
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Targeting immune checkpoint receptors expressed in the T cell synapse induces active and long-lasting antitumor immunity in preclinical tumor models and oncology patients. However, traditional nonhuman primate (NHP) studies in healthy animals have thus far demonstrated little to no pharmacological activity or toxicity for checkpoint inhibitors (CPIs), likely due to a quiescent immune system. We developed a NHP vaccine challenge model in Mauritius cynomolgus monkey (MCMs) that elicits a strong CD8+ T cell response to assess both pharmacology and safety within the same animal. MHC I-genotyped MCMs were immunized with three replication incompetent adenovirus serotype 5 (Adv5) encoding Gag, Nef and Pol simian immunodeficiency virus (SIV) proteins administered 4 weeks apart. Immunized animals received the anti-PD-L1 atezolizumab or an immune checkpoint-targeting bispecific antibody (mAbX) in early development. After a single immunization, Adv5-SIVs induced T-cell activation as assessed by the expression of several co-stimulatory and co-inhibitory molecules, proliferation, and antigen-specific T-cell response as measured by a Nef-dependent interferon-γ ELIspot and tetramer analysis. Administration of atezolizumab increased the number of Ki67+ CD8+ T cells, CD8+ T cells co-expressing TIM3 and LAG3 and the number of CD4+ T cells co-expressing 4–1BB, BTLA, and TIM3 two weeks after vaccination. Both atezolizumab and mAbX extended the cytolytic activity of the SIV antigen-specific CD8+ T cell up to 8 weeks. Taken together, this vaccine challenge model allowed the combined study of pharmacology and safety parameters for a new immunomodulatory protein-based therapeutic targeting CD8+ T cells in an NHP model.
Introduction
The emergence of cancer immunotherapies (CITs) and the demonstration that immune stimulatory antibodies can be used to induce potent and durable antitumor activity has transformed the way patients with cancer are treated. Extensive research in this area has notably revealed that CD8+ T cells are a key player of antitumor immunity, with the infiltration of CD8+ T cells into the tumor microenvironment seen as a favorable prognosis for numerous malignancies.Citation1 New therapeutic antibodies preferentially target co-stimulatory or co-inhibitory proteins expressed on T cells, rather than the tumor itself, to reinvigorate CD8+ effector functions and prevent T cell exhaustion.Citation2 These therapies have the advantage of generating an active and long-lasting antitumor immunity, with a broad and polyclonal immune response capable of managing the heterogeneity of cancers and reducing the chances of immune escape.Citation2–5
These new therapies have substantially increased the efficacy of cancer treatment. However, the unwanted and sometimes severe consequences associated with exaggerated immune activation have often not been well predicted by standard nonclinical toxicology studies in healthy animals.Citation6,Citation7 Hence, there is a need for translational animal models that allow pharmacology and safety to be concomitantly monitored within the same test system. Ideally, these animal models should be predictive of efficacy, have clinical applicability, possess the targeted cell populations and antigen, and mimic the expected pharmacodynamics and exaggerated pharmacology seen in patients. In some cases, the standard naïve rodent and nonrodent species used in toxicological studies, as well as the mouse tumor models routinely used to test efficacy of cancer immunotherapeutics, are not sufficient to evaluate the safety and the pharmacologic activity of cancer therapeutic antibodies.Citation8–11 Indeed, syngeneic mouse tumor models are relatively affordable and simple, but poorly recapitulate the steps that result in the development of the disease. They have limited investigational potential with immune modulatory drugs because the immunomodulatory effect of these drugs cannot be properly studied.Citation11–14 Moreover, they have inadequately predicted human clinical outcomes, particularly in the case of targeted therapeutics.Citation15–17
In larger animal species, such as nonhuman primates (NHPs), studying the safety and efficacy of CITs presents its own challenges. These are mainly linked to the fact that healthy, non-tumor-bearing animals do not recapitulate the immune status associated with oncology patients, or CIT-related changes associated with tumor-specific immune responses. However, most monoclonal antibodies targeting co-stimulatory/co-inhibitory checkpoint molecules are pharmacologically active in NHPs, making them a suitable species for nonclinical safety testing if a translational model were available that could recapitulate the immune status of the oncology patient in the animal.Citation18–23 Depending on the immunomodulatory target and the mechanism of action of the therapeutic tested, increases in immune cell activation markers may be demonstrated upon dosing in a healthy animal. However, activation of the immune system via an antigenic challenge or vaccination will provide an immune stimulus and upregulation of co-stimulatory and co-inhibitory molecules targeted for cancer immunotherapeutics. Furthermore, depending on how the antigen challenge model is conducted, it may allow the determination of immune suppression or enhancement. Keyhole limpet hemocyanin (KLH) immunization and the subsequent measure of a T cell-dependent antibody response (TDAR), is one of several methods used in NHP to primarily study immune suppression. Historically, many of the antigens used to challenge the immune system and assess immune functions were used for the detection of immunosuppression rather than immunostimulation; thus, the antigen was administered at a high dose to induce a robust immune response. Since the desired pharmacological effect of cancer immunotherapy is to enhance the immune response, the antigenic dose may need to be adjusted to induce a sub-maximal response that would allow detection of the cancer immunotherapy-mediated enhancement.
Consistent with this approach, a modification to the KLH model may allow for the detection of immune stimulation.Citation24 This approach is considered most suitable to assess functional characteristics of CD4+ T cells, antigen-presenting cells (APCs) and B cells, resulting in humoral immune responses. The US Food and Drug Administration has requested safety studies that included an antigenic challenge for several recently approved PD-1/PD-L1-targeting CITs to demonstrate pharmacodynamic activity in vivo. TDAR testing has proven to be informative with some CITs, such as anti-CTLA4 and anti-OX-40 antibodies.Citation24–29 However, for others, such as the anti-PD-1 antibodies nivolumab and the anti-PD-L1 antibodies durvalumab and atezolizumab, either no changes or a reduction in the anti-KLH antibody response was observed in animals receiving the therapeutic antibody (unpublished data). This contrasts with the clear beneficial effects reported in cancer patients, highlighting the limitations of this TDAR model.Citation30,Citation31 One of the hypotheses that has been put forward to explain why a reduction in the antibody response rather than an increase may have been measured in this context relates to the specific immune cell types stimulated following a KLH challenge. KLH primarily drives strong CD4+ helper T lymphocyte and antibody responses, yet most CITs currently under development primarily target CD8+ cytotoxic T lymphocytes (CTLs) to achieve a strong anti-tumor activity. Therefore, having an antigen challenge model where the CD8+ T cell is the primary immune effector cell would better assess the effect of CITs on the T cell population important in the anti-tumor response and help to understand the pharmacology and safety of CITs.Citation18,Citation32
The goal of this study was to build upon an existing NHP vaccination model,Citation33 which would have scientific advantages over the classical TDAR model. This model would specifically elicit a CTL response and allow the simultaneous evaluation of the pharmacology and safety of molecules targeting CD8+ T cells. To achieve this, Mauritian cynomolgus macaques (MCMs) expressing major histocompatibility complex (MHC) I-alleles Mafa-A1*063 alone, or in combination with Mafa-B*104:01, were immunized with 3 replication incompetent recombinant adenovirus serotype 5 (Adv5) vectors, each containing the coding sequence for Gag, Nef or Pol simian immunodeficiency virus (SIV) proteins, Adv5-SIV, which are known to drive a CD8+ T cell response.Citation26,Citation33–35 MCMs were distributed into three groups receiving two intramuscular injections of adenoviruses 4 weeks apart (). Group 1 served as the control and only received the Adv5-SIV; Group 2 was dosed once weekly with a bispecific antibody targeting PD-1 and a second undisclosed immune checkpoint molecule that is currently in early development (mAbX), and Group 3 was dosed once weekly with atezolizumab (anti-PD-L1). The effects of atezolizumab and mAbX on the Adv5-SIV responses were characterized via immunophenotyping of the different T cell sub-populations and their activation status, and functional activity as assessed via ex vivo intracellular cytokine recall response, interferon (IFN)γ enzyme-linked immunospot (ELIspot) and CTL killing assay. The results demonstrated that Adv5-SIV vaccination was well tolerated and could be conducted within the context of a standard safety toxicology study without compromising standard safety endpoints, including clinical pathology. The Adv5-SIV -related response was mainly characterized by the modulation of activation markers, including co-stimulatory and co-inhibitory molecules, proliferation and the SIV antigen-specific responses of the CD8+ T cells (ELIspot, CTL activity). Moreover, the cynomolgus monkeys that received atezolizumab had an increased number of CD4 and/or CD8+ T cells two weeks after the primary vaccination that expressed other costimulatory/coinhibitory molecules, such as 4–1BB, BTLA4, and TIM3. Interestingly, cynomolgus monkeys given mAbX, which also targets the PD-1/PD-L1 pathway, had an increase in FoxP3+/CD25+ CD4+ T cells one week after vaccination. Both mAbX and atezolizumab increased the Nef-specific IFNg ELISpot response two weeks after vaccination and their Nef antigen-specific cytolytic activity out to 8 weeks, which was the latest timepoint measured. Altogether, the data suggest that the Adv5-SIV vaccination model in Mauritius cynomolgus macaques could be used to evaluate pharmacological activity and the safety of new drug candidates targeting the cytotoxic T cell population.
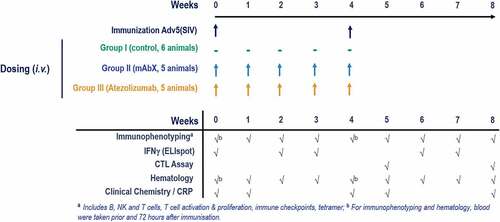
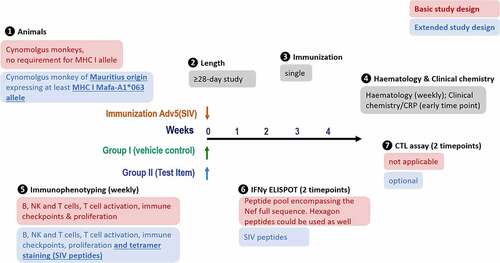
Figure 1. Study design. MHC I-genotyped Mauritian cynomolgus macaques (MCMs) were immunized with 3 replication incompetent adenovirus serotype 5 (Ad5) encoding Gag, Nef or Pol SIV (Simian immunodeficiency virus) proteins. One group received the anti-PD-L1 atezolizumab and the second group received an immune checkpoint-targeting bi-specific antibody mAbX in early development. Blood samples, from all animals, were taken on a weekly basis to characterize the T cell phenotype (Immunophenotyping) and function (IFNγ ELIspot and cytolytic T cell assay)
Results
Adv5-SIV-related clinical pathology changes were limited to transient C-reactive protein and globulins increases
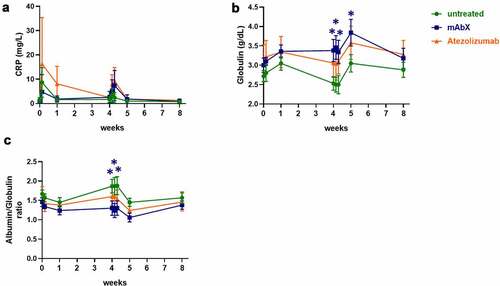
To determine whether vaccination of Adv5-SIV could be incorporated into a general nonclinical safety study, standard safety endpoints, including body weights, general observations and clinical pathology assessment were performed at various timepoints following vaccination. Based on clinical signs, the administration of Adv5-SIV was well tolerated, with no redness or inflammation observed at the site of immunization. Similarly, administration of mAbX or atezolizumab was well tolerated throughout the study, with no clinical signs, or any other effect observed on parameters, such as body weight or food consumption. Administration of Adv5-SIV resulted in a mild to moderate increase in C-reactive protein (CRP) concentration on Day 2 following the first injection and on Days 30 and 31 following the second injection ()). These increases were generally above historical or pre-treatment range. Moreover, a minimal increase in globulin concentration on Days 8 and 36 with a concurrent decrease in albumin/globulin ratio was observed, suggestive of a transient inflammatory response to the Adv5-SIV vaccine (), which is consistent with previous reports demonstrating an increase in serum levels of proinflammatory cytokines.Citation36–38 The increase in CRP and globulin, as well as the decrease in the globulin/albumin ratio, were further amplified in animals dosed with mAbX and atezolizumab, which was attributed to the pharmacological effect of the antibodies. However, no clinical signs were observed, indicating that the addition of the immunomodulatory therapeutics only had limited effects on clinical pathology parameters. Importantly, the effects on clinical pathology parameters were transient and returned to baseline by the end of the 8-week study.
Figure 2. Changes in C-reactive proteins (a), globulin (b), and in the globulin/albumin ratio (c) in the peripheral blood following the immunization of Cynomolgus macaques (on weeks 0 and 4) for up to eight weeks. For each condition, * (p < .05) indicates statistical significance of the selected condition, compared to the untreated group
Adv5-SIV-administration alone had no effect on hematology parameters, while administration of mAbX and/or atezolizumab following Adv5-SIV had minimal effects on neutrophil, monocyte, and lymphocyte counts
Administration of Adv5-SIV had no direct effect on hematology parameters. However, in vaccinated animals subsequently administered mAbX, an increase in neutrophil, monocyte, eosinophil, and white blood cell counts was observed. A decrease in lymphocyte counts was also noted during the first weeks of this study (data not shown). This decrease remained within historical data.
In animals given atezolizumab, hematology changes consisted of increases in neutrophil counts between Days 4 and 57, a decrease in lymphocyte counts on Day 8 followed by increases between Days 15 and 22, and an increase in eosinophil counts on Day 22 (data not shown).
The administration of CIT further potentiates the effects of Adv5-SIV on peripheral lymphocyte populations
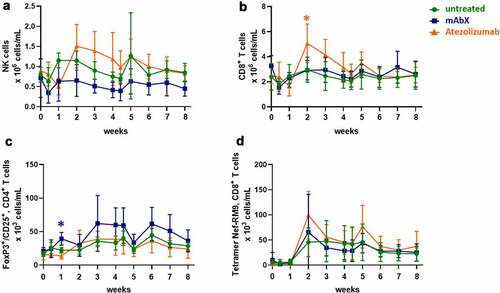
Administration of a single dose of the Adv5-SIV alone did not result in any significant changes in the circulating numbers of B cells, CD4+ lymphocytes, or naïve, central memory and effector memory CD4+ and CD8+ T cells in whole blood (data not shown). However, an increase in the total numbers of natural killer (NK) cells, CD8+ T cells, and SIV Nef-specific CD8+ T cells was observed between 1 and 2 weeks following the immunization, (). In animals given atezolizumab, a slight increase in the number of NK cells was noted between weeks 2–5, whereas the number of CD8+ cells was significantly increased 2 weeks post first immunization (), while administration of mAbX increased the number of FoxP3+CD25+CD4+ Treg cells 1 week after the first immunization ()).
Figure 3. Changes in the numbers of NK (a), CD8+ (b), Tregs (c), and RM9-specific CD8+ T cells (d) in the peripheral blood following the immunization of Cynomolgus macaques (on weeks 0 and 4) for up to eight weeks. Depending on the amount of cells detected, data are expressed as 106 cells/mL or 103 cells/mL. For each condition, * (p < .05) indicates statistical significance of the selected condition, compared to the untreated group
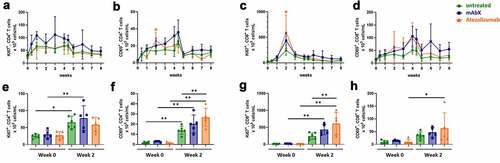
To assess the activation and proliferation profiles in CD4+ and CD8+ T cells, CD69 and Ki67 were used as markers. A significant increase in activated (CD69+) and proliferating (Ki67+) CD4+ T cells was observed within one week after the first administration of the Adv5-SIV. Moreover, administration of mAbX and atezolizumab increased the number of CD69-expressing CD4+ T cells (). Similarly, there was a general increase in the number of Ki67+ and CD69+ CD4+ T cells between weeks 2 to 4 in animals that received mAbX. For CD8+ T cells, the peak proliferative response was observed on week 2 in all groups, which was increased following administration of mAbX and atezolizumab (). Activation of CD8+ T cells also appeared to be potentiated in the presence of either mAbX beginning on week two after the primary vaccination ().
Figure 4. Proliferation and activation profiles of CD4+ and CD8+ T cells for up to 8 weeks (A to D). Comparison of the proliferation and activation profiles at pre-dose and 2 weeks post first administration of Ad5(SIV) (E to H). For each group shown data are presented as the percentage of these subsets out of the total T cell subsets. For each condition, * (p < .05), ** (p < .01), *** (p < .001), & **** (p < .0001) indicates statistical significance of the selected condition, compared to the untreated group
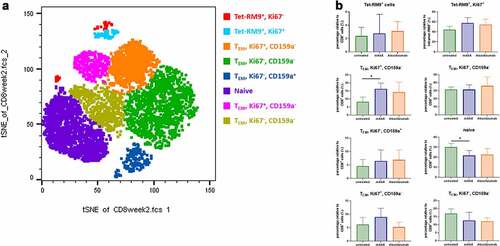
To assess whether dosing with mAbX or atezolizumab might result in the emergence of unique T cells subsets, we analyzed CD45, CD3, CD4, CD8, CD20, CD159a, CD69, CD152, CD28, CD95, CD25, CD14, Ki67, and FoxP3 markers at week 2, using the t-distributed stochastic neighbor embedding (tSNE) algorithm that reduces all data to two dimensions. As shown in ), unique populations not identified by conventional flow cytometry analysis appeared in all the animals tested. Notably, cells were clustered depending on their profile as naïve, central (TCM) or effector (TEM) memory cells, and on the detection of Tetramer-RM9-, Ki67- and CD159a-positive cells ()). Administration of mAbX or atezolizumab further increased the subsets corresponding to TEM cells expressing either Ki67 or CD159a and decreased the level of naïve cells. Although differences were relatively small, this analysis allowed the detection of a trend toward a CIT-related enhancement of Adv5-SIV-induced T-cell responses.
Figure 5. Frequency and phenotype of CD8 + T-cell subsets in NHPs. (a) Representative tSNE map of total CD8+ cells in NHP (untreated group), using CD45, CD3, CD4, CD8, CD20, CD159a, CD69, CD152, CD28, CD95, CD25, CD14, Ki67, and FoxP3 as markers. (b) Box plots showing the frequency of CD8 + T cells in each significantly enriched cluster. *p < .05
Adv5-SIV induces the expression of immunomodulatory molecules, which are further altered by mAbX and atezolizumab
Expression of other immunomodulatory proteins that are currently being investigated as potential CIT target receptors was assessed following Adv5-SIV challenge in the presence or absence of atezolizumab or mAbX. The expression profile of three co-stimulatory (4–1BB, OX40, and ICOS) and seven co-inhibitory receptors (B7-H4, BTLA, CTLA-4, LAG3, PD-1, TIM3, and VISTA) on the surface of CD4+ and CD8+ T cells was evaluated.
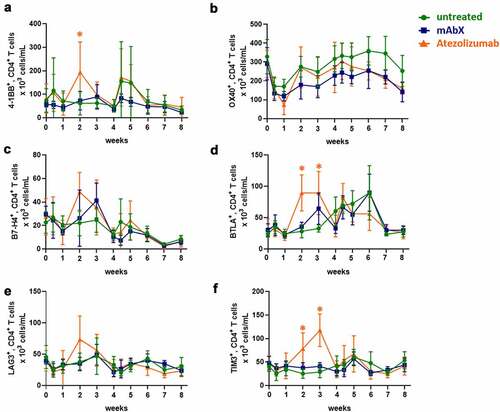
On CD4+ T cells, immunization with Adv5-SIV increased the number of 4–1BB+ cells within 3 days of both primary and secondary administrations and the number of BTLA+ cells as of 4 weeks after the first immunization. In contrast, the number of OX-40+ CD4+ T cells decreased within 3 days of the first immunization and recovered to baseline levels by week 2 (). Compared to pre-dose values, the administration of atezolizumab increased the number of CD4+ T cells 2–3 weeks after the primary vaccination that co-expressed 4–1BB, BTLA or TIM3. This observation was also made when the atezolizumab-treated group was compared to the untreated group for the same timepoint. In contrast, administration of mAbX did not alter the number of CD4+ T cells that co-expressed any of the immunomodulatory receptors following the primary immunization. Similarly, no changes were noted after the secondary immunization in any of the treatment groups. No clear modulation of ICOS, CTLA-4, and VISTA on CD4+ T cells was observed (data not shown).
Figure 6. Modulation of immune checkpoints expressed on CD4+ T cells up to 8 weeks after administration of Ad5(SIV) (Weeks 0 and 4): (a), 4–1BB; (b), OX40; (c), B7-H4; (d), BTLA; (e), LAG3; and (f), TIM3. Results are represented as 103 cells/mL in blood. For each condition, * (p < .05) indicates statistical significance of the selected condition, compared to the untreated group
On CD8+ T cells, Adv5-SIV administration resulted in an increase in the number of BTLA+ cells beginning on day 3 after the primary immunization and continued to increase out to 3 weeks post immunization. A similar increase in the number of TIM3+CD8+ was noted between 1 and 3 weeks following the primary Adv5-SIV administration (). Following the secondary immunization with Adv5-SIV, there was an increase in the number of CD8+ T cells that co-expressed BTLA, LAG3, TIM3, and VISTA within one week. No clear modulation of OX40 and ICOS on CD8+ T cells were observed (data not shown).
Figure 7. Modulation of immune checkpoint molecules expressed on CD8+ T cells up to 8 weeks after administration of Ad5(SIV) (Weeks 0 and 4): (a), 4–1BB; (b), B7-H4; (c), BTLA; (d), LAG3; (e), PD-1; (f), CTLA-4; (g), TIM3; and (h), VISTA. Results are represented as 103 cells/mL in blood. For each condition, * (p < .05) indicates statistical significance of the selected condition, compared to the untreated group
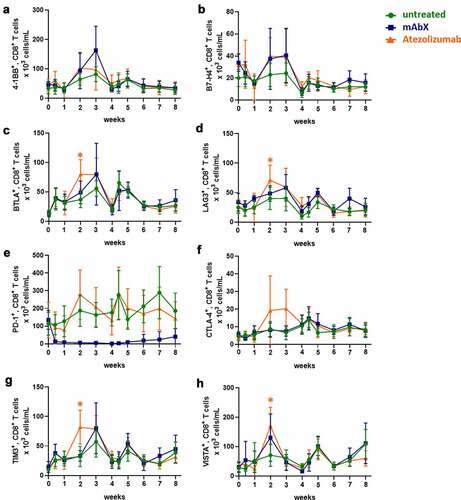
In the presence of atezolizumab, the number of CD8+ T cells that co-expressed BTLA, LAG3, TIM3, and VISTA significantly increased, mainly 2 weeks following the first administration of the Adv5-SIV, in comparison to the untreated group (). In contrast, mAbX decreased the number of PD-1+CD8+ T cells throughout the duration of the study. Given that one arm of the bispecific mAbX antibody targets PD-1, the decrease in PD-1 staining observed in CD4+ T cells (data not shown) and CD8+ T cells ()) was most likely due to competition with the anti-PD-1 detection reagent, rather than decreased PD-1 expression. There were no mAbX or atezolizumab-related changes in the expression of these markers following the second Ad5(SIV) immunization.
Adv5-SIV induces Nef-RM9-specific cytotoxic T lymphocytes, which is further enhanced and sustained with the administration CITs
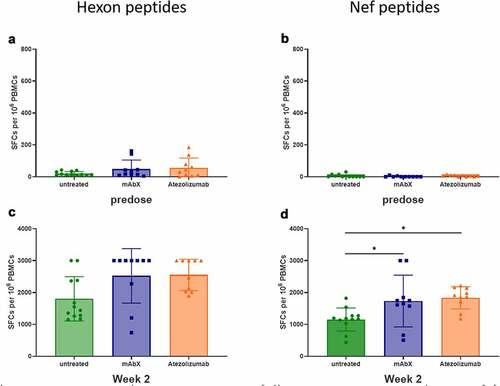
To assess whether Adv5-SIV administration modulated the functional activity of CD8+ T cells, the secretion of IFN-γ, as measured by ELIspot, was evaluated after stimulation of freshly isolated peripheral blood mononuclear cells (PBMCs) with Nef peptides known to induce an immune response or a peptide pool covering the adenovirus hexon protein.Citation39–41 Since macaques may have been exposed to adenoviruses throughout their lifetime,Citation42 a low response was observed at predose upon stimulation with the hexon peptide pool ()). This suggests that the animals had previously been in contact with adenoviruses, but not with any SIV proteins. However, no IFN-γ cellular response was observed at predose after ex vivo stimulation with the Nef peptides (RM9, LT9, and HW8) in the ELIspot assay ()). Following Adv5-SIV immunization, ex vivo stimulation of PBMCs elicited a robust IFN-γ response in all animals from week 2 with the Hexon and Nef peptides () and (d)), up to the end of this study (data not shown). Since the immunization had already reached a maximum response after a primary vaccination, we did not follow the ex vivo stimulation in the presence of the hexon peptides after a second vaccination. Moreover, the IFN-γ cellular response was further enhanced on week 2 in animals dosed with mAbX and atezolizumab.
Figure 8. Detection of cellular IFNγ response by ELISPOT assay following ex-vivo stimulation of freshly isolated PBMCs (sixteen immunized MCMs) with a peptide pool covering the complete sequence of the Adenovirus hexon, at 0.6 µM (total peptide concentration, A and C) or with a pool of three Nef peptides (RM9, HW8 and LT9 at 10 µM each, B and D) or Conditions for which spots were too numerous to count were arbitrary set at 3000 SFCs per 106 PBMCs. *, p < .05 indicates statistical significance compared to the untreated group
Confirmation of the enhanced activity of Nef-specific CD8+ T cells with an ex vivo recall response flow cytometry assay measuring intracellular IFN-γ, tumor necrosis factor (TNF) and interleukin (IL)-2 in CD4+ and CD8+ T cells was not achieved given that the results obtained after stimulation with an anti-CD3 antibody positive control were generally weak, suggesting that this assay was not optimal in our model (data not shown).
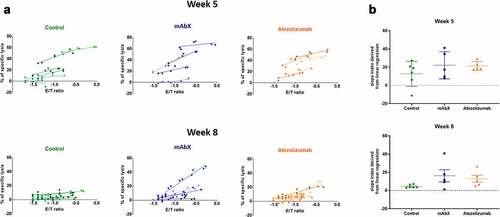
The functionality of CD8+ T cells was further evaluated via the measurement of Nef-RM9 specific cytotoxic activity. Nef-RM9 specific CTL activity was observed on week 5, in 4 of 6 control animals, 4 of 5 animals given mAbX and 5 of 5 animals given atezolizumab (). No clear differences between each group were observed, beside the fact that the inter-animal variability was greater in the control- and mAbX-dosed animals, compared to the atezolizumab dosed animals, similar to what was observed with the IFN-γ ELIspot assay. On week 8, the Nef-RM9-specific CTL activity was maintained in animals given mAbX or atezolizumab, whereas in the control group, little to no activity was seen in most animals. Altogether, the ELIspot and CTL data correlated well with the activation and proliferation profiles assessed by flow cytometry and demonstrated that the activity of the Nef-RM9-specific CD8+ T cells was maintained by the CITs.
Figure 9. Determination of the cytolytic capacity of the Nef RM9-specific CD8+ T cells for each animal at week 5 and 8. (a) Each linear regression corresponds to one specific NHP. The steepness of the slope is indicative of the CTL activity. (b), Schematic representation of the coefficient slope derived from the linear regression depending on the dosing regimen
Discussion
The lack of nonrodent preclinical models that show pharmacodynamic effects of immunomodulatory drugs designed to primarily affect the activity of CD8+ CTLs has been a challenge for drug development. Although preclinical toxicology studies in NHP are widely conducted in the pharmaceutical industry to support entry into clinical trials, they rely on testing drug candidates in the context of naïve healthy animals, leading to the potential to overlook safety liabilities because of the quiescent nature of the NHP immune system. Using a model that activates the immune system and upregulates the expression of targets could, at least partly, help identify potential toxicities that are associated with exaggerated pharmacology, including T cell activation and function. Moreover, it allows the direct assessment of the targeted cell population instead of an indirect antibody response. However, a well-characterized model that incorporates an antigen that preferentially activates CD8+ T cells within cynomolgus monkeys commonly used in nonclinical safety assessment has proven to be challenging. In this study, we describe the T cell immune response of non-human primates immunized with non-replicative adenoviruses encoding SIV proteins in the presence or absence of the approved anti-PD-L1 mAb, atezolizumab, or an early development bispecific molecule, mAbX, that also targets co-inhibitory molecules.
Administration of Adv5-SIV resulted in a mild acute-phase response and activation of the immune system. No signs of illness or distress were observed in all the animals, suggesting that the immunization with replication-deficient Adv5-SIV was well tolerated. Adv5-SIV-related clinical pathology changes were limited to transient mild to moderate CRP, and minimal increases in globulin concentration after each Adv5-SIV injection. These increases reflected an acute-phase reaction, which is encountered after most vaccine administrations,Citation43–46 and did not compromise the readout of the standard safety parameters. Indeed, animals administered mAbX or atezolizumab presented an increase in CRP and globulin concentrations, and/or a decrease in albumin concentration of higher magnitude compared to animals receiving Adv5-SIV alone. The animals that received mAbX or atezolizumab also presented increases in neutrophil, monocyte and/or eosinophil counts, and decreases in lymphocyte counts that were not observed in animals administered Adv5-SIV only. Changes in neutrophil and monocytes reflected a mild inflammatory response and appeared to be mAbX or atezolizumab-related and not Adv5-SIV-related.
Following an initial challenge with the Adv5-SIV, a broad T-cell response was induced, with the activation and proliferation of total CD4+ and CD8+ T cells, the generation of SIV-specific CD8+ T cells, and an increase in their functionality, as measured by IFN-γ secretion and cytolytic activity, that was modulated by both mAbX and atezolizumab. Furthermore, we demonstrate that immunization with Adv5-SIV induced the expression of numerous immunomodulatory receptors, which may help overcome the current challenges of assessing parameters such as receptor occupancy of costimulatory/co-inhibitory molecules in naive NHP models, where the animals have a quiescent immune system and limited target expression.Citation47
Of the lymphocyte populations analyzed, CD8+ T cells showed the strongest activation following immunization with Adv5-SIV. Within this population, a potentiation of the immune response to Adv5-SIV administration was observed in the animals dosed with the two CITs tested, as compared to the control animals. This enhancement was generally seen between 1 and 3 weeks following the first Adv5-SIV administration and trended toward baseline levels by the 4th week. The absence of a strong SIV-specific immune response following the boost administration was likely due to the generation of an immune response against the adenoviruses, preventing a proper activation of the T cells. Indeed, adenoviruses are known to induce a strong production of anti-adenovirus antibodies, thereby reducing their immunogenicity.Citation48–53 Despite the fact that a boost of the immune response was not observed following a second administration of the Adv5-SIV, cytotoxic T cells in the animals dosed with CITs remained functionally superior compared to the control animals, keeping their ability to recognize and kill target cells until the end of the study.
Animals treated with atezolizumab showed the most notable changes in the expression of immunomodulatory molecules within the first 2 weeks of the study. Changes in the number of CD8+ T cells expressing BTLA, LAG3, TIM3, or VISTA and of CD4+ T cell expressing 4–1BB, BTLA or TIM3 were statistically higher than in animals of the two other groups ( and , respectively). At the end of the 8-week study, most of the co-stimulatory and co-inhibitory receptors had returned to basal level, except for OX40, which was lower on CD4+ T cells in animals dosed with both CIT drugs ()) and VISTA, which remained higher on CD8+ T cells in animals dosed with mAbX and in untreated animals ()). Thus, a single immunization with the Adv5-SIV vectors stimulates the expression of immunomodulatory molecules, which may increase the ability to evaluate pharmacological effects of other CIT molecules.
Importantly, it should be noted that the intent of this study was to evaluate whether it was possible to detect changes in T-cell (primarily CD8+ T cell) activation, proliferation, and/or function, following administration of immunomodulatory molecules in the context of an immunization that is predominantly CD8+-dependent. Atezolizumab and mAbX were selected because they represented different formats and target different immune modulating receptors that have been extensively studied in healthy nonhuman primates with quiescent immune systems and not demonstrated the expected pharmacology of enhanced T-cell responses. A direct comparison of the mAbX- and atezolizumab-dependent responses is challenging due to factors such as the different dose/exposure levels, targets, molecular format, and affinity/avidity. However, we believe it is fair to compare the response of each molecule to the control group. To overcome potential exposure variability, the dose levels of mAbX and atezolizumab used in this study were previously shown to be well tolerated and maintain complete receptor occupancy for the entire duration of this study. Furthermore, we wanted to highlight that this model does appear to identify differences in the Adv5-SIV T cell response between mAbX and atezolizumab and could serve as a useful model to better characterize pharmacodynamics effects of immunomodulatory therapeutics in NHPs.
Interestingly, although similarities in the T cell immunophenotypic profiles were observed between the animals dosed with the bispecific mAbX and atezolizumab, the Adv5-SIV model highlighted some differences. Notably, animals dosed with the bispecific mAbX presented a transient increase in the number of peripheral Tregs one-week post immunization ()). Although both targets of the bispecific mAbX are expressed on effector T cells and Tregs, no increase in Tregs was observed in pharmacology studies in tumors of humanized mice or in the periphery of cynomolgus monkeys in the repeat-dose safety studies. While the observed Treg increase with mAbX in this study has not been further examined, it may be related to the co-inhibition of both pathways targeted by mAbX in the context of a viral-based immunization. In comparison, animals dosed with atezolizumab had more of an activating effect on the T cell phenotype compared to mAbX, including expression of several co-stimulatory and co-inhibitory molecules (), the highest levels of circulating proliferating CD8+ T cells ()) and the highest number of IFNγ-secreting RM9-Specific CD8+ T cells compared to the other groups ()).
MHC genotyping was performed in this study to permit the characterization of the antigen-specific T-cell immune response by tetramer staining, ELIspot and cytolytic assays. These peptide-specific responses required MCMs to express the MHC Class I alleles Mafa-A1*063 with or without Mafa-B*104:01. If, however, the antigen-specific CD8+ T-cell immune response is not required, looking at the broad CD8 response will not need to identify animals carrying a specific MHC allele (). As such, a basic study design could include cynomolgus monkeys from diverse origin in a study of at least 4-week duration with a single immunization. The parameters tested would be hematology, clinical chemistry/CRP, immunophenotyping (excluding Tetramer staining), and ELIspot assay with an overlapping peptide pool covering the entire Nef sequence. Alternatively, hexon peptides could be used in place of a Nef peptide pool, as long as NHPs only show a basal level of response during predose.
Figure 10. Proposed study design
The goal of this study was to develop an NHP vaccination model with enhanced translational potential to humans that specifically elicits a CD8+ T-cell response and allows simultaneous evaluation of safety and pharmacology of immunomodulatory therapies targeting CD8+ T cells. We demonstrated that, following Adv5-SIV immunization, atezolizumab enhanced the expression of several co-stimulatory and co-inhibitory receptors on CD4+ and CD8+ T cells and increased the SIV antigen-specific IFNγ and CTL response of the CD8+ T cells when compared to control Adv5-SIV immunized NHPs. These data are consistent with clinical data demonstrating that treatment with atezolizumab increases neoantigen-specific T cells in oncology patients. For example, Fehlings et al. demonstrated that late-differentiated neoantigen-specific CD8+ T cells are enriched in the peripheral blood of non-small cell lung carcinoma patients responding to atezolizumab.Citation54 Furthermore, recent data from a Phase 1 study using an RNA-based personalized neoantigen-vaccine and atezolizumab was presented at the 2020 American Association for Cancer Research Virtual Annual Meeting II. Results from this study demonstrated that the combination was well tolerated and importantly showed that neoantigen-specific CD4+ and CD8+ T cell responses were detected in the periphery in 73% of patients.Citation55 While it is difficult to directly compare the results of this study with clinical T-cell responses, the enhanced CD8+ T cell response observed in response to Adv5-SIV immunization suggests that this model may be more translational to humans with regards to expected pharmacology than testing in naïve NHPs. This contrasts with previous reports, which showed no effect or even a reduction in the anti-KLH antibody response in a TDAR model with KLH for some PD-1 or PD-L1 blocking antibodies.Citation30,Citation31 As a result, this Adv5-SIV vaccine model may not only be useful to assess the safety and pharmacology of new biotherapeutics, but could also provide additional information on the mode of action of new compounds, particularly when the target is expressed on CD8+ T cells.
In summary, this NHP Adv5-SIV vaccine model allowed the evaluation of biotherapeutics targeting immunomodulatory proteins expressed on T cells, in nonhuman primates. It has the advantage of combining safety and pharmacology readouts within the same animal, hence reducing the use of NHP animals by performing one single study in which information on both aspects can be gathered. The study design presented can be performed without compromising the assessments included in standard toxicology studies and allows immunology readouts to be performed within 4 weeks following a single Adv5-SIV administration (). In fact, some responses may be blunted after a second vaccination, possibly due to the development of a strong neutralizing anti-adenovirus response over time. Moreover, this model can address questions related to primary T-cell response, such as those involving APC function, primary T-cell expansion and T cell differentiation. Although this model cannot provide information on the mode of action of CITs within a tumor microenvironment, it can further increase the relevance of safety studies potentially linked to exaggerated pharmacology for targets which are not highly expressed and may otherwise not be detected in un-challenged healthy animals. In conclusion, this Adv5-SIV model may help to better predict the outcome of human therapeutics intended to enhance CD8+ T-cell function, filling a need in the nonclinical development of immunomodulatory therapeutics.
Materials and methods
Selection, immunization and dosing of Mauritius cynomolgus macaques
Since MCMs express only seven MHC haplotypes, identification of an antigen-specific response does not require large-scale screening of animals.Citation39,Citation56 Sixteen adult male cynomolgus macaques (Macaca fascicularis), each weighing between 3.4 and 4.5 kg, imported from Mauritius islands and housed according to European guidelines, were used in this study. The protocol of this study was reviewed and approved by our Institutional Animal Care and Use Committee (IACUC) before conduct. Moreover, the care and use of animals were conducted in accordance with the guidelines of the USA National Research Council and the Canadian Council on Animal Care (CCAC). Study animals were confirmed to carry the transcriptionally abundant MHC Class I allele Mafa-A1*063 with or without Mafa-B*104:01 by MiSeq sequence-based genotyping, based on previous studies demonstrating the prevalence of the Mafa-A1*063 allele in approximately 80% of macaques of Mauritius origin and their relevance to NHP SIV-Specific CD8+ T-cell responses.Citation56 Following an acclimation period of 9 weeks, each animal was immunized intramuscularly 4 weeks apart with 3 × 1010 pfu of three recombinant, non-replicative adenoviruses expressing the SIVmac239 Gag, Pol, and Nef under the control of the CMV promoter. Of these 16 animals, six were concomitantly given vehicle (0.9% Saline) (untreated Group 1), five administered 100 mg/kg mAbX (Group 2), a bispecific antibody in early development, and five administered 50 mg/kg atezolizumab (anti-PD-L1) (Group 3), by bolus intravenous dosing at a dose volume of 3 mL/kg once weekly for 5 doses (Days 1, 8, 15, 22, and 29) (). When falling on days of immunization (Days 1 and 29), the antibody administrations were performed approximately 10 minutes after immunization. The dose levels selected for this study corresponded to the highest dose tested in previous repeat-dose toxicity studies in NHP that were well tolerated and maintained 100% receptor occupancy for the duration of the study (unpublished data). Animals were observed twice daily for the duration of the study. Group sizes were selected to provide reliable results based on potential variability of parameters evaluated; no power calculation was performed.
Hematology and clinical chemistry parameters
Animals were fasted for at least 6 hours prior to blood collection. Blood samples were collected by venipuncture at the corresponding time points. Serum chemistry was evaluated on Days 1 (predose), 2, 8 (predose), 29 (predose), 30, 31, 36, and 57. Blood samples were processed to serum and analyzed for the following clinical chemistry parameters on a Cobas® 6000 chemistry analyzer (Roche, Canada): alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, gamma-glutamyltransferase and creatine kinase activities, total bilirubin, urea nitrogen, creatinine, calcium, phosphorus, C-reactive protein, total protein, albumin, globulin, albumin/globulin ratio, glucose, cholesterol, triglycerides, potassium, and chloride concentrations.
Hematology was evaluated at pre-treatment and on Days 1 (predose), 4, 8 (predose), 15, 22 (predose), 29 (predose), 32, 43, 50, and 57. Whole blood samples were collected in K2EDTA tubes and analyzed for the following hematology parameters on an Advia® 120 hematology analyzer (Siemens, Canada): red blood cell count, hemoglobin concentration, hematocrit, mean corpuscular volume, red blood cell distribution width, mean corpuscular hemoglobin concentration, mean corpuscular hemoglobin, reticulocyte count (absolute and percent), platelet count, white blood cell count and differential (absolute and percent).
Immunophenotyping, antibodies, and flow cytometry analysis
Samples were collected (on the same occasions as hematology), and stained for cell surface and intracellular markers, using the following antibodies: CD45-APC-R700 (D058-1283, BD Biosciences, San Jose, CA), CD3-BV510 (SP34-2, BD Biosciences), CD4-BV711 (L200, BD Biosciences), CD8-FITC (SK1, BD Biosciences), CD20-APC (2H7, BD Biosciences), CD159a-PC7 (Z199, Beckman Coulter, Indianapolis, IN), CD69-PerCP-Cy5.5 (FN50, BD Biosciences), CD152-BV786 (BNI3, BD Biosciences), CD28-PE (CD28.2, BD Biosciences), CD95-PE-Cy5 (DX2, BD Biosciences), CD25-BV605 (M-A251, BD Biosciences), CD14-APC-H7 (M5E2, BD Biosciences), Ki67-BV650 (B56, BD Biosciences), FoxP3-PE-Cy5.5 (PCH101, ThermoFisher, Waltham, MA), LAG3-BV650 (11C3C65, Biolegend, San Diego, CA), TIM3-BV605 (F38-2E2, Biolegend), OX40-PE (L106, BD Biosciences), 4–1BB-PE-Cy5 (4B4-1, Biolegend), PD-1-PerCP-Cy5.5 (EH12.2H7, Biolegend), VISTA-Alexa647 (730804, R&D Systems, Minneapolis, MN), BTLA-APC-Cy7 (MIH26, Biolegend), CTLA-4 (BNI3, BD Biosciences), ICOS-PE-Cy7 (C398.4A, Biolegend), B7-H4/Dazzle594 (MIH43, Biolegend), Tetramer-Nef-RM9 (MBL International, Woburn, MA). Samples were acquired on a BD LSRFortessa flow cytometer (BD Biosciences), with approximately 500,000 to 1,000,000 events were collected per sample. Samples were then analyzed using FloJo software (Tree Star, Ashland, OR). The immunophenotyping panels were analyzed by excluding doublets by forward scatter (FSC) area versus FSC height and gating the lymphocytes population based on side scatter (SSC) and CD45+. Total T cells (CD3+), CD8+ cytotoxic T cells, CD4+ helper T cells, B cells (CD20+) and NK cells (CD159a+) were assessed. From the CD8+ and the CD4+ population, additional subpopulations were analyzed as follows: central memory (TCM), effector memory (TEM) and naïve T cells using CD28/CD95 markers; regulatory T cells using CD25/FoxP3 markers; activated T cells using CD69, Ki67, 4–1BB, OX40, and ICOS markers; exhausted T cells using B7-H4, BTLA, CTLA-4, LAG3, PD-1, TIM3, and VISTA; and lastly Nef-specific T cells using a Tetramer to Nef-RM9 peptide (MBL International). Results were reported as relative percentages of their respective parent population and as calculated absolute counts in blood (cells/µL).
Dimensionality reduction was performed on the flow cytometry panel containing the following markers: CD45-APC-R700, CD3-BV510, CD4-BV711, CD8-FITC, CD20-APC, CD159a-PC7, CD69-PerCP-Cy5.5, CD152-BV786, CD28-PE, CD95-PE-Cy5, CD25-BV605, CD14-APC-H7, Ki67-BV650, and FoxP3-PE-Cy5.5. For each set of analyses, individual cell subsets of the CD8+ T cell population from each animal were randomly down-sampled to an equal number of events (6000 events) and then analyzed.
For identification of CD8+ T cell clusters, t-distributed stochastic neighbor embedding (t-SNE) were performed on the t-SNE function from FloJo software using the following default parameters: iterations, 1000; perplexity, 30; learning rate, 6720. For each cluster identified, the differences between the percentage in each treatment groups were calculated.
IFN-γ ELIspot assay
ELIspot assays were conducted according to the Primate IFN-gamma ELISpot Kit manufacturer’s protocol (R&D Systems). Briefly, PBMCs were freshly isolated before immunization and at weeks 2, 3, 6 and 7, using lympholyte®. Mammal cell separation medium (Cedarlane) and density centrifugation from whole blood containing sodium heparin. Subsequently, 2 × 105 cells per well (in 100 µL of AIM-V medium) were added to a primate IFN-γ pre-coated ELIspot plate with a Nef RM9, LT9, and HW8 peptide pool (10 µM) or with the PepTivator AdV5 Hexon at 0.6 µM (Miltenyi). The selection of the Nef peptides was made based on the literature and the results of a pilot study that confirmed that, of the three Gag, Pol, and Nef, the stimulation was the strongest with these three Nef peptides.Citation39,Citation40 Cells incubated with phorbol myristate acetate and ionomycin were used as the positive control and cells with AIM-V medium were used as the negative control. Plates were read using an AID ELIspot reader, and spots were counted using an automated program with fixed parameters. Results were reported as spot forming units per million PBMCs after subtracting the spots obtained from the negative controls.
Ex vivo recall response assay
PBMCs were freshly isolated (see ELIspot section) before immunization and at weeks 2, 3, 6 and 7. PBMCs were resuspended in RPMI medium (1 × 106 cells per well) containing anti-CD28 (L293, BD Biosciences) and anti-CD49d antibodies (9F10, BD Biosciences) as co-stimulants. PBMCs were incubated for 4 hours, in a humidified incubator set to maintain 37°C, 5% CO2 in the presence of the Nef RM9, LT9, and HW8 peptide pool (10 µM/peptide) (MBL International), PepTivator AdV5 Hexon peptide pool (0.6 µM/peptide) (Miltenyi) or medium only (as negative control). As positive control, PBMCs were incubated in the presence of an anti-CD3 antibody (CD3-1, Mabtech, Stockholm, Sweden). After adding BD GolgiPlug™ Protein T Inhibitor (BD Biosciences) and incubating the cells for an additional 16–20 hours in a humidified incubator set to maintain 37°C, 5% CO2, the intracellular level of TNFα (MAb11, BD Biosciences), IFNγ (B27, BD Biosciences) and IL-2 (MQ1-17 H12, Biolegend) as relative percentages of CD4+ T cells and CD8+ T cells was measured by flow cytometry (BD LSRFortessa).
Cytotoxic T lymphocytes assays
Target cells (autologous B cells) were isolated from the 16 study animals prior to the beginning of the study using CD20 microbeads (Miltenyi) according to the manufacturer’s instruction and kept in liquid nitrogen until use. The day before the experiment, primary B cells were thawed and incubated for 18 hours with IFN-γ (R&D System). On the day of the assay, half of the target B cells were pulsed with 10 µg/mL of Nef RM9 peptide (MBL international) and stained with 0.2 µM carboxyfluorescein succinimidyl ester (CFSE; ThermoFisher), while the other half of the target B cells were left un-pulsed and stained with CellTrace Yellow diluted 1/50,000 (ThermoFisher). A portion of un-pulsed target B cells were stained with 0.2 µM CFSE. Cells were then mixed at a ratio of 1:1 using CellTrace Yellow cells (un-pulsed):CFSE cells (pulsed or un-pulsed) target B cells. On week 5 and 8, primary CD8+ T cells (effector cells) were freshly isolated from PBMCs using the CD8+ T cell Isolation kit (Miltenyi) by negative selection. Target (T) cells and effector (E) cells were then mixed at different T:E cell ratio, with equal numbers of pulsed and un-pulsed target B cells. After 18 hours of incubation at 37°C, 5% CO2, cells were stained with tetramer-Nef-RN9-BV421 (MBL International), anti-CD8-FITC (SK1, BD bioscience), LIVE/DEAD™ Fixable Far Red Dead Cell Stain Kit (ThermoFisher) and analyzed by flow cytometry (BD LSRFortessa). Live target B cells were gated on CellTrace Yellow (un-pulsed) and CFSE (pulsed or un-pulsed) intensities. Total autologous target B cells were gated on CFSE and effector antigen-specific CD8+ T cells were gated on pMHC tetramer-Nef-RM9/CD8 double positive cells.Citation57 Counts for the four populations: CellTrace Yellow, CFSE (pulsed and non-pulsed) target cells, and effector cells were extracted from the analysis and used to plot the percentage of specific lysis in function of the effector/target ratio.
First, the E/T ratio was calculated using the following equation:
Then, the ratio TCSFE-pulsed/TCellTrace Yellow between Nef RM9-pulsed CFSE B cells (TCFSE-pulsed) and un-pulsed CellTrace Yellow B cells (TCellTrace Yellow) after an incubation with effector T cells was calculated using the live or combined live and dead cells:
The median between wells containing only un-pulsed target cells (TCFSE-unpulsed and TCellTrace Yellow) was used to calculate the ratio (TCSFE-unpulsed/TCellTrace Yellow):
For each E/T ratio, the percentage of specific lysis was finally calculated using the following formula:
Disclosure statement
The authors report no conflict of interest.
References
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–15. doi:10.1016/j.immuni.2013.07.012.
- van Meer PJK, Graham ML, Schuurman HJ. The safety, efficacy and regulatory triangle in drug development: impact for animal models and the use of animals. Eur J Pharmacol. 2015;759:3–13. doi:10.1016/j.ejphar.2015.02.055.
- Chen DS, and Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1-10.
- Lesterhuis WJ, Haanen JBAG, and Punt CJA. Cancer immunotherapy – revisited. Nat Rev Drug Discov. 2011;10(8):591-600.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252-64.
- Herzyk DJ, and Haggerty HG. Cancer immunotherapy: factors important for the evaluation of safety in nonclinical studies. AAPS J. 2018;20(2):28.
- Lebrec H, Molinier B, Boverhof D, Collinge M, Freebern W, Henson K, Mytych DT, Ochs HD, Wange R, Yang Y, et al. The T-cell-dependent antibody response assay in nonclinical studies of pharmaceuticals and chemicals: study design, data analysis, interpretation. Regul Toxicol Pharmacol. 2014;69(1):7–21. doi:10.1016/j.yrtph.2014.02.008.
- Olson B, Li Y, Lin Y, Liu ET, Patnaik A. Mouse models for cancer immunotherapy research. Cancer Discov. 2018;8(11):1358–65. doi:10.1158/2159-8290.CD-18-0044.
- Overgaard NH, Fan TM, Schachtschneider KM, Principe DR, Schook LB, Jungersen G. Of mice, dogs, pigs, and men: choosing the appropriate model for immuno-oncology research. ILAR J. 2018;59:247–62.
- Schachtschneider KM, Schwind RM, Newson J, Kinachtchouk N, Rizko M, Mendoza-Elias N, Grippo P, Principe DR, Park A, and Overgaard NH, et al. The oncopig cancer model: an innovative large animal translational oncology platform. Front Oncol. 2017;7:190.
- Zitvogel L, Pitt JM, Daillère R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. 2016;16(12):759–73. doi:10.1038/nrc.2016.91.
- Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28(6):690–714. doi:10.1016/j.ccell.2015.10.012.
- Curran M, Mairesse M, Matas-Céspedes A, Bareham B, Pellegrini G, Liaunardy A, Powell E, Sargeant R, Cuomo E, and Stebbings R, et al. Recent advancements and applications of human immune system mice in preclinical immuno-oncology. Toxicol Pathol. 2019Feb;48(2):302-316.
- De La Rochere P, Guil-Luna S, Decaudin D, Azar G, Sidhu SS, Piaggio E. Humanized mice for the study of immuno-oncology. Trends Immunol. 2018;39(9):748–63. doi:10.1016/j.it.2018.07.001.
- Gillet J-P, Calcagno AM, Varma S, Marino M, Green LJ, Vora MI, Patel C, Orina JN, Eliseeva TA, Singal V, et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc Natl Acad Sci U S A. 2011;108(46):18708–13. doi:10.1073/pnas.1111840108.
- Mak IW, Evaniew N, Ghert M Review article lost in translation: animal models and clinical trials in cancer treatment [Internet]. 2014. Available from: www.ajtr.org
- Sanmamed MF, Chester C, Melero I, and Kohrt H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies downloaded from [internet]. Ann Oncol. 2016 Jul;27(7):1190-8. . Available from: http://annonc.oxfordjournals.org/
- Fisher TS, Kamperschroer C, Oliphant T, Love VA, Lira PD, Doyonnas R, Bergqvist S, Baxi SM, Rohner A, Shen AC, et al. Targeting of 4-1BB by monoclonal antibody PF-05082566 enhances T-cell function and promotes anti-tumor activity. Cancer Immunol Immunother. 2012;61(10):1721–33. doi:10.1007/s00262-012-1237-1.
- Herzyk DJ, Haggerty HG. Cancer immunotherapy: factors important for the evaluation of safety in nonclinical studies. AAPS J. 2018;20(2). doi:10.1208/s12248-017-0184-3.
- Iwasaki K, Uno Y, Utoh M, Yamazaki H. Importance of cynomolgus monkeys in development of monoclonal antibody drugs. Drug Metab Pharmacokinet. 2019;34(1):55–63. doi:10.1016/j.dmpk.2018.02.003.
- Messaoudi I, Estep R, Robinson B, Wong SW Nonhuman primate models of human immunology.
- van Meer PJK, Kooijman M, Brinks V, Gispen-De Wied CC, Silva-Lima B, Moors EHM, Schellekens H. Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. mAbs. 2013;5(5):810–16. doi:10.4161/mabs.25234.
- Weinberg AD, Thalhofer C, Morris N, Walker JM, Seiss D, Wong S, Axthelm MK, Picker LJ, Urba WJ Anti-OX40 (CD134) administration to nonhuman primates: immunostimulatory effects and toxicokinetic study. 2006.
- Satterwhite CM, Comba RW, Oldendorp A, and Prell R Investigation of KLH (Keyhole Limpet Hemocyanin) antigen dose response in male and female cynomolgus monkeys to establish an optimal response to study immune enhancement. SOT 55th Annual Meeting and ToxExpo. New Orleans, LA. 2016.
- Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, Garcia C, Wu Y, Kuhne M, Srinivasan M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014;2(9):846–56. doi:10.1158/2326-6066.CIR-14-0040.
- Loffredo J, Vuyyuru R, Spires V, Beyer S, Fox M, Ehrmann J, Taylor K, Engelhardt J, Korman A, and Graziano R Non-fucosylated anti-CTLA-4 antibody enhances vaccine-induced T cell responses in a non-human primate pharmacodynamic vaccine model-32nd Annual Meeting and Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2017). In: Journal for ImmunoTherapy of Cancer. National Harbor, Maryland. 2017.
- Keler T, Halk E, Vitale L, O’Neill T, Blanset D, Lee S, Srinivasan M, Graziano RF, Davis T, and Lonberg N, et al. Activity and safety of CTLA-4 blockade combined with vaccines in cynomolgus macaques. J Immunol. 2003;171(11):6251-9.
- Gonzalez AM, Manrique ML, Swiech L, Horn T, Breous E, Waight J, Savitsky D, Liu Y, Lin S, Clarke C, et al. Abstract 4703: INCAGN1949, an anti-OX40 antibody with an optimal agonistic profile and the ability to selectively deplete intratumoral regulatory T cells. In: Clinical Research (Excluding Clinical Trials). American Association for Cancer Research; 2017.
- Macoin J, Blein S, Monney T, Sancheti P, Reddy V, Back J GBR830, a true OX40 antagonist antibody with potent suppressive effects on T cell-mediated pathological responses [abstract]. In: Arthritis & Rheumatology. American College of Rheumatology; 2018.
- U. S. Food and Drug Administration. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761069Orig1s000PharmR.pdf, December 2020.
- European Medicines Agency: EMA assessment report EMA/CHMP/392114/2015, https://www.ema.europa.eu/en/documents/assessment-report/nivolumab-bms-epar-public-assessment-report_en.pdf, Access date 01 Dec 2020 .
- Kamperschroer C, O’Donnell LM, Schneider PA, Li D, Roy M, Coskran TM, Kawabata TT. Measuring T-cell responses against LCV and CMV in cynomolgus macaques using ELISPOT: potential application to non-clinical testing of immunomodulatory therapeutics. J Immunotoxicol. 2014;11(1):35–43. doi:10.3109/1547691X.2013.766287.
- Finnefrock AC, Tang A, Li F, Freed DC, Feng M, Cox KS, Sykes KJ, Guare JP, Miller MD, Olsen DB, et al. PD-1 blockade in rhesus macaques: impact on chronic infection and prophylactic vaccination. J Immunol. 2009;182(2):980–87. doi:10.4049/jimmunol.182.2.980.
- Tatsis N, Lasaro MO, Lin S-W, Xiang ZQ, Zhou D, DiMenna L, Li H, Bian A, Abdulla S, Li Y, et al. Adenovirus vector-induced immune responses in nonhuman primates: responses to prime boost regimens. J Immunol. 2009;182(10):6587–99. doi:10.4049/jimmunol.0900317.
- Zak DE, Andersen-Nissen E, Peterson ER, Sato A, Hamilton MK, Borgerding J, Krishnamurty AT, Chang JT, Adams DJ, Hensley TR, et al. Merck Ad5/HIV induces broad innate immune activation that predicts CD8+ T-cell responses but is attenuated by preexisting Ad5 immunity. Proc Natl Acad Sci U S A. 2012;109(50):E3503–12. doi:10.1073/pnas.1208972109.
- Sakurai F, Nakamura S, Akitomo K, Shibata H, Terao K, Kawabata K, Hayakawa T, and Mizuguchi H. Transduction properties of adenovirus serotype 35 vectors after intravenous administration into nonhuman primates. Mol Ther. 2008;16(4):726-733.
- Wu J, Chen G, Zhuang F-C, Gao M, Wu C-D, He Z-L, Jiang Y-S, Li J-B, Bao J-Y, Mao Z-A. Long-term toxicity, pharmacokinetics and immune effects of a recombinant adenovirus vaccine expressing human papillomavirus 16 E6 and E7 proteins (HPV16 E6E7-Ad5 Vac) in primates. Am J Transl Res. 2018;10:1539–51.
- Ni S, Bernt K, Gaggar A, Li Z-Y, Kiem H-P, Lieber A. Evaluation of biodistribution and safety of adenovirus vectors containing group B fibers after intravenous injection into baboons. Hum Gene Ther. 2005;16.
- Budde ML, Greene JM, Chin EN, Ericsen AJ, Scarlotta M, Cain BT, Pham NH, Becker EA, Harris M, Weinfurter JT, et al. Specific CD8+ T cell responses correlate with control of simian immunodeficiency virus replication in Mauritian cynomolgus macaques. J Virol. 2012;86(14):7596–604. doi:10.1128/JVI.00716-12.
- Burwitz BJ, Pendley CJ, Greene JM, Detmer AM, Lhost JJ, Karl JA, Piaskowski SM, Rudersdorf RA, Wallace LT, Bimber BN, et al. Mauritian cynomolgus macaques share two exceptionally common major histocompatibility complex class I alleles that restrict simian immunodeficiency virus-specific CD8 + T cells. J Virol. 2009;83(12):6011–19. doi:10.1128/JVI.00199-09.
- Greene JM, Chin EN, Budde ML, Lhost JJ, Hines PJ, Burwitz BJ, Broman KW, Nelson JE, Friedrich TC, and O’Connor DH. Ex vivo SIV-specific CD8 T cell responses in heterozygous animals are primarily directed against peptides presented by a single MHC haplotype. Plos One. 2012;7(8):e43690.
- Calcedo R, Vandenberghe LH, Roy S, Somanathan S, Wang L, Wilson JM. Host immune responses to chronic adenovirus infections in human and nonhuman primates. J Virol. 2009;83(6):2623–31. doi:10.1128/JVI.02160-08.
- del Giudice G, Rappuoli R, Didierlaurent AM. Correlates of adjuvanticity: a review on adjuvants in licensed vaccines. Semin Immunol. 2018;39:14–21.
- Jeyanathan M, Shao Z, Yu X, Harkness R, Jiang R, Li J, Xing Z, and Zhu T. AdHu5Ag85A respiratory mucosal boost immunization enhances protection against pulmonary tuberculosis in bcg-primed non-human primates. Plos One. 2015;10(8):e0135009.
- Kim JR, Holbrook BC, Hayward SL, Blevins LK, Jorgensen MJ, Kock ND, De Paris K, D’Agostino RB, Aycock ST, Mizel SB, et al. Inclusion of flagellin during vaccination against influenza enhances recall responses in nonhuman primate neonates. J Virol. 2015;89(14):7291–303. doi:10.1128/JVI.00549-15.
- McDade TW, Borja JB, Kuzawa CW, Perez TLL, Adair LS. C-reactive protein response to influenza vaccination as a model of mild inflammatory stimulation in the Philippines. Vaccine. 2015;33(17):2004–08. doi:10.1016/j.vaccine.2015.03.019.
- Sabins NC, Harman BC, Barone LR, Shen S, and Santulli-Marotto S. Differential expression of immune checkpoint modulators on in vitro primed CD4+ and CD8+ T cells. Front Immunol. 2016;7:221.
- Bottermann M, Foss S, van Tienen LM, Vaysburd M, Cruickshank J, O’Connell K, Clark J, Mayes K, Higginson K, Hirst JC, et al. TRIM21 mediates antibody inhibition of adenovirus-based gene delivery and vaccination. Proc Natl Acad Sci U S A. 2018;115(41):10440–45. doi:10.1073/pnas.1806314115.
- Choi JH, Jonsson-Schmunk K, Qiu X, Shedlock DJ, Strong J, Xu JX, Michie KL, Audet J, Fernando L, Myers MJ, et al. A single dose respiratory recombinant adenovirus-based vaccine provides long-term protection for non-human primates from lethal Ebola infection. Mol Pharm. 2015;12(8):2712–31. doi:10.1021/mp500646d.
- Lawrence M, Foellmer H, Elsworth J, Kim J, Leranth C, Kozlowski D, Bothwell A, Davidson B, Bohn M, and Redmond JD Inflammatory responses and their impact on-galactosidase transgene expression following adenovirus vector delivery to the primate caudate nucleus [Internet]. Gene Ther.1999;6(8):1368-79. Available from: http://www.stockton-press.co.uk/gt
- Schirmbeck R, Reimann J, Kochanek S, Kreppel F. The immunogenicity of adenovirus vectors limits the multispecificity of CD8 T-cell responses to vector-encoded transgenic antigens. Mol Ther. 2008;16(9):1609–16. doi:10.1038/mt.2008.141.
- t’Hart BA, Vervoordeldonk M, Heeney JL, Tak PP. Gene therapy in nonhuman primate models of human autoimmune disease. Gene Ther. 2003;10(10):890–901. doi:10.1038/sj.gt.3302017.
- Young LS, Mautner V. The promise and potential hazards of adenovirus gene therapy. Gut. 2001;48(48):733–36. doi:10.1136/gut.48.5.733.
- Fehlings M, Jhunjhunwala S, Kowanetz M, O’Gorman WE, Hegde PS, Sumatoh H, Lee BH, Nardin A, Becht E, Flynn S, et al. Late-differentiated effector neoantigen-specific CD8+ T cells are enriched in peripheral blood of non-small cell lung carcinoma patients responding to atezolizumab treatment. J ImmunoTher Cancer. 2019;7.
- Lopez JS, Camidge R, Iafolla M, Rottey S, Schuler M, Hellmann M, Balmanoukian A, Dirix L, Gordon M, Sullivan R, et al. Abstract CT301: a phase Ib study to evaluate RO7198457, an individualized Neoantigen Specific immunoTherapy (iNeST), in combination with atezolizumab in patients with locally advanced or metastatic solid tumors. In: Bioinformatics, Convergence Science, and Systems Biology. American Association for Cancer Research; 2020.
- Budde ML, Lhost JJ, Burwitz BJ, Becker EA, Burns CM, O’Connor SL, Karl JA, Wiseman RW, Bimber BN, Zhang GL, et al. Transcriptionally abundant major histocompatibility complex class I alleles are fundamental to nonhuman primate simian immunodeficiency virus-specific CD8 + T cell responses. J Virol. 2011;85(7):3250–61. doi:10.1128/JVI.02355-10.
- Mbitikon-Kobo FM, Bonneville M, Sekaly RP, Trautmann L. Ex vivo measurement of the cytotoxic capacity of human primary antigen-specific CD8 T cells. J Immunol Methods. 2012;375(1–2):252–57. doi:10.1016/j.jim.2011.09.008.