
ABSTRACT
Therapeutic monoclonal antibodies and their derivatives are key components of clinical pipelines in the global biopharmaceutical industry. The availability of large datasets of antibody sequences, structures, and biophysical properties is increasingly enabling the development of predictive models and computational tools for the “developability assessment” of antibody drug candidates. Here, we provide an overview of the antibody informatics tools applicable to the prediction of developability issues such as stability, aggregation, immunogenicity, and chemical degradation. We further evaluate the opportunities and challenges of using biopharmaceutical informatics for drug discovery and optimization. Finally, we discuss the potential of developability guidelines based on in silico metrics that can be used for the assessment of antibody stability and manufacturability.
1 Introduction
Monoclonal antibodies (mAbs) and antibody-based biotherapeutics represent a unique class of biologics that have greatly reshaped our modern biopharmaceutical industry since the first mAb drug, muromonab (Orthoclone®), was approved by the Food and Drug Administration in June 1986. The global mAb market is currently valued at 152.5 billion USD and is projected to exhibit an annual growth rate of 14.6% in the next decade.Citation1 Antibody therapeutics currently in late-stage clinical studies have more than tripled to 88 compared to 2010 and over 550 novel antibody therapeutics are currently in the early-stage commercial clinical pipeline.Citation1,Citation2 Antibody therapeutics are anticipated to be the key treatments in a broad range of disease areas, such as cancer, cardiovascular, inflammation, neurological, autoimmune, and infectious diseases.
Biopharmaceutical informatics is the application of computational methods and bioinformatics tools toward addressing challenges in biopharmaceutical drug development. It also includes development of databases containing biophysical data, molecular modeling and simulations, and statistical analysis of biopharmaceutical datasets. The term “Biopharmaceutical Informatics” was first introduced by Kumar et al.Citation3 as the umbrella term for applications of computational approaches in drug discovery and development. Here, we present different aspects of computational applications to antibody-based biopharmaceutical drug development by highlighting key scientific advances in the developability assessment of antibody-based biologic drug candidates.
One of the first practical applications of software relevant to antibody informatics was the antigenic index,Citation4 which was a program to generate surface contour profiles and predict antigenic sites from the linear amino acid sequence of proteins including antibodies. These techniques were the precursors of modern sequence- and structure-based bioinformatics tools used in biopharmaceutical discovery and development. The multitude of computational tools and algorithms now available have ushered in an era of high-throughput biopharmaceutical informatics.
This review is organized into four main sections. The first section outlines the databases and tools available for biopharmaceutical informatics relevant to antibody-based drugs. In the second section, we discuss the role of developability at early-stage development and computational developability assessment of antibody therapeutics. The third section describes the application of biopharmaceutical informatics to identify key developability issues in antibody-based drug discovery and design. The final section summarizes emerging trends in the use of biopharmaceutical informatics for antibody therapeutics. While we discuss antibody informatics tools and approaches for evaluating developability issues, comprehensive review of every developability issue was not possible within this article. We have, however, cited previously published reviews that include more details for each developability issue in the respective sections below.
1.1 Creation of databases and data mining for comparison of biophysical attributes
The availability of larger datasets with new high-throughput experimental methods has improved the predictions made by biopharmaceutical informatics tools. The challenge of data scarcity is now being resolved by open-source libraries and public databases of biopharmaceutical data. Data in biopharmaceutical informatics are highly heterogeneous and interrelated. Consequently, it is not possible to capture these broad ranges of properties in a single algorithm. Datasets currently used to assess the biophysical properties of antibodies are curated from internal releases by pharmaceutical companies or data points from scientific papers.Citation5–7 Experimental data sourced from scientific papers might not be comparable with one another because of differences in experimental setups, the plethora of developability assays, and different antibody formats tested. Additional data sources that potentially contain much antibody-engineering knowledge are patents, where one needs to scan the documentation for primary sequence information.Citation8 Altogether, there is currently much yet-untapped data in the public domain, but these are often hard to curate and not immediately compatible and useful without much earlier pre-processing.
Further advantages from curating antibody databases to learn biophysical properties of antibodies can be obtained by linking information from heterogeneous sources. Current predictive approaches typically use either structural or sequence data rarely linking information from different sources (e.g., structural, and next-generation sequencing (NGS)). Collating information from different sources, however, can augment information available in heterogeneous sources. For instance, structural modeling can provide a conformational dimension to millions of sequences drawn from NGS,Citation9 whereas contrasting naturally sourced and therapeutically developed molecules can provide insights on commonalities and divergences between the two sources.Citation10 A good example of such an integrated approach is the INDI database,Citation11 which contains data for antibody-cognate nanobodies (single-domain antibodies VHH) collected from all major public sources, encompassing patents,Citation8 NCBI GenBank, Protein Data Bank (PDB), and NGS/AIRRCitation12 supplemented by manual curation from the scientific literature. The sequences and structures of antibodies from these heterogeneous sources are linked with textual information into an antibody-specific database. Integrating the heterogeneous sources in this manner facilitates searching and creation of custom datasets of nanobodies. Extrapolating such data integration approaches to antibodies should allow researchers to focus more on the machine learning/statistical approaches addressing the prediction of biophysical properties of these molecules.
Norman et al.Citation13 have previously provided an overview of available databases and tools for computational antibody analysis. However, our specific focus here is on computational developability assessment tools and databases. provides a list of relevant databases and datasets for antibody-based drugs that can be used for training, validation, and assessment of biopharmaceutical informatics tools.
Table 1. Relevant databases and datasets for biopharmaceutical informatics
1.2 Relevance of biopharmaceutical informatics tools
Biopharmaceutical informatics tools are widely used for in silico screening of biophysical properties in an antibody library. These antibody informatics approaches have been used to evaluate key biochemical and biophysical properties such as solubility, stability, viscosity, charge profiles, posttranslational modifications (PTMs), pharmacokinetic and pharmacodynamic (PK/PD) profiles, and hydrophobicity to rank the candidates. The prediction of protein tertiary structure is accomplished by either homology modeling approaches, fold recognition, or ab initio modeling approaches when similar sequences with known structures are absent. Several studies have implemented homology modeling to calculate the biochemical and biophysical properties of a mAb library.Citation16–18 Specific homology modeling algorithms for antibodies have been developed for better accuracy and representation.Citation19–21 In general, antibody sequences and structures are well conserved except for the complementarity-determining regions (CDRs). The CDRs, except for CDR-H3, can be classified into a set of limited conformations called canonical structuresCitation22–24 that can be predicted from sequence key residues, enabling sub-ångström accuracy in structure prediction. However, predicting conformations of CDR-H3 is still challenging because it is the most diverse both in sequence and structure.Citation25 Sequence-structure correlations identified for CDR-H3 have been used as geometric constraints in simulations for structure prediction.Citation26,Citation27
The antibody modeling tools provide an integrated computer-aided molecular design platform that can be used to access liabilities and optimize the affinity, solubility, and stability of antibody-based drug candidates. Several other biopharmaceutical informatics tools for various developability issues depend on protein sequence features that are based on amino acid physicochemical properties. There have been increasing efforts to compile these tools for integrated antibody sequence and structure management, analysis, and prediction. For instance, a large number of tools for antibody informatics are compiled under the abYsis database, abYmod antibody modeling program, and abYbank database. abYsisCitation28 incorporates a wide-ranging species-specific analysis of residue frequencies that can be combined with residue clustering to identify either hydrophobic or unusual patches that are likely to be important for the stability and immunogenicity of antibodies. The Scratch suite of predictorsCitation29 also provides a set of comprehensive tools to evaluate the physicochemical properties of mAbs, such as the solvent accessibility, secondary structure, tertiary structure, contact maps, protein antigenicity, and domain locations. The Oxford Protein Informatics Group (OPIG) also maintains several webservers and databases relevant to antibody informatics. An up-to-date list of antibody-related resources is maintained at http://naturalantibody.com/tools. provides a list of biopharmaceutical informatics tools for the developability assessment of antibody therapeutics.
Table 2. Relevant biopharmaceutical informatics tools
2 Computational developability assessment using biopharmaceutical informatics
Novel criteria based on biochemical and biophysical properties of mAbs are being increasingly used to select a mAb candidate from the early discovery to the development stage. Computational developability assessment approaches are now becoming a routine step in the drug discovery and development process. Developability assessments at the early stage of development can significantly de-risk development pipelines, thus saving valuable time and resources. Incorporating developability assessments in early-stage development provides an opportunity to re-engineer the molecule to mitigate any sequence or structural liabilities, or to select alternative molecules of similar potency, but with more favorable developability profiles. Previous studies have summarized various experimental platforms and computational tools to identify developability issues in therapeutic antibodies and antibody-like molecules.Citation35,Citation36
In the past decade, applications of techniques such as phage display, cell surface display, yeast display, hybridoma, and NGS have revolutionized biomedical research with the successful discovery of several therapeutic antibodies. Although most antibody libraries focus on maximizing library diversity, there are growing concerns regarding the developability of the selected antibodies for successful commercialization.Citation6 Therefore, frameworks and procedures are being developed for the design of antibody libraries with improved developability and manufacturability.Citation37 In silico engineering and design of biologics using rational design principles has emerged as a faster and economic alternative to traditional methods of lead generation such as hybridoma and phage display. provides a visual representation of the recommended biopharmaceutical informatics tools for computational developability assessment of antibody therapeutics and antibody-based drugs.
Figure 1. Biopharmaceutical informatics tools for computational developability assessment of antibody therapeutics. These tools have been selected by authors from several other available antibody informatics tools for general proteins.
. Venn diagram of tools listed under different developability categories.
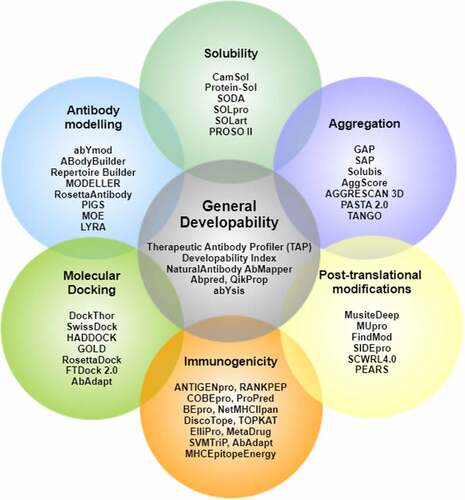
2.1 De-risking biopharmaceutical development using developability assessments
Developability assessment is used to systematically evaluate mAb candidates that have the lowest risks for development to the final product. Previous studies have demonstrated the utility of the developability assessment of mAb lead candidates for screening out mAbs with low solubility and stability, low potency, high aggregation propensity, and high immunogenicity risk.Citation38 Any such predictions will inevitably reject some antibodies that could have made excellent drugs, but not using such approaches comes with huge financial risk.
The general biophysical properties of approved mAbs can serve as a reference for the design of new mAb candidates. Several databases of biophysical properties of these approved mAb candidates have been reported such as the Jain datasetCitation6 and TheraSabDab.Citation14 The Jain dataset provides biophysical characterization across 12 different platforms for 137 clinical-stage and approved antibodies.Citation6 This benchmarking with approved mAbs provides an estimate of the acceptable ranges of the biophysical properties that can be considered in the developability assessments for new antibody candidates. Xu et al. have outlined some generally preferred quality attributes of a panel of approved and clinical stage mAb products.Citation39 The general concept of examining the properties of successful antibody-based drugs has been exploited by Raybould et al.Citation5 resulting in Therapeutic Antibody Profiler (TAP) developability guidelines that are derived from the values of 377 post-Phase 1 clinical-stage antibody therapeutics. It relies on the hypothesis that antibodies that have deviating biophysical properties from clinically tested therapeutic mAbs are likely to have poor developability profiles. TAP can be used to analyze several properties linked to poor developability for any candidate mAb with known heavy and light chain variable domain sequences.
In addition, AbpredCitation40 can be used to predict the biophysical performance on 12 commonly used developability assessment assays with just the amino acid sequence input. In Abpred, machine learning methods have been trained on heavy and light chain variable domain sequences from the Jain dataset using the amino acid composition and 15 sequence-derived features to represent physicochemical properties of antibodies. Other developability assessments using machine learning approaches have been used to predict and select the antibodies with optimal pH and thermal stabilities from 77 antibodies in development at Pfizer.Citation41 Lonza Biologics has also demonstrated the use of aggregation propensity screening along with other computational approaches during early drug development to select molecules with reduced risk of aggregation and optimal developability properties for screening several anti-interferon γ antibody variants.Citation7 Pfizer has implemented in vitro assays that correlate with in vivo human studies to differentiate mAbs at high risk for rapid clearance from those with favorable PK.Citation42 Finally, molecular dynamics simulation has also implemented a high-throughput developability workflow on a panel of 152 human or humanized mAbs.Citation43 Here, physicochemical properties of these 152 mAbs were evaluated from multiple biophysical assays – size exclusion chromatography for aggregation, reverse-phase chromatography and sodium dodecyl sulfate capillary electrophoresis for purity, differential scanning fluorimetry for thermostability, hydrophobic interaction chromatography (HIC) for hydrophobicity, affinity-capture self-interaction nanoparticle spectrometry for self-interaction and capillary isoelectric focusing for isoelectric point (pI) and charge variant analysis. These examined biophysical properties and key assay endpoints were also predictive of key downstream process parameters in development and clinical manufacturing.Citation43
2.2 Design of antibody libraries with improved developability
Screening libraries of antibodies is a commonly used strategy in antibody drug discovery. There are two main approaches to library design: creation of (1) a highly diverse library potentially containing binders to varied targets or (2) a library focused on potential binders to a specific antigen or set of antigens. The ideal library contains genetically varied antibodies with the potential for high affinity and activity, but this can result in the generation of increasingly large libraries to achieve high diversity. With the huge amount of available sequence data and increased understanding of developability prediction, methods are being investigated for the optimal design of antibody libraries with high functionality and desired biophysical properties.
2.2.1 Natural
Methods using B-cell receptor (BCR), i.e., antibody repertoires from antigen-exposed animals or humans (“immune libraries”, to generate antigen-specific libraries) or from non-exposed humans (“naïve libraries”, to generate functionally diverse libraries) try to capture the capabilities of the natural immune response in making functional, highly expressed and low immunogenicity antibodies. However, not all naturally occurring antibodies are suitable drug candidates owing to other developability concerns, such as aggregation.Citation5 Libraries can aim to combat this by selecting for genes with known favorable characteristics using native heavy and light chains for improved specificity.Citation44–46 Limitations to natural-repertoire approaches also include the inherently biased nature, meaning diverse antibodies may be missed owing to sequence space restrictions. Nevertheless, available sequence space might not be as constrained as previously expected, as multiple clinical-stage therapeutics have high sequence-identity matches in naturally sourced antibody repertoires.Citation10 Another way to select antibodies is by considering the “structural space”. For example, a library of antibody structures identified in the repertoires of multiple individuals was found to contain structures highly similar to clinical-stage therapeutic antibodiesCitation47 and may suggest antibodies with functionality and low likelihood of immunogenicity.
2.2.2 Synthetic
Synthetic libraries introduce diversity, often at defined regions of an antibody, to generate novel and varied sequences. Such methods can produce antibodies with higher affinity than natural repertoires,Citation48 but a proportion of the library may be non-folding or immunogenic. To reduce nonfunctionality, methods such as position frequency analysis (PFA) and deep learning have been applied. PFA introduces mutations based on the amino acid frequencies found at each CDR position in natural antibody repertoires, often using identical or only a small number of framework regions.Citation49,Citation50 Such methods do not account for correlations between residues at different sites. A different approach has used a database of antibodies with known functionality and interchanged CDR regions, assuming CDR regions are modular and can be interchanged without negative impact. In doing so, they achieved high functionality.Citation51
2.2.3 Deep learning
Deep learning models aim to utilize the stability of natural repertoires and capture higher-order dependencies, missed by PFA, to avoid producing nonfunctional proteins. However, current limitations of deep learning approaches include a focus on only CDR regions or heavy chains, with a lack of experimental validation of predicted properties. For instance, 74% antigen binding was achieved in a mouse library designed by a variational autoencoder that generated novel CDR-H regions, but such an approach ignores non-CDR region contributions to the paratope, and the diversity of the sequences in this library is unknown.Citation52 Other generative approaches such as Generative Adversarial Networks can be trained on natural human antibody repertoires and biased via transfer learning (further training on antibodies with known properties such as solubility, stability and predicted immunogenicity) to generate sequences predicted to have the desired biophysical properties.Citation53 However, more information is needed to understand how such properties influence the overall developability of the antibody. Additionally, experimental validation of the predicted properties is necessary, as has been conducted for an enzymatically active protein libraryCitation54 and a nanobody library created by a generative deep neural network-powered autoregressive model trained on a native llama repertoire.Citation55
Previous work has demonstrated the use of mammalian display libraries for the selection of antibody variants with optimal biophysical properties, reduced polyreactivity, and immunogenicity.Citation56 Here, they have described the use of a nuclease-directed integration system to generate antibody variants with differing biophysical properties based only on the display level achieved on the mammalian cell surface. Other studies have demonstrated the use of machine learning-guided directed evolution on the combinatorial sequence space.Citation57 Recently, a machine learning pipeline has been formulated to predict the developability of a library of 2400 antibodies from sequence alone.Citation58 These advances in bioinformatics and in silico methods have enabled the efficient development of commercially viable antibodies. Thus, antibody library variants of an antibody candidate are designed to exhibit better developability than the parent molecule.
2.3 Mitigating aggregation and post-translational modifications in biopharmaceuticals
2.3.1 Aggregation
Aggregation of antibody-based drugs can lead to precipitation and decreased shelf-life of drugs before administration, while aggregation in vivo can increase the immunogenicity of the drug. The aggregation propensity is a critical attribute correlated with product failure.Citation59 Indeed, aggregate levels in the final drug product are key quality indicators.Citation60,Citation61 Seeliger et al. have highlighted four key factors that must be avoided to minimize aggregation, many of which can be predicted computationally: (1) the number of “reactive sites”, such as those susceptible to oxidation, deamidation, or proteolysis, should be minimized; (2) thermodynamic stability should be high to minimize protein unfolding; (3) the structure should not contain hydrophobic or charged surface patches; and (4) the sequence should not contain cross-beta-sheet aggregation hotspots.Citation62
Van der Kant et al. showed that mutating residues in predicted aggregation hotspots could reduce aggregation and found that those hotspots having the largest impact on thermodynamic instability are frequently found in the CDRs.Citation63 The solubility can be improved in mAbs having aggregation-prone regions (APRs) by inserting glycosylation sites near these APRs.Citation64,Citation65 Several other studies have used protein-engineering approaches to reduce self-association and aggregation to achieve high solubility and low viscosity.Citation66–69 A specific prediction of the tendency to aggregation is the AggScore,Citation70 which uses structural modeling to identify patches at risk of driving aggregation. Several methods have been developed to create so-called “developability indices” for antibodies and these tend to focus on aggregation propensity. For example, Lauer et al. used data from the storage of 12 IgG antibodies for periods of up to 2 y to examine aggregation. They then combined net charge (at a given pH using a calculated pKa) with a “spatial aggregation propensity” (SAP) score (derived from accessibility and residue hydrophobicity and calculated over a molecular dynamics simulation) to create their developability index and correlated this with the experimental aggregation propensity.Citation34 Developability IndexCitation34 is a well-known tool for estimating the developability of a candidate antibody. However, a potential drawback of the Developability Index is that it is based only on the full-length antibody’s net charge and the SAP of the CDR region, and, therefore, may ignore other indicators of developability.
The Therapeutic Antibody Profiler (TAP) has been demonstrated to be very useful in selectively highlighting antibodies with expression or aggregation issues.Citation5 Further, Lonza’s aggregation prediction toolCitation7 has been instrumental in the selection of lead antibody candidates from combinatorial libraries with improved developability. abYsisCitation28 incorporates a wide-ranging species-specific analysis of residue frequencies that can be combined with residue clustering to identify either hydrophobic or unusual patches that are likely to be important for the stability and immunogenicity of biopharmaceuticals. Therefore, using these computational aggregation prediction tools can identify aggregation issues early in biopharmaceutical development and avoid expensive late-stage product failures.
2.3.2 Post-translational modifications
PTMs can lead to several issues encountered with the development of antibodies. By their nature, PTMs lead to heterogeneity, something that generally concerns regulators since variants must be considered in risk assessments and during characterization to assess the impact on product quality, safety, and efficacy. This includes potential effects on antigen binding, immunogenicity, and Fc-mediated effector functions.
In antibodies, the N-terminal glutamate or glutamine is frequently cyclized by nucleophilic attack of the lone pair of electrons from the backbone terminal NH2 onto the sidechain carboxy or amide, forming a five-membered lactam ring known variously as pyroglutamic acid (pyroGlu), pyrrolidone carboxylic acid (PCA), 5-oxoproline, or pidolic acid, and this has been shown to occur in vitro.Citation71–73 The N-terminus is comparatively close to the antigen binding site, so the difference in charge could have an effect on antigen binding, particularly for large antigens that may approach close to this part of the antibody. In addition to N-terminal heterogeneity, “clipping” frequently occurs at the C-terminus of the heavy chain. The last three residues of the heavy chain are Pro-Gly-Lys; the proline is the last residue of the CH3 domain, and the glycine and lysine form the CHS region. The C-terminal lysine is mostly clipped posttranslationally by endogenous carboxypeptidases during cell culture, or by endogenous serum carboxypeptidase B once the antibody is administered to a patient.Citation74 However, this PTM is unlikely to have any serious effect on the in vivo performance of antibody-based drugs since the C-terminus is remote from any functional sites. That said, C-terminal clipping has been shown to be required for optimal complement activation and the presence of the lysine can affect the blood circulation time.Citation75 The third major PTM in antibodies is the N-linked glycosylation present in the CH2 domain. While these are the three best-known PTMs present in the vast majority of antibodies, many other sequence-specific PTMs are also observed, all of which lead to heterogeneity potentially affecting charge, pI, aggregation, and binding. Heterogeneity as a result of PTMs and their effects are reviewed by Liu et al.,Citation76 while a comprehensive analysis of charge heterogeneity in adalimumab (Humira®) was performed by Füssl et al.Citation77
Asparagine and aspartate residues form hot spots susceptible to deamidation and isomerization.Citation39,Citation78 In addition to the effect of antibody deamidation, there have been reports of deamidation in protein antigens in severe diseases such as anthrax.Citation79 Oxidation of methionine and tryptophan residues is another sequence liability that can lead to low potency, decreased thermal stability, and high aggregation propensity.Citation80,Citation81 Disulfide scrambling due to cysteine residues is another phenomenon causing configurational changes in the hinge region of antibodies, thus impeding antigen binding and mAb functionalities.Citation82,Citation83 The variable domains of mAbs may also contain N-glycosylation sites, which may cause variable domain glycosylation that results in the formation of Fab-associated oligosaccharides with α1,3-galactose that are known to cause immunogenicity.Citation84–86 These PTMs often lead to low potency, immunogenicity, and instability of circulating mAbs.Citation87 Consequently, suitable developability assessment protocols must be designed to capture these sequence liabilities.
abYsisCitation28 (http://www.abysis.org) provides screens for a number of these PTMs for optimization of therapeutic antibodies. It also annotates residues as being exposed, buried, or intermediate based on averaged information from several hundred known structures and can be used in concert with abYmod (http://abymod.abysis.org) to build an antibody model from which more detailed exposure information can be obtained. As described, PTMs could seriously hamper the safety or efficacy of therapeutic antibodies and this safety concern calls for an immediate need for appropriate tools to relate a biophysical property to a single, or a set of, molecular sequence-structural motifs in biologic drugs. In summary, biopharmaceutical informatics tools are used to locate the amino acids critical for certain biophysical properties that are in undesirable ranges.
2.4 Biopharmaceutical informatics for drug safety and in vivo performance
2.4.1 Drug safety
A strategic framework for using computational tools for predicting chemical degradation sites in biologic drugs has been presented in a previous study by Sandeep et al.Citation3 Several computational tools for predicting the toxicity of antibody-based drugs are now available.Citation88 A critically important step in drug development for establishing clinical safety is the identification of adverse drug reactions (ADRs). Computer-aided prediction of ADRs provides an alternative to recognize ADRs before clinical trials. Kuang et al. have reviewed and compared the computational models available for predicting ADRs.Citation89 Here, among the topological features of drug-ADR association networks, the Jaccard coefficient (a measure of the relationship between the neighborhood set of homology nodes) was the most important feature for the prediction of drug-ADR associations. Consequently, the Jaccard coefficient of drug-ADR association networks is an important topological feature that should be used in models designed for prediction of antibody drug safety.
Previous computational approaches have estimated in vivo performance descriptors such as the PK, PD, and immunogenicity of biologics.Citation42,Citation90–93 Avery et al. have demonstrated a combinatorial triage approach on in vitro assay parameters and categories for screening therapeutic mAb candidates with desirable PK properties and minimal non-target-related PK risk.Citation42 Here, threshold values of in vitro assays reflecting nonspecific interactions and self-association were established to define criteria for avoiding the selection of mAbs with rapid in vivo clearance. Grinshpun et al. have also analyzed biophysical and sequence-based in silico properties that are predictive of PK properties such as clearance for a panel of 64 clinical-stage mAbs.Citation94 They have concluded that experimental poly-specificity assay results and in silico estimated pIs were the best predictors to estimate clearance in therapeutic antibodies.
2.4.2 Antigen–antibody interactions
General protein–protein interaction prediction tools for proteins frequently do not work well for antigen–antibody interactions because antibody–antigen binding is a rather distinct mechanism. Unlike normal protein interfaces, the epitope on an antigen has evolved to be an exposed region rather than to be involved in a protein–protein interface. Consequently, other computational techniques such as epitope mapping are used to identify the regions of an antigen likely to form the epitope before docking. B-cell Epitope (BCE) mapping tools can broadly be divided into linear epitope predictors, which attempt to identify epitopes consisting of continuous amino acid primary sequences, and conformational epitope predictors, predicting discontinuous epitopes in three-dimensional (3D) space.Citation13 However, like other protein–protein interfaces, antibody–antigen interactions involve a combination of nonpolar van der Waals interactions, hydrogen bonding, charge interactions, and the hydrophobic effect. Consequently, along with these epitope prediction tools, several docking algorithms such as Megadock, Haddock, RosettaDock, and Piper are being actively used to understand the binding between an antibody and the target. However, their performance is often poor compared with general protein–protein docking.
2.4.3 Immunogenicity
The presence of T-cell and B-cell epitopes influences the immunogenicity of antibody therapeutics, and, therefore, bioinformatics approaches to avoid immunogenicity fall into two major categories: T-cell epitope prediction and B-cell epitope prediction. Computational tools for immunogenicity risk assessment provide an alternative to in vitro or in vivo immunogenicity assays. The use of in silico tools to identify lead candidates with a reduced risk of immunogenicity is an important step in biologic drug development.
T-cell epitope prediction, which is relatively well established, requires predicting linear peptides within a protein sequence that will bind to the Major Histocompatibility Complex (MHC). MHC molecules present peptides to T cells, which trigger T-cell immune responses. MHC molecules can be classified into class I and class II. MHC class I molecules present peptides derived from intracellular proteins, whereas MHC class II presents peptides from extracellular proteins. Since antibodies are extracellular, the focus is on the prediction of peptide binding to MHC class II molecules. These tools usually examine the primary sequences of candidate antibodies to identify binding motifs of MHC class II allotypes or for similarity to epitopes known to elicit an immune response. Several MHC class II binding predictors are available and the overall prediction performance is generally good.Citation95,Citation96
For example, some tools such as RANKPEP,Citation97 Propred,Citation98 Tepitope,Citation99 and NetMHCIICitation100 make predictions based on algorithms trained on MHC class II binding assay data. Other tools such as NetMHCIIpan and IEDB (Consensus)Citation101 are based on sequence alignments with MHC class II binding peptide databases. Overall, studies have established that NetMHCIIpan, Propred, IEDB (Consensus), and MULTIPREDCitation102 were the best predictors of MHC class II binding and these are the most commonly used tools in the industry for the prediction of MHC class II binding. Other previous studies compared nine different MHC class II binding prediction tools and six different methods showing that NetMHCIIpan was the best method to predict peptide binding to MHC class II epitopes with an updated version, having improved predictions, now available.Citation103,Citation104 While less important for antibody-based drugs, computational tools for determining binding to MHC class I molecules require locating motifs that bind to the binding groove. Prediction methods for interrogating peptide binding to MHC class I alleles include NetMHC-3.0,Citation105 NetMHCpan-1.0, the Kernel-based Inter-allele peptide binding prediction system,Citation106 and Adaptive Double Threading.Citation107 Based on this predicted T-cell epitope information, Yachnin et al. recently developed a Rosetta-based platform to deimmunize therapeutic proteins.Citation108 They incorporated a new score term utilizing predicted or experimentally identified T-cell epitope information into the scoring function so that computational protein design calculations can be guided based on the epitope information as well as the energetic stability.
In contrast to the prediction of T-cell epitopes, a much harder task is B-cell epitope (BCE) prediction – predicting sites where the patient antibodies will bind to the drug. Such approaches have not been very successful, mostly owing to the discontinuity of antigen binding sites. As mentioned above, the problem is made harder by the fact that B-cell epitopes are, by their nature, regions of a protein surface that have not evolved to be involved in protein–protein interactions. Consequently, they do not have clearly recognizable features that are bound by antibodies.Citation109 Nonetheless, some regions will be more likely to interact with an antibody than others, but making mutations to remove a dominant B-cell epitope can simply result in the immune response switching to a less dominant epitope.
Several predictors have been produced that work at either the sequence level or the level of 3D structure. The earliest BCE prediction methods attempted to predict linear epitopes (i.e., a continuous stretch of amino acid sequence) using sequence features such as hydrophilicity,Citation110 amino acid composition,Citation111 and predicted accessibility and mobility.Citation112 An early evaluation showed that no single sequence feature performed well, leading to attempts to combine features.Citation113 However, machine learning effortsCitation114 and additional features such as sequence conservationCitation115 have provided limited improvements to BCE prediction. In general, conformational epitope predictors such as CBTOPE, BETOPE, CEP, and DISCOTOPE are more accurate than linear epitope predictors such as LBTope, SYMTriP, and ABCored.Citation116–118
The performance of computational epitope prediction tools and tools for predicting immunogenicity has been reviewed previously to establish guidelines for the deimmunization of protein therapeutics.Citation119 It is worth nothing that epitope databases are not exhaustive because of the heterogeneity of proteins involved in the immune response across the human population.Citation90 This variability of immune response for the same antigen limits the utility of in silico immunogenicity assessment methods as stand-alone tools. Therefore, this key limitation of immune response diversity needs to be captured by the forthcoming immunogenicity prediction tools.
2.5 Guidelines for the design of developability assessment protocols
Assessment of developability by biopharmaceutical informatics protocols at an early stage in a development pipeline reduces the costs of development failures. Companies using transgenic mice to produce antibodies can generate as many as a million sequences a week (after cleaning the high-throughput sequence data) and it is impractical to take all these through to experimental validation. Even computational evaluation requires significant computing resources and optimization. If each sequence takes 1 s to analyze, a million sequences will require ~11.5 days of computer time. Consequently, it makes sense to use a triaging pipeline that performs evaluations that can be done quickly first and leave more computer-intensive evaluations to be performed only on those sequences that have survived the initial rapid triages.
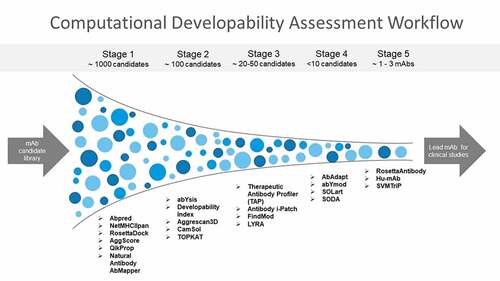
A screening paradigm used in the industry for selecting mAbs with desirable PK properties during mAb discovery and lead selection has been demonstrated in a previous study.Citation42 This staged approach for developability assessment involves using the high-throughput assays first when hundreds of mAbs are available for screening. Here, mAbs scoring above assay thresholds or having results outside the acceptable range are deprioritized because they have unfavorable physicochemical properties. Next, additional physicochemical properties such as thermal stability are evaluated for only the mAbs that have passed the previous stage. These additional screens include assays measuring properties such as biological activity, expression, stability that are often low-throughput and need higher quantities of mAbs. Finally, a combinatorial triage approach is used that ranks and classifies the mAbs based on the aggregate result of all the assays. It is very important to combine results of multiple assays together since individual developability assays can have some false-positive results. This ensures that mAbs with desirable physicochemical properties advance to scale-up and costly preclinical and clinical development. A Computational Developability Assessment (CDA) workflow should follow a similar strategy where a panel of high-throughput computationally undemanding tools is applied first to a mAb library followed by specific computationally intensive antibody informatics tools as per the required objective, such as those for immunogenicity assessment. The final step in the CDA workflow as shown in is to use a combinatorial triage approach to combine scores and rankings from multiple tools together and classify the mAbs based on the aggregate result of all the informatics tools.
Figure 2. Computational developability assessment workflow for screening mAbs with optimal biophysical properties. An orthogonal combination of conceptually different algorithms is used to reduce method-specific biases. High-throughput antibody informatics tools are implemented first to an antibody library. mAbs scoring above assay thresholds or having results outside the acceptable range are deprioritized. Next, more computationally intensive antibody informatics tools are applied to evaluate additional developability issues. The final step in the CDA workflow is to use a combinatorial triage approach to combine scores and rankings from multiple tools together and classify the mAbs based on the aggregate result of all tools.
. Funnel flowchart with mAbs shown in blue dots and tools listed for each stage of developability assessment.
Together with previously discussed approaches to assessing developability, Raybould et al. have described five computational developability guidelines for therapeutic antibody profiling: (1) total CDR length, (2) patches of surface hydrophobicity (PSH) metric across the CDR vicinity, (3) patches of positive charge (PPC) metric across the CDR vicinity, (4) patches of negative charge (PNC) metric across the CDR vicinity, and (5) structural Fv charge symmetry parameter.Citation5 Overall, local charge and global charge asymmetry between the CDR and the framework have been correlated with higher aggregation and poor developability. Here, the approach was to look at the characteristics of clinically successful antibodies and rank candidate antibodies by ensuring they stay within these bounds. This is conceptually similar to Lipinski’s rules used in small-molecule drug design.Citation120 An efficient high-throughput developability workflow was also demonstrated by Bailly et al. on a panel of 152 mAbs for rank ordering of molecules during early-stage discovery screening.Citation43 Here, they have demonstrated that key physicochemical properties from multiple biophysical assays correlated well with major downstream process parameters.
As above, most types of analysis performed for developability assessment include identification of PTM sites, analysis of likely aggregation propensity (largely through examining surface hydrophobicity), pI, prediction of stability, and identification of T-cell epitopes/B-cell epitopes together with humanness scoring or unusual surface patches. Other considerations that can be included early in the pipeline (as they are fast to evaluate) include checking for the presence of the standard two cysteines present in antibody variable domains, the Trp-Gly motif present immediately after CDR-H3, and the length of CDR-H3 since unusually long CDR-H3 loops have been correlated with poor developability.Citation121 However, each tool relies on different interpretations and weighting of the essential features that determine developability. Therefore, an orthogonal combination of conceptually different algorithms should be used in computational developability assessment protocols to reduce method-specific biases.
3 Applications of biopharmaceutical informatics
3.1 Biopharmaceutical informatics for solubility predictions
Solubility is one of the key biophysical properties that underpins developability potential, as high solubility typically translates into high expression yields, low aggregation, and provides the opportunity of formulating products at high concentrations while retaining a good shelf life.
Identifying antibodies with low solubility and high aggregation propensity from combinatorial libraries remains a hurdle for antibody development. Several in silico predictors have been reported that are now able to predict solubility or aggregation propensity accurately in many cases, a feature that makes them highly competitive with experiments.Citation37,Citation122–124 These solubility predictors include CamSol, Protein-Sol, SOLpro, SODA, Aggrescan, SAP, and Solubis. They have been shown to be effective at predicting solubility and aggregation propensity of diverse antibody libraries.Citation122,Citation124
As an example, the CamSol method of predicting solubility relies on a combination of physicochemical properties of amino acids. These include charge, hydrophobicity, and propensity to form secondary structure elements, which are first considered at the individual residue level, then averaged locally across sequence regions, and finally considered globally to yield a solubility score.Citation124,Citation125 In particular, while a structural model is necessary to identify aggregation hotspots, the solubility prediction itself is performed using only the amino acid sequence. This aspect makes computational calculation significantly faster and makes the method readily applicable to the screening of antibody libraries without the need for structural modeling, and thus it is fully independent of model accuracy. For example, CamSol was used to rank the solubility of hits from a phage-display library from MedImmune.Citation125 The mAbs that were analyzed differed by up to 32 mutations in the Fv region, and the correlation between prediction and experiments of PEG-precipitation was R ~ 0.97 after one outlier was removed (p < 10–Citation4), which is fully consistent with the R ~ 0.98 reported for a nanobody in the original report.Citation124 Similarly, a statistically significant correlation (R ~ 0.71 to 0.93) between CamSol predictions and solubility measurements was also reported for mutational variants of a troublesome mAb.Citation126 In a study on a library of 17 mAbs from Novo Nordisk, CamSol predictions were compared with a battery of commonly used developability assays and one measurement of relative solubility, and the correlations between CamSol and these experimental readouts were on a par with those seen between the assays.Citation127 Notably, all these measurements were carried out with different experimental techniques, on widely different molecules, and in different laboratories. Taken together, these strong correlations suggest that CamSol predictions can greatly facilitate the screening of solubility and hence of developability potential. In particular, at the initial stages of antibody discovery campaigns, when numbers of candidates can be very high while yield and purity are often low, such predictions may entirely replace experiments.
Kingsbury et al. have previously predicted the solution behavior of a diverse dataset of 59 mAbs, including 43 approved antibodies, using a comprehensive array of 23 molecular descriptors categorized as colloidal, electrostatic, conformational, hydrodynamic, and hydrophobic.Citation128 They have shown that the diffusion interaction parameter (kD), a measure of colloidal self-interaction is the key parameter that is most predictive of solution viscosity and opalescence for mAbs. So, they have postulated that computational developability assessment protocols should use a threshold value of the diffusion interaction parameter, kD (10 mM histidine-HCl buffer at pH 6.0) to screen antibodies with optimal antibody solution behavior.
3.2 Biopharmaceutical informatics for predicting protein stability and interactions
There can be opportunities to address the underlying balance of biophysical forces that drive interactions when developing models to predict the properties of biopharmaceutical candidates. Two such examples are discussed here, one relating to the measurement of hydrophobic interactions and the other to the protein structural basis of hydrophobic interaction between proteins. Several machine learning methods to predict the HIC retention time from antibody sequence input have been reported previously in the literature.Citation33,Citation40,Citation129 Assessment of aggregation propensity using HIC was the best-predicted biophysical property across 12 models produced using Abpred (www.protein-sol.manchester.ac.uk/abpred), one for each of the 12 biophysical properties measured across a set of antibodies.Citation40 Even so, there was a marked reduction in performance of the model for antibodies with higher retention times in HIC, leading to a model in which the salt gradient that is used to modulate hydrophobic interaction strength also affects interactions between charged proteins. A revised scheme was derived in which charge interactions play a role alongside hydrophobic effects in the HIC method. In this scheme, proteins with higher net charge repel more within the column when salt concentration (ionic strength) is lower, and are eluted faster, than proteins with lower net charge but the same hydrophobicity.
In this second example, another set of HIC data for 24 antibodies was used.Citation130 Here, aromatic sidechain content of CDRs correlated well with the experimental data, but the equivalent correlation was much lower for the solvent-accessible surface area calculated for nonpolar atoms in the CDRsCitation131 and it was concluded that hydrophobic interaction strength may be dependent on nonpolar surface shape as well as surface area, consistent with thermodynamic measurements made for mutations in an antibody–antigen interface.Citation132 These examples demonstrate that models rooted in biophysical descriptions of protein stability and interactions, and benchmarked against experimental data, can both provide predictive insight for biopharmaceuticals and further the understanding of the underlying biophysical mechanisms.
3.3 Biopharmaceutical informatics for preclinical immunogenicity risk assessment
A key concern with any biologic drug is immunogenicity, the effects of which range from simply having an immune response, meaning that the drug is rapidly cleared from the body when administered, through to the possibility of anaphylactic shock. As described above, methods can be applied to predict T-cell epitopes and (to some extent) B-cell epitopes, but a more practical approach has been to ensure that antibody-based drugs are as human as possible and this has become one of the main aims in producing antibody-based drugs. As described earlier, the first monoclonal antibody-based drug to be approved was a mouse antibody (muromonab). However, since then, efforts have gone into making antibody-based drugs less immunogenic, first by producing chimerics (where the variable domains are from the donor species while constant domains are human) and then by “humanization” (where the CDR loops that form the antibody combining site are from the donor species and the rest of the variable domains is predominantly human, as well as human constant domains).Citation133 A halfway step between chimerics and humanization to reduce immunogenicity has been “resurfacing” of chimeric antibodies in which surface residues of the variable domain, away from the CDRs, are mutated to human residues.Citation134 This is done to remove primarily B-cell epitopes on the antibody surface. Many antibody-based drugs are now “fully human”, being produced by phage display, using transgenic mice, or by identifying antibodies from recovering patients. However, antibodies produced by such methods can still be immunogenic. For example, adalimumab (Humira®, the world’s top-grossing drug), while “fully human” (produced by guided phage display), elicits an immune response in >25% of patients with only 4% of these patients having sustained remission, compared with 34% of patients who did not have antibodies against adalimumab.Citation135
Thus, even with fully human antibodies, computational BCE and TCE predictors can be used to predict B-cell epitopes and T-cell epitopes, which can also be experimentally identified through proteomic assays.Citation136,Citation137 It is then desirable to remove these potentially immunogenic regions in advance of clinical trials. As well as the application of BCE and TCE predictors, various “humanness” scores have been proposed based on sequence information of human antibodies, enabling the in silico assessment of human-likeness given sequences of antibodies.Citation138–140 Recently, Schmitz et al. developed a computational method that maps the sequence of a given antibody onto human B-cell repertoires comprising 326 million sequences of human antibodies.Citation141 Chin et al. built a machine learning-based predictive model that distinguishes human antibody sequences from non-human ones, which was trained on large-scale repertoire dataset.Citation142 These human-likeness scoring approaches will be useful when assessing how much given antibodies are close to human repertoires; the more human-like antibodies are, the less immunogenic they are expected to be. As described above, another approach is to identify patches of unusual residues on the protein surface that may lead to an immune response.
4 Future perspectives in biopharmaceutical informatics
4.1 Decoding human antibody gene repertoires and their role in target validation and drug discovery
New high-throughput sequencing methods have generated a vast amount of antibody sequence data, with over one billion antibody sequences publicly accessible in repositories.Citation12,Citation28,Citation143–148 A sequenced human B-cell receptor (BCR, i.e., antibody) repertoire provides a snapshot of the BCRs present, typically those circulating in the blood, at a given time. BCR sequence and structure datasets can be used to investigate immune system mechanisms for improved library design, understand disease pathogenesis and identify antibodies for potential therapeutic development.Citation149
4.1.1 Immune system mechanisms
The diversity of BCR repertoires can be used to develop an understanding of the mechanisms underlying the immune system. Typical BCR repertoire profiling includes sequence-based analysis, such as clonotyping. Clonotyping involves clustering sequences into clones, usually based on identical V and J genes and high CDR-H3 identity.Citation150 Such analysis can reveal dominant antibody sequences, potentially indicative of a response to an antigen, e.g., after vaccination. The availability of large datasets has been useful in characterizing the response to antigens and estimating true antibody genetic diversity,Citation151 though these are still far from fully understood. Sequence-based analysis has revealed that the immune systems of unrelated individuals have similarities; an estimated 0.02% of clones are “public” – shared across multiple individuals.Citation152 However, differences identified between identical twins indicate the complexity of the immune response and the importance of epigenetics and environmental factors.Citation153 Understanding such mechanisms is useful for antibody drug development, for example, to design antibody libraries for drug discovery.
4.1.2 Understanding disease pathogenesis
Immune responses to disease, and also therapies, can be profiled using BCR repertoires to investigate B cell subtype involvement and levels of antibody response. Using such analysis, we can distinguish between healthy and disease repertoires and learn about disease mechanisms, particularly those associated with B cells, such as autoimmune diseases, chronic lymphoid leukemia, and other cancers.Citation154,Citation155 In the future, such information will hopefully be used to improve patient outcomes by identifying the most at-risk patients, tracking disease progression and monitoring response to therapies. A better understanding of the immune system involvement in disease may also indicate targets for potential therapeutic intervention, and even suggest antibody drug candidates present in the BCR repertoires of patients with the disease.
4.1.3 Therapeutic antibody candidate identification – using sequence information
BCR sequence repertoires can be used to suggest suitable candidates for drug development. A previous study has contextualized the sequence and structural properties of clinical-stage antibodies with human immunoglobulin datasets (Ig-seq) to evaluate the extent of humanness/originality of antibodies in clinical investigation.Citation5 While not all naturally occurring antibodies make good drug candidates, 29 clinical-stage therapeutic antibodies were found to share 100% CDR-H3 identity with a BCR sequence from a healthy human repertoire.Citation10,Citation152 By looking for antibody sequences frequently found after exposure to an antigen, we can identify those that might bind specifically to that particular antigen. When assessing individuals with the same disease or who have been exposed to the same antigen (either through infection or vaccination), these sequence-convergent responses can be a useful starting point for a potential therapeutic. Evidence to support this approach for drug discovery comes from vaccine studiesCitation156 and more recently SARS-CoV-2-infected individuals, where convergent antibodies had sequence similarity with identified SARS-CoV-2-binding antibodies.Citation157 In addition to being potential binders, public clones may also have low immunogenicity, making them attractive as drug candidates.Citation47
If existing binders are already known, likely drug candidates can be identified from a BCR repertoire by comparing with known antibodies binding to the desired antigen. Identification can be based on sequence identity, such as clonotyping,Citation158 or prediction of similar binding properties.Citation159 As such, sequence data from BCR repertoires can be useful starting points for suggested therapeutic antibody candidates, with or without knowledge of existing binding antibodies.
4.1.4 Therapeutic antibody candidate identification – incorporating structural information
While most examination of immune repertoires focuses on sequence analysis, utilizing available structural information may also be important when identifying potential therapeutic antibody candidates. Conventional antibody modeling tools are inefficient for building 3D models of entire repertoires of BCRs, with the fastest taking seconds per antibody model via homology modeling methodsCitation20,Citation160 or ~285 CPU hoursCitation161 with ab initio methods. Therefore, structural modeling methods have been developed specifically for large-scale BCR or TCR repertoire data analysis. Incorporating structural information from models can allow prediction of antibody properties in a repertoire and we may be able to predict antibody domain binding by performing structural clustering of antibody models with known-function antibody datasets, such as CoV-AbDab.Citation162,Citation163
A high-throughput alternative to modeling utilizes structural annotation to rapidly predict antibody CDR loop shapes, based on sequence identity matching to a template.Citation164 Repertoires can be evaluated based on predicted CDR structures, for example, to identify over-represented CDR-H3 templates or clusters of templates that may represent a response to an antigen, and therefore be a useful starting point in therapeutic antibody design. Using structural prediction tools with BCR repertoire sequence data can reveal antibody drug candidates not seen using sequence-only analysis.
Current limitations for utilizing BCR repertoire data in drug development include the major challenges of predicting antibody–antigen binding and affinity. In addition, existing BCR sequencing datasets often contain only heavy chain information, and methods for obtaining BCR repertoires and binding affinities are varied and lack standardized protocols and analysis pipelines. With the development of high throughput methods for single-cell sequencing and antigen specificity mapping,Citation165 increased amounts of high quality, antigen-labeled antibody data might enable new accurate and reliable computational methods for drug discovery.
4.2 Biopharmaceutical informatics for design and optimization of next-generation biotherapeutics
The spectrum of biological activities accessible to antibody therapeutics is being expanded by exploring novel mechanisms of action. For example, bispecific antibodies can be created by engineering different specificities into each arm of the antibody, and multi-specific antibodies can be created by adding further VH/VL domains on the heavy and light chains or as a single-chain Fv (scFv) appended on the N- or C-terminus. In addition, novel binding functions can be created using scFvs or nanobodies (heavy-chain only), often combined in tandem for higher avidity or multi-specificity. Other technologies include antibody–drug conjugates (ADCs) created by conjugating cytotoxic drugs (payloads) for site-specific delivery. These novel antibody constructs are often collectively referred to as “next-generation antibodies”Citation166 and are emerging as potential therapeutics with unique properties.
The sequences of these antibody formats may differ substantially from those of immune-system-derived immunoglobulins, as extensive engineering is typically required to bring about the desired functionality. It is often the case that engineering additional functionality comes at the expense of other important properties that underpin developability, including conformational and colloidal stability, solubility, immunogenicity, and PK. Therefore, the successful development of next-generation biotherapeutics presents additional challenges, which are usually system-specific. For example, ADCs are complex molecules that require careful attention to various components, including the mAb, the engineered drug conjugation sites, the selected linker, the payload, and the drug load distribution.Citation167–169 Similarly, multi-specific antibodies require the selection of multiple binding domains that must be successfully combined to ultimately yield a homogeneous product with the desired functionality and suitable developability profile.Citation166
In general, the computational prediction of the developability potential of these novel antibody-based formats presents two overarching challenges. The first is that there is no guarantee that combining together components with suitable properties will translate into a final therapeutic that has desirable characteristics. For example, a bispecific antibody obtained by combining two Fvs with good developability profiles may present unexpected liabilities, such as increased oligomerization brought about by cross interactions between its components. Therefore, while the tools described here may be used to pre-select or engineer binding domains and mAbs with optimal characteristics, when these are combined in a multi-specific format the resulting construct may not necessarily be well behaved. The second challenge lies in the combinatorial nature of combining multiple constructs, which amplifies prediction errors and hence the risk of failure, even assuming that different components behave independently. As an example, consider a computational predictor of a “good” characteristic (such as having good solubility) with precision, or “positive predictive value” (PPV) of 0.9 that implies a false discovery rate (FDR = 1-PPV) of 0.1 (i.e., of the positive predictions, 90% of them are correct, or in other words, one in ten antibodies that are predicted as good are actually poor). If we apply this method to select two distinct Fvs for a bispecific antibody, then the probability of introducing at least one liability in this construct is given by 1-(PPV),Citation2 i.e., 0.19 or 19%. Similarly, for a tri-specific construct, such as nanobodies in tandem, the probability of introducing a “poor” binding domain becomes 27.1%. Therefore, even when neglecting the first challenge and considering the different components as fully independent of each other, the accurate prediction of the developability profile of next-generation biotherapeutics will require exceedingly precise methods.
Some of the databases that can be used for the analysis of nanobody-derived therapeutics are the Single Domain Antibody DatabaseCitation170 (sdAb-DB), Integrated Nanobody Database for ImmunoinformaticsCitation11 (INDI Nanobodies DB), Non-redundant Nanobody databaseCitation171 and database of Institute Collection and Analysis of NanobodiesCitation172 (iCAN). These databases host large collections of natural and synthetic camelid single-domain antibody sequences from literature sources and other online repositories. Each of these databases further provides unified annotation and integrative analysis tools for describing various single-domain antibodies. Overall, computational predictions of developability potential can already be used to aid the development of next-generation biotherapeutics. However, further developments are required before these methods will become highly competitive with experimental readouts in terms of accuracy and reliability. To accelerate innovation in this area, it will be essential that experimental data of developability are published together with the antibody sequences used in the experiments, including any engineered modifications. We anticipate that, just as the Jain et al. studyCitation6 and othersCitation43,Citation173 spurred the development of several computational predictors,Citation5,Citation40 similar investigations using next-generation biotherapeutics will enable such methods to be refined, or new approaches to be developed, to yield accurate predictions of the developability profiles of these constructs.
4.3 Applications of artificial intelligence and machine learning toward antibody discovery, development, and manufacturing
Machine learning algorithms have been used for classification, regression, or clustering of biopharmaceutical experimental datasets. Machine learning models have been used for the prediction of protein secondary structure,Citation174,Citation175 relative solvent accessibility,Citation176–179 protein folding,Citation180–183 protein–protein interactions,Citation184–188 and PTMs.Citation189–192 Machine learning methods have also been applied to the prediction of aggregation using a classification tree ensemble with sequence-derived physicochemical properties.Citation7,Citation193 Other machine learning approaches such as gradient-boosting machines have been used for the prediction of CDR structure from protein sequence, particularly CDR-H3.Citation194,Citation195 The most common strategy used by these algorithms is the use of biophysical propensity scales as input features for machine learning methods to characterize the structural and functional properties of proteins.Citation196
Narayanan et al. have reviewed the application of machine learning approaches in predicting the developability of antibody-based biologics.Citation197 A machine learning algorithm has been shown to predict antibody developability solely by sequence using a dataset of 2400 antibodies.Citation58 Here, a support vector machine model trained on physicochemical features with multiple sequence alignment emerged as the best machine learning pipeline combination to capture antibody developability from the sequence.
Deep learning approaches for antibody design and engineering are also becoming popular.Citation198 Several deep learning models have been described for predicting paratope regions in antibody sequences,Citation199 epitope-specific paratope identification,Citation200 predicting antibody/antigen binding,Citation201 CDR-H3 region optimization,Citation202 and virtual screening for therapeutic antibody optimization.Citation203 Deep learning algorithms offer the ability to capture key biophysical features and properties for any developability objective without the need to create complex theoretical functions. Consequently, deep learning approaches are ideal for cases where mechanistic understanding of the underlying developability issue is not fully understood. However, deep learning algorithms generally require large amounts of data, and so can be unsuitable for smaller datasets.
The choice of the machine learning algorithm is decided by the dataset availability and the objectives of the application. Supervised machine learning methods such as support vector machines, random forests, and conditional random fields are usually more appropriate for balanced datasets.Citation191 Although machine learning-based methods lack the physical transparency of other approaches, their practical application is remarkably successful. Therefore, given that the amount of available training data across biological and structural databases is rapidly increasing, and that machine-learning algorithms are constantly improving, these methods are destined to play key roles in shaping the future of biopharmaceutical informatics.
5 Conclusion
The past two decades have seen transformational advances in the biomedical sciences. In particular, the Human Genome Project has triggered the development of NGS technologies, which are enriching biological databases with millions of sequences of proteins, including antibodies from myriad different sources. Furthermore, improvements in the pace and accuracy of protein structure determination techniques are contributing unprecedented amounts of high-quality structural data, comprising large numbers of antibody–antigen complexes.Citation204–206 The increasing use of quantitative methods in biology has gradually transformed the way biological observations are made, and it is now possible to assemble large datasets of highly accurate measurements of antibody biophysical properties. Finally, computers able to perform complex calculations quickly are available, and extremely powerful algorithms for data mining and machine learning are constantly being developed. Taken together, these advances are enabling the antibody community to address questions that were essentially intractable a decade ago, including the development of highly accurate computational methods to streamline the development of biotherapeutics.
Here, we have described numerous metrics for computational developability assessment and established that no single tool or biophysical parameter can be used for predicting the developability potential of a biotherapeutic. The orthogonal combination of conceptually different algorithms should be used in developability assessment protocols to reduce method-specific biases. However, as stated by Narayaram et al.,Citation197 “one common disadvantage of such in silico tools is that they use only protein sequences or structure-based information as input and usually do not consider the impact of formulation conditions”. The biophysical solution behavior is also influenced by the excipients and solution conditions of the formulated product. Therefore, the developability assessment algorithms will have more real-life practical applications if they also consider the solution conditions and formulation parameters in the algorithms. In addition, minimal information has been provided in the available literature on the validation of these tools in the industrial setting. Therefore, it is important that biopharmaceutical informatics approaches are uniformly applied across the industry to expand and accelerate their potential for biotherapeutics development.
Biopharmaceutical informatics can also be a valuable guide for the commercialization and licensing of antibody-based drugs. The insights from computational developability assessments can aid the due-diligence activities performed during licensing and acquisition transactions.Citation207 The application of biopharmaceutical informatics tools is likely to increase in the future as new accurate and faster software are becoming available for generating antibody structure from the sequence for mAbs. Recently, AlphaFold,Citation208 a neural-network-based algorithm that was recognized as the optimal solution to the protein-folding problem at the Critical Assessment of protein Structure Prediction (CASP) competition, has received wide media attention, but its efficacy in modeling antibodies remains unproven. The recent success of AlphaFold at predicting protein structures demonstrates the power of bioinformatics applications. With increasing efforts devoted to data curation and method development as described here, biopharmaceutical informatics holds the potential to play a leading role in selection and engineering of safe therapeutics.
6 Abbreviations
Author contributions
Conceptualization – RK, RC, JW, SR; Original draft Preparation – RK, RC, JW, SR, KK, JH, AM, DK, UK, PS; Review and editing – RK, KK, RC, JW, AM, CD, PS; Figures and visualizations – RK, KK. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC) via UK EPSRC grant EP/N024796/1. PS is a Royal Society University Research Fellow (URF\R1\201461). We gratefully acknowledge Max Hebditch for his scientific discussions and critically reading of the manuscript.
Disclosure statement
AM is the developer of abYsis and abYmod, which are available for free use over the web or for local use through commercial licenses. PS is the developer of CamSol method, which is available as a free web server but also through commercial licenses. KK is the founder of NaturalAntibody.
Additional information
Funding
References
- Kaplon H, Reichert JM. Antibodies to watch in 2021. MAbs. 2021;13(1):1860476. doi:10.1080/19420862.2020.1860476.
- Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to watch in 2020. MAbs. 2020;12(1):1703531. doi:10.1080/19420862.2019.1703531.
- Kumar S, Plotnikov NV, Rouse JC, Singh SK. Biopharmaceutical informatics: supporting biologic drug development via molecular modelling and informatics. J Pharm Pharmacol. 2018;70(5):595–22. doi:10.1111/jphp.12700.
- Jameson B, Wolf H. The antigenic index: a novel algorithm for predicting antigenic determinants. Bioinformatics. 1988;4(1):181–86. doi:10.1093/bioinformatics/4.1.181.
- Raybould MI, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, Bujotzek A, Shi J, Deane CM. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci U.S.A. 2019;116(10):4025–30. doi:10.1073/pnas.1810576116.
- Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U.S.A. 2017;114(5):944–49. doi:10.1073/pnas.1616408114.
- Obrezanova O, Arnell A, de La Cuesta RG, Berthelot ME, Gallagher TR, Zurdo J, Stallwood Y. Aggregation risk prediction for antibodies and its application to biotherapeutic development. MAbs. 2015;7(2):352–63. doi:10.1080/19420862.2015.1007828.
- Krawczyk K, Buchanan A, Marcatili P. Data mining patented antibody sequences. MAbs. 2021;13(1):1892366. doi:10.1080/19420862.2021.1892366.
- Krawczyk K, Kelm S, Kovaltsuk A, Galson JD, Kelly D, Trück J, Regep C, Leem J, Wong WK, Nowak J. Structurally mapping antibody repertoires. Front Immunol. 2018;9:1698. doi:10.3389/fimmu.2018.01698.
- Krawczyk K, Raybould MI, Kovaltsuk A, Deane CM. Looking for therapeutic antibodies in next-generation sequencing repositories. MAbs. 2019;11(7):1197–205. doi:10.1080/19420862.2019.1633884.
- Deszynski P, Mlokosiewicz J, Volanakis A, Jaszczyszyn I, Castellana N, Bonissone S, Ganesan R, Krawczyk K. INDI-Integrated Nanobody Database for Immunoinformatics. medRxiv. 2021. doi:10.1101/2021.08.04.21261581.
- Kovaltsuk A, Leem J, Kelm S, Snowden J, Deane CM, Krawczyk K. Observed antibody space: a resource for data mining next-generation sequencing of antibody repertoires. J Immunol. 2018;201(8):2502–09. doi:10.4049/jimmunol.1800708.
- Norman RA, Ambrosetti F, Bonvin AM, Colwell LJ, Kelm S, Kumar S, Krawczyk K. Computational approaches to therapeutic antibody design: established methods and emerging trends. Brief Bioinform. 2020;21(5):1549–67. doi:10.1093/bib/bbz095.
- Raybould MI, Marks C, Lewis AP, Shi J, Bujotzek A, Taddese B, Deane CM. Thera-SAbDab: the therapeutic structural antibody database. Nucleic Acids Res. 2020;48(D1):D383–D8. doi:10.1093/nar/gkz827.
- Sirin S, Apgar JR, Bennett EM, Keating AE. AB‐bind: antibody binding mutational database for computational affinity predictions. Protein Sci. 2016;25(2):393–409. doi:10.1002/pro.2829.
- Yadav S, Laue TM, Kalonia DS, Singh SN, Shire SJ. The influence of charge distribution on self-association and viscosity behavior of monoclonal antibody solutions. Mol Pharm. 2012;9(4):791–802. doi:10.1021/mp200566k.
- Li L, Kumar S, Buck PM, Burns C, Lavoie J, Singh SK, Warne NW, Nichols P, Luksha N, Boardman D. Concentration dependent viscosity of monoclonal antibody solutions: explaining experimental behavior in terms of molecular properties. Pharm Res. 2014;31(11):3161–78. doi:10.1007/s11095-014-1409-0.
- Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc Natl Acad Sci U.S.A. 2015;112(19):5997–6002. doi:10.1073/pnas.1408766112.
- Marcatili P, Olimpieri PP, Chailyan A, Tramontano A. Antibody modeling using the Prediction of ImmunoGlobulin Structure (PIGS) web server. Nat Protoc. 2014;9(12):2771. doi:10.1038/nprot.2014.189.
- Leem J, Dunbar J, Georges G, Shi J, Deane CM. ABodyBuilder: automated antibody structure prediction with data–driven accuracy estimation. MAbs. 2016;8(7):1259–68. doi:10.1080/19420862.2016.1205773.
- Weitzner BD, Jeliazkov JR, Lyskov S, Marze N, Kuroda D, Frick R, Adolf-Bryfogle J, Biswas N, Dunbrack JRL, Gray JJ. Modeling and docking of antibody structures with Rosetta. Nat Protoc. 2017;12(2):401. doi:10.1038/nprot.2016.180.
- North B, Lehmann A, Dunbrack JRL. A new clustering of antibody CDR loop conformations. J Mol Biol. 2011;406(2):228–56. doi:10.1016/j.jmb.2010.10.030.
- Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol. 1997;273(4):927–48. doi:10.1006/jmbi.1997.1354.
- Martin AC, Thornton JM. Structural families in loops of homologous proteins: automatic classification, modelling and application to antibodies. J Mol Biol. 1996;263(5):800–15. doi:10.1006/jmbi.1996.0617.
- Weitzner BD, Dunbrack JRL, Gray JJ. The origin of CDR H3 structural diversity. Structure. 2015;23(2):302–11. doi:10.1016/j.str.2014.11.010.
- Kuroda D, Shirai H, Kobori M, Nakamura H. Structural classification of CDR‐H3 revisited: a lesson in antibody modeling. Proteins Struct Funct Bioinf. 2008;73(3):608–20. doi:10.1002/prot.22087.
- Weitzner BD, Kuroda D, Marze N, Xu J, Gray JJ. Blind prediction performance of RosettaAntibody 3.0: grafting, relaxation, kinematic loop modeling, and full CDR optimization. Proteins Struct Funct Bioinf. 2014;82(8):1611–23. doi:10.1002/prot.24534.
- Swindells MB, Porter CT, Couch M, Hurst J, Abhinandan K, Nielsen JH, Macindoe G, Hetherington J, Martin AC. abYsis: integrated antibody sequence and structure—management, analysis, and prediction. J Mol Biol. 2017;429(3):356–64. doi:10.1016/j.jmb.2016.08.019.
- Cheng J, Randall AZ, Sweredoski MJ, Baldi P. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res. 2005;33(suppl_2):W72–W6. doi:10.1093/nar/gki396.
- Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci U.S.A. 2009;106(29):11937–42. doi:10.1073/pnas.0904191106.
- Van Durme J, De Baets G, Van Der Kant R, Ramakers M, Ganesan A, Wilkinson H, Gallardo R, Rousseau F, Schymkowitz J. Solubis: a webserver to reduce protein aggregation through mutation. Protein Eng Des Sel. 2016;29(8):285–89. doi:10.1093/protein/gzw019.
- Gil-Garcia M, Bano-Polo M, Varejao N, Jamroz M, Kuriata A, Díaz-Caballero M, Lascorz J, Morel B, Navarro S, Reverter D. Combining structural aggregation propensity and stability predictions to redesign protein solubility. Mol Pharm. 2018;15(9):3846–59. doi:10.1021/acs.molpharmaceut.8b00341.
- Jain T, Boland T, Lilov A, Burnina I, Brown M, Xu Y, Vásquez M. Prediction of delayed retention of antibodies in hydrophobic interaction chromatography from sequence using machine learning. Bioinformatics. 2017;33(23):3758–66. doi:10.1093/bioinformatics/btx519.
- Lauer TM, Agrawal NJ, Chennamsetty N, Egodage K, Helk B, Trout BL. Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J Pharm Sci. 2012;101(1):102–15. doi:10.1002/jps.22758.
- Jarasch A, Koll H, Regula JT, Bader M, Papadimitriou A, Kettenberger H. Developability assessment during the selection of novel therapeutic antibodies. J Pharm Sci. 2015;104(6):1885–98. doi:10.1002/jps.24430.
- Kohli N, Jain N, Geddie ML, Razlog M, Xu L, Lugovskoy AA. A novel screening method to assess developability of antibody-like molecules. MAbs. 2015;7(4):752–58. doi:10.1080/19420862.2015.1048410.
- Sormanni P, Aprile FA, Vendruscolo M. Third generation antibody discovery methods: in silico rational design. Chem Soc Rev. 2018;47(24):9137–57. doi:10.1039/C8CS00523K.
- Kumar S, Singh SK. Developability of biotherapeutics: computational approaches. Boca Raton (FL), CRC Press; 2015.
- Xu Y, Wang D, Mason B, Rossomando T, Li N, Liu D, Cheung JK, Xu W, Raghava S, Katiyar A. Structure, heterogeneity and developability assessment of therapeutic antibodies. MAbs. 2019;11(2):239–64. doi:10.1080/19420862.2018.1553476.
- Hebditch M, Warwicker J. Charge and hydrophobicity are key features in sequence-trained machine learning models for predicting the biophysical properties of clinical-stage antibodies. PeerJ. 2019;7:e8199. doi:10.7717/peerj.8199.
- King AC, Woods M, Liu W, Lu Z, Gill D, Krebs MR. High‐throughput measurement, correlation analysis, and machine‐learning predictions for pH and thermal stabilities of Pfizer‐generated antibodies. Protein Sci. 2011;20(9):1546–57. doi:10.1002/pro.680.
- Avery LB, Wade J, Wang M, Tam A, King A, Piche-Nicholas N, Kavosi MS, Penn S, Cirelli D, Kurz JC. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. MAbs. 2018;10(2):244–55. doi:10.1080/19420862.2017.1417718.
- Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, Uchida M, Kofman E, Raoufi F, Motlagh S. Predicting antibody developability profiles through early stage discovery screening. MAbs. 2020;12(1):1743053. doi:10.1080/19420862.2020.1743053.
- Erasmus MF, D’Angelo S, Ferrara F, Naranjo L, Teixeira AA, Buonpane R, Stewart SM, Nastri HG, Bradbury AR. A single donor is sufficient to produce a highly functional in vitro antibody library. Commun biol. 2021;4(1):1–16. doi:10.1038/s42003-021-01881-0.
- Tiller T, Schuster I, Deppe D, Siegers K, Strohner R, Herrmann T, Berenguer M, Poujol D, Stehle J, Stark Y. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. MAbs. 2013;5(3):445–70. doi:10.4161/mabs.24218.
- Adler AS, Bedinger D, Adams MS, Asensio MA, Edgar RC, Leong R, Leong J, Mizrahi RA, Spindler MJ, Bandi SR. A natively paired antibody library yields drug leads with higher sensitivity and specificity than a randomly paired antibody library. MAbs. 2018;10(3):431–43. doi:10.1080/19420862.2018.1426422.
- Raybould MI, Marks C, Kovaltsuk A, Lewis AP, Shi J, Deane CM. Public baseline and shared response structures support the theory of antibody repertoire functional commonality. PLoS Comput Biol. 2021;17(3):e1008781. doi:10.1371/journal.pcbi.1008781.
- Adams JJ, Sidhu SS. Synthetic antibody technologies. Curr Opin Struct Biol. 2014;24:1–9. doi:10.1016/j.sbi.2013.11.003.
- Prassler J, Thiel S, Pracht C, Polzer A, Peters S, Bauer M, Nörenberg S, Stark Y, Kölln J, Popp A. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J Mol Biol. 2011;413(1):261–78. doi:10.1016/j.jmb.2011.08.012.
- Zhai W, Glanville J, Fuhrmann M, Mei L, Ni I, Sundar PD, Van Blarcom T, Abdiche Y, Lindquist K, Strohner R. Synthetic antibodies designed on natural sequence landscapes. J Mol Biol. 2011;412(1):55–71. doi:10.1016/j.jmb.2011.07.018.
- Zhao Q, Buhr D, Gunter C, Frenette J, Ferguson M, Sanford E, Holland E, Rajagopal C, Batonick M, Kiss MM. Rational library design by functional CDR resampling. N Biotechnol. 2018;45:89–97. doi:10.1016/j.nbt.2017.12.005.
- Friedensohn S, Neumeier D, Khan TA, Csepregi L, Parola C, de Vries ARG, Erlach L, Mason DM, Reddy ST. Convergent selection in antibody repertoires is revealed by deep learning. bioRxiv. 2020. doi:10.1101/2020.02.25.965673.
- Amimeur T, Shaver JM, Ketchem RR, Taylor JA, Clark RH, Smith J, Van Citters D, Siska CC, Smidt P, Sprague M. Designing feature-controlled humanoid antibody discovery libraries using generative adversarial networks. bioRxiv. 2020. doi:10.1101/2020.04.12.024844.
- Repecka D, Jauniskis V, Karpus L, Rembeza E, Rokaitis I, Zrimec J, Poviloniene S, Laurynenas A, Viknander S, Abuajwa W. Expanding functional protein sequence spaces using generative adversarial networks. Nat Mach Intell. 2021;3(4):324–33. doi:10.1038/s42256-021-00310-5.
- Shin J-E, Riesselman AJ, Kollasch AW, McMahon C, Simon E, Sander C, Manglik A, Kruse AC, Marks DS. Protein design and variant prediction using autoregressive generative models. Nat Commun. 2021;12(1):1–11. doi:10.1038/s41467-021-22732-w.
- Dyson MR, Masters E, Pazeraitis D, Perera RL, Syrjanen JL, Surade S, Thorsteinson N, Parthiban K, Jones PC, Sattar M. Beyond affinity: selection of antibody variants with optimal biophysical properties and reduced immunogenicity from mammalian display libraries. MAbs. 2020;12(1):1829335. doi:10.1080/19420862.2020.1829335.
- Wu Z, Kan SJ, Lewis RD, Wittmann BJ, Arnold FH. Machine learning-assisted directed protein evolution with combinatorial libraries. Proc Natl Acad Sci U.S.A. 2019;116(18):8852–58. doi:10.1073/pnas.1901979116.
- Chen X, Dougherty T, Hong C, Schibler R, Zhao YC, Sadeghi R, Matasci N, Wu Y-C, Kerman I. Predicting antibody developability from sequence using machine learning. bioRxiv. 2020. doi:10.1101/2020.06.18.159798.
- Starr CG, Tessier PM. Selecting and engineering monoclonal antibodies with drug-like specificity. Curr Opin Biotechnol. 2019;60:119–27. doi:10.1016/j.copbio.2019.01.008.
- Vázquez‐Rey M, Lang DA. Aggregates in monoclonal antibody manufacturing processes. Biotechnol Bioeng. 2011;108(7):1494–508. doi:10.1002/bit.23155.
- Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of therapeutic proteins: influence of aggregation. J Immunotoxicol. 2014;11(2):99–109. doi:10.3109/1547691X.2013.821564.
- Seeliger D, Schulz P, Litzenburger T, Spitz J, Hoerer S, Blech M, Enenkel B, Studts JM, Garidel P, Karow AR. Boosting antibody developability through rational sequence optimization. MAbs. 2015;7(3):505–15. doi:10.1080/19420862.2015.1017695.
- van der Kant R, Karow-Zwick AR, Van Durme J, Blech M, Gallardo R, Seeliger D, Aßfalg K, Baatsen P, Compernolle G, Gils A. Prediction and reduction of the aggregation of monoclonal antibodies. J Mol Biol. 2017;429(8):1244–61. doi:10.1016/j.jmb.2017.03.014.
- Kuhn AB, Kube S, Karow-Zwick AR, Seeliger D, Garidel P, Blech M, Schäfer LV. Improved solution-state properties of monoclonal antibodies by targeted mutations. J Phys Chem B. 2017;121(48):10818–27. doi:10.1021/acs.jpcb.7b09126.
- Casaz P, Boucher E, Wollacott R, Pierce BG, Rivera R, Sedic M, Ozturk S, Thomas JWD, Wang Y. Resolving self-association of a therapeutic antibody by formulation optimization and molecular approaches. MAbs. 2014;6(6):1533–39. doi:10.4161/19420862.2014.975658.
- Wu S-J, Luo J, O’Neil KT, Kang J, Lacy ER, Canziani G, Baker A, Huang M, Tang QM, Raju TS. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng Des Sel. 2010;23(8):643–51. doi:10.1093/protein/gzq037.
- Nichols P, Li L, Kumar S, Buck PM, Singh SK, Goswami S, Balthazor B, Conley TR, Sek D, Allen MJ. Rational design of viscosity reducing mutants of a monoclonal antibody: hydrophobic versus electrostatic inter-molecular interactions. MAbs. 2015;7(1):212–30. doi:10.4161/19420862.2014.985504.
- Pindrus M, Shire SJ, Kelley RF, Demeule B, Wong R, Xu Y, Yadav S. Solubility challenges in high concentration monoclonal antibody formulations: relationship with amino acid sequence and intermolecular interactions. Mol Pharm. 2015;12(11):3896–907. doi:10.1021/acs.molpharmaceut.5b00336.
- Yadav S, Sreedhara A, Kanai S, Liu J, Lien S, Lowman H, Kalonia DS, Shire SJ. Establishing a link between amino acid sequences and self-associating and viscoelastic behavior of two closely related monoclonal antibodies. Pharm Res. 2011;28(7):1750–64. doi:10.1007/s11095-011-0410-0.
- Sankar K, Krystek JSR, Carl SM, Day T, Maier JK. AggScore: prediction of aggregation‐prone regions in proteins based on the distribution of surface patches. Proteins Struct Funct Bioinf. 2018;86(11):1147–56. doi:10.1002/prot.25594.
- Liu YD, Goetze AM, Bass RB, Flynn GC. N-terminal glutamate to pyroglutamate conversion in vivo for human IgG2 antibodies. J Biol Chem. 2011;286(13):11211–17. doi:10.1074/jbc.M110.185041.
- Chelius D, Jing K, Lueras A, Rehder DS, Dillon TM, Vizel A, Rajan RS, Li T, Treuheit MJ, Bondarenko PV. Formation of pyroglutamic acid from N-terminal glutamic acid in immunoglobulin gamma antibodies. Anal Chem. 2006;78(7):2370–76. doi:10.1021/ac051827k.
- Yu L, Vizel A, Huff MB, Young M, Remmele JRL, He B. Investigation of N-terminal glutamate cyclization of recombinant monoclonal antibody in formulation development. J Pharm Biomed Anal. 2006;42(4):455–63. doi:10.1016/j.jpba.2006.05.008.
- Tang L, Sundaram S, Zhang J, Carlson P, Matathia A, Parekh B, Zhou Q, Hsieh M-C. Conformational characterization of the charge variants of a human IgG1 monoclonal antibody using H/D exchange mass spectrometry. MAbs. 2013;5(1):114–25. doi:10.4161/mabs.22695.
- van Den Bremer ET, Beurskens FJ, Voorhorst M, Engelberts PJ, de Jong RN, van der Boom BG, Cook EM, Lindorfer MA, Taylor RP, van Berkel PH. Human IgG is produced in a pro-form that requires clipping of C-terminal lysines for maximal complement activation. MAbs. 2015;7(4):672–80. doi:10.1080/19420862.2015.1046665.
- Liu H, Nowak C, Shao M, Ponniah G, Neill A. Impact of cell culture on recombinant monoclonal antibody product heterogeneity. Biotechnol Prog. 2016;32(5):1103–12. doi:10.1002/btpr.2327.
- Füssl F, Trappe A, Cook K, Scheffler K, Fitzgerald O, Bones J. Comprehensive characterisation of the heterogeneity of Adalimumab via charge variant analysis hyphenated on-line to native high resolution Orbitrap mass spectrometry. MAbs. 2019;11(1):116–28. doi:10.1080/19420862.2018.1531664.
- Sydow JF, Lipsmeier F, Larraillet V, Hilger M, Mautz B, Mølhøj M, Kuentzer J, Klostermann S, Schoch J, Voelger HR. Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. PloS One. 2014;9(6):e100736. doi:10.1371/journal.pone.0100736.
- Verma A, Ngundi MM, Burns DL. Mechanistic analysis of the effect of deamidation on the immunogenicity of anthrax protective antigen. Clin vaccine immunol. 2016;23(5):396. doi:10.1128/CVI.00701-15.
- Mo J, Yan Q, So CK, Soden T, Lewis MJ, Hu P. Understanding the impact of methionine oxidation on the biological functions of IgG1 antibodies using hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2016;88(19):9495–502. doi:10.1021/acs.analchem.6b01958.
- Dashivets T, Stracke J, Dengl S, Knaupp A, Pollmann J, Buchner J, Schlothauer T. Oxidation in the complementarity-determining regions differentially influences the properties of therapeutic antibodies. MAbs. 2016;8(8):1525–35. doi:10.1080/19420862.2016.1231277.
- Liu-Shin LP-Y, Fung A, Malhotra A, Ratnaswamy G. Evidence of disulfide bond scrambling during production of an antibody-drug conjugate. MAbs. 2018;10(8):1190–99. doi:10.1080/19420862.2018.1521128.
- Moritz B, Stracke JO. Assessment of disulfide and hinge modifications in monoclonal antibodies. Electrophoresis. 2017;38(6):769–85. doi:10.1002/elps.201600425.
- Wright A, Tao M, Kabat E, Morrison S. Antibody variable region glycosylation: position effects on antigen binding and carbohydrate structure. EMBO J. 1991;10(10):2717–23. doi:10.1002/j.1460-2075.1991.tb07819.x.
- Leibiger H, Wüstner D, Stigler R-D, Marx U. Variable domain-linked oligosaccharides of a human monoclonal IgG: structure and influence on antigen binding. Biochem J. 1999;338(2):529–38. doi:10.1042/bj3380529.
- van Bueren JJL, Rispens T, Verploegen S, van der Palen-merkus T, Stapel S, Workman LJ, James H, van Berkel PH, van de Winkel JG, Platts-Mills TA. Anti-galactose-α-1, 3-galactose IgE from allergic patients does not bind α-galactosylated glycans on intact therapeutic antibody Fc domains. Nat Biotechnol. 2011;29(7):574–76. doi:10.1038/nbt.1912.
- Jefferis R. Posttranslational modifications and the immunogenicity of biotherapeutics. J Immunol Res. 2016;2016:1–15. doi:10.1155/2016/5358272.
- Muster W, Breidenbach A, Fischer H, Kirchner S, Müller L, Pähler A. Computational toxicology in drug development. Drug Discov Today. 2008;13(7–8):303–10. doi:10.1016/j.drudis.2007.12.007.
- Kuang Q, Wang M, Li R, Dong Y, Li Y, Li M. A systematic investigation of computation models for predicting Adverse Drug Reactions (ADRs). PloS One. 2014;9(9):e105889. doi:10.1371/journal.pone.0105889.
- Bryson CJ, Jones TD, Baker MP. Prediction of immunogenicity of therapeutic proteins. BioDrugs. 2010;24(1):1–8. doi:10.2165/11318560-000000000-00000.
- Baker M, Reynolds HM, Lumicisi B, Bryson CJ. Immunogenicity of protein therapeutics: the key causes, consequences and challenges. Self/nonself. 2010;1(4):314–22. doi:10.4161/self.1.4.13904.
- Diao L, Meibohm B. Tools for predicting the PK/PD of therapeutic proteins. Expert Opin Drug Metab Toxicol. 2015;11(7):1115–25. doi:10.1517/17425255.2015.1041917.
- Wang J, Iyer S, Fielder PJ, Davis JD, Deng R. Projecting human pharmacokinetics of monoclonal antibodies from nonclinical data: comparative evaluation of prediction approaches in early drug development. Biopharm Drug Dispos. 2016;37(2):51–65. doi:10.1002/bdd.1952.
- Grinshpun B, Thorsteinson N, Pereira JN, Rippmann F, Nannemann D, Sood VD, Fomekong Nanfack Y. Identifying biophysical assays and in silico properties that enrich for slow clearance in clinical-stage therapeutic antibodies. MAbs. 2021;13(1):1932230. doi:10.1080/19420862.2021.1932230.
- Desai DV, Kulkarni-Kale U. T-cell epitope prediction methods: an overview. Immunoinformatics. 2014:333–64. doi:10.1007/978-1-4939-1115-8_19.
- Peters B, Nielsen M, Sette A. T cell epitope predictions. Annu Rev Immunol. 2020;38:123–45. doi:10.1146/annurev-immunol-082119-124838.
- Reche PA, Glutting J-P, Zhang H, Reinherz EL. Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics. 2004;56(6):405–19. doi:10.1007/s00251-004-0709-7.
- Singh H, Raghava G. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 2001;17(12):1236–37. doi:10.1093/bioinformatics/17.12.1236.
- Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, Sahin U, Braxenthaler M, Gallazzi F, Protti MP, Sinigaglia F. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol. 1999;17(6):555–61. doi:10.1038/9858.
- Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinform. 2007;8(1):1–12. doi:10.1186/1471-2105-8-238.
- Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4(4):e1000048. doi:10.1371/journal.pcbi.1000048.
- Zhang GL, Khan AM, Srinivasan KN, August JT, Brusic V. MULTIPRED: a computational system for prediction of promiscuous HLA binding peptides. Nucleic Acids Res. 2005;33(suppl_2):W172–W9. doi:10.1093/nar/gki452.
- Andreatta M, Trolle T, Yan Z, Greenbaum JA, Peters B, Nielsen M. An automated benchmarking platform for MHC class II binding prediction methods. Bioinformatics. 2018;34(9):1522–28. doi:10.1093/bioinformatics/btx820.
- Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48(W1):W449–W54. doi:10.1093/nar/gkaa379.
- Nielsen M, Lundegaard C, Blicher T, Peters B, Sette A, Justesen S, Buus S, Lund O. Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Comput Biol. 2008;4(7):e1000107. doi:10.1371/journal.pcbi.1000107.
- Jacob L, Vert J-P. Efficient peptide–MHC-I binding prediction for alleles with few known binders. Bioinformatics. 2008;24(3):358–66. doi:10.1093/bioinformatics/btm611.
- Zhang H, Lundegaard C, Nielsen M. Pan-specific MHC class I predictors: a benchmark of HLA class I pan-specific prediction methods. Bioinformatics. 2009;25(1):83–89. doi:10.1093/bioinformatics/btn579.
- Yachnin BJ, Mulligan VK, Khare SD, Bailey-Kellogg C. MHCEpitopeEnergy, a flexible Rosetta-based biotherapeutic deimmunization platform. J Chem Inf Model. 2021;61(5):2368–82. doi:10.1021/acs.jcim.1c00056.
- Peng H-P, Lee KH, Jian J-W, Yang A-S. Origins of specificity and affinity in antibody–protein interactions. Proc Natl Acad Sci U.S.A. 2014;111(26):E2656–E65. doi:10.1073/pnas.1401131111.
- Hopp TP, Woods KR. Prediction of protein antigenic determinants from amino acid sequences. Proc Natl Acad Sci U.S.A. 1981;78(6):3824–28. doi:10.1073/pnas.78.6.3824.
- Welling GW, Weijer WJ, van der Zee R, Welling-Wester S. Prediction of sequential antigenic regions in proteins. FEBS Lett. 1985;188(2):215–18. doi:10.1016/0014-5793(85)80374-4.
- Van Regenmortel M, De Marcillac GD. An assessment of prediction methods for locating continuous epitopes in proteins. Immunol Lett. 1988;17(2):95–107. doi:10.1016/0165-2478(88)90076-4.
- Pellequer J, Westhof E. PREDITOP: a program for antigenicity prediction. J Mol Graph. 1993;11(3):204–10. doi:10.1016/0263-7855(93)80074-2.
- Saha S, Raghava GPS. Prediction of continuous B‐cell epitopes in an antigen using recurrent neural network. Proteins Struct Funct Bioinf. 2006;65(1):40–48. doi:10.1002/prot.21078.
- Gao J, Faraggi E, Zhou Y, Ruan J, Kurgan L. BEST: improved prediction of B-cell epitopes from antigen sequences. PLoS One. 2012;7(6):e40104. doi:10.1371/journal.pone.0040104.
- Singh H, Ansari HR, Raghava GP. Improved method for linear B-cell epitope prediction using antigen’s primary sequence. PLoS One. 2013;8(5):e62216. doi:10.1371/journal.pone.0062216.
- Yao B, Zhang L, Liang S, Zhang C. SVMTriP: a method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity. PloS One. 2012;7(9):e45152. doi:10.1371/journal.pone.0045152.
- Kulkarni-Kale U, Bhosle S, Kolaskar AS. CEP: a conformational epitope prediction server. Nucleic Acids Res. 2005;33(suppl_2):W168–W71. doi:10.1093/nar/gki460.
- Zinsli LV, Stierlin N, Loessner MJ, Schmelcher M. Deimmunization of protein therapeutics–Recent advances in experimental and computational epitope prediction and deletion. Comput Struct Biotechnol J. 2020;19:315–29. doi:10.1016/j.csbj.2020.12.024.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1–3):3–25. doi:10.1016/S0169-409X(96)00423-1.
- Lecerf M, Kanyavuz A, Lacroix-Desmazes S, Dimitrov JD. Sequence features of variable region determining physicochemical properties and polyreactivity of therapeutic antibodies. Mol Immunol. 2019;112:338–46. doi:10.1016/j.molimm.2019.06.012.
- Hebditch M, Carballo-Amador MA, Charonis S, Curtis R, Warwicker J. Protein–Sol: a web tool for predicting protein solubility from sequence. Bioinformatics. 2017;33(19):3098–100. doi:10.1093/bioinformatics/btx345.
- Hou Q, Kwasigroch JM, Rooman M, Pucci F. SOLart: a structure-based method to predict protein solubility and aggregation. Bioinformatics. 2020;36(5):1445–52. doi:10.1093/bioinformatics/btz773.
- Sormanni P, Aprile FA, Vendruscolo M. The CamSol method of rational design of protein mutants with enhanced solubility. J Mol Biol. 2015;427(2):478–90. doi:10.1016/j.jmb.2014.09.026.
- Sormanni P, Amery L, Ekizoglou S, Vendruscolo M, Popovic B. Rapid and accurate in silico solubility screening of a monoclonal antibody library. Sci Rep. 2017;7(1):1–9. doi:10.1038/s41598-017-07800-w.
- Shan L, Mody N, Sormani P, Rosenthal KL, Damschroder MM, Esfandiary R. Developability assessment of engineered monoclonal antibody variants with a complex self-association behavior using complementary analytical and in silico tools. Mol Pharm. 2018;15(12):5697–710. doi:10.1021/acs.molpharmaceut.8b00867.
- Wolf Pérez A-M, Sormanni P, Andersen JS, Sakhnini LI, Rodriguez-Leon I, Bjelke JR, Gajhede AJ, De Maria L, Otzen DE, Vendruscolo M. In vitro and in silico assessment of the developability of a designed monoclonal antibody library. MAbs. 2019;11(2):388–400. doi:10.1080/19420862.2018.1556082.
- Kingsbury JS, Saini A, Auclair SM, Fu L, Lantz MM, Halloran KT, Calero-Rubio C, Schwenger W, Airiau CY, Zhang J. A single molecular descriptor to predict solution behavior of therapeutic antibodies. Sci Adv. 2020;6(32):eabb0372. doi:10.1126/sciadv.abb0372.
- Hanke AT, Klijn ME, Verhaert PD, van der Wielen LA, Ottens M, Eppink MH, van de Sandt EJ. Prediction of protein retention times in hydrophobic interaction chromatography by robust statistical characterization of their atomic‐level surface properties. Biotechnol Prog. 2016;32(2):372–81. doi:10.1002/btpr.2219.
- Goyon A, D’Atri V, Colas O, Fekete S, Beck A, Guillarme D. Characterization of 30 therapeutic antibodies and related products by size exclusion chromatography: feasibility assessment for future mass spectrometry hyphenation. J Chromatogr B. 2017;1065:35–43. doi:10.1016/j.jchromb.2017.09.027.
- Hebditch M, Roche A, Curtis RA, Warwicker J. Models for antibody behavior in hydrophobic interaction chromatography and in self-association. J Pharm Sci. 2019;108(4):1434–41. doi:10.1016/j.xphs.2018.11.035.
- Sundberg EJ, Urrutia M, Braden BC, Isern J, Tsuchiya D, Fields BA, Malchiodi EL, Tormo J, Schwarz FP, Mariuzza RA. Estimation of the hydrophobic effect in an antigen− antibody protein− protein interface. Biochemistry. 2000;39(50):15375–87. doi:10.1021/bi000704l.
- Almagro JC, Fransson J. Humanization of antibodies. Front Biosci. 2008;13(1):1619–33. doi:10.2741/2786.
- Roguska MA, Pedersen JT, Keddy CA, Henry AH, Searle SJ, Lambert JM, Goldmacher VS, Blättler W, Rees AR, Guild BC. Humanization of murine monoclonal antibodies through variable domain resurfacing. Proc Natl Acad Sci U.S.A. 1994;91(3):969–73. doi:10.1073/pnas.91.3.969.
- Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, Dijkmans BA, Aarden L, Wolbink GJ. Development of antidrug antibodies against Adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305(14):1460–68. doi:10.1001/jama.2011.406.
- Sekiguchi N, Kubo C, Takahashi A, Muraoka K, Takeiri A, Ito S, Yano M, Mimoto F, Maeda A, Iwayanagi Y. MHC-associated peptide proteomics enabling highly sensitive detection of immunogenic sequences for the development of therapeutic antibodies with low immunogenicity. MAbs. 2018;10(8):1168–81. doi:10.1080/19420862.2018.1518888.
- Karle AC. Applying MAPPs assays to assess drug immunogenicity. Front Immunol. 2020;11:698. doi:10.3389/fimmu.2020.00698.
- Abhinandan K, Martin AC. Analyzing the “degree of humanness” of antibody sequences. J Mol Biol. 2007;369(3):852–62. doi:10.1016/j.jmb.2007.02.100.
- Gao SH, Huang K, Tu H, Adler AS. Monoclonal antibody humanness score and its applications. BMC Biotechnol. 2013;13(1):1–12. doi:10.1186/1472-6750-13-55.
- Thullier P, Huish O, Pelat T, Martin AC. The humanness of macaque antibody sequences. J Mol Biol. 2010;396(5):1439–50. doi:10.1016/j.jmb.2009.12.041.
- Schmitz S, Soto C, Crowe JJE, Meiler J. Human-likeness of antibody biologics determined by back-translation and comparison with large antibody variable gene repertoires. MAbs. 2020;12(1):1758291. doi:10.1080/19420862.2020.1758291.
- Chin M, Marks C, Deane CM. Humanization of antibodies using a machine learning approach on large-scale repertoire data. bioRxiv. 2021. doi:10.1093/bioinformatics/btab434.
- Chailyan A, Tramontano A, Marcatili P. A database of immunoglobulins with integrated tools: DIGIT. Nucleic Acids Res. 2012;40(D1):D1230–D4. doi:10.1093/nar/gkr806.
- Christley S, Scarborough W, Salinas E, Rounds WH, Toby IT, Fonner JM, Levin MK, Kim M, Mock SA, Jordan C. VDJServer: a cloud-based analysis portal and data commons for immune repertoire sequences and rearrangements. Front Immunol. 2018;9:976. doi:10.3389/fimmu.2018.00976.
- Corrie BD, Marthandan N, Zimonja B, Jaglale J, Zhou Y, Barr E, Knoetze N, Breden FM, Christley S, Scott JK. iReceptor: a platform for querying and analyzing antibody/B‐cell and T‐cell receptor repertoire data across federated repositories. Immunol Rev. 2018;284(1):24–41. doi:10.1111/imr.12666.
- DeWitt WS, Lindau P, Snyder TM, Sherwood AM, Vignali M, Carlson CS, Greenberg PD, Duerkopp N, Emerson RO, Robins HS. A public database of memory and naive B-cell receptor sequences. PLoS One. 2016;11(8):e0160853. doi:10.1371/journal.pone.0160853.
- Rosenfeld AM, Meng W, Luning Prak ET, Hershberg U. Immunedb, a novel tool for the analysis, storage, and dissemination of immune repertoire sequencing data. Front Immunol. 2018;9:2107. doi:10.3389/fimmu.2018.02107.
- Zhang W, Wang L, Liu K, Wei X, Yang K, Du W, Wang S, Guo N, Ma C, Luo L. PIRD: pan immune repertoire database. Bioinformatics. 2020;36(3):897–903. doi:10.1093/bioinformatics/btz614.
- Marks C, Deane CM. How repertoire data are changing antibody science. J Biol Chem. 2020;295(29):9823–37. doi:10.1074/jbc.REV120.010181.
- Galson JD, Clutterbuck EA, Trück J, Ramasamy MN, Münz M, Fowler A, Cerundolo V, Pollard AJ, Lunter G, Kelly DF. BCR repertoire sequencing: different patterns of B‐cell activation after two Meningococcal vaccines. Immunol Cell Biol. 2015;93(10):885–95. doi:10.1038/icb.2015.57.
- Miho E, Yermanos A, Weber CR, Berger CT, Reddy ST, Greiff V. Computational strategies for dissecting the high-dimensional complexity of adaptive immune repertoires. Front Immunol. 2018;9:224. doi:10.3389/fimmu.2018.00224.
- Briney B, Inderbitzin A, Joyce C, Burton DR. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature. 2019;566(7744):393–97. doi:10.1038/s41586-019-0879-y.
- Slabodkin A, Chernigovskaya M, Mikocziova I, Akbar R, Scheffer L, Pavlović M, Bashour H, Snapkov I, Mehta BB, Weber CR. Individualized VDJ recombination predisposes the available Ig sequence space. bioRxiv. 2021. doi:10.1101/2021.04.19.440409.
- Bashford‐Rogers RJ, Smith KG, Thomas DC. Antibody repertoire analysis in polygenic autoimmune diseases. Immunology. 2018;155(1):3–17. doi:10.1111/imm.12927.
- Liu J, Yang X, Lu X, Zhang L, Luo W, Cheng Y, Zhang L, Yang Y, Dai N, Xu Y. Impact of T-cell receptor and B-cell receptor repertoire on the recurrence of early stage lung adenocarcinoma. Exp Cell Res. 2020;394(2):112134. doi:10.1016/j.yexcr.2020.112134.
- Galson JD, Pollard AJ, Trück J, Kelly DF. Studying the antibody repertoire after vaccination: practical applications. Trends Immunol. 2014;35(7):319–31. doi:10.1016/j.it.2014.04.005.
- Galson JD, Schaetzle S, Bashford-Rogers RJ, Raybould MI, Kovaltsuk A, Kilpatrick GJ, Minter R, Finch DK, Dias J, James LK. Deep sequencing of B cell receptor repertoires from COVID-19 patients reveals strong convergent immune signatures. Front Immunol. 2020;11:3283. doi:10.3389/fimmu.2020.605170.
- Richardson E, Galson JD, Kellam P, Kelly DF, Smith SE, Palser A, Watson S, Deane CM. A computational method for immune repertoire mining that identifies novel binders from different clonotypes, demonstrated by identifying anti-Pertussis toxoid antibodies. MAbs. 2021;13(1):1869406. doi:10.1080/19420862.2020.1869406.
- Wong WK, Robinson SA, Bujotzek A, Georges G, Lewis AP, Shi J, Snowden J, Taddese B, Deane CM. Ab-Ligity: identifying sequence-dissimilar antibodies that bind to the same epitope. MAbs. 2021;13(1):1873478. doi:10.1080/19420862.2021.1873478.
- Schritt D, Li S, Rozewicki J, Katoh K, Yamashita K, Volkmuth W, Cavet G, Standley DM. Repertoire builder: high-throughput structural modeling of B and T cell receptors. Mol Syst Des Eng. 2019;4(4):761–68. doi:10.1039/C9ME00020H.
- DeKosky BJ, Lungu OI, Park D, Johnson EL, Charab W, Chrysostomou C, Kuroda D, Ellington AD, Ippolito GC, Gray JJ. Large-scale sequence and structural comparisons of human naive and antigen-experienced antibody repertoires. Proc Natl Acad Sci U.S.A. 2016;113(19):E2636–E45. doi:10.1073/pnas.1525510113.
- Raybould MI, Marks C, Kovaltsuk A, Lewis AP, Shi J, Deane C. Evidence of antibody repertoire functional convergence through public baseline and shared response structures. bioRxiv. 2020. doi:10.1101/2020.03.17.993444.
- Robinson SA, Raybould MI, Marks C, Schneider C, Wong WK, Deane CM. Epitope profiling of coronavirus-binding antibodies using computational structural modelling. bioRxiv. 2021. doi:10.1101/2021.04.12.439478.
- Kovaltsuk A, Raybould MI, Wong WK, Marks C, Kelm S, Snowden J, Trück J, Deane CM. Structural diversity of B-cell receptor repertoires along the B-cell differentiation axis in humans and mice. PLoS Comput Biol. 2020;16(2):e1007636. doi:10.1371/journal.pcbi.1007636.
- Setliff I, Shiakolas AR, Pilewski KA, Murji AA, Mapengo RE, Janowska K, Richardson S, Oosthuysen C, Raju N, Ronsard L. High-throughput mapping of B cell receptor sequences to antigen specificity. Cell. 2019;179(7):1636–46. e15. doi:10.1016/j.cell.2019.11.003.
- Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the’high-hanging fruit’. Nat Rev Drug Discov. 2018;17(3):197. doi:10.1038/nrd.2017.227.
- Beck A, D’atri V, Ehkirch A, Fekete S, Hernandez-Alba O, Gahoual R, Leize-Wagner E, François Y, Guillarme D, Cianférani S. Cutting-edge multi-level analytical and structural characterization of antibody-drug conjugates: present and future. Expert Rev Proteomics. 2019;16(4):337–62. doi:10.1080/14789450.2019.1578215.
- Khongorzul P, Ling CJ, Khan FU, Ihsan AU, Zhang J. Antibody–drug conjugates: a comprehensive review. Mol Cancer Res. 2020;18(1):3–19. doi:10.1158/1541-7786.MCR-19-0582.
- Leung D, Wurst JM, Liu T, Martinez RM, Datta-Mannan A, Feng Y. Antibody conjugates-recent advances and future innovations. Antibodies. 2020;9(1):2. doi:10.3390/antib9010002.
- Wilton EE, Opyr MP, Kailasam S, Kothe RF, Wieden H-J. sdAb-DB: the single domain antibody database. ACS Synth Biol. 2018;7(11):2480–84. doi:10.1021/acssynbio.8b00407.
- Zavrtanik U, Hadži S. A non-redundant data set of nanobody-antigen crystal structures. Data Brief. 2019;24:103754. doi:10.1016/j.dib.2019.103754.
- Zuo J, Li J, Zhang R, Xu L, Chen H, Jia X, Su Z, Zhao L, Huang X, Xie W. Institute collection and analysis of Nanobodies (iCAN): a comprehensive database and analysis platform for nanobodies. BMC Genom. 2017;18(1):1–5. doi:10.1186/s12864-017-4204-6.
- Gentiluomo L, Svilenov HL, Augustijn D, El Bialy I, Greco ML, Kulakova A, Indrakumar S, Mahapatra S, Morales MM, Pohl C. Advancing therapeutic protein discovery and development through comprehensive computational and biophysical characterization. Mol Pharm. 2019;17(2):426–40. doi:10.1021/acs.molpharmaceut.9b00852.
- Pollastri G, Przybylski D, Rost B, Baldi P. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins Struct Funct Bioinf. 2002;47(2):228–35. doi:10.1002/prot.10082.
- Magnan CN, Baldi P. SSpro/ACCpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics. 2014;30(18):2592–97. doi:10.1093/bioinformatics/btu352.
- Adamczak R, Porollo A, Meller J. Accurate prediction of solvent accessibility using neural networks–based regression. Proteins Struct Funct Bioinf. 2004;56(4):753–67. doi:10.1002/prot.20176.
- Yuan Z, Burrage K, Mattick JS. Prediction of protein solvent accessibility using support vector machines. Proteins Struct Funct Bioinf. 2002;48(3):566–70. doi:10.1002/prot.10176.
- Nepal R, Spencer J, Bhogal G, Nedunuri A, Poelman T, Kamath T, Chung E, Kantardjieff K, Gottlieb A, Lustig B. Logistic regression models to predict solvent accessible residues using sequence-and homology-based qualitative and quantitative descriptors applied to a domain-complete X-ray structure learning set. J Appl Crystallogr. 2015;48(6):1976–84. doi:10.1107/S1600576715018531.
- Joo K, Lee SJ, Lee J. Sann: solvent accessibility prediction of proteins by nearest neighbor method. Proteins Struct Funct Bioinf. 2012;80(7):1791–97. doi:10.1002/prot.24074.
- Noé F, De Fabritiis G, Clementi C. Machine learning for protein folding and dynamics. Curr Opin Struct Biol. 2020;60:77–84. doi:10.1016/j.sbi.2019.12.005.
- Raimondi D, Orlando G, Pancsa R, Khan T, Vranken WF. Exploring the sequence-based prediction of folding initiation sites in proteins. Sci Rep. 2017;7(1):1–11. doi:10.1038/s41598-017-08366-3.
- Tan AC, Gilbert D, Deville Y. Multi-class protein fold classification using a new ensemble machine learning approach. Genome Inf. 2003;14:206–17. doi:10.11234/gi1990.14.206.
- Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Žídek A, Nelson AW, Bridgland A. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577(7792):706–10. doi:10.1038/s41586-019-1923-7.
- Liu S, Liu C, Deng L. Machine learning approaches for protein–protein interaction hot spot prediction: progress and comparative assessment. Molecules. 2018;23(10):2535. doi:10.3390/molecules23102535.
- Zhang M, Su Q, Lu Y, Zhao M, Niu B. Application of machine learning approaches for protein-protein interactions prediction. Med Chem (Los Angeles). 2017;13(6):506–14. doi:10.2174/1573406413666170522150940.
- Sarkar D, Saha S. Machine-learning techniques for the prediction of protein–protein interactions. J Biosci. 2019;44(4):1–12. doi:10.1007/s12038-019-9909-z.
- Melo R, Fieldhouse R, Melo A, Correia JD, Cordeiro MND, Gümüş ZH, Costa J, Bonvin AM, Moreira IS. A machine learning approach for hot-spot detection at protein-protein interfaces. Int J Mol Sci. 2016;17(8):1215. doi:10.3390/ijms17081215.
- Wang W, Yang Y, Yin J, Gong X, Wang Z, Tamada K, Takumi T, Hashimoto R, Otani H, Pazour GJ. Different protein-protein interface patterns predicted by different machine learning methods. Sci Rep. 2017;7(1):1–13. doi:10.1038/s41598-017-16397-z.
- Wang D, Liu D, Yuchi J, He F, Jiang Y, Cai S, Li J, Xu D. MusiteDeep: a deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res. 2020;48(W1):W140–W6. doi:10.1093/nar/gkaa275.
- Xu Y, Chou K-C. Recent progress in predicting posttranslational modification sites in proteins. Curr Top Med Chem. 2016;16(6):591–603. doi:10.2174/1568026615666150819110421.
- Bao W, Yuan C-A, Zhang Y, Han K, Nandi AK, Honig B, Huang D-S. Mutli-features prediction of protein translational modification sites. IEEE/ACM Trans Comput Biol Bioinf. 2017;15(5):1453–60. doi:10.1109/TCBB.2017.2752703.
- Sankar K, Hoi KH, Yin Y, Ramachandran P, Andersen N, Hilderbrand A, McDonald P, Spiess C, Zhang Q. Prediction of methionine oxidation risk in monoclonal antibodies using a machine learning method. MAbs. 2018;10(8):1281–90. doi:10.1080/19420862.2018.1518887.
- Lai P-K, Fernando A, Cloutier TK, Kingsbury JS, Gokarn Y, Halloran KT, Calero-Rubio C, Trout BL. Machine learning feature selection for predicting high concentration therapeutic antibody aggregation. J Pharm Sci. 2021;110(4):1583–91. doi:10.1016/j.xphs.2020.12.014.
- Long X, Jeliazkov JR, Gray JJ. Non-H3 CDR template selection in antibody modeling through machine learning. PeerJ. 2019;7:e6179. doi:10.7717/peerj.6179.
- Wong WK, Georges G, Ros F, Kelm S, Lewis AP, Taddese B, Leem J, Deane CM. SCALOP: sequence-based antibody canonical loop structure annotation. Bioinformatics. 2019;35(10):1774–76. doi:10.1093/bioinformatics/bty877.
- Raimondi D, Orlando G, Vranken WF, Moreau Y. Exploring the limitations of biophysical propensity scales coupled with machine learning for protein sequence analysis. Sci Rep. 2019;9(1):1–11. doi:10.1038/s41598-019-53324-w.
- Narayanan H, Dingfelder F, Butté A, Lorenzen N, Sokolov M, Arosio P. Machine learning for biologics: opportunities for protein engineering, developability, and formulation. Trends Pharmacol Sci. 2021;42(3):151–65. doi:10.1016/j.tips.2020.12.004.
- Graves J, Byerly J, Priego E, Makkapati N, Parish SV, Medellin B, Berrondo M. A review of deep learning methods for antibodies. Antibodies. 2020;9(2):12. doi:10.3390/antib9020012.
- Liberis E, Veličković P, Sormanni P, Vendruscolo M, Liò P. Parapred: antibody paratope prediction using convolutional and recurrent neural networks. Bioinformatics. 2018;34(17):2944–50. doi:10.1093/bioinformatics/bty305.
- Deac A, Veličković P, Sormanni P. Attentive cross-modal paratope prediction. J Comput Biol. 2019;26(6):536–45. doi:10.1089/cmb.2018.0175.
- Akbar R, Robert PA, Pavlović M, Jeliazkov JR, Snapkov I, Slabodkin A, Weber CR, Scheffer L, Miho E, Haff IH. A compact vocabulary of paratope-epitope interactions enables predictability of antibody-antigen binding. Cell Rep. 2021;34(11):108856. doi:10.1016/j.celrep.2021.108856.
- Liu G, Zeng H, Mueller J, Carter B, Wang Z, Schilz J, Horny G, Birnbaum ME, Ewert S, Gifford DK. Antibody complementarity determining region design using high-capacity machine learning. Bioinformatics. 2020;36(7):2126–33. doi:10.1093/bioinformatics/btz895.
- Mason DM, Friedensohn S, Weber CR, Jordi C, Wagner B, Meng S, Gainza P, Correia BE, Reddy ST. Deep learning enables therapeutic antibody optimization in mammalian cells by deciphering high-dimensional protein sequence space. bioRxiv. 2019:617860. doi:10.1101/617860.
- Dunbar J, Krawczyk K, Leem J, Baker T, Fuchs A, Georges G, Shi J, Deane CM. SAbDab: the structural antibody database. Nucleic Acids Res. 2014;42(D1):D1140–D6. doi:10.1093/nar/gkt1043.
- Allcorn LC, Martin AC. SACS—self-maintaining database of antibody crystal structure information. Bioinformatics. 2002;18(1):175–81. doi:10.1093/bioinformatics/18.1.175.
- Ferdous S, Martin AC. AbDb: antibody structure database—a database of PDB-derived antibody structures. Database. 2018:2018. doi:10.1093/database/bay040.
- Khetan R. Biopharma licensing and M&A trends in the 21st-century landscape. J Commer Biotechnol. 2020;25(3). doi:10.5912/jcb943.
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A. Highly accurate protein structure prediction with AlphaFold. Nature. 2021:1–11. doi:10.1038/s41586-021-03819-2.