
- Tan MP, Dolton GM, Gerry AB, Brewer JE, Bennett AD, Pumphrey NJ, Jakobsen BK, Sewell AK. Human leucocyte antigen class I-redirected anti-tumour CD4(+) T cells require a higher T cell receptor binding affinity for optimal activity than CD8(+) T cells. Clin Exp Immunol. 2017;187:124–37. doi:https://doi.org/10.1111/cei.12828.
- Krummey SM, Martinez RJ, Andargachew R, Liu D, Wagener M, Kohlmeier JE, Evavold BD, Larsen CP, Ford ML. Low-affinity memory CD8+ T cells mediate robust heterologous immunity. J Immunol. 2016;196:2838–46. doi:https://doi.org/10.4049/jimmunol.1500639.
- Gee MH, Sibener LV, Birnbaum ME, Jude KM, Yang X, Fernandes RA, Mendoza JL, Glassman CR, Garcia KC. Stress-testing the relationship between T cell receptor/peptide-MHC affinity and cross-reactivity using peptide velcro. Proc Natl Acad Sci USA. 2018;115:E7369–E78. doi:https://doi.org/10.1073/pnas.1802746115.
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–45. doi:https://doi.org/10.1038/nmeth.1318.
- Giry-Laterriere M, Verhoeyen E, Salmon P. Lentiviral vectors. Methods Mol Biol. 2011;737:183–209.
- Schmittnaegel M, Hoffmann E, Imhof-Jung S, Fischer C, Drabner G, Georges G, Klein C, Knoetgen H. A new class of bifunctional major histocompatibility class i antibody fusion molecules to redirect CD8 T cells. Mol Cancer Ther. 2016;15:2130–42. doi:https://doi.org/10.1158/1535-7163.MCT-16-0207.
- Joglekar AV, Leonard MT, Jeppson JD, Swift M, Li G, Wong S, Peng S, Zaretsky JM, Heath JR, Ribas A, et al. T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat Methods. 2019;16:191–98. doi:https://doi.org/10.1038/s41592-018-0304-8.
- Kisielow J, Obermair FJ, Kopf M. Deciphering CD4(+) T cell specificity using novel MHC-TCR chimeric receptors. Nat Immunol. 2019;20:652–62. doi:https://doi.org/10.1038/s41590-019-0335-z.
- van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14:499–509.
- Davis SJ, van der Merwe PA. The structure and ligand interactions of CD2: implications for T-cell function. Immunol Today. 1996;17:177–87. doi:https://doi.org/10.1016/0167-5699(96)80617-7.
- Alonso-Camino V, Sanchez-Martin D, Compte M, Nunez-Prado N, Diaz RM, Vile R, Alvarez-Vallina L. CARbodies: human antibodies against cell surface tumor antigens selected from repertoires displayed on T cell chimeric antigen receptors. Mol Ther Nucleic Acids. 2013;2:e93. doi:https://doi.org/10.1038/mtna.2013.19.
- Rydzek J, Nerreter T, Peng H, Jutz S, Leitner J, Steinberger P, Einsele H, Rader C, Hudecek M. Chimeric antigen receptor library screening using a novel NF-kappaB/NFAT reporter cell platform. Mol Ther. 2019;27:287–99. doi:https://doi.org/10.1016/j.ymthe.2018.11.015.
- Darowski D, Jost C, Stubenrauch K, Wessels U, Benz J, Ehler A, Freimoser-Grundschober A, Brünker P, Mössner E, Umaña P, Kobold S, Klein C. P329G-CAR-J: a novel Jurkat-NFAT-based CAR-T reporter system recognizing the P329G Fc mutation. Protein Eng Des Sel. 2019;32:207–18. doi: https://doi.org/10.1093/protein/gzz027.
- Darowski D, Kobold S, Jost C, Klein C. Combining the best of two worlds: highly flexible chimeric antigen receptor adaptor molecules (CAR-adaptors) for the recruitment of chimeric antigen receptor T cells. MAbs. 2019;11:621–31. doi:https://doi.org/10.1080/19420862.2019.1596511.
- Gopalakrishnan R, Matta H, Choi S, Natarajan V, Prins R, Gong S, Zenunovic A, Narasappa N, Patel F, Prakash R, Chaudhary V. A novel luciferase-based assay for the detection of chimeric antigen receptors. Sci Rep. 2019;9:1957. doi:https://doi.org/10.1038/s41598-018-38258-z.
- Barrett J. Expanding the antigenic repertoire of CAR-T cells with “TCR-like” antibody specificity. Cytotherapy. 2016;18(8):929–30. doi:https://doi.org/10.1016/j.jcyt.2016.06.007.
- Akatsuka Y. TCR-like CAR-T cells targeting MHC-bound minor histocompatibility antigens. Front Immunol. 2020;11:257. doi:https://doi.org/10.3389/fimmu.2020.00257.
- Rafiq S, Purdon TJ, Daniyan AF, Koneru M, Dao T, Liu C, Scheinberg DA, Brentjens RJ. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms tumor 1 antigen. Leukemia. 2017;31(8):1788–97. doi:https://doi.org/10.1038/leu.2016.373.
- Maus MV, Plotkin J, Jakka G, Steward-Jones G, Riviere I, Merghoub T, Wolchok J, Renner C, Sadelain M. A MHC-restricted antibody-based chimeric receptor requires TCR-like affinity to maintain antigen specificity. Mol Ther Oncol. 2016;3:1–9. doi:https://doi.org/10.1038/mto.2016.23.
- Sun L, Guo H, Jiang R, Lu L, Liu T, He X. Engineered cytotoxic T lymphocytes with AFP-specific TCR gene for adoptive immunotherapy in hepatocellular carcinoma. Tumour Biol. 2016;37(1):799–806. doi:https://doi.org/10.1007/s13277-015-3845-9.
- Liu H, Xu Y, Xiang J, Long L, Green S, Yang Z, Zimdahl B, Lu J, Cheng N, Horan LH, et al. Targeting Alpha-Fetoprotein (AFP)-MHC complex with CAR T-cell therapy for liver cancer. Clin Cancer Res. 2017;23(2):478–88. doi:https://doi.org/10.1158/1078-0432.CCR-16-1203.
- Zhang G, Wang L, Cui H, Wang X, Zhang G, Ma J, Han H, He W, Wang W, Zhao Y, et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci Rep. 2014;4:3571. doi:https://doi.org/10.1038/srep03571.