
ABSTRACT
A major impediment to successful use of therapeutic protein drugs is their ability to induce anti-drug antibodies (ADA) that can alter treatment efficacy and safety in a significant number of patients. To this aim, in silico, in vitro, and in vivo tools have been developed to assess sequence and other liabilities contributing to ADA development at different stages of the immune response. However, variability exists between similar assays developed by different investigators due to the complexity of assays, a degree of uncertainty about the underlying science, and their intended use. The impact of protocol variations on the outcome of the assays, i.e., on the immunogenicity risk assigned to a given drug candidate, cannot always be precisely assessed. Here, the Non-Clinical Immunogenicity Risk Assessment working group of the European Immunogenicity Platform (EIP) reviews currently used assays and protocols and discusses feasibility and next steps toward harmonization and standardization.
Introduction
The concept of “predicting immunogenicity risk” in the pharmaceutical industry is currently understood as the ability to identify a therapeutic protein’s intrinsic and extrinsic factors that may contribute to the mounting of an anti-drug antibody (ADA) response by a patient’s immune system (see for a list of terminologies). Key steps in driving this process are: 1) the priming of innate cells such as macrophages and dendritic cells (DCs), followed by 2) the recruitment of T cells to develop a mature immune response, leading to 3) the production of ADAs by plasma cells. Uptake and processing of therapeutic proteins by antigen-presenting cells (APCs) is believed to be one of the initial and critical steps in driving an immune response. APCs are a heterogenous group of immune cells (including dendritic cells, macrophages, and B cells) that can take up and process antigens that are then presented as linear peptides in the context of class II MHC molecules to naïve T lymphocytes in peripheral lymphoid organs. Priming of naïve T cells requires three distinct signals:Citation1 1) activation of the T cell receptor (TCR) by a specific interaction with a distinct Major Histocompatibility Complex (MHC)II:peptide complex, 2) survival and clonal expansion of the activated T cell due to CD28 receptor interaction with CD80/CD86 co-stimulatory molecules, and 3) differentiation into various effector cells depending on the cytokines produced by the APCs. For example, activated naïve CD4 T cells may differentiate into distinct T effector cell types, such as Th1, Th2, Th9, Th17, T follicular helper cells, T regulatory cells, or into T follicular regulatory cells depending on variations in the third signal (cytokines produced by the DCs).Citation2 Activated T cells express numerous surface receptors (e.g., CD25, CD69, CD70, CD71, CD95, HLA-DR-DP-DQ), co-stimulatory molecules (e.g., CD26, CD27, CD28, CD30, CD40L, CD134), chemokine receptors (e.g., CXCR3, CCR5), and adhesion molecules (e.g., ICAM-1, LFA-1)Citation3 that all can modulate and direct further activation of the immune response, in particular the activation of naïve B cells leading to their differentiation to antibody-secreting plasma cells. In the late phase of the immune response, the majority of the antigen-specific T cells die while a pool of memory T cells remain at 10- to 1000-fold higher frequency than the naïve precursors activated during the primary response.Citation4,Citation5 In particular, central memory T cells, which are located in secondary lymphoid organs, retain an elevated proliferative capacity and may rapidly re-create a new pool of effector T cells after activation by MHC-II:peptide complexes presented by APCs (including resting B cells), even in the absence of APC co-stimulatory signals.Citation6
Table 1. Terminology of pre-clinical immunogenicity assessment
While a therapeutic protein’s intrinsic and extrinsic properties () may initiate the immune response cascade, other factors, such as patient-related factors (Human Leukocyte Antigen (HLA) haplotype, immune status), therapeutic context, routes of drug delivery, and mode of action (MoA), also contribute to immunogenicity risks.Citation7 Thus, the overall contribution of each of these components to the global immunogenic response in the clinic may not become apparent in the absence of large, systematic studies. From a pre-clinical perspective, drug developers have over the years set up a series of assays that recapitulate parts of the immune system, with the aim of capturing critical steps in the immune activation reflecting the in vivo situation. However, because pre-clinical assays are reductionist in nature, an immunogenicity risk assessment represents a combination of probabilities and consequences, with the risk level influenced by the degree of confidence in predicting these consequences based on the knowledge and representability of these assays.
The pre-clinical tools and assays discussed here () reflect our current understanding of the immune response as described above and can be broadly summarized in three categories: 1) in silico tools, which aim at identifying potential T cell epitopes on the sole basis of predicted HLA affinity; 2) in vitro assays, which report the response of a specific population of cell(s) toward a controlled stimulus; and 3) in vivo systems, which attempt to replicate a humanized immune response in an animal model. A common challenge and complication met by all pre-clinical immunogenicity assessment tools is MHC-II polymorphism, with currently more than 7300 allelesCitation8 known and expressed with different prevalence across the human population. Thus, an important consideration for translation of data to the general population or targeted sub-populations is to ensure that MHC-II diversity is appropriately represented within each assay. The complexity of the immunogenic process and the many factors that contribute to it highlight the need for a standardized approach for methodologies and metrics to attribute commonly accepted risk scores and trigger effective strategies for mitigation.
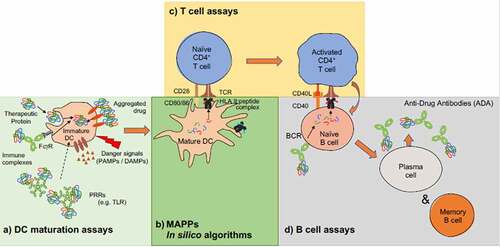
Figure 1. Schematic overview of an immunogenic signal cascade resulting in the production of anti-drug antibodies. Currently available tools and assays cover some of the key areas that drive an anti-drug antibody response to a biotherapeutic. Integrating information from multiple of such tools and assays informs about the intrinsic antigenicity potential of a biotherapeutic. (a) Antigen up-take and processing can be evaluated in dendritic cell uptake and activation assays. (b) In silico peptide binding prediction tools and MAPPs help reveal potential T cell epitopes (c) and in vitro T cell assays, with multiple different formats available, confirm previously identified potential T cell epitopes and help estimate prevalence of pre-cursor T cell responses in a given population. More recently, B cell assays (d) emerged aiming at identifying and cloning of B cell clones from patients who developed and anti-drug antibody response or to reveal B cell clones which produce cross-reacting antibodies to biotherapeutics, while in vivo systems attempt to replicate a humanized immune response in animal models and include all required steps of an anti-drug antibody response
The risk of eliciting innate and adaptive immune responses from next-generation modalities like nucleic acids, viral vectors and cellular therapies is not in scope of this review. Although immunogenicity risk can be assessed to a certain extent with current methods described here, some additional adaptations may be needed to better understand innate responses that would include viral- and nucleic acid-mediated innate receptor-specific assessments, complement activation and adaptive phase responses, such as cytotoxic T lymphocyte (CTL) responses. The transgenes associated with viral gene therapies and chimeric antigen components associated with CAR-T therapies can use existing adaptive response assays, including cellular and humoral response assays. Additional risk assessment considerations and relevant assays would include virology, preexisting serotype reactivity assessments, tissue-specific tropism and associated immune responses. Although concepts discussed in here apply to the field of the new modalities as well, different controls and additional standardization approaches that go beyond the scope of this review are required, and are therefore not discussed here.
This work, conducted by the Non-Clinical Immunogenicity Risk Assessment working group of the European Immunogenicity Platform (EIP), aims to provide an overview of the current approaches and poses the question of whether criteria can be established that can be systematically applied by industry and academic groups that use these risk assessment strategies.
In silico algorithms for potential T cell epitope risk assessment
In silico approaches used for the assessment of immunogenicity risks focus on the identification of potential T cell epitopes based on the requirement for antigen presentation (in the form of linear peptides bound to MHC class II molecules) to trigger an amino acid sequence-based immune response. The primary sequence of the protein drug is scanned in a systematic manner, moving along the sequence, and a binding score is calculated for each 9–15ʹmer for each HLA molecule included in the analysis. The results of these in silico assessments can be used to triage molecules during the discovery phase, as hits are reduced to leads, and during optimization of leads to reduce the number of potential T cell epitopes by the time a clinical candidate is identified. Most drug developers use in vitro assays described herein alongside in silico algorithms to increase confidence that immunogenicity risk has been reduced through this design process.
Multiple factors contribute to a 9–15ʹmer amino-acid sequence becoming a functional T cell epitope, including stability of the complexes formed with the HLA molecules, natural processing of the epitope by the APC, and the existence of a specific CD4 T cell repertoire that may recognize the sequence within an MHC-II groove. Therefore, algorithms based solely on HLA affinity tend to be over-predictive and filtering is further required to provide context to the flagged amino acid sequences. For monoclonal antibody (mAb)-based products for instance, framework amino acid sequences found at high frequency across populations are removed from the list of potential epitopes due to presumed immunologic tolerance. Other reasons that HLA binding sequences might not become epitopes include deletion of specific T cell repertoire during negative selection, or because of an avidity that is too low to trigger a specific response. Additional mechanisms include peripherally induced tolerance, also caused by the presence of so-called Tregitopes, which might induce a T regulatory response.Citation9 Likewise, sequences with similarity to previously exposed disease agents as well as microbiomes can further develop a more tolerant T-cell repertoire, leading to lack of response to some of the presented sequences.Citation10 Clinical experience gained with antibodies derived from common frameworks and isotypes may also provide additional confidence that certain sets of presented epitopes do not elicit long-term immune responses.
Peptide elution-based algorithms of MHC class II epitope prediction have been developed to address factors pertaining to the making of an epitope beyond HLA affinity, incorporating processing and stability data that were experimentally generated using mass spectrometry (MS). Also, much larger input datasets have been used to further improve performance and accuracy. For instance, MixMHC2pred integrates allele-specific motifs, peptide length and binding core offset preferences determined using over 99,000 unique eluted peptides analyzed with MoDec, a novel peptide deep motif deconvolution probabilistic framework.Citation11 Similarly, the multimodal recurrent neural network-based algorithm MARIA includes peptide sequence, cleavage scores based on predictive information from flanking residues, and HLA binding scores, both obtained from new pre-trained neural networks. MARIA also includes gene expression level, which is unique to this approach.Citation12 Likewise, the Neonmhc2 ligandome predictor was developed and trained on input data obtained from MS analysis generated with a new technology named MAPTAC (mono-allelic purification with tagged allele constructs).Citation13 Likewise, another predictor, NNAlign_MAC, showed that the integration of MHC-II associated peptide proteomics (MAPPs) results with conventional affinity data such as collected in the Immune Epitope Database (IEDB; http://www.iedb.org) provided context information and HLA binding promiscuity scores, resulting in superior performance in predicting CD4 T cell epitopes in the well-studied infliximab and rituximab molecules.Citation14 In direct comparison studies, MixMHC2pred, MARIA, neonmhc2, and NNAlign_MAC were found to outperform existing prediction methods.Citation11–14 However, these studies are usually limited to very specific scenarios, e.g., prediction of binding to certain HLA-DR alleles, prediction of T cell epitopes identified from certain cell lines, or peptides recovered by MAPPs. It is not surprising that these new methods outperform existing prediction approaches in settings that were used to train and are thus optimized for these novel algorithms, but not for the methods they to which were compared. Therefore, it is essential to fully understand what the in silico algorithm was designed for and how it was trained for meaningful use in the context of immunogenicity risk assessment. Applying these methods blindly to “predict immunogenicity” is not recommended. Likewise, prediction of clinical ADA incidence from a pure in silico risk assessment is not advised due to the multitude of other contributing factors not included in this assessment.
In vitro assays
Assays described in this section attempt to measure experimentally the ability of an exogenous stimulus to trigger an explicit immune response in an enclosed cellular system correlating directly or indirectly with part(s) of the human immune response cascade. While it is difficult to fully replicate the tissue environment in vitro, the properties of some immune cells have been exploited in the design of assays to evaluate whether therapeutic proteins contain CD4 T cell epitopes capable of driving a memory T cell response. Current in vitro assays encompass each step of an immune activation (generation and presentation of an antigen): activation of APCs, presentation of MHC-II peptides, activation of CD4 T cells, activation of B cells, and combination thereof.
Assays developed in the past few decades have been reported with a diversity of setups and conditions reflecting each group’s emphasis, core knowledge, and historical experience, which render inter-assay comparison difficult. Also, cell availability and quality (and methods of isolation) are of primary importance, and factors such as genetics, lineage, activation status may contribute to the variability of the final output. For example, T and B cells are either purified in bulk from peripheral blood mononuclear cells (PBMC) or positively isolated using magnetic bead selection, and assay readouts very much depend on the exact lineage composition and activation status. In contrast, while several protocols may be used for the generation of monocyte-derived dendritic cell (moDC), assays based on these immune cells are robust, and outcomes do not seem to depend on which differentiation strategy is used. A common hurdle to all in vitro assays is the multiple MHC-II allelic composition, which in turn directs MHC-II peptide restriction. As a consequence, assays must be performed with multiple sample sets, using a wide variety of donors to ensure a coverage representative for a population’s HLA composition. This in turn requires assay designs with reasonable throughput and analyses that are performed according to standardized metrics for a statistical evaluation of a protein therapeutics’ immunogenicity profile.
Finally, therapeutic proteins’ MoA (in particular, those with immunomodulatory functions) and preparation (purity, aggregation state, excipients composition) affect assay designs and readouts. For example, therapeutics containing a CD3-binding domainCitation15 may not be analyzed in a PBMC-based T cell assay, as activation of these cells is part of their MoA. Similarly, preparations containing elevated levels of host cell proteinsCitation16 or with degraded, aggregated, or post-translationally-modified formsCitation17 of the therapeutic protein may elicit false positive responses in certain assays. In conclusion, there is no definite consensus on a minimal set of assays for the evaluation of pre-clinical immunogenicity risk. Rather, each drug developer is encouraged to develop an array of tests informative for each therapeutic in consideration to the intended use and treated population.
DC maturation assay
While the amino acid sequence of a protein directly contributes to the genesis of a T cell epitope, external factors might alert the immune system to the presence of aggravating factors that may activate APCs through cross-linking or via damage/pathogen-associated molecular patterns (DAMPs/PAMPs).Citation18 DCs, as the paradigm APC (), play a key role in the induction of an adaptive immune response. Activation of these cells combined with their ability to present antigens is a useful indicator for the potential of biotherapeutics to induce ADAs. Monitoring of DC maturation can also be used as a tool to determine whether external factors, such as formulation or protein aggregation, contribute to the immunogenicity of therapeutics.Citation19
Table 2. Antigen presenting cell types that have antigen-presenting function in vitro
moDCs may be generated using several protocols. After isolation of PBMCs from human blood, monocytes are isolated with magnetic bead selection or by plastic adherence. Purified monocytes are then differentiated to immature moDCs by culturing the cells in the presence of granulocyte-macrophage colony-stimulating factor and interleukin (IL)-4.Citation20 In the immature state, DCs are challenged with the test biotherapeutics and their maturation status may be monitored from 15 minutes to 48 hours after challenge.Citation21 Parameters of interest may include: 1) cell surface-marker expression (; CD83, CD80, CD86 and CD40 are recommended as minimal panel) measured 4–48 hours after stimulation; 2) cytokine secretion (IL-1β, IL-6, IL-8, IL-10, IL-12 and tumor necrosis factor (TNF)) measured 24–48 hours after stimulation in cell culture supernatant; 3) phosphorylation state of proteins involved in signaling pathways possibly playing a role in DC maturation such as Akt, ERK1/2 and Syk, measured 15 to 30 minutes after immature DC (iDC) challenge with therapeutic,Citation19 and 4) mRNA quantification (upregulation) of cytokines and/or chemokines at earlier timepoints, measured 6–24 hours after stimulation of DCs with biotherapeutics.Citation19
Table 3. DC markers
Use of relevant assay controls within the assay is essential to assess both the validity of the assay and to allow comparison among different laboratories; we suggest including a negative (medium alone), a positive control (such as keyhole limpet hemocyanin (KLH), a lipopolysaccharide (LPS) titration, or a maturation cocktail) and ideally, one low-immunogenic (e.g., bevacizumab) and one medium-high (e.g., ATR-107) benchmark controls. Evaluation of the results depends on the methodology used and typically involves a statistical analysis over a minimum of 10 donors. When cell surface markers expression is the chosen readout, DC activation is reported as median fluorescence intensity values (% increase per marker for the test condition compared to the unstimulated condition) and % of donors showing an increase of 50% or 100% per marker.Citation22
MHC-II associated peptide proteomics
In the context of an immunogenicity risk assessment, MAPPs assays have represented the method of choice for the identification of peptides presented by class II MHC molecules on moDCs. The assay generates a repertoire of proteins that have been taken up by the APC, processed, loaded and presented as peptides on MHC-II receptors through the exogenous antigen processing pathway. Thus, MAPPs provide a biologically driven snapshot complementing currently over-predictive T cell epitope bioinformatic platforms. While this assay cannot provide an immunogenicity risk scale for identified MHC-II peptides, it enables the identification of more “authentic” T cell epitopes compared to an overlapping peptide scanning approach.Citation23 However, as T cell recognition is not tested in this assay, MAPPs will also identify antigen-derived peptides that are not recognized by T cells.
MAPPs assays have been generally performed according to the following protocol using moDCs cell cultures generated as delineated in the previous section. Immature moDC are pulsed with a test compound followed by maturation with LPS or TNF to increase the antigen processing and presentation of MHC-II-peptides complexes on the moDC cell surface. Adherent cells are then pelleted and lysed in the presence of various protease inhibitors. MHC-II-peptide complexes are subsequently immunoprecipitated, typically using anti-MHC-II antibodies (specific for the MHC-II isotype of interest) immobilized to a resin or magnetic nanoparticle beads. After extensive washing to remove impurities and nonspecific peptides, the MHC-II bound peptides are eluted with acid and separated and sequenced using a liquid chromatography-tandem mass spectrometric approach.Citation24,Citation25 Database searching is typically performed against a global protein database containing the amino acid sequence(s) of the biotherapeutics of interest, making sure to omit protease cleavage specificity and including pertinent post-translational modifications (PTMs; for example, methionine sulfoxide or deamidation), as some of the peptides might be modified during the antigen processing or during the MS analysis process. In recent years, the use of increasingly more sensitive (and reliable) mass spectrometers has contributed to the ability of MAPPs assays to accurately characterize MHC-II associated peptides isolated from a number of moDCs compatible with low-throughput screening formats.
There are important contributing factors to the performance and sensitivity of a MAPPs assay, including the use of fresh or frozen cells, the number of cells used per assay, the quantity of test material and the analysis of the mass spectrometric data in terms of threshold and effective false discovery rate settings. Interestingly, while protocols for generating moDCs and performing MAPPs assays remain fairly generic, there is mounting evidence that MAPPs are a fairly robust assay and results compare well across studies.Citation26,Citation27 Comparative data from the analysis of infliximab and rituximab using only 15 donors demonstrated a very close alignment with the peptide clusters characterized by Hamze et al. who used more than 30 donors for their MAPPs study.Citation26 Moreover, in a separate unpublished study (C.B., Abzena, personal communication) peptide clusters identified by MAPPs and putative T cell epitopes in Factor VII and vatreptacog alfa aligned directly with the point mutation regions shown to associate with MHC-II molecules as published by Lamberth et al.Citation28 Due to the relative output of the assay, negative and positive controls are typically included to determine whether the pattern of peptide presentation is consistent and whether the assay has performed as expected. Assuming consistent culture and supplement conditions for the generation of DCs, endogenous reference peptides can be used to evaluate assay performance, such as MHC-II peptides derived from supplements (e.g., from supplemental serum proteins), from tracer proteins added to the assay, or from test proteins such as antibodies that have known germline sequences consistently presented by MHC-II receptors.Citation27 The systematic monitoring of these peptides derived from either media or cell-associated proteins, such as serum albumin, lysosome-associated membrane glycoproteins, and cathepsins, serves as a good measure of MS performance and consistency of the assay with respect to antigen uptake and presentation across conditions and donors.
To date, most of the MHC-II MAPPs studies have been performed to study HLA-DR-presented peptides as the dominant class II genotype, partly because of the availability of well-performing immunoprecipitating pan HLA-DR antibodies, such as the mAb clone L243. Few studies have reported the identification of MHC-II peptides presented on HLA-DPCitation29 or HLA-DQCitation30 receptors. In particular, HLA-DP is expressed only at low levels (~10 fold) compared to HLA-DR and DQ receptors, and therefore it has been considered less important with respect to immunogenicity risk. Recently, two independent studies using MAPPs on FVIII and dengue virus, respectively, showed that ~80% of the MHC-II identified peptides were HLA-DR-specific, while ~20% were DQ- and 5–10% DP-specific, indicating the major role of HLA-DR in initiating a CD4 T cell response in these models.Citation31,Citation32 With regards to the MAPPs assays’ sensitivity, the low expression of HLA-DP, and to a lesser degree HLA-DQ alleles have implications in the purification process and subsequently in the MS identification of peptides binding to HLA-DP and DQ receptors. The necessity and requirement to monitor MHC-II peptides binding to these molecules with respect to immunogenicity risk assessment remain to be demonstrated.
Presently, the descriptive nature of the MAPPs assay (the outcome is a list of presented MHC-II peptides that may contain potential T cell epitopes) has limited its use as an in vitro immunogenicity risk assessment tool. So far, most published studies have been conducted retrospectively on known immunogenic therapeutics. Also, it remains to be shown that the MHC-II peptide repertoire presented by moDC is the same as that of in vivo DCs. However, emerging data show that MHC-II peptides presented by moDCs from healthy donors are similar to these presented by moDCs isolated from patients; moreover, some of these peptides overlapped with reactive T cell epitopes identified in both healthy donors and patients.Citation32,Citation33
The rich nature of the data it generates added to increasing indications of its robustness are contributing to the growing popularity of the MAPPs platform to support immunogenicity profiling. For example, MAPPs are an optimal tool for the de-immunization of therapeutic peptides and proteins. Efforts to reduce the binding and presentation of peptides on APCs can easily be assessed by analyzing parent molecule and de-immunized variants hereof. There are, however, a number of important aspects of the assay to standardize. A minimum of 10 donors should be used for early sequence assessment, while a minimum of 15–30 genotyped donors ought to be probed to account for MHC-II diversity across the global population. Currently, there are no anti-HLA-DP or -DQ antibodies with appropriate performance available for the MAPPs assay. Therefore, the antibody used for MHC-II-peptide complex purification should be a standard antibody specific for HLA-DR, for example, clone L243, until satisfactory HLA-DR, DP, and DQ antibodies, which could be used in a mixture, or pan MHC-II antibodies may become available and allow a comprehensive analysis to be performed. The total number of identified mammalian endogenous peptides and proteins should be in the range of 5,000 and 1,000, respectively, as evidence for a process with good MHC-II peptide purification and high sensitivity MS detection. Lastly, clear information on identified target molecule peptide clusters and associated common sequence for each cluster (including the MHC-II binding core sequence) should be provided to enable harmonization between laboratories.Citation27 Standardization will also benefit from routine reporting of MAPPs datasets on qualified and easily accessible control protein(s), such as the well-studied infliximab and rituximab.
T cell functional analysis
In vitro T cell activation assays attempt to evaluate the immunogenic risk of therapeutic proteins and peptides by measuring the activation of CD4 T cells in response to drug-derived epitope(s) (). However, the presence of and/or requirement for many different immune players led to the development of several assay formats to probe various effector functions and outcomes (). In addition, the nature of the evaluated biologics and MoA, such as immune-modulatory vs metabolic nonimmune modulatory target, may also dictate which T cell activation assay must be used.Citation34–38 Each assay format presents discrete advantages and disadvantages with regards to sample preparation, ease of use, and interpretation of activation signal(s). For example, whole blood-based assays require less sample manipulation and ensure a rapid turnaround, but need to be conducted within 4–6 hours of draw since the immune cells function in blood samples cannot be preserved over long periods of time. Also, activation signals detected from such assays lack specificity since immune cells have not been previously sorted. Similarly, for whole PBMC-based assays, activation signals cannot be attributed to a particular subset of cells and usually cannot be used to assess immune modulatory drugs. PBMCs also lack the functional mature and professional APCs matching T cell subsets with different effector functions. Therefore, the use of peptides instead of intact proteins as challenge antigens can lead to over-representation of the HLA-binding peptides presented by the APCs.Citation39 Some approaches to address these limitations include depleting PBMCs of CD8 T cells to reduce interference and to assess more specifically the CD4 T effector response. DC-CD4 T cell or DC-PBMC co-culture-based assays may also prove advantageous to evaluate therapeutic proteins with immune modulatory MoA, in particular those containing T cell binding domains. Staggered cultures, wherein the intact biologic is only administered to DCs prior to co-culture, or co-culture with adjusted T cells to DCs ratios, may so avoid interference with the assay’s readout.Citation40 A further expansion of PBMC-based assays may lead to the generation of enriched T cell clones with antigen specificity. Such assays were originally intended to be high throughput, but require multiple rounds of antigen stimulation and cytokine challenge. Alternatively, individual peptides or peptide pools can also be used to elicit antigen-specific T cell clones while considering that not all peptides will be naturally presented by APCs.Citation41
Table 4. Overview of effector cell activation assays
T-cell assays are typically composed of four distinct components and sequential steps: 1) antigen challenge, 2) cell culture components and stimulus, 3) assay readout, and 4) data analysis; some essentials of these steps are discussed below and summarized in (). The amount of antigen needed for activation depends on the assay format, on the nature of the cells being challenged and of the target, on the time and frequency of such challenges, and whether the assay’s primary aim is to elicit a naïve vs. a memory response. Similarly, the setup of cell cultures and the required nutrients and growth factors may vary depending on whether the investigated cells have an effector or an antigen-presenting role. Accordingly, T cell activation assay outputs may range from an antigen-specific modulation of a cytokine, as reported by an enzyme-linked immune absorbent spot (ELISPOT) assay, to complex readouts, such as the profiling of secreted proteins using MS. It is therefore of importance to carefully design and execute such experiments following a formal analytical plan to enable a statistical interpretation of the data, since activation is usually based on an arbitrary activation threshold, as discussed below.
Table 5. Phases of T-cell assays and key factors for consideration
Data analysis
A positive T cell activation response to a protein therapeutic is determined by statistical analysis of the data based on an empirical threshold determined within the experiment. Several models are being used to carry out this analysis, such as an unpaired Student’s t-test to compare the means of two independent groups (usually the T cell response for a test compound against that of the medium blank and/or a known non-immunogenic control).Citation48,Citation52,Citation53 The Distribution Free Resampling (DFR) method and its derivative, the DFR(2x) method,Citation54 have also been used in this context, especially for the analysis of ELISPOT-based assays. The p value is usually set at a significance of 0.05 (p < .05) below which a T cell activation signal will be considered as true. Overall, an empirical T cell stimulation index (SI) >2 has been a widely accepted threshold to report a positive response based on data acquired from known immunogenic test compounds,Citation55 while some authors have used lower SI, such as SI>1.9,Citation53 to increase sensitivity of the assay. T cell activation response based on proliferation-based assays is interpolated from dose-response curves at concentrations of KLH inducing 50% of maximum proliferation (EC50). For these specific experiments, a response is considered positive when an antigen induces a (mean+2 standard deviation) proliferation rate greater than the response obtained in the absence of antigen or a non-immunogenic control.
Harmonization of T cell assays
The large diversity of existing T cell assays to accommodate different purposes is likely to prevent the adoption of a universal format, which raises the question whether results and data can be effectively compared across approaches. However, and possibly more importantly, there is also a growing need to establish commonly accepted quality controls (QC) and to define the minimal set of data to report from T cell assays.
The first harmonization we propose here is to set QC criterion for the PBMCs used in the assays. The QC should include post-thaw viability and recovery assessment when using frozen PMBCs. To confirm that T cells are functional and not pre-activated, the QC should include assessment of baseline or background responses as well as the cells capability of responding to polyclonal and neo- or recall antigens. Acceptable QC criteria for a PBMC preparation include a viability >85% and an SI for polyclonal activation ≥ 2. Baseline responses may differ between donors and sometimes even between samples taken at different time points from the same donor due to biological variability. Ideally, a baseline response should be recorded for each PBMC preparation to establish sample-specific cut-points. It is important to stress that calculations to set SI thresholds are highly dependent on assay types and on the statistical method used to evaluate the results, in particular, on how measurement replicates are taking into account to report significance; it is therefore desirable that reports on T cell assays provide a comprehensive summary of the experimental procedure and measurements and describe the statistical models used to report results for better inter-laboratory comparability.
A second harmonization we suggest is to define a minimal number of donors to include in a study. This number might depend on the stage of development of the drug candidate, on the type of T cell assay that is performed, but also on prior experience and data collected in past experiments. At early discovery stages, where assays are typically run in an investigational manner, we consider that studies including 10 to 25 donors representative for HLA allele frequencies of a target population may be appropriate to report on PBMC-based assays results. This number might be lower for a DC-T cell assay due to the time and labor required to obtain results. A pool of non-self-peptides may be designed to flag sequences potentially binding to prevalent MHC-II allele supertypes. Similarly, pools spanning multiple epitopes found in high-risk regions of a biologic amino acid sequence would enable identification of a potentially highly immunogenic molecule at an early stage of development. Still, such relatively small donor panels might not be sufficiently representative of a genetically diverse population. At a later stage, we recommend confirming early findings using a larger pool (50) of HLA-characterized donors to fit the HLA allele distribution of the intended target population.
A further harmonization we suggest is to add a panel of control molecules/drugs with well-characterized naïve as well as memory T cell responses to calibrate and monitor the assay performance over time. The frequency of antigen-specific T cell responses for control molecules should be sufficient at the cellular and population levels to be run with a standard average number of cells in tested individuals. One such universally accepted assay control is KLH because of its high neo-antigenic content, which elicits a positive response in >90% of the tested cells. However, assay consistency and performance require a careful examination of the origin and batch-to-batch quality of the KLH protein; it is therefore recommended to titrate every new batch of KLH and to avoid changes during a screening assay. Similar considerations apply for other essential reagents and materials to keep the experimental variability within one assay as low as possible.
Although T cell assays are less robust in their performance compared to DC activation and MAPPs assays, and their outcome depends on the setup and the MoA of the tested biologics, relative ranking is possible using control molecules with known immunogenic outcomes in each of the assay format. For example, bococizumab is known to elicit a positive response in 50–70% of the donors tested in a T cell assay, whereas it is lower for ATR-107 (40–60%) and minimal (<5%) for bevacizumab. Assays investigating peptides as challenge antigens should include relevant positive controls such as a CD3/CD28 cross-linker, which elicits a positive response in >90% of the donors and positive peptide controls such as PADRE for CD4 T cell peptide assaysCitation56 and a CEF/CEFA/CEFT pool for MHC Class I and Class II -restricted T cell epitope assays.Citation57,Citation58
To achieve full harmonization, we propose that any laboratory using such assays and willing to participate should establish jointly a common QC protocol for in vitro T cell assays immunogenicity risk assessment. A central lab would provide PBMCs from a central bank of donors and relevant controls to participating laboratories. These would run T cell assays using previously agreed parameters around assay setups, cell numbers and viability, time of incubation, and readouts. Raw data would be sent anonymously to designated biostatisticians who could provide feedback to all participating labs and identify sources of variability that affect consistency across assays and laboratories. Results may lead to the drafting of a guidance document stating general considerations for performing T cell assays, including QC acceptance criteria for cells used in each type of assay, number of donors to be included at different stages of drug development, the coverage of the MHC-II allele supertypes distribution in a target population, and a selection of relevant controls for innate vs adaptive immune response assays. Such an initiative could also set more standardized guidelines on statistical methods to be used for the data analysis from T cell assays, accounting for the nature of the data (for example, to take into consideration the high variability of results when working with primary cells and/or the low activation or proliferation signals due to the rare T cell receptor frequency), and for the calculation of robust activation thresholds.
B cell assays
Commonly used immunogenicity risk assessment tools focus mainly on the potential of a given biologic to activate CD4 T cells, due to the pivotal role they play in activating B cells and leading to their differentiation to ADA-producing plasma cells. Biologics also need to be directly recognized and internalized by specific populations of B cells for subsequent peptide presentation to T cells. This interaction is required for the activation of B cells, which leads to B cell differentiation into memory and plasma B cells, which are ultimately responsible for the production of high-affinity IgG (ADA) against the biologic. Hence, the B cell epitope content and the number of B cells producing IgG that are specific for a given biologic might serve as an important ranking criterion for immunogenicity risk. However, the B cell component of the immunogenicity risk assessment is currently greatly overlooked, due in part to technical limitations to assess the B cell response in vitro.
Predictive in silico methods have been developed, such as SEPIa, an algorithm combining 13 different sequence-derived features, including amino acid composition, hydrophilicity, solvent accessibility, and backbone flexibility. SEPIa was found to outperform both sequence-based and structure-based preexisting methods, but still exhibited limited performance compared to T cell epitope predictors.Citation59
To date, only one ELISpot/FluoroSpot-based assay has been reported to assess cellular B cell response to a biotherapeutic in vitro.Citation60 It is performed with PBMCs and reports the number of B cells secreting antibodies specific to the target antigen. The assay is initiated by treating B cells with a polyclonal activation cocktail (typically containing at least a Toll-like receptor agonist and IL-2), which differentiates memory B cells into plasma cells. These plasma cells secrete large amounts of antibody that can be captured on a polyvinylidene fluoride (PVDF) membrane. The antibody capture step can be carried out in two different ways: 1) The target antigen can be directly immobilized onto the PVDF membrane and is available to bind to the secreted antibodies, which can be detected and quantified using an anti-Fc antibody labeled for colorimetric or fluorescent detection; or 2) an anti-Fc antibody is used to capture all antibodies secreted by the B-cell derived plasma cells; detection and quantification is performed using the directly labeled antigen by colorimetric or fluorescent detection. The latter method provides the advantage that the total number of antibody-secreting cells can be determined alongside the number of antigen-specific secreting cells and enables the calculation of the frequency or % of antigen-secreting cells in the memory B cell population. The assay procedure can also be modified to detect various antibody isotypes (IgA, IgE, IgG1-4 and IgM) while the use of a FluoroSpot-based instrumentation enables the multiplexed detection of multiple antigens or different antibody isotypes (e.g., IgM and IgG) in the same well. In this modality, when run on healthy donor PBMCs, the assay will primarily detect memory B cells specific for an unrelated antigen that cross-react with the biologic of interest. In a recent study, Liao and colleagues demonstrated that the B cell response generated in vitro against a proprietary compound originated solely from memory B cells, with antibody-secreting cells detectable in treatment naïve subjects with preexisting ADA. When the assay was performed with peripheral blood from ADA+ subjects, ADA titers and the number of ADA-secreting B cells correlated, further demonstrating the specificity of the response detected by the assay.Citation61
Although currently limited to assessing the memory B cell response, the B cell ELISpot/FluoroSpot assay could become a tool to assess and compare the risk of preexisting cross-reactive antibodies present in drug-naïve healthy donors during the early stages of biotherapeutic development. In these settings, an optimal positive control would recall a B cell response in a high percentage of the donor panel and could take the form, for example, of an influenza-derived protein antigen mix representative of the main serotypes used in flu vaccines over the past decades. Harmonization of the assay would involve the use of a common protocol in terms of starting cells and readouts, and the use of a standard positive control for which a range of B cell memory response frequency would have been established on an extended panel of healthy donor PBMCs.
There is currently no broadly available in vitro strategy shown to induce a robust IgG-based response in naïve B cells. This is due to the complex processes involved in class switching, wherein B cells switch from low-affinity IgM production to high-affinity IgG antibodies associated with ADA responses in the clinic. Potential in vitro strategies to assess the naïve B cell repertoire rather than the cross-reactive memory repertoire have been evaluated and include measurement of B cell activation and proliferation in response to the biologic and B cell receptor-specific binding and internalization. However, these assays are yet to be optimized and validated.
In vivo models
The complexity and the challenges to capture relevant parts of the immune system through in silico and in vitro systems have led researchers to investigate in vivo models that consider the entire immune system, as it is arguably better suited to trigger ADA generation when tolerance is breached. Standard animal models are not appropriate because any human therapeutic protein might be recognized by the animal as foreign. Therefore, numerous non-clinical animal models have been generated in an effort to recapitulate the entire ADA response. Here, we review both transgenic models expressing human transgenes such as HLA and immunoglobulins,Citation62 and models in which the human immune system has been incorporated into a mouse to diverse extents, with varying levels of success in generating the expected response to a known antigen.
Human HLA transgenic mice express a human MHC transgene and consequently can have a T cell repertoire that reflects more closely the human counterpart. These models are largely used to study autoimmune conditions with known risk alleles,Citation63 but they show limited value for immunogenicity prediction since they do not reflect the diversity of MHC alleles in the human population and the TCR repertoire remains entirely murine. Transgenic models expressing large fractions of the human germline V gene repertoire of the immunoglobulin family are more widely used to study immunogenicity because their expression provides immune tolerance to a broad range of human antibodies.Citation64 Mice made tolerant to certain subclasses of human immunoglobulin, such as the Xeno-het mouse, have been used to evaluate the IgG2 mAbs attribute-related immunogenicity risk. Aggregates made by mechanical stirring were able to induce an ADA response, although weak and transient.Citation65 Despite expression of a large repertoire of human IgG antibodies, the membrane form of human antibodies lead to allelic exclusion of murine antibody repertoire, thus forcing a laborious crossing of the transgenic line with knock-out Ig alleles and one wild-type allele. In contrast, other groups have generated transgenic mice bearing only the five most used human VH and VL (kappa and lambda) genes. These mice rearrange the human VDJ genes and, importantly, bear an intact murine repertoire as the transgenic Cγ1 constant region encodes only the secretory form of human antibodies.Citation66 These transgenic mice have been shown to tolerate antibody aggregatesCitation66 and sub-visible particlesCitation67 devoid of new T cell epitopes, but they mount an immune response against antibody preparations that contained chemical modifications of primary amino acid residues.
As the aforementioned transgenic animals bear T and B cell repertoires as well as murine MHC alleles, they cannot be used for the identification of potential immunogenic human T cell epitopes; only mice carrying a human MHC II allele may be used for the determination of an immunodominant CD4 T cell epitopeCitation68,Citation69 (see also below). However, protein sequence is only one factor contributing to ADA formation. Other product-related factors such as MoA and compound format can also influence the immunogenicity of biologics. Thus, infliximab, a chimeric anti-TNF antibody, elicits similar or even lower ADA rates in human than adalimumab, a fully human anti-TNF.Citation70 Also, work performed by Reipert’s group has shown that the route of administration and degree of PEGylation influenced the immune response against Factor VIII in immune-tolerant mice.Citation68,Citation69 Thus, these transgenic mouse models are useful tools to study product-related factors causing a break of immune tolerance against human antibodies and other proteins.
Currently, the human immune response is best modeled in humanized mice (i.e., Human Immune System “HIS”), created by myeloablation of immunodeficient mice and reconstitution with human hematopoietic stem cells obtained either from fetal liver or (more commonly) from umbilical cord blood. Three models have shown potential in this respect: 1) the bone marrow, liver, thymus (BLT) mouse, 2) the Balb/c Rag2−/−IL2Rγ−/−SirpaNOD TSLPTg (BRGSF/T) mouse, and 3) the NOD.SCID.IL2Rγ−/− (NSG) mouse.Citation71,Citation72 Since their introduction, these models have consistently achieved high rates of reconstitution, averaging 60–90% HIS (as calculated by the % of hCD45+/ (total mCD45+ + hCD45+). These humanized mice have historically been tested in the context of infectious disease; in recent years, however, providers of HIS mice have focused their efforts on generating models for the burgeoning immuno-oncology field. provides an overview of the three different HIS models and their potential for immunogenicity testing.
Table 6. Overview of transgenic animal models
To date, the use of humanized mice has not entered the pre-clinical immunogenicity risk assessment toolkit as a standard go-to platform when evaluating biotherapeutics. Commitment to such an approach is non-trivial due to costs and time to reach an appropriate decision point, and therefore the use of such in vivo models will be focused on the study/assessment of biotherapeutics deemed to have high immunogenicity risk. In the absence of other pre-clinical models replicating all steps required for ADA formation (i.e., lymph node on a chip), transgenic immune tolerant and humanized animals represent the only available tool to understand better the likelihood of biologics to trigger ADA formation in the clinic. Future studies using next-generation sequencing to interrogate deeply the T and B cell repertoires combined with single cell RNA sequencing in in vivo models, as well as the systematic study of ADA-positive patients, will be critical to allow researchers to identify immunogenicity biomarkers and provide a better understanding of translatability of individual models.
Immunogenicity modeling
Mechanistic modeling is used across drug development to support our understanding of complex systems and to provide simulations in predicting outcomes in experiments and in clinical trials. The integration of data from assays that investigate key mechanisms of the immune response in mathematical models is expected to result in better prediction of immunogenicity outcomes in clinical trials.Citation87 Data that can be used for such modeling may be obtained from DC activation, protein uptake by DCs, T cell epitopes (in silico predicted and/or identified using MAPPs), and T cell proliferation assays. Model sensitivity analysis identifies inputs on DC activation, number of naïve T cells, and the affinity of HLA-peptide interactions as being most influential for ADA incidence simulations, indicating the high value of generating these data. Additional variables that are currently investigated (but which will require additional assays to refine modeling outputs) include B cell activation, B cell epitopes determination, B cell proliferation, and rate of ADA production. Also, extrinsic variables derived from ADA determination and PK/PD assays or from patient-centric parameters, such as individualized T and B cell epitopes and HLA genotyping, may also be included in modeling.
At present, immune response modeling has been shown to predict appropriately the clinical outcome of drugs such as adalimumab.Citation88 Simulations were able to describe the overall ADA generation in a population, its impact on pharmacokinetics, and the likelihood that patients will lose efficacy due to reduced exposure. Additionally, sub-models can be used to interrogate the assays and specific biological components in order to improve our understanding of how an individual assayCitation89 or individual risk factorsCitation18 contribute to immunogenicity prediction. The model contains multiple components that are targets of, or would-be biomarkers for, immunomodulatory therapeutics. Therefore, the immunomodulatory pharmacology of a therapeutic can be modeled directly or indirectly, for example by reducing the number of B cells for B cell depleting therapies or enhancing T cell activation for cancer immunotherapies. Future developments for immunogenicity modeling will include the ability to distinguish the effects of neutralizing ADA from clearing ADA, which could be achieved using modeling concepts already in use. For example, ADAs can be modeled as if there were bona fide compounds, with clearing and neutralizing functions with respect to the investigative drug as the target. A longer-term aim would be to include mechanisms of hypersensitivity to investigate whether safety endpoints (in particular cytokine release syndromes) could be simulated.
Conclusions and future developments
The aim of pre-clinical immunogenicity risk assessment is to estimate the likelihood of a biotherapeutic to induce an ADA response in treated patients. In this review, we gathered pre-clinical methods, platforms and strategies that we believe are currently used to interrogate the potential immunogenic liabilities of protein drug candidates. While the data generated by these assays and platform is becoming increasingly informative and valuable, the interpretation and translation into an overall immunogenicity risk of a given therapeutic protein remains challenging due to lack of assay standardization and harmonization. As of today, in vitro-based pre-clinical risk assessments do not (yet) enable accurate prediction of clinical immunogenicity. This is in part because most methods and assays focus on sequence-related risk factors, leaving out additional risk factors, such as MoA of the drug, route of administration, dosing regimen, and patient-related risk factors (e.g., a possible interaction with concomitant medication). The addition of approaches such as in silico modeling and in vivo models, which recapitulate most of the multiple factors pertaining to immunogenicity development, might increase the predictive power of pre-clinical risk assessments. However, validation of such outcomes through post-hoc analyses is challenging due to the use of disparate, non-standardized bioanalytical methods to report ADA occurrence.Citation90 While incidence is typically reported, kinetics, magnitude of response, the presence of neutralizing species and cross-reactivity to endogenous analyte or other therapeutic drug(s) should also be stated to provide a more complete picture of the immunogenic response. Recent regulatory guidance for therapeutic proteins development (FDA 2019 https://www.fda.gov/media/119788/download; EMA 2017 https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therap eutic-proteins-revision-1_en.pdf) and peptides (FDA 2017 https://www.fda.gov/files/drugs/published/ANDAs-for-Cert ain-Highly-Purified-Synthetic-Peptide-Drug-Products-That-Refer-to-Listed-Drugs-of-rDNA-Origin-Guidance-for-Ind ustry.pdf) recommend that intrinsic and extrinsic risks identified during pre-clinical and clinical development should guide the development of a robust bioanalytical and clinical strategy. An example of roadmaps for conducting and summarizing such risk-based strategies has been recently published for fusion proteins, pegylated proteins and multi-domain therapeutics.Citation91
Selecting the most appropriate pre-clinical immunogenicity risk assessment tool(s) among the many available might be a delicate exercise, as no universal strategy currently exists (see () for an overview of the assays discussed in this review). It may help, however, to consider first the following points to select an initial approach: 1) in which development stage is the drug candidate? and 2) what is the pharmacology of the drug candidate?
Table 7. Assay overview
In silico approaches are the most high-throughput and cost-effective way of filtering out candidates with high sequence-associated risks, and can provide results even before the availability of the biologics of interest. They are therefore most successful during discovery and early drug design stages. Cell-based assays are amenable to relatively large sample sets (several tens of donors). However, they should be conducted with control molecules to allow suitable benchmarking and sample ranking. In cases where biotherapeutics have an immunomodulatory component, assay formats should be selected with caution so as not to generate false positive or negative results. T cell activation assays (using isolated DCs or PBMCs) in the presence of whole antigens are usually the most biologically relevant approach to assess overall immunogenicity risk. Depending on the drug development stage, the test molecule will also have different characteristics (e.g., expression cell line, formulation buffer, contaminants, post-translational modifications) and may not fully recapitulate the final drug product. Finally, due to costs and sample throughput, in vivo models may only be used in the selected cases where the recapitulation of the entire immune system is required for assessing immunogenicity risks in a comprehensive manner.
Further, selection of an assay (or a set of assays) should be guided by the nature of the data that will be generated by the study and its intended use in the evaluation of a potential immunogenicity risk, for example: 1) To inform a clinical safety management plan, e.g., if data suggests a moderate immunogenicity risk, then dosing and ADA monitoring strategy may be adjusted; 2) To compare or rank against similar product candidates or biosimilar molecules to select the best candidate to take forward for clinical development; and 3) To identify sequence liabilities and remove them using protein engineering techniques.
Different assays will provide different types of data that may be better suited to one objective compared to another. For example, the growing utilization of peptide proteomics (MHC-II MAPPs assays) has generated a wealth of data enabling the interrogation of peptides that are physically presented in the context of MHC class II molecules. While these are not necessarily T cell epitopes, it is a highly sensitive and robust method to investigate potential liabilities within a protein sequence or to compare antigen presentation profiles between very similar biologics, as exemplified in the investigation of therapeutic Factor VIII proteins for the treatment of hemophilia A.Citation32,Citation92 Therefore, it is reasonable to expect the MAPPs platform could also be used to profile biosimilars or to assess the effect of formulation on the immunogenic profile of a lead biotherapeutic.
Reported ADA data from clinical studies represent an important source of information in understanding the potential for pre-clinical assessment to predict immunogenicity risk of biologics. Reverse translational analysis of clinical data enables a more detailed analysis of a biotherapeutic’s features informing whether an ADA response might be expected and how it might occur. For example, a number of studies have validated the use of MAPPs assays as a method to identify native T cell epitopes correlating with a positive CD4 T cell response in ADA positive patients.Citation33 A systematic comparative analysis may lead to the identification of reactive epitopes that can be subsequently modified to dampen the initial T cell induction, therefore reducing the overall immunogenicity risk.Citation32 However, despite the many putative T cell epitopes that can be characterized in a MAPPs assay, recognition by T cells remains highly patient-dependent and may hinder the identification of the one dominant epitope inducing ADA production.Citation93 Efforts are ongoing to streamline the analytical process and to identify parameters predicting high-risk sequences with better confidence.Citation94,Citation95 In another example, Walsh et al. reported a post-hoc analysis of three clinically immunogenic antibodies that differ by the mechanism of T cell activation, and demonstrated that a biotherapeutics’ intrinsic properties influence immunogenicity risk.Citation96 Interestingly, the authors mention that a T cell proliferation assay performed with PBMCs devoid of CD8 T cells provided the most predictive immunogenicity risk assessment if the results were correlated to the data collected in the Phase 1 clinical trial, although the assay data in isolation was not sufficient to explain all the immunogenic events.
The inherent complexity of some of the assays described in this review makes it challenging to harmonize and standardize; in , we attempt to summarize, for all in vitro assays, what we believe to be a minimal set of conditions and controls to better understand the nature of the generated data and to facilitate its replication if needed. For example, T cell activation assays have been developed and used by many different laboratories for over 20 years without common directives on how to perform such assays and report data. In light of an increasing reliance on these assays, we believe that standardization and definition of common standardized platforms that enable cross-validation and data comparability would increase our confidence in conducting pre-clinical risk assessments. Concomitantly, minimal quality requirements for cells, samples, and standards need to be established. The ability of T cells to be activated and DC to mature in the presence of known stimuli should be monitored systematically for every donor where practical, and when changes are applied to the process (e.g., when using a new source of blood cells). Universal controls and benchmarks from centralized sources should ideally be used by all laboratories, with stringent acceptance criteria for purity. Controls and benchmarks should also be routinely batch-tested, bridging studies conducted, and trend analysis performed to ensure consistent assay performance. Most laboratories use bevacizumab as a low immunogenicity benchmark in T cell-based assays, which has the advantage of being commercially available in a Good Manufacturing Practices (GMP)-quality grade. In contrast, a collaborative effort is most likely needed to identify a suitable universal ‘high immunogenicity’ benchmark for T cell activation assays. With the support of drug developers, a set of biologics with no direct immunomodulatory activity but known clinical immunogenicity, either marketed, under development, or withdrawn from development could be identified, manufactured to agreed specifications, and tested in multiple assay formats at various locations. This may lead to the election of an approved benchmark, which can then be manufactured to GMP grade and made commercially available to all for general use.
Table 8. Recommendations for harmonization or standardization of human in vitro cellular assaysa
In summary, we foresee that significant improvement and refinement of pre-clinical immunogenicity assessment tools will come through common agreements across drug makers on assay QC acceptance criteria (despite possible hurdles such as confidentiality, legal, and commercial considerations), as well as from learnings from reverse translation of data collected on compounds found to be immunogenic in the clinic.
Abbreviations
Acknowledgments
We would like to thank Sofie Pattijn for critically reading the manuscript.
Disclosure statement
Tim Hickling, Juliana Bessa and Axel Ducret are full-time employeesof Roche Pharmaceutical Research and Early Development.
Chloé Ackaert is part-time employee of ImmunXperts SA, a Nexelis company.
Campbell Bunce is full time employee of and shareholder in Abzena.
Vibha Jawa is full time employee of and shareholder in BMS.
Hweixian L Penny and Mark Kroenke are a full time employees of and shareholders in Amgen.
Kasper Lamberth is full time employeeof and shareholder in Novo Nordisk A/S.
Noel Smith is full time employee of LONZA BIOLOGICS.
Grzegorz Terszowski is full time employee of Novartis Pharma AG.
Sophie Tourdot is full time employee of and shareholder in Pfizer, Inc.
Sebastian Spindeldreher is full time employee of Integrated Biologix GmbH.
Additional information
Funding
References
- Goral S. The three-signal hypothesis of lymphocyte activation/targets for immunosuppression. Dial Transplant. 2011;40(1):14–17. doi:10.1002/dat.20527.
- Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM. T cell responses: naive to memory and everything in between. Adv Physiol Educ. 2013;37(4):273–83. doi:10.1152/advan.00066.2013.
- Shipkova M, Wieland E. Surface markers of lymphocyte activation and markers of cell proliferation. Clin Chim Acta. 2012;413(17–18):1338–49. doi:10.1016/j.cca.2011.11.006.
- Geiger R, Duhen T, Lanzavecchia A, Sallusto F. Human naive and memory CD4+ T cell repertoires specific for naturally processed antigens analyzed using libraries of amplified T cells. J Exp Med. 2009;206(7):1525–34. doi:10.1084/jem.20090504.
- Kwok WW, Tan V, Gillette L, Littell CT, Soltis Ma, LaFond RB, Yang J, James EA, DeLong JH. Frequency of epitope-specific naive CD4(+) T cells correlates with immunodominance in the human memory repertoire. J Immunol (Baltimore, Md: 1950). 2012;188(6):2537–44.doi:10.4049/jimmunol.1102190.
- Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22(1):745–63. doi:10.1146/annurev.immunol.22.012703.104702.
- Fernandez L, Bustos RH, Zapata C, Garcia J, Jauregui E, Ashraf GM. Immunogenicity in protein and peptide based-therapeutics: an overview. Curr Protein Pept Sci. 2018;19(10):958–71. doi:10.2174/1389203718666170828123449.
- Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SG. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 2015;43(D1):D423–31. doi:10.1093/nar/gku1161.
- De Groot A, Cousens L, Mingozzi F, Martin W. Tregitope peptides: the active pharmaceutical ingredient of IVIG?. Clin Dev Immunol. 2013;2013:493138. doi:10.1155/2013/493138.
- Bresciani A, Paul S, Schommer N, Dillon MB, Bancroft T, Greenbaum J, Sette A, Nielsen M, Peters B. T-cell recognition is shaped by epitope sequence conservation in the host proteome and microbiome. Immunology. 2016;148(1):34–39. doi:10.1111/imm.12585.
- Racle J, Michaux J, Rockinger GA, Arnaud M, Bobisse S, Chong C, Guillaume P, Coukos G, Harari A, Jandus C, et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat Biotechnol. 2019;37(11):1283–86. doi:10.1038/s41587-019-0289-6.
- Chen B, Khodadoust MS, Olsson N, Wagar LE, Fast E, Liu CL, Muftuoglu Y, Sworder BJ, Diehn M, Levy R, et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat Biotechnol. 2019;37(11):1332–43. doi:10.1038/s41587-019-0280-2.
- Abelin JG, Harjanto D, Malloy M, Suri P, Colson T, Goulding SP, Creech AL, Serrano LR, Nasir G, Nasrullah Y, et al. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity. 2019;51(4):766–779.e17. doi:10.1016/j.immuni.2019.08.012.
- Barra C, Ackaert C, Reynisson B, Schockaert J, Jessen LE, Watson M, Jang A, Comtois-Marotte S, Goulet J-P, Pattijn S, et al. Immunopeptidomic data integration to artificial neural networks enhances protein-drug immunogenicity prediction. Front Immunol. 2020;11:1304. doi:10.3389/fimmu.2020.01304.
- Kuhn C, Weiner HL. Therapeutic anti-CD3 monoclonal antibodies: from bench to bedside. Immunotherapy. 2016;8(8):889–906. doi:10.2217/imt-2016-0049.
- Hogwood CE, Bracewell DG, Smales CM. Measurement and control of host cell proteins (HCPs) in CHO cell bioprocesses. Curr Opin Biotechnol. 2014;30:153–60. doi:10.1016/j.copbio.2014.06.017.
- Kuriakose A, Chirmule N, Nair P. Immunogenicity of biotherapeutics: causes and association with posttranslational modifications. J Immunol Res. 2016;2016:1298473. doi:10.1155/2016/1298473.
- Yin L, Chen X, Vicini P, Rup B, Hickling TP. Therapeutic outcomes, assessments, risk factors and mitigation efforts of immunogenicity of therapeutic protein products. Cell Immunol. 2015;295(2):118–26. doi:10.1016/j.cellimm.2015.03.002.
- Morgan H, Tseng SY, Gallais Y, Leineweber M, Buchmann P, Riccardi S, Nabhan M, Lo J, Gani Z, Szely N, et al. Evaluation of in vitro assays to assess the modulation of dendritic cells functions by therapeutic antibodies and aggregates. Front Immunol. 2019;10:1–14. doi:10.3389/fimmu.2019.00601.
- Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179(4):1109–18. doi:10.1084/jem.179.4.1109.
- Turbica I, Gallais Y, Gueguen C, Tharinger H, Al Sabbagh C, Gorges R, Gary-Gouy H, Kerdine-ro¨mer S, Pallardy M, Mascarell L, et al. Ectosomes from neutrophil-like cells down-regulate nickel-induced dendritic cell maturation and promote Th2 polarization. J Leukoc Biol. 2015;97(4):737–49. doi:10.1189/jlb.3A0314-132RR.
- Xue L, Hickling T, Song R, Nowak J, Rup B. Contribution of enhanced engagement of antigen presentation machinery to the clinical immunogenicity of a human interleukin (IL)-21 receptor-blocking therapeutic antibody. Clin Exp Immunol. 2016;183(1):102–13. doi:10.1111/cei.12711.
- Spindeldreher S, Karle A, Correia E, Tenon M, Gottlieb S, Huber T, Maillere B, Kolbinger F. T cell epitope mapping of secukinumab and ixekizumab in healthy donors. mAbs. 2020;12(1):1707418. doi:10.1080/19420862.2019.1707418.
- Kropshofer H, Vogt AB, Hämmerling GJ. Structural features of the invariant chain fragment CLIP controlling rapid release from HLA-DR molecules and inhibition of peptide binding. Proc Natl Acad Sci U S A. 1995;92(18):8313–17. doi:10.1073/pnas.92.18.8313.
- Sekiguchi N, Kubo C, Takahashi A, Muraoka K, Takeiri A, Ito S, Yano M, Mimoto F, Maeda A, Iwayanagi Y, et al. MHC-associated peptide proteomics enabling highly sensitive detection of immunogenic sequences for the development of therapeutic antibodies with low immunogenicity. mAbs. 2018;10(8):1168–81. doi:10.1080/19420862.2018.1518888.
- Karle AC. Applying MAPPs assays to assess drug immunogenicity. Front Immunol. 2020;11:1–11. doi:10.3389/fimmu.2020.00698.
- Steiner G, Marban-Doran C, Langer J, Pimenova T, Duran-Pacheco G, Sauter D, Langenkamp A, Solier C, Singer T, Bray-French K, et al. Enabling routine MHC-II-associated peptide proteomics for risk assessment of drug-induced immunogenicity. J Proteome Res. 2020;19(9):3792–806. doi:10.1021/acs.jproteome.0c00309.
- Lamberth K, Reedtz-Runge SL, Simon J, Klementyeva K, Pandey GS, Padkjær SB, Pascal V, León IR, Gudme CN, Buus S, et al. Post hoc assessment of the immunogenicity of bioengineered factor VIIa demonstrates the use of preclinical tools. Sci Transl Med. 2017;9(372):1–12. doi:10.1126/scitranslmed.aag1286.
- Sidney J, Steen A, Moore C, Ngo S, Chung J, Peters B, Sette A. Five HLA-DP molecules frequently expressed in the worldwide human population share a common HLA supertypic binding specificity. J Immunol (Baltimore, Md: 1950). 2010;184(5):2492–503. doi:10.4049/jimmunol.0903655.
- Hrdinová J, Verbij FC, Kaijen PHP, Hartholt RB, Van Alphen F, Lardy N, Ten Brinke A, Vanhoorelbeke K, Hindocha PJ, De Groot AS, et al. Mass spectrometry-assisted identification of ADAMTS13-derived peptides presented on HLA-DR and HLA-DQ. Haematologica. 2018;103(6):1083–92. doi:10.3324/haematol.2017.179119.
- Grifoni A, Moore E, Voic H, Sidney J, Phillips E, Jadi R, Mallal S, De Silva AD, De Silva AM, Peters B, et al. Characterization of magnitude and antigen specificity of HLA-DP, DQ, and DRB3/4/5 restricted DENV-specific CD4+ T cell responses. Front Immunol. 2019;10:1568. doi:10.3389/fimmu.2019.01568.
- Jankowski W, Park Y, McGill J, Maraskovsky E, Hofmann M, Diego VP, Luu BW, Howard TE, Kellerman R, Key NS, et al. Peptides identified on monocyte-derived dendritic cells: a marker for clinical immunogenicity to FVIII products. Blood Adv. 2019;3(9):1429–40. doi:10.1182/bloodadvances.2018030452.
- Hamze M, Meunier S, Karle A, Gdoura A, Goudet A, Szely N, Pallardy M, Carbonnel F, Spindeldreher S, Mariette X, et al. Characterization of CD4 T cell epitopes of infliximab and rituximab identified from healthy donors. Front Immunol. 2017;8:500. doi:10.3389/fimmu.2017.00500.
- Deenadayalan A, Maddineni P, Raja A. Comparison of whole blood and PBMC assays for T-cell functional analysis. BMC Res Notes. 2013;6(1):120. doi:10.1186/1756-0500-6-120.
- Jawa V, Joubert MK, Zhang Q, Deshpande M, Hapuarachchi S, Hall MP, Flynn GC. Evaluating immunogenicity risk due to host cell protein impurities in antibody-based biotherapeutics. AAPS J. 2016;18(6):1439–52. doi:10.1208/s12248-016-9948-4.
- Joubert MK, Deshpande M, Yang J, Reynolds H, Bryson C, Fogg M, Baker MP, Herskovitz J, Goletz TJ, Zhou L, et al. Use of in vitro assays to assess immunogenicity risk of antibody-based biotherapeutics. PLoS ONE. 2016;11(8):1–22. doi:10.1371/journal.pone.0159328.
- Seyfert-Margolis V, Gisler TD, Asare AL, Wang RS, Dosch HM, Brooks-Worrell B, Eisenbarth GS, Palmer JP, Greenbaum CJ, Gitelman SE, et al. Analysis of T-cell assays to measure autoimmune responses in subjects with type 1 diabetes: results of a blinded controlled study. Diabetes. 2006;55(9):2588–94. doi:10.2337/db05-1378.
- Wullner D, Zhou L, Bramhall E, Kuck A, Goletz TJ, Swanson S, Chirmule N, Jawa V. Considerations for optimization and validation of an in vitro PBMC derived T cell assay for immunogenicity prediction of biotherapeutics. Clin Immunol. 2010;137(1):5–14. doi:10.1016/j.clim.2010.06.018.
- Meunier S, Menier C, Marcon E, Lacroix-Desmazes S, Maillère B. CD4 T cells specific for factor VIII are present at high frequency in healthy donors and comprise naïve and memory cells. Blood Adv. 2017;1(21):1842–47. doi:10.1182/bloodadvances.2017008706.
- Moser JM, Sassano ER, Leistritz DC, Eatrides JM, Phogat S, Koff W, Drake DR. Optimization of a dendritic cell-based assay for the in vitro priming of naïve human CD4+ T cells. J Immunol Methods. 2010;353(1–2):8–19. doi:10.1016/j.jim.2009.11.006.
- Jahn-Schmid B, Radakovics A, Luttkopf D, Scheurer S, Vieths S, Ebner C, Bohle B. Bet v 1142-156 is the dominant T-cell epitope of the major birch pollen allergen and important for cross-reactivity with Bet v 1-related food allergens. J Allergy Clin Immunol. 2005;116(1):213–19. doi:10.1016/j.jaci.2005.04.019.
- Henrickson SE, Mempel TR, Mazo IB, Liu B, Artyomov MN, Zheng H, Peixoto A, Flynn MP, Senman B, Junt T, et al. T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for T cell activation. Nat Immunol. 2008;9(3):282–91. doi:10.1038/ni1559.
- Rothoeft T, Gonschorek A, Bartz H, Anhenn O, Schauer U. Antigen dose, type of antigen-presenting cell and time of differentiation contribute to the T helper 1/T helper 2 polarization of naive T cells. Immunology. 2003;110(4):430–39. doi:10.1111/j.1365-2567.2003.01758.x.
- Wölfl M, Greenberg PD. Antigen-specific activation and cytokine-facilitated expansion of naive, human CD8+ T cells. Nat Protoc. 2014;9(4):950–66. doi:10.1038/nprot.2014.064.
- Castelli FA, Leleu M, Pouvelle-Moratille S, Farci S, Zarour HM, Andrieu M, Auriault C, Ménez A, Georges B, Maillere B, et al. Differential capacity of T cell priming in naive donors of promiscuous CD4+ T cell epitopes of HCV NS3 and Core proteins. Eur J Immunol. 2007;37(6):1513–23. doi:10.1002/eji.200636783.
- Delluc S, Ravot G, Maillere B. Quantitative analysis of the CD4 T-cell repertoire specific to therapeutic antibodies in healthy donors. FASEB J off Publ Fed Am Soc Exp Biol. 2011;25(6):2040–48. doi:10.1096/fj.10-173872.
- Von Baehr V, Mayer W, Liebenthal C, Von Baehr R, Bieger W, Volk HD. Improving the in vitro antigen specific T cell proliferation assay: the use of interferon-alpha to elicit antigen specific stimulation and decrease bystander proliferation. J Immunol Methods. 2001;251(1–2):63–71. doi:10.1016/S0022-1759(01)00297-6.
- Schultz HS, Reedtz-Runge SL, Bäckström BT, Lamberth K, Pedersen CR, Kvarnhammar AM, Akatsuka Y. Quantitative analysis of the CD4+ T cell response to therapeutic antibodies in healthy donors using a novel T cell: PBMC assay. PloS One. 2017;12(5):e0178544. doi:10.1371/journal.pone.0178544.
- Navarrete MA, Bertinetti-Lapatki C, Michelfelder I, Veelken H. Usage of standardized antigen-presenting cells improves ELISpot performance for complex protein antigens. J Immunol Methods. 2013;391(1–2):146–53. doi:10.1016/j.jim.2013.03.004.
- Möller JF, Möller B, Wiedenmann B, Berg T, Schott E. CD154, a marker of antigen-specific stimulation of CD4 T cells, is associated with response to treatment in patients with chronic HCV infection. J Viral Hepat. 2011;18(7):e341–9. doi:10.1111/j.1365-2893.2010.01430.x.
- Kutscher S, Dembek CJ, Deckert S, Russo C, Körber N, Bogner JR, Geisler F, Umgelter A, Neuenhahn M, Albrecht J, et al. Overnight resting of PBMC changes functional signatures of antigen specific T- cell responses: impact for immune monitoring within clinical trials. PloS One. 2013;8(10):e76215. doi:10.1371/journal.pone.0076215.
- Joubert MK, Hokom M, Eakin C, Zhou L, Deshpande M, Baker MP, Goletz TJ, Kerwin BA, Chirmule N, Narhi LO, et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J Biol Chem. 2012;287(30):25266–79. doi:10.1074/jbc.M111.330902.
- Karle A, Spindeldreher S, Kolbinger F. Secukinumab, a novel anti–IL-17A antibody, shows low immunogenicity potential in human in vitro assays comparable to other marketed biotherapeutics with low clinical immunogenicity. mAbs. 2016;8(3):536–50. doi:10.1080/19420862.2015.1136761.
- Moodie Z, Price L, Gouttefangeas C, Mander A, Janetzki S, Löwer M, Welters MJP, Ottensmeier C, Van Der Burg SH, Britten CM, et al. Response definition criteria for ELISPOT assays revisited. Cancer Immunol Immunother CII. 2010;59(10):1489–501. doi:10.1007/s00262-010-0875-4.
- Ten BA, Marek-Trzonkowska N, Mansilla MJ, Turksma AW, Piekarska K, Iwaszkiewicz-Grzes D, Passerini L, Locafaro G, Puñet-Ortiz J, Van Ham SM, et al. Monitoring T-Cell responses in translational studies: optimization of dye-based proliferation assay for evaluation of antigen-specific responses. Front Immunol. 2017;8:1870. doi:10.3389/fimmu.2017.01870.
- Alexander J, Sidney J, Southwood S, Ruppert J, Oseroff C, Maewal A, Snoke K, Serra HM, Kubo RT, Sette A, et al. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity. 1994;1(9):751–61. doi:10.1016/S1074-7613(94)80017-0.
- Currier JR, Kuta EG, Turk E, Earhart LB, Loomis-Price L, Janetzki S, Ferrari G, Birx DL, Cox JH. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J Immunol Methods. 2002;260(1–2):157–72. doi:10.1016/S0022-1759(01)00535-X.
- Grenga I, Donahue RN, Lepone LM, Richards J, Schlom J. A fully human IgG1 anti-PD-L1 MAb in an in vitro assay enhances antigen-specific T-cell responses. Clin Transl Immunol. 2016;5(5):e83. doi:10.1038/cti.2016.27.
- Dalkas GA, Rooman M. SEPIa, a knowledge-driven algorithm for predicting conformational B-cell epitopes from the amino acid sequence. BMC Bioinform. 2017;18(1):95. doi:10.1186/s12859-017-1528-9.
- Hwai H, Chen YY, Tzeng SJ. B-cell ELISpot assay to quantify antigen-specific antibody-secreting cells in human peripheral blood mononuclear cells. Methods Mol Biol. 2018;1808:133–41.
- Liao K, Derbyshire S, Wang K-F, Caucci C, Tang S, Holland C, Loercher A, Gunn GR. Detection of memory B activity against a therapeutic protein in treatment-naïve subjects. AAPS J. 2018;20(3):51. doi:10.1208/s12248-018-0198-5.
- Jiskoot W, Kijanka G, Randolph TW, Carpenter JF, Koulov AV, Mahler H-C, Joubert MK, Jawa V, Narhi LO. Mouse models for assessing protein immunogenicity: lessons and challenges. J Pharm Sci. 2016;105(5):1567–75. doi:10.1016/j.xphs.2016.02.031.
- Mangalam AK, Rajagopalan G, Taneja V, David CS. HLA class II transgenic mice mimic human inflammatory diseases. Adv Immunol. 2008;97:65–147.
- Chen WC, Murawsky CM. Strategies for generating diverse antibody repertoires using transgenic animals expressing human antibodies. Front Immunol. 2018;9:460.doi: 10.3389/fimmu.2018.00460.
- Bi V, Jawa V, Joubert MK, Kaliyaperumal A, Eakin C, Richmond K, Pan O, Sun J, Hokom M, Goletz TJ, et al. Development of a human antibody tolerant mouse model to assess the immunogenicity risk due to aggregated biotherapeutics. J Pharm Sci. 2013;102(10):3545–55. doi:10.1002/jps.23663.
- Bessa J, Boeckle S, Beck H, Buckel T, Schlicht S, Ebeling M, Kiialainen A, Koulov A, Boll B, Weiser T, et al. The immunogenicity of antibody aggregates in a novel transgenic mouse model. Pharm Res. 2015;32(7):2344–59. doi:10.1007/s11095-015-1627-0.
- Boll B, Bessa J, Folzer E, Ríos Quiroz A, Schmidt R, Bulau P, Finkler C, Mahler H-C, Huwyler J, Iglesias A, et al. Extensive chemical modifications in the primary protein structure of IgG1 subvisible particles are necessary for breaking immune tolerance. Mol Pharm. 2017;14(4):1292–99. doi:10.1021/acs.molpharmaceut.6b00816.
- Steinitz KN, Van Helden PM, Binder B, Wraith DC, Unterthurner S, Hermann C, Schuster M, Ahmad RU, Weiller M, Lubich C, et al. CD4+ T-cell epitopes associated with antibody responses after intravenously and subcutaneously applied human FVIII in humanized hemophilic E17 HLA-DRB1*1501 mice. Blood. 2012;119(17):4073–82. doi:10.1182/blood-2011-08-374645.
- Van Helden PM, Unterthurner S, Hermann C, Schuster M, Ahmad RU, Schiviz AN, Weiller M, Antoine G, Turecek PL, Muchitsch EM, et al. Maintenance and break of immune tolerance against human factor VIII in a new transgenic hemophilic mouse model. Blood. 2011;118(13):3698–707. doi:10.1182/blood-2010-11-316521.
- Dingman R, Balu-Iyer SV. Immunogenicity of protein pharmaceuticals. J Pharm Sci. 2019;108(5):1637–54. doi:10.1016/j.xphs.2018.12.014.
- Walsh NC, Kenney LL, Jangalwe S, Aryee K-E, Greiner DL, Brehm MA, Shultz LD. Humanized mouse models of clinical disease. Annu Rev Pathol. 2017;12(1):187–215. doi:10.1146/annurev-pathol-052016-100332.
- Yong KSM, Her Z, Chen Q. Humanized mice as unique tools for human-specific studies. Arch Immunol Ther Exp (Warsz). 2018;66(4):245–66. doi:10.1007/s00005-018-0506-x.
- Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature. 2011;481(7379):81–84. doi:10.1038/nature10660.
- Brainard DM, Seung E, Frahm N, Cariappa A, Bailey CC, Hart WK, Shin H-S, Brooks SF, Knight HL, Eichbaum Q, et al. Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J Virol. 2009;83(14):7305–21. doi:10.1128/JVI.02207-08.
- Choudhary SK, Rezk NL, Ince WL, Cheema M, Zhang L, Su L, Swanstrom R, Kashuba AD, Margolis DM. Suppression of human immunodeficiency virus type 1 (HIV-1) viremia with reverse transcriptase and integrase inhibitors, CD4+ T-cell recovery, and viral rebound upon interruption of therapy in a new model for HIV treatment in the humanized Rag2-/-{gamma}c-/- mouse. J Virol. 2009;83:8254–58.
- Denton PW, Estes JD, Sun Z, Othieno FA, Wei BL, Wege AK, Powell DA, Payne D, Haase AT, Garcia JV, et al. Antiretroviral pre-exposure prophylaxis prevents vaginal transmission of HIV-1 in humanized BLT mice. PLoS Med. 2008;5(1):e16. doi:10.1371/journal.pmed.0050016.
- Frias-Staheli N, Dorner M, Marukian S, Billerbeck E, Labitt RN, Rice CM, Ploss A. Utility of humanized BLT mice for analysis of dengue virus infection and antiviral drug testing. J Virol. 2014;88(4):2205–18. doi:10.1128/JVI.03085-13.
- Greenblatt MB, Vrbanac V, Tivey T, Tsang K, Tager AM, Aliprantis AO, Stoddart Ca. Graft versus host disease in the bone marrow, liver and thymus humanized mouse model. PloS One. 2012;7(9):e44664. doi:10.1371/journal.pone.0044664.
- Joseph A, Zheng JH, Chen K, Dutta M, Chen C, Stiegler G, Kunert R, Follenzi A, Goldstein H. Inhibition ofIn VivoHIV infection in humanized mice by gene therapy of human hematopoietic stem cells with a lentiviral vector encoding a broadly neutralizing Anti-HIV antibody. J Virol. 2010;84(13):6645–53. doi:10.1128/JVI.02339-09.
- Sun C-M, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204(8):1775–85. doi:10.1084/jem.20070602.
- Legrand N, Huntington ND, Nagasawa M, Bakker AQ, Schotte R, Strick-Marchand H, De Geus SJ, Pouw SM, Böhne M, Voordouw A, et al. Functional CD47/signal regulatory protein alpha (SIRPα) interaction is required for optimal human T- and natural killer- (NK) cell homeostasis in vivo. Proc Nat Acad Sci. 2011;108(32):13224–29. doi:10.1073/pnas.1101398108.
- Li Y, Masse-Ranson G, Garcia Z, Bruel T, Kök A, Strick-Marchand H, Jouvion G, Serafini N, Lim AI, Dusseaux M, et al. A human immune system mouse model with robust lymph node development. Nat Methods. 2018;15(8):623–30. doi:10.1038/s41592-018-0071-6.
- Chen Q, He F, Kwang J, Chan JKY, Chen J. GM-CSF and IL-4 stimulate antibody responses in humanized mice by promoting T, B, and dendritic cell maturation. J Immunol. 2012;189(11):5223–29. doi:10.4049/jimmunol.1201789.
- Gunawan M, Her Z, Liu M, Tan SY, Chan XY, Tan WWS, Dharmaraaja S, Fan Y, Ong CB, Loh E, et al. A novel human systemic lupus erythematosus model in humanised mice. Sci Rep. 2017;7(1):16642. doi:10.1038/s41598-017-16999-7.
- Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, Watanabe T, Akashi K, Shultz LD, Harada M, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood. 2005;106(5):1565–73. doi:10.1182/blood-2005-02-0516.
- Zhao Y, Shuen TWH, Toh TB, Chan XY, Liu M, Tan SY, Fan Y, Yang H, Lyer SG, Bonney GK, et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut. 2018;67(10):1845–54. doi:10.1136/gutjnl-2017-315201.
- Chen X, Hickling TP, Vicini P. A mechanistic, multiscale mathematical model of immunogenicity for therapeutic proteins: part 1—theoretical model. CPT Pharmacometrics Syst Pharmacol. 2014;3:133.
- Kierzek AM, Hickling TP, Figueroa I, Kalvass JC, Nijsen M, Mohan K, Veldman GM, Yamada A, Sayama H, Yokoo S, et al. A quantitative systems pharmacology consortium approach to managing immunogenicity of therapeutic proteins. CPT Pharmacometrics Syst Pharmacol. 2019;8:773–76.
- Yogurtcu ON, Yang H, Chancey C, Forshee RA, Eder AF. Predictive model for Zika virus RNA minipool nucleic acid testing in outbreak scenarios. Transfusion. 2019;59(7):2211–17. doi:10.1111/trf.15296.
- Rup B, Pallardy M, Sikkema D, Albert T, Allez M, Broet P, Carini C, Creeke P, Davidson J, De Vries N, et al. Standardizing terms, definitions and concepts for describing and interpreting unwanted immunogenicity of biopharmaceuticals: recommendations of the innovative medicines initiative ABIRISK consortium. Clin Exp Immunol. 2015;181(3):385–400. doi:10.1111/cei.12652.
- Gorovits B, Clements-Egan A, Birchler M, Liang M, Myler H, Peng K, Purushothama S, Rajadhyaksha M, Salazar-Fontana L, Sung C, et al. Pre-existing antibody: biotherapeutic modality-based review. AAPS J. 2016;18(2):311–20. doi:10.1208/s12248-016-9878-1.
- Diego VP, Luu BW, Hofmann M, Dinh LV, Almeida M, Powell JS, Rajalingam R, Peralta JM, Kumar S, Curran JE, et al. Quantitative HLA-class-II/factor VIII (FVIII) peptidomic variation in dendritic cells correlates with the immunogenic potential of therapeutic FVIII proteins in hemophilia A. J Thromb Haemost. 2020;18(1):201–16. doi:10.1111/jth.14647.
- Cassotta A, Mikol V, Bertrand T, Pouzieux S, Le Parc J, Ferrari P, Dumas J, Auer M, Deisenhammer F, Gastaldi M, et al. A single T cell epitope drives the neutralizing anti-drug antibody response to natalizumab in multiple sclerosis patients. Nat Med. 2019;25(9):1402–07. doi:10.1038/s41591-019-0568-2.
- Dhanda SK, Karosiene E, Edwards L, Grifoni A, Paul S, Andreatta M, Weiskopf D, Sidney J, Nielsen M, Peters B, et al. Predicting HLA CD4 immunogenicity in human populations. Front Immunol. 2018;9:1369. doi:10.3389/fimmu.2018.01369.
- Paul S, Grifoni A, Peters B, Sette A. Major histocompatibility complex binding, eluted ligands, and immunogenicity: benchmark testing and predictions. Front Immunol. 2019;10:3151. doi:10.3389/fimmu.2019.03151.
- Walsh RE, Lannan M, Wen Y, Wang X, Moreland CA, Willency J, Knierman MD, Spindler L, Liu L, Zeng W, et al. Post-hoc assessment of the immunogenicity of three antibodies reveals distinct immune stimulatory mechanisms. mAbs. 2020;12(1):1764829. doi:10.1080/19420862.2020.1764829.