
ABSTRACT
Targeting PD1/PDL1 with blocking antibodies for cancer therapy has shown promising benefits in the clinic, but only approximately 20–30% of patients develop durable clinical responses to the treatment. Bispecific antibodies (BsAbs) that combine PD1/PDL1 blockade with the modulation of another immune checkpoint target may have greater potential to enhance immune checkpoint blockade therapy. In this study, we identified an anti-PD1 monoclonal antibody, 609A, whose heavy chain can pair with a variety of light chains from different antibodies while maintaining its PD1 binding/blocking activity. Taking advantage of this property and using a linear F(ab’)2 format, we successfully produced a series of tetravalent IgG-like BsAbs that simultaneously target PD1 and other immune checkpoint targets, including PDL1 and CTLA4. The BsAbs exhibited superior bioactivities in vitro and in vivo compared to their respective parental mAbs. Importantly, the BsAbs demonstrated the desired IgG-like physicochemical properties in terms of high-level expression, ease of purification to homogeneity, good stability and in vivo pharmacokinetics. In summary, we describe a novel and flexible plug-and-play platform to engineer IgG-like BsAbs with excellent development potential for clinical applications.
Introduction
Bispecific antibodies (BsAbs) are a class of antibodies comprising two antigen-targeting domains that can simultaneously act on two distinct targets or two different epitopes of the same target to provide additive benefits that cannot be achieved by a monoclonal antibody (mAb) or a simple combination of mAbs. Because of the high potential of BsAbs in therapeutic applications, dozens of BsAbs are currently under preclinical development or are being evaluated in the clinic.Citation1
Since the first BsAb was described almost half a century ago, only five molecules, catumaxomab, blinatumomab, emicizumab, amivantamab, and faricimab have been successfully launched into the market.Citation2 One of the obstacles to broad BsAb application is the complexity of developing BsAbs. Unlike naturally occurring bivalent IgGs that consist of two symmetrical heavy and light chains, most BsAbs are engineered hybrid molecules that do not assume a natural form.Citation2,Citation3 For example, in the construction of an IgG-like format BsAb that combines two distinct heavy (H) and light (L) chain pairs (H-L) for targeting two different antigens into a single IgG format,Citation3 the pairing of the two asymmetrical H-L pairs is a substantial engineering obstacle, as only 1 of 10 of the pairings can generate correct asymmetrical H-L pairs. Strategies for the heterodimerization of heavy chains, such as knobs-into-holes,Citation4 duobody,Citation5,Citation6 electrostatic steeringCitation7 or strand-exchange engineered domains (SEEDs),Citation8 partially circumvent the mispairing of heavy chains, while strategies with common light chains,Citation9,Citation10 crossover of CH1 and CL domainsCitation11 and species-restricted H-L pairingCitation12,Citation13 improve the pairing of light chains with heavy chains. Despite these efforts, BsAbs created with such techniques generally require further optimization for good developability and scale-up production. For BsAbs whose IgG is appended with additional binding domains, including single-chain Fvs (scFv), VH and VL or antigen-binding fragments (Fabs), additional efforts are often required to improve the physicochemical properties or bioactivities of BsAbs to meet the need for drug developability.Citation3
Targeting PD1/PDL1 for therapeutic application has been extensively investigated in the immuno-oncology (I/O) space.Citation14,Citation15 PD1/PDL1 blockade by mAbs restores the antitumor functions of T cells in the tumor microenvironment.Citation16–18 Crystal structures of the complexes of PD1 with nivolumab or pembrolizumab, which are both marketed anti-PD1 mAbs, were resolved recently.Citation19–21 Nivolumab appears to bind predominantly to the N-loop of PD1 with additional-binding sites situated at the FG and BC loops. The overlap in the binding area of nivolumab and PDL1 is mainly located at the FG loop of PD1, and thus, nivolumab relies on the heavy chain to establish contacts with PD1 and on the light chain to compete with PDL1 for PD1 binding.Citation19,Citation22 Similarly, pembrolizumab predominantly binds to the C’D loop of PD1 with its heavy chain while making extra contacts with the CC’ strands of PD1, where it clashes with the PDL1 binding regions.Citation20 Thus, the two are equally effective in blocking the association of PDL1 with PD1, but the binding surfaces of the two antibodies on PD1 do not overlap. Despite the broad antitumor efficacies in numerous cancers in the clinic,Citation15 only 20%–30% of patients developed durable clinical responses after anti-PD1 or anti-PDL1 monotherapy, and most patients either showed no responses at all or became refractory later.Citation14,Citation23,Citation24 Thus, the combination of anti-PD1 or anti-PDL1 mAb with mAbs targeting other immune checkpoint targets, such as LAG3, TGFβ, CTLA4, TIGIT, is being pursued very actively in an attempt to provide new therapies with enhanced antitumor efficacies.Citation25–28
609A is a new anti-PD1 mAb that is currently being investigated in clinical studies. Interestingly, we discovered that the heavy chain of 609A has great flexibility to pair with a variety of light chains from different mAbs directed against a variety of unrelated targets while retaining the original binding and biological activities of 609A. Based on this property of 609A, together with a linear Fab format,Citation29 we developed the Common Light Chain Linear Fab x2 (CLF2) platform to construct anti-PD1-based BsAbs for therapeutic applications. Using these techniques, we successfully produced a number of BsAbs with IgG-like physicochemical properties and the ability to target PD1 along with other immune checkpoint molecules and tumor-associated targets. These BsAbs have been manufactured on a large scale with ease and exhibit good developability and strong potential for therapeutic use in the clinic.
Results
The heavy chain of 609A, a novel clinical-stage anti-PD1 mAb, plays a dominant role in binding to PD1
609A is a novel clinical-stage anti-PD1 mAb selected and produced from hybridomas. The mAb binds to PD1 with high affinity in enzyme-linked immunosorbent assays (ELISA) and cell-based assays and effectively blocks the interaction of PD1 with PDL1. To further characterize the interaction of 609A and PD1, we obtained the X-ray crystal structure of the PD1/609A Fab complex (, PDB ID: 7VUX). X-ray diffraction revealed that the total buried surface areas (BSAs) of the interface between PD1 and 609A Fab heavy and light chains are 567.6A2 and 378.5A2, respectively. Within the interface, the 609A Fab heavy-chain forms 12 noncovalent bonds with PD1, including 10 hydrogen bonds and 2 salt bridges (, ), whereas the light chain forms two hydrogen bonds ().
Table 1. Data collection and refinement statistics
Table 2. Hydrogen bond interactions between PD1 and 609A Fab
Figure 1. The crystal structure of the PD1/609A Fab complex showed that the heavy chain of 609A plays a dominant role in establishing the interaction of the antibody with PD1. (a,b) Close-up views of the hydrogen bond interaction between PD1 and the heavy chain of 609A Fab (left) or the light chain of 609A Fab (right). The residues that formed hydrogen bonds are shown as sticks. PD1, the Fab heavy chain and light chain are colored blue, green and pink, respectively. The key hydrogen bonds are highlighted as yellow dash lines. (c) A ribbon representation of the PD1 structure. The residues that contact with the 609A Fab are shown in sticks. Salt bridge interactions, hydrogen bond interactions and water-bridged hydrogen bond interactions are colored Orange, pink and green, respectively. The residues that are involved in both hydrogen bond interactions and water-bridged hydrogen bond interactions are colored marine. The N-loop (N-ter) is colored red. (d) A close-up view of the salt bridge interaction between PD1 and the heavy chain of 609A Fab. The residues are shown as sticks. PD1 and the Fab heavy chain are colored blue and green. The ionic bonding is highlighted as yellow dash lines.
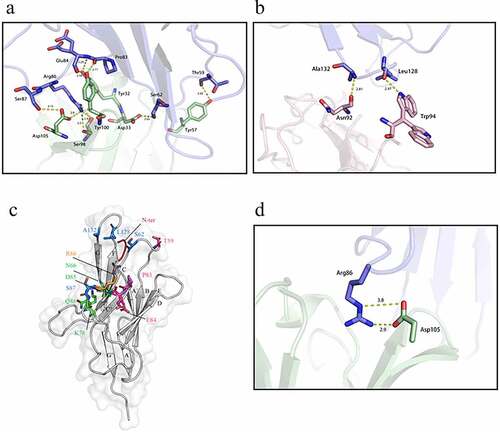
The structure revealed that 609A binds to residues located at the FG and C’D loops of PD1, partially overlapping with the binding interface of PDL1Citation22 (). Indeed, the backbone amide of Ala132, which resides in the FG loop of PD1, formed a hydrogen bond with the carboxyl group of Asn92 in the 609A light chain. A similar contact was established between the backbone carboxyl of the nearby Leu128 and the sidechain NE1 of Trp94 of the antibody light chain (). The side-chain NH of Arg86 located at the C’D loop of PD1 made four hydrogen-bond contacts with the side chain of Asp105 and the carboxyl and hydroxyl groups of Ser98 of the antibody heavy chain. The nearby residue Ser87, situated in the same loop, forms a second hydrogen bond with the side chain of Asp105. Additionally, within the C’D loop, the backbone amide and carboxyl of Glu84 and the Pro83 of PD1 formed three more hydrogen bonds with the side-chain hydroxyl of Tyr32 and Tyr100 of the 609A heavy chain, respectively, further strengthening the interaction of the two proteins. Beyond the C’D loop, residues Ser62 and Thr59 located at the BC loop of PD1 established two additional hydrogen bonds with the side-chain carboxyl of Asp33 and the hydroxyl of Tyr57 of the 609A heavy chain (). In addition, the side-chain Nε and NH1 of Arg86 were also capable of forming two salt bridges with the two side-chain carbonyl oxygens of Asp105 of the antibody heavy chain (). Taken together, this crystal structure of the complex suggests that the heavy chain of 609A plays a dominant role in establishing the interaction of the antibody with PD1.
Alanine scanning mutagenesis of the 609A light chain confirms the dominance of the heavy chain in binding to PD1
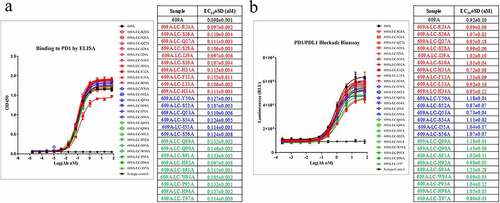
Given that the crystal structure of the PD1/609A Fab complex showed that the number of bonds established between 609A VH and PD1 (12 in total) is sixfold greater than that of 609A VL (2 in total), we reasoned that the light chain of 609A might play only a supportive role relative to the heavy chain in terms of the binding of the antibody to PD1. To test this hypothesis, we individually substituted all residues within the 3 complementarity-determining regions (CDRs) of the 609A light chain with alanine to identify the ones that may be critical for the binding of the antibody to PD1. The mutagenesis scanning showed that almost all residues in the CDRs of the 609A light chain could be replaced individually by alanine without significantly affecting the binding efficiencies of the antibody to PD1. The EC50 values of all variants were approximately 0.1 nM, on par with that of the parental 609A (EC50 = 0.098 nM). Interestingly, variant F32A, an alanine substitution at residue Phe32, which does not seem to form any bonds with PD1, was the only variant that showed a reduced binding affinity for PD1, showing a slightly increased EC50 value (0.16 nM) and a lower maximum binding than all the other variants. On the other hand, variants with alanine substitution at Asn92 or Trp94 (N92A or W94A), which contact PD1 directly, retained their binding to PD1 with EC50 values of 0.097 nM and 0.105 nM, respectively (). In line with the binding results, the potency of these 609A variants for T cell activation appeared to be comparable to that of the parental 609A when assessed using a cell-based PD1/PDL1 blockade system from Promega (). These findings suggest that the 609A light chain is relatively unlikely to play an essential role in specifying the interaction of 609A with PD1.
Figure 2. The alanine scanning of the CDRs in 609A light chain demonstrated the dominance of the heavy chain in binding to PD1. (a) Residues in the CDRs (CDR1: red, CDR2: blue, CDR3: green) of the 609A light chain were substituted to Alanine by site-directed mutagenesis individually. The binding abilities of the resulting 609A variants to PD1 were measured in triplicate by ELISA (n = 2) and compared with that of the parental mAb, 609A. PD1 protein was coated on the plate. The 609A variants, 609A and the isotype control mAb were serially diluted and added to the plate. (b) The abilities of these 609A variants to activate T cells were assayed in triplicate using a cell based PD1/PDL1 blockade system (Promega) and compared to that of 609A (n = 2). N = the number of independent experiments. The luciferase expression under the control of the NFAT response elements in response to PD1/PDL1 blockade were measured and plotted as the Luminescence readout.
Pairings of the 609A heavy chain with light chains from a variety of unrelated antibodies were capable of specifically binding to PD1 and blocking the PD1/PDL1 interaction
Based on the above observation, we tested whether the light chain of 609A can be replaced with a different light chain while retaining its specific binding to PD1 by swapping the 609A light chain with those from a variety of other unrelated antibodies. We first aligned the sequences of light chains from eight publicly available (from various databases and published patents) and four in-house generated antibodies directed against tumor-associated antigens and IO-related targets with the sequence of the 609A light chain. The results showed that within the 12 antibody light chains, the sequence homology to the 609A light chain ranged from 84.11% to 59% ().
Table 3. Characterization of the pairings of the 609A heavy chain with a variety of light chains
We then paired the heavy chain of 609A with each of the 12 individual light chains and expressed the hybrid molecules in mammalian cell culture. All the light chains paired well with the 609A heavy chain and were expressed as the correct IgG format molecules. Seven of the 12 hybrid molecules were capable of binding to PD1 efficiently, with EC50 values ranging from 0.15 to 0.24 nM, compared to EC50 values of 0.11 nM for 609A and 0.27 nM for nivolumab. Four hybrid molecules appeared to have a modest reduction in the binding affinities for PD1, with EC50 values ranging from 0.62 to 1.04 nM (). Interestingly, the binding affinity of these hybrid molecules for PD1 did not appear to correlate directly with the degree of sequence homology of the individual light chains to the 609A light chain. For example, the hybrid molecule comprising the light chain with the highest sequence homology to that of 609A (609HC-1LC, 84.11%) bound more weakly to PD1 (EC50 = 0.62 nM) than one with a lower sequence homology (609A-9LC, 65.77%). In one case, pairing the 609A heavy chain with the light chain of mAb10 (64% sequence homology to the light chain of 609A) resulted in a hybrid molecule (609HC-10LC) with complete loss of PD1 binding. We speculate that pairing the light chain of mAb10 with the 609A heavy chain might have induced a change in the configuration of the heavy chain that resulted in the loss of the binding ability (, ). To verify that the hybrid molecules were also able to bind to PD1 expressed from cells, we generated a PD1-overexpressing TF1 cell line. Using this cell line, we confirmed that 11 of the 12 hybrid molecules were indeed capable of binding to PD1 expressed on the surface of the cells. Consistent with the ELISA findings, 609HC-10LC failed to bind to PD1-overexpressing TF1 cells ().
Figure 3. All 12 light chains were able to dimerize with the heavy chain of 609A. (a) 11 of the 12 pairings were capable of binding to PD1 with EC50ʹs ranging from 0.15 nM to 1.05 nM. The pairing of 609A heavy chain with the light chain of mAb10, 609HC-10LC, failed to bind to PD1. PD1 protein was coated on the plate. The 609A hybrid pairings, 609A, Opdivo and an isotype control were serially diluted and added to the plate in triplicate (n = 2). (b) 11 of the 12 pairings were capable of binding to TF1 cells overexpressing PD1. The pairing of 609A heavy chain with the light chain of mAb10, 609HC-10LC, failed to bind to PD1-expressing TF1 cells. The 609A hybrid pairings, 609A and an isotype control were serially diluted and added to the cells, followed by measurement by FACS. (c) All pairings were able to equally block the interaction of PD1 and PDL1 as effectively as 609A by ELISA in triplicate (n = 2).
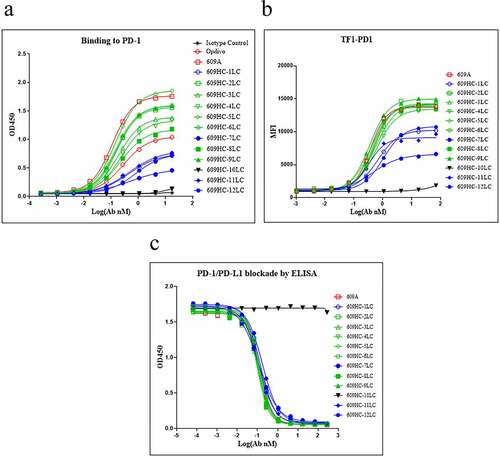
All 12 hybrid molecules were further assessed for their ability to block PD1/PDL1 interaction. All except one were able to potently block the interaction of PD1 and PDL1 with similar IC50 values near 0.1 nM (, ). Not surprisingly, 609HC-10LC lacked any ability to block the PD1/PDL1 interaction.
We next confirmed the PD1-binding specificity of the hybrid molecules by testing their cross-reactivity to a variety of other protein targets, including a number of tumor-associated antigens, e.g., HER2 and EGFR; immune checkpoint molecules, e.g., PDL1 and LAG3; and growth factors, such as VEGF. Several light chain sequences from functional (neutralizing) antibodies to these targets were used in the construction of the hybrid molecules paired with the heavy chain of 609A (). None of the 12 hybrid molecules showed any binding activity to protein targets other than PD1, suggesting that the specificity of 609A for PD1 is well preserved for all hybrid heavy/light chain pairings (Supplementary Fig. 1).
Development of a CLF2 platform for the efficient construction of tetravalent BsAbs that exhibit IgG-like physicochemical properties
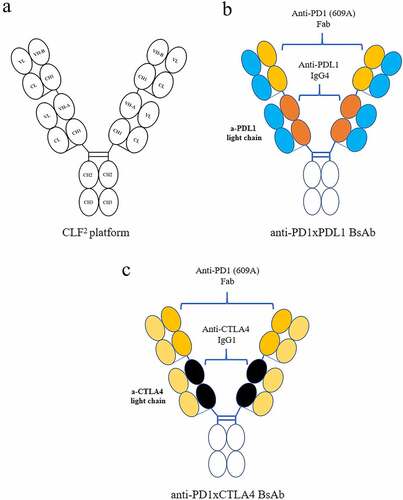
Given that the light chain of 609A can be replaced with a variety of other antibody light chains without sacrificing its ability to bind to PD1 and block PD1/PDL1 interaction, we developed the CLF2 platform for the efficient construction of tetravalent BsAbs that exhibit IgG-like physicochemical properties. In this strategy, the Fds of two different antibodies, namely, antibody 1 and antibody 2, are first joined together linearly via a flexible (G4S)3 linker. The resultant VH1-CH1-(G4S)3-VH2-CH1 is then fused to an IgG Fc. Coexpression of the linear Fd-Fc with a common light chain led to the formation of a homogenous tetravalent IgG-like BsAb molecule. This approach can overcome the concern of mispairing between an antibody heavy chain and a noncognate light chain (). Using the CLF2 platform, we successfully constructed anti-PD1 (609A)-based BsAbs, including BsAbs cotargeting PD1/PDL1 and PD1/CTLA4, the two BsAb molecules characterized in detail here.
Figure 4. Construction of novel common-light-chain linear-Fab-based (CLF2) bispecific antibodies. (A) schematics show the structure of the CLF2 platform, (b) the anti-PD1xPDL1 BsAb and (c) the anti-PD1xCTLA4 BsAb. Horizontal lines represent disulfide bonds.
The BsAbs made from the CLF2 platform exhibited IgG-like physicochemical properties comparable to those of conventional mAbs
To construct the anti-PD1xPDL1 BsAb, which simultaneously targets PD1 and PDL1, we fused the Fd of 609A via a (G4S)3 linker to the N-terminus of the heavy chain of the anti-PDL1 mAb, 9C10, and coexpressed the fusion with the light chain of 9C10 (). With respect to the anti-PD1xCTLA4 BsAb, the Fd of 609A was fused to the N-terminus of the heavy chain of the anti-CTLA4 antibody ipilimumab, and the light chain of ipilimumab was used as the common light chain (). The light chains of 9C10 and ipilimumab share 73.8% (609HC-3LC) and 81.5% (609HC-2LC) sequence homology with the light chain of 609A, respectively ().
The two BsAbs were produced in Chinese hamster ovary (CHO) cells and purified via single-step Protein A chromatography. The purified BsAbs were subjected to size exclusion chromatography (SEC), capillary electrophoresis (CE) and ion exchange chromatography (IEC) to determine the protein integrity. SEC showed that both BsAbs have >99% monomeric fraction, which is on par with that of the original 609A mAb (: Panel i). When measured by the reducing CE (R-CE), the purity of the total heavy and light chains of the BsAbs, anti-PD1xPDL1 and anti-PD1xCTLA4, were 97.87% and 98.68% with a heavy chain to light chain ratio of 1.55 (59.54%: 38.33%) and 1.56 (60.07%: 38.61%), respectively, consistent with the theoretical molecular weights of the heavy and light chain of the BsAbs. Similarly, the purity of the total heavy and light chains of 609A was 98.38% with a heavy to light chain ratio of 2.18 (67.48%: 30.90%), closely matching the molecular weights of the heavy and light chains of the mAb (: panel ii). With respect to the charge variants, the purity of the charge variants of the BsAbs was also comparable to that of 609A, with main peaks of 92.7%, 88.0%, and 83.0% for anti-PD1xPDL1 BsAb, anti-PD1xCTLA4 BsAb and 609A, respectively (: panel iii).
Figure 5. The BsAbs generated from the CLF2 platform exhibited IgG-like physicochemical properties without the need for engineering. (a) SEC Chromatograms of 609A, anti-PD1xPDL1 BsAb and anti-PD1xCTLA4 BsAb after single-step protein A purification (panel i); Reducing CE (R-CE) graphs of 609A, anti-PD1xPDL1 BsAb and anti-PD1xCTLA4 (panel ii); IEC Chromatograms of 609A, anti-PD1xPDL1 BsAb and anti-PD1xCTLA4, respectively (panel iii). (b) DSC graphs of 609A (left) and the anti-PD1xPDL1 BsAb, respectively (right). (c) the purity (%) of 609A and the anti-PD1xPDL1 BsAb was analyzed using SEC (top) and IEC (bottom) after the accelerated stability assay and plotted as a function of time (month). 609A (red line and star), the anti-PD1xPDL1 BsAb (blue line and circle). (d) Pharmacokinetics of the two BsAbs was analyzed in rats.
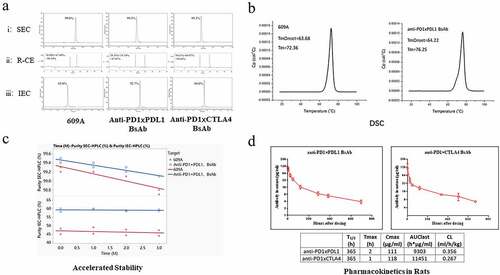
Consistent with their physicochemical properties, the melting temperature (Tm) of the anti-PD1xPDL1 BsAb was approximately 4°C (76.25°C) higher than that of 609A (72.36°C), although the onset temperature (Tonset) of the BsAb was only approximately 0.5°C (64.22°C) higher than that of 609A (63.68°C), as measured by differential scanning calorimetry (DSC) (). The accelerated stability analysis by SEC and IEC further demonstrated that the anti-PD1xPDL1 BsAb was stable at room temperature (25°C) for up to 3 months, on par with the stability of 609A (). Not only did the BsAb keep the protein properties intact after incubation at an elevated temperature for a month, but the abilities of the BsAb to bind to their targets also remained unchanged (Supplementary Fig. 2A, B, C and D). Similarly, the anti-PD1xCTLA4 BsAb also showed impressive thermostability, with a Tonset at 66.48°C and a Tm at 74.49°C, and like the anti-PD1xPDL1 BsAb, it did not lose its target-binding affinity after accelerated stability testing (Supplementary Fig. 2C, D and E).
Consistent with the favorable thermostability, both BsAbs also exhibited favorable pharmacokinetics (PK) in rats with T1/2 at approximately 365 h (), comparable to the T1/2 of 358 h of 609A and longer than the T1/2 of 259 h of a reported BsAb.Citation30
BsAbs made from the CLF2 platform exhibited bioactivities superior to those of their mAb counterparts both in vitro and in vivo
The anti-PD1xPDL1 BsAb effectively bound to PD1 at an EC50 of 0.42 nM, which is comparable to that of the monospecific hybrid molecule consisting of the 609A heavy chain and the light chain of 9C10 (609HC-3LC/609HC-9C10LC, EC50 = 0.38 nM). Similarly, the BsAb effectively bound to PDL1 with an EC50 of 0.13 nM, on par with the EC50 of 0.12 nM of the parental anti-PDL1 antibody, 9C10. To confirm that the BsAb can bind simultaneously to its two targets, we showed that the BsAb was able to bridge PD1 and PDL1 with an EC50 of 0.19 nM, whereas the monospecific anti-PDL1 mAb, 9C10, failed to do so (: Panels i, ii, and iii). Importantly, the anti-PD1xPDL1 BsAb was more potent in stimulating IL-2 (EC50 = 0.03 nM) and IFNγ (EC50 = 0.05 nM) secretion from T cells than the monospecific 609HC-9C10LC, whose EC50 was 0.23 nM for IL2 secretion and 1.29 nM for IFNγ secretion. In this regard, the BsAb was also more potent than the parental mAb counterparts 609A (EC50 = 0.09 nM for IL2, EC50 = 0.08 nM for IFNγ) and 9C10 (EC50 = 0.18 nM for IL2, EC50 = 0.15 nM for IFNγ; : Panels iv and v). Ligation of PD1 on T cells with PDL1 expressed on CHO (Promega) also led to direct cell–cell engagement and induced PD1 synapse formation at the cell–cell contact points,Citation31–33 indicating that although simultaneous blocking of PD1 and PDL1 seemed redundant, crosslinking PD1 on T cells and PDL1 on target cells (e.g., cancer cells) with BsAbs may lead to physical cell–cell engagement, resulting in enhanced activation of T cells via immune cell synapse formation (, and Supplemental Movies 1, 2 and 3). Furthermore, the BsAb also retained Fc functions, as the binding of the BsAb to either FcγRI or FcRn was intact relative to that of two of our in-house produced mAbs (Supplementary Fig. 3). Encouraged by the promising in vitro data, we next studied the antitumor effects of the anti-PD1xPDL1 BsAb in animal tumor models. Since the BsAb does not cross-react with murine PD1 and PDL1, we co-injected human peripheral blood mononuclear cells (PBMCs) with human H292 lung cancer cells (ratio of 1:5) subcutaneously into immune-compromised NOD scid gamma (NSG) mice. Treatment of tumor-bearing mice with the anti-PD1xPDL1 BsAb resulted in 96% inhibition of tumor growth on Day 27 after treatment, whereas nivolumab (Opdivo), a marketed mAb targeting PD1, resulted in 50% inhibition of tumor growth. Thus, the anti-PD1xPDL1 BsAb outperformed the mAb counterpart in preventing tumor growth, demonstrating a synergistic antitumor effect compared to the individual mAb ().
Figure 6. The BsAbs exhibited superior bioactivities to the mAb counterparts in vitro and in vivo. (a) Binding abilities of the anti-PD1xPDL1 BsAb, an isotype antibody and (i) the parental pairing, 609HC-9C10LC or (ii) the parental anti-PDL1 mAb, 9C10 to PD1 or PDL1 were measured in triplicate by ELISA (n = 3). PD1 or PDL1 proteins were coated on the plates, respectively. The indicated proteins were serially diluted and added to the corresponding plates. iii) A bridging ELISA was done in a way that PDL1 was coated on the plate, followed by detection of biotinylated PD1 captured by the BsAb in the solution. iv–v) The abilities of the anti-PD1xPDL1 BsAb, an isotype antibody, 609HC-9C10LC, 9C10 and 609A to stimulate the secretion of IL2 (left) and IFNγ (right) from T cells in the presence of Dendritic cells (DCs) were measured in triplicate by mixed lymphocyte reaction (MLR) (n = 3). (b) PD1-overexpressing Jurkat T cells were stimulated to enhance PD1 expression. The activated T cells were labeled with Alexa Fluor 488 (488)-conjugated anti-PD1xPDL1 BsAb. PDL1-expressing CHO cells were stained with cell proliferation Dye eFluorTM 670 (Thermo fisher). The pre-stained T and CHO cells were then co-cultured and filmed on an Operetta CLS high-content analysis system (PerkinElmer). (i–ii) are representative Time-lapse movie frames showing the progress of the formation of PD1 immunological synapses. Green: T cells, Red: PDL1-expressing CHO cells. (c) A control (close black circle), Opdivo (red circle) and the anti-PD1xPDL1 BsAb (blue triangle) were injected into M-NSG mice bearing NCI-H292 tumors in the presence of human PBMCs at the indicated doses. Tumor volumes (mm3) were measured at the indicated time points. T/C means the ratio of tumor volumes between the treated group and the control group. CR means complete response. **** P < .0001 calculated by two-way ANOVA for the comparison of all indicated groups with the control group. (d) Binding abilities of the anti-PD1xCTLA4 BsAbs with two opposite orientations (609Fab-IpiIgG1 and IpiFab-609IgG1), 609A and ipilimumab to (i) PD1 or (ii) CTLA4 were measured in triplicate by ELISA (n = 3). PD1 or CTLA4 proteins were coated on the plates, followed by adding serial dilutions of indicated antibodies to the plates. iii) The bridging ELISA was setup as above, except that CTLA4 was coated on the plate. iv) The amount of IL2 secreted from SEB-activated human PBMCs in the presence of the anti-PD1xCTLA4 BsAbs (609Fab-IpiIgG1 and IpiFab-609IgG1), an isotype antibody, ipilimumab or 609A were measured in triplicate by ELISA, respectively (n = 3).
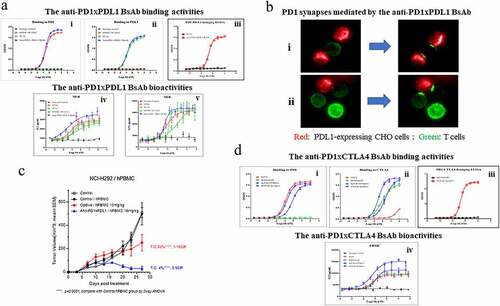
For the anti-PD1xCTLA4 BsAb, we found that the orientation of the N-terminal (outer) and C-terminal (inner) Fds, VH(o)-CH1-(linker)-VH(i)-CH1, have a clear effect on binding to the respective targets of the two antibodies. When the pairing of the 609A Fd with the light chain of ipilimumab (the outer Fab) was fused to the N-terminus of ipilimumab (namely, 609Fab-IpiIgG1), the binding of the inner Fabs from ipilimumab to CTLA4 was reduced approximately 3-fold compared to that of ipilimumab (EC50: 2.12 nM/0.84 nM), whereas the outer 609A hybrid Fabs exhibited a similar binding affinity for PD1 (EC50 = 0.17 nM) to that of the parental 609A (EC50 = 0.12 nM). Conversely, when the Fabs of ipilimumab were placed outside (namely, IpiFab-609IgG1), the inner 609A hybrid Fabs bound to PD1 at an EC50 of 0.79 nM, approximately eightfold weaker than that of 609A, whereas the affinity of the outer Fabs of ipilimumab for CTLA4 (EC50 = 0.34 nM) was comparable to that of ipilimumab (EC50 = 0.84 nM). As expected, the BsAb was also capable of binding to PD1 and CTLA4 simultaneously (EC50 = 0.19 nM), which cannot be achieved by the mAb counterpart ipilimumab (: Panels i, ii and iii). In a cell-based assay, 609Fab-IpiIgG1 was more potent in stimulating IL-2 secretion from human PBMCs with an EC50 of 0.06 nM than did IpiFab-609IgG1, which had an EC50 of 0.12 nM. Importantly, 609Fab-IpiIgG1 was capable of inducing much higher IL-2 production (~2-fold) at concentrations over 1 nM than its mAb counterparts, 609A and ipilimumab (: panel iv). Thus, 609Fab-IpiIgG1 was chosen as the lead candidate for further development. Taken together, the tetravalent BsAbs made from the CLF2 platform not only showed excellent physicochemical properties comparable to those of mAbs, but also retained the desired antitumor bioactivities.
Discussion
By analyzing the structure of 609A with PD1, we showed that the major residues in 609A that contact PD1 differ from those of nivolumab and pembrolizumab, but the binding interface covered by 609A overlaps with that of both marketed antibodies.Citation19–21 The heavy chain of nivolumab forms 10 hydrogen bonds with the N-loop and 3 hydrogen bonds with the FG and BC loops of PD1, while the light chain forms 3 additional hydrogen bonds with the FG loop of PD1.Citation19 Similarly, pembrolizumab is associated mainly with the C’D loop and C’ strands of PD1, with the heavy chain contributing most of the contacts.Citation20 In the case of 609A, the antibody made multiple contacts with the FG, C’D and BC loops of PD1, among which 12 of 14 noncovalent bonds were formed by the heavy chain. Interestingly, both 609A and nivolumab used Ala132 on the FG loop and Thr59 on the BC loop of PD1 to establish bonding, while both 609A and pembrolizumab used Glu84 and Ser87 on the C’D loop to form hydrogen bonds with PD1. Thus, the light chain and heavy chain of 609A share epitopes with nivolumab and pembrolizumab, respectively. In contrast, nivolumab and pembrolizumab do not overlap with each other.Citation19 In this regard, the binding of 609A to PD1 is expected to interrupt the interaction of PD1 with PDL1 as effectively as nivolumab and pembrolizumab, as the binding interfaces of all three antibodies obstruct the binding surface of PD1 with its ligands PDL1 and PDL2.Citation22,Citation34–36 Nevertheless, the interaction of 609A and PD1 is different from that of either nivolumab or pembrolizumab, since 609A did not interact with the N loop of PD1 as nivolumab does, nor bind to the CC’ strands of PD1 where pembrolizumab binds.
It is known that the antibody heavy chain generally plays a dominant role in the formation of bonds with antigens.Citation4,Citation37,Citation38 Indeed, the two marketed anti-PD1 mAbs, nivolumab and pembrolizumab, both rely on their heavy chains to form multiple contacts with the residues located in the well-exposed loops of PD1. Based on this finding, we hypothesized that it might be possible for an anti-PD1 mAb to rely on its heavy chain to exert its functions, and we may be able to find a new anti-PD1 mAb that relies on the heavy chain for its bioactivities and thus can be used as a foundation for the development of an anti-PD1-based BsAb platform. In support of this idea, we confirmed that 609A also relies on its heavy chain for its binding and blocking activities.
It is intriguing that many light chains from various unrelated antibodies can pair with the heavy chain of 609A to form hybrid molecules without altering the specificity of 609A for PD1. We showed that the various hybrid molecules bind specifically to PD1, but not to the antigens targeted by the original antibodies from which the light chains were derived. This feature allowed us to use the 609A heavy chain as a scaffold, along with the linear Fab IgG formatCitation29 and a diverse source of common light chains, for the development of an anti-PD1-based bispecific platform, which we denoted CLF2. Each CLF2 consists of four identical light chains and two identical heavy chains, each of which contains two linearly arranged Fd via a G4S linker, VHa-CH1-(G4S)3-VHb-CH1, derived from two antibodies with different antigen specificities. Within the format, all heavy/light chain pairings are symmetrical and thus are expected to be produced as a homogenous population and to behave similarly to a conventional IgG molecule (). To our knowledge, the CLF2 format is different from other BsAb formats published thus far. Even though some formats, such as FIT-Ig®, also consist of two Fabs, which requires the cotransfection of three constructs and thus has a possibility of mispairing the two different light chains, they generally need additional steps to circumvent the producibility issues.Citation30,Citation39 In contrast, the CLF2 format uses the common light chain and thus assembles the light and heavy chains in a way closely resembling that of an IgG. Therefore, BsAbs in the CLF2 format not only avoid mispairing issues, but also have IgG-like physicochemical properties (). Although the common light chain has been used for the construction of a BsAb for decades, searching common light chains for BsAb construction generally involves the screening of sizable librariesCitation40,Citation41 or engineering for the stable interaction of heavy and light chainsCitation13,Citation42,Citation43 due to pairing limitations. In contrast, with our approach, one can focus on the selection of antibodies that can pair and function synergistically with 609A, with less concern about the tedious work generally needed for the conventional application of the common light chain. Nevertheless, it is worth noting that steric hindrance does occur between the two noncognate light chains in some scenarios, which emphasizes that, although the homology of the second light chain to the light chain of 609A was not strictly associated with the affinity of the pair and could retain affinity in a range as low as 60–70% homology, the specificity of a sequence plays an important role in the pairing.
Using the CLF2 platform, we successfully produced a series of BsAbs, including an anti-PD1xPDL1 and an anti-PD1xCTLA4 BsAb. These BsAbs exhibited superior physicochemical properties and thermostability on par with those of conventional mAbs, e.g., 609A. This streamlined the workflow by eliminating the need for additional engineering for the heterodimerization of heavy chains or the concern of mispairing between the heavy and light chains. Along with good thermostability, the BsAbs showed favorable PK in rats. Furthermore, the BsAbs not only retained comparable binding and blocking activities to their parental mAb counterparts, but also activated T cells more effectively in both mixed lymphocyte reaction assays and human PBMC-based assays. These data suggest that the BsAbs were likely capable of exerting synergistic effects, boosting immune responses by simultaneously engaging both immune checkpoint molecules. It is worth noting that the orientation of the two Fabs in CLF2 is important for retaining the activity of its component Fabs. Generally, it is optimal to place the anti-PD1 Fab at the N-termini to avoid reducing the binding affinity for PD1 when constructing anti-PD1-based CLF2, possibly because the inner Fabs might experience steric hindrance to gain access to the target protein on a target-to-target basis. Furthermore, it was intriguing to see a low-level cross-reactivity of 609A for CTLA4 at high concentrations in an ELISA assay (ii). Although we cannot completely rule out a possibility of contaminants or impurity of the samples in the system causing nonspecific signals, it is also possible that 609A may cross react with CTLA4 at high concentrations since PD1 and CTLA4 both belong to the CD28 family.
Consistent with the cell-based assays where the BsAbs stimulated cytokine production more potently than the parental mAbs, the BsAbs also exhibited superior antitumor effects in vivo. To measure the synergistic effects of the BsAb on tumor growth inhibition, we chose a model system in which we subcutaneously introduced human PBMCs into NSG mice along with human H292 lung cancer cells to circumvent the issue that the anti-PD1xPDL1 BsAb does not cross-react with murine PD1 and PDL1. One caveat with this model is that the introduced human PBMCs might have natural killing effects on the engrafted tumor cells, and the animal might merely act as a vessel for hosting the grafts. In this study, however, we did not observe significant inhibition of tumor growth directly induced by the human PBMCs (). Importantly, with this model, we demonstrated that the BsAb inhibited H292 tumor growth more efficiently than nivolumab, the marketed anti-PD1.
Taken together, we developed a BsAb platform, CLF2, and generated anti-PD1-based BsAbs using the specific feature of 609A. These BsAbs not only demonstrated favorable IgG-like manufacturability, but also exhibited potent additive antitumor effects that cannot be achieved by their mAb counterparts. Whereas other formats generally require in silico high-throughput screening, as in the case of Sanofi’s CODV-Ig,Citation44 structure modeling to avoid steric hindrance, as in the case of AbbVie’s DVD-Ig,Citation45 or multiple rounds of screening from large-scale libraries to overcome developability issues, as in the making of emicizumab by Chugai,Citation40 our approach, although it sometimes requires certain adjustments, provides a rapid and effective way to generate BsAbs competent for manufacturing at large scales with qualities as good as those of regular mAbs using the standard production protocol designed for mAb production.
Thus, we provide a new approach to effectively produce stable and active BsAbs that are suitable for large-scale production and meet the needs of clinical applications.
Materials and methods
Antibodies
609A is a humanized anti-PD1 IgG4 mAb. 609A light chain variants, each of which contains one point mutation, were generated by site-directed mutagenesis as described in Molecular Cloning (3rd edition). The high-fidelity PrimeSTAR® HS DNA Polymerase (Takara, Cat# R010A) was used in all PCR reactions. Amino acid sequences of heavy and light chain variable regions of a variety of mAbs (mAb1, 2, 4, 5, 7 and 8) were retrieved from the RSCB protein data bank. The sequence of mAb12 was retrieved from the KEGG database (Entry: D09013). The sequences of mAb9 were retrieved from US20100136021A1 (SEQ ID NO: 80 and 81). MAb3, mAb6, mAb10 and mAb11 are humanized mAbs generated in-house. DNA fragments encoding the variable and constant regions of the above mAbs were generated by commercial gene synthesis (Sangon Biotech, China). To construct expression vectors, the genes of full-length heavy chains or light chains were produced using conventional techniques and then cloned into pcDNA3.4. The sequences of all genes were verified by sequencing at Genewiz (Shuzou, China).
Antibody expression and purification
FreeStyle™ 293-F cells (Thermo Fisher Scientific, Cat#R79007) were cultured in serum-free medium. Transient transfection was performed by co-transfection of expression vectors encoding a heavy chain or a light chain individually into FreeStyle™ HEK293-F cells using 1 μg/ml 25 KDa linear polyethylenimine (Polysciences, Inc.). One day after transfection, valproic acid (Sigma) was added to cell culture at a final concentration of 3 mM. On day 2 post-transfection, medium comprising 10% GlutaMAX, 10% 400 g/L glucose and 80% freestyle 293 medium was added to the cell culture at 10% of the total volume. Conditioned medium was collected 5–6 days after transient transfection. Antibodies in the culture media were purified by MabSelect SuRe affinity columns (GE Healthcare) on an AKTA Avant 25 fast protein liquid chromatography (FPLC) System. The columns were equilibrated with buffer A (25 mM sodium phosphate, 150 mM sodium chloride, pH = 7.0) prior to use. The culture media containing antibodies were then applied to the columns followed by elution with Buffer B (100 mM sodium citrate, pH 3.5) to collect the desired proteins. Collected proteins were neutralized with 1 M Tris (pH 9.0), which were then dialyzed against phosphate-buffered saline (PBS). Finally, the purity of the samples was analyzed on a SEC-high performance liquid chromatography (SEC-HPLC).
Complex preparation and crystallization
The E. coli-expressed human PD1 protein and 609A-Fab were mixed at a molar ratio of 1:1. The mixed sample was incubated for 30 min on ice and then purified by gel filtration (Superdex200, GE Healthcare). The crystal screening of the PD1/609A-Fab complex (approximately 10 mg/mL) was carried out at 18°C using the vapor-diffusion sitting-drop method. Diffracting crystals were grown in a buffer composed of 0.2 M ammonium formate (pH 6.6) and 20% (w/v) PEG 3350. The resulting crystals were stored in the reservoir solution comprising a cryoprotectant and 20% (v/v) glycerol and flash-frozen in liquid nitrogen.
Data collection and structure determination
Diffraction images were collected at the Shanghai Synchrotron Radiation Facility (SSRF), beamline BL18U1. Raw images were collected through a Pilatus detector and were indexed, integrated and scaled using XDS. All diffraction experiments were carried out at 100 K. Data were collected at a wavelength of 0.97915 Å for PD1/609A-Fab complexes. Resolution limits were cut off at I/σ(I) = 2.2. Phase was determined by molecular replacement with Phaser using the human PD1 structure (PDB ID: 5WT9) and the nivolumab/Fab structure (PDB ID: 5WT9) as search ensembles (models). Structure refinement was carried out using Refmac5. Structure inspection, building, and validation were performed by COOT. Analysis for interaction within complexes was carried out by ccp4.PISA, and figures were generated using PyMOL. The atomic coordinates and sequences of the PD1/609A-Fab complex have been deposited in the Protein Data Bank with an accession code of 7VUX.
Chromatography and physicochemical property analysis
Chromatography was performed using a HPLC system (Thermo Dionex Ultimate 3000, USA) equipped with a TSK G3000SWxl (Tosoh Bioscience, Japan) gel filtration column for SEC or a ProPac™ WCX-10, 4 × 250 mm, Thermo Dionex 054993 column for IEC, respectively. Twenty ul of samples were injected onto the columns at a final concentration of 1 mg/ml. A mobile phase containing 200 mM sodium phosphate (pH 6.8) was then applied at a flow rate of 0.5 mL/min for SEC. In the case of IEC, the samples were eluted by an elution buffer containing 20 mM MES plus 200 mM sodium chloride at a flow rate of 1.0 ml/min.
Reduced capillary electrophoresis sodium dodecyl sulfate (CE-SDS) was performed on a PA800 plus instrument equipped with an ultraviolet detector (AB Sciex, USA). All samples were denatured and reduced by β-mercaptoethanol prior to injection into capillary electrophoresis system. Samples were separated in 30.7 cm long precut capillaries (diameter = 50 μm) with a constant voltage of 15 kV. Detection was positioned 20.5 cm from the point of sample injection. The detection wavelength was set to 214 nm and the 32 Karat software package was used for data acquisition and analysis.
Thermostability testing
The long-term thermostability of the anti-PD1× PDL1 BsAb was determined in parallel with 609A using the accelerated stability analysis. Briefly, 609A and the anti-PD1× PDL1 BsAb were prepared in solution at a concentration of 25 mg/ml and 40 mg/ml, respectively. 609A and the anti-PD1× PDL1 BsAb (two batches each) solutions were then placed in an incubator at 25°C for accelerated stability testing. The sampling points were set at 0, 1, 2, and 3 months, and the integrity of samples was measured by SEC-HPLC and IEC-HPLC separately.
DSC was performed on a MicroCalTM VP-Capillary DSC system (Malvern). In short, samples were adjusted to a final concentration of 1 mg/ml along with a blank excipient solution, prior to the test. Four hundred μl of the test sample and a blank excipient control were simultaneously added to the sample plates separately. The experiment was carried out by gradually increasing the temperature with the following parameters: Temperature range: 10 ~ 100°C; Heating rate: 100°C/h; Filtering period: 8s; Nitrogen pressure: 50 ~ 60psi.
PD1/PDL1 blockade bioassay
Blockade of PD1/PDL1 interaction was assessed using a commercially available bioassay system (Promega, Cat# J1250 and J1255) and following the manufacturer’s instruction. Briefly, PDL1/CHO (an artificial APC, or aAPC) cells were seeded at 4 × 104 cells/well in 100 μl in white 96-well plates, which were cultured overnight at 37°C. The next day, the PDL1/CHO aAPC cells were incubated with serial dilutions of antibodies and Jurkat-PD1-NFAT-luc T cells (5x104/well) that express human PD1 and luciferases under the control of NFAT response elements in 80 μl RPMI-1640 supplemented with L-glutamine and 1% fetal bovine serum for 6 h. Bio-GloTM Reagent (80 μl) was added to each well and the plates were incubated at room temperature for 5–30 min. Luminescence was read on a SpectraMax i3x. The expression of luciferases was measured as readouts in response to PD1/PDL1 blockade.
Binding to cell surface PD1 by flow cytometry
TF-1 cells were purchased from the American Type Culture Collection (ATCC, Cat#CRL-2003) and cultured according to ATCC’s instructions. TF-1 cells were stably transfected with full-length human PD1 gene using a lentiviral vector. The resulting cell line was designated TF1-PD1. To measure the binding affinity of antibody variants for cell surface PD1, TF1-PD1 cells were incubated with serial dilutions of the antibodies for 1 h in PBS containing 1% bovine serum albumin. Cells were washed three times with PBS and then incubated with the fluorescein isothiocyanate-conjugated goat anti-human IgG (Sigma, Cat#F9512) at 4°C for 1 h. Cells were washed three times and then analyzed on the CytoFLEX Cytometer System (Beckman Coulter).
Allogeneic mixed lymphocyte reaction
Monocytes were enriched from PBMCs (AllCells, Cat# PB002-C-300) by adherence on tissue culture treated flasks. Dendritic cells (DC) were then generated by culturing the monocytes in vitro for 7 days with 25 ng/ml interleukin-4 (IL-4, R&D Systems) and 25 ng/ml GM-CSF (Xiamen Amoytop Biotech). CD4+ T cells were positively selected from PBMCs using Magnetic Activated Cell Sorting (MACS) and anti-CD4-conjugated microbeads (Miltenyi Biotec, Cat# 130–045-101). CD4+ T cells (1 × 105/well) and allogeneic DCs (1 × 104/well) were cocultured in the presence of serially diluted antibodies. After 3 days, the amount of IL-2 and IFNγ in the culture media was determined by ELISA (BD Biosciences, Cat# 555051and 555040 for IL2; Cat# 551221 and 554550 for IFNγ).
PBMC activation assay
Freshly isolated human PBMC (Allcells, Cat#PB005-C) were washed and then resuspended in RPMI 1640 medium (Gibco™, Cat#22400089) supplemented with 10% fetal bovine serum (Gibco™, Cat#10091148), 1% MEM non-essential amino acids solution (Gibco™, Cat#11140050), 1% sodium pyruvate (Gibco™, Cat#111360070), 1% penicillin–streptomycin (Gibco™, Cat#15140122), 1% GlutaMAX (Gibco™, Cat#35050061), 50 µM 2-mercaptoethanol (Gibco™, Cat#21985023) and 100 ng/ml Staphylococcus aureus enterotoxin B (SEB) (prepared in-house). The PBMCs (2 × 105 cells/150 µL/well) were incubated with 50 μl of serially diluted antibodies in round-bottomed 96-well microplates in a humidified CO2 incubator for 4 days. The amount of IL2 in the medium was determined by standard sandwich ELISA (BD Biosciences).
Fluorescence time-lapse movie
PD1-overexpressing Jurkat T cells (Promega) were stimulated to enhance PD1 expression. The activated T cells were labeled with 200 nM Alexa Fluor 488 (488)-conjugated anti-PD1× PDL1 BsAb (T1) or 488-conjugated anti-PD1 mAb (609A) (T2) for 1 h at room temperature, respectively. PDL1-overexpressing CHO cells (Promega) were stained with cell proliferation Dye eFluorTM 670 (Thermo fisher) in the presence (A1) or absence (A2) of the anti-PDL1 mAb, 9C10 for 1 h at room temperature, respectively. The 488-Labeled T cells were then co-cultured with pre-stained PDL1-overexpressing CHO cells as the following formats T1+ A2 or T2+ A1 and filmed on an Operetta CLS high-content analysis system (PerkinElmer).
Pharmacokinetic study in rats
Sprague-Dawley (SD) rats were randomized into groups (n = 4) and antibodies were administered by single intravenous injection at a dose of 5 mg/kg. Blood samples were collected by retro-orbital vein puncture using heparinized capillary tubes at indicated times. Serum was separated after centrifugation at 2000 g for 10 min and stored at −80°C until analysis. Antibody serum levels were determined by standard ELISA. The half-life was calculated with Phoenix WinNonlin (Pharsight Corporation). All studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC).
Animal tumor models
Animal care and in vivo experiments were approved by the IACUC of Sunshine Guojian Pharmaceutical (Shanghai) Co. Ltd. and performed under approved protocols (approval code for NCI-H292/hPBMC xenograft model: AS-2020-103). NCI-H292/hPBMC xenograft tumor models were established in M-NSG mice (NOD.Cg-PrkdcscidIl2rgem1/Smoc) (Model Organism, Cat# NM-NSG-001) by subcutaneous co-injection of 5 × 106 H292 tumor cells mixed with 50% Matrigel and 1 × 106 human PBMCs into the right back of the mice. After injection, the animals were randomly divided into groups (10 mice/group) and intraperitoneally injected twice a week (4 weeks in total). Tumor volume was measured twice per week and calculated using the formula V = LW2/2 (where V = volume, L = length and W = width).
Statistical analysis and alignment
IC50 and EC50 values were determined using GraphPad Prism 7 (GraphPad Software). Unless otherwise noted, all numerical data were presented as mean ± standard deviation (SD). P values were calculated using a two-way ANOVA multiple-comparison test. In all tests, differences with p values < .05(*) were considered statistically significant. Amino acid sequences were aligned, and the homology was analyzed using MEGA6.
Abbreviations
| CDRs | = | Complementarity-determining Regions |
| CE-SDS | = | Capillary Electrophoresis Sodium Dodecyl Sulfate |
| CH | = | Constant region of heavy chain |
| CL | = | Constant region of light chain |
| CLF2 | = | Common Light Chain Linear Fab x2 |
| DSC | = | Differential Scanning Calorimetry |
| EC50 | = | Half maximal effective concentration |
| Fab | = | Antigen Binding Fragment |
| FPLC | = | Fast Protein Liquid Chromatography |
| HC | = | Heavy Chain |
| HPLC | = | High Performance Liquid Chromatography |
| IC50 | = | Half maximal inhibitory concentration |
| IEC | = | Ion Exchange Chromatography |
| LC | = | Light Chain |
| NOD | = | Non-obese Diabetic |
| SEC | = | Size Exclusion Chromatography |
| SEQ ID NO | = | Sequence Identification Number |
| VH | = | Variable region of heavy chain |
| VL | = | Variable region of light chain |
Supplemental Material
Download MS Word (481.2 KB)Acknowledgments
The authors thank Weihua Li, Mingfei Wu, Junyan Chen, Fan Jiang and Wenyang Jiao for their excellent crystallography services at Viva Biotech.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Nie S, Wang Z, Moscoso-Castro M, D’Souza P, Lei C, Xu J, Gu J. Biology drives the discovery of bispecific antibodies as innovative therapeutics. Antib Ther. 2020;3(1):18–14. doi:10.1093/abt/tbaa003.
- Labrijn AF, Janmaat ML, Reichert JM, Parren PW. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18(8):585–608. doi:10.1038/s41573-019-0028-1.
- Brinkmann U, and Kontermann RE. The making of bispecific antibodies. In: MAbs. Taylor & Francis; 2017. p. 182–212.
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, and Carter P. An efficient route to human bispecific IgG. Nat Biotechnol. 1998;16:677–81. doi:10.1038/nbt0798-677.
- Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, Strumane K, Verploegen S, Kundu A, and Gramer MJ. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci. 2013;110:5145–50.
- Labrijn AF, Meesters JI, Priem P, De Jong RN, Van Den Bremer ET, Van Kampen MD, Gerritsen AF, Schuurman J, Parren PWHI. Controlled Fab-arm exchange for the generation of stable bispecific IgG1. Nat Protoc. 2014;9(10):2450–63. doi:10.1038/nprot.2014.169.
- Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, Ng SB, Born T, Retter M, Manchulenko K, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285(25):19637–46. doi:10.1074/jbc.M110.117382.
- Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, K-m L, Huston JS. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel. 2010;23(4):195–202. doi:10.1093/protein/gzp094.
- Van Blarcom T, Lindquist K, Melton Z, Cheung WL, Wagstrom C, McDonough D, Valle Oseguera C, Ding S, Rossi A, and Potluri, S. Productive common light chain libraries yield diverse panels of high affinity bispecific antibodies. In: MAbs. Taylor & Francis; 2018. p. 256–68.
- Shiraiwa H, Narita A, Kamata-Sakurai M, Ishiguro T, Sano Y, Hironiwa N, Tsushima T, Segawa H, Tsunenari T, Ikeda Y, et al. Engineering a bispecific antibody with a common light chain: identification and optimization of an anti-CD3 epsilon and anti-GPC3 bispecific antibody, ERY974. Methods. 2019;154:10–20. doi:10.1016/j.ymeth.2018.10.005.
- Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, Gassner C, Georges G, Kettenberger H, and Imhof-Jung S. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci. 2011;108:11187–92.
- Lindhofer H, Mocikat R, Steipe B, Thierfelder S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J Immunol. 1995;155:219–25.
- Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, Guntas G, Leaver-Fay A, Smith EM, Ho C, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotechnol. 2014;32(2):191–98. doi:10.1038/nbt.2797.
- Chen L, Han X. Anti–PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125(9):3384–91. doi:10.1172/JCI80011.
- Yu JX, Hodge JP, Oliva C, Neftelinov ST, Hubbard-Lucey VM, Tang J. Trends in clinical development for PD-1/PD-L1 inhibitors. Nat Rev Drug Discov. 2020;19(3):163–64. doi:10.1038/d41573-019-00182-w.
- Wei F, Zhong S, Ma Z, Medvec A, Ahmed R, Freeman GJ, Krogsgaard M, and Riley JL. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci. 2013;110:E2480–E9.
- Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36(4):265–76. doi:10.1016/j.it.2015.02.008.
- Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. Rescue of exhausted CD8 T cells by PD-1–targeted therapies is CD28-dependent. Science. 2017;355(6332):1423–27. doi:10.1126/science.aaf0683.
- Tan S, Zhang H, Chai Y, Song H, Tong Z, Wang Q, Qi J, Wong G, Zhu X, Liu WJ, et al. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat Commun. 2017;8(1):1–10. doi:10.1038/ncomms14369.
- Na Z, Yeo SP, Bharath SR, Bowler MW, Balıkçı E, Wang C-I, Song H. Structural basis for blocking PD-1-mediated immune suppression by therapeutic antibody pembrolizumab. Cell Res. 2017;27(1):147–50. doi:10.1038/cr.2016.77.
- Lee JY, Lee HT, Shin W, Chae J, Choi J, Kim SH, Lim H, Won Heo T, Park KY, Lee YJ, et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat Commun. 2016;7(1):1–10. doi:10.1038/ncomms13354.
- Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Dömling A, Dubin G, Holak TA. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure. 2015;23(12):2341–48. doi:10.1016/j.str.2015.09.010.
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–29. doi:10.1056/NEJMoa1604958.
- Wang Q, Wu X. Primary and acquired resistance to PD-1/PD-L1 blockade in cancer treatment. Int Immunopharmacol. 2017;46:210–19. doi:10.1016/j.intimp.2017.03.015.
- Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7(1):1–9. doi:10.1038/ncomms10501.
- Ascierto PA, Melero I, Bhatia S, Bono P, Sanborn RE, Lipson EJ, Callahan MK, Gajewski T, Gomez-Roca CA, Hodi FS, et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. Am Soc Clin Oncol. 2017;35(15_suppl):9520–9520. doi:10.1200/JCO.2017.35.15_suppl.9520.
- Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, Yu H, Qin G, Sircar A, Hernández VM, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci Transl Med. 2018;10(424). doi:10.1126/scitranslmed.aan5488.
- Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob -J-J, Rutkowski P, Cowey CL, Lao CD, Schadendorf D, Wagstaff J, Dummer R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19(11):1480–92. doi:10.1016/S1470-2045(18)30700-9.
- Miller K, Meng G, Liu J, Hurst A, Hsei V, Wong W-L, Ekert R, Lawrence D, Sherwood S, DeForge L, et al. Design, construction, and in vitro analyses of multivalent antibodies. J Immunol. 2003;170(9):4854–61. doi:10.4049/jimmunol.170.9.4854.
- Gong S, Ren F, Wu D, Wu X, Wu C. Fabs-in-tandem immunoglobulin is a novel and versatile bispecific design for engaging multiple therapeutic targets. In: MAbs. Taylor & Francis; 2017. p. 1118–28.
- Li L, Deng L, Meng X, Gu C, Meng L, Li K, Zhang X, Meng Y, Xu W, Zhao L, et al. Tumor-targeting anti-EGFR x anti-PD1 bispecific antibody inhibits EGFR-overexpressing tumor growth by combining EGFR blockade and immune activation with direct tumor cell killing. Transl Oncol. 2021;14(1):100916. doi:10.1016/j.tranon.2020.100916.
- Gu C-L, Zhu H-X, Deng L, Meng X-Q, Li K, Xu W, Zhao L, Liu Y-Q, Zhu Z-P, and Huang H-M. Bispecific antibody simultaneously targeting PD1 and HER2 inhibits tumor growth via direct tumor cell killing in combination with PD1/PDL1 blockade and HER2 inhibition. Acta Pharmacol Sin. 2021:1–9.
- Kotanides H, Li Y, Malabunga M, Carpenito C, Eastman SW, Shen Y, Wang G, Inigo I, Surguladze D, Pennello AL, et al. Bispecific targeting of PD-1 and PD-L1 enhances T-cell activation and antitumor immunity. Cancer Immunol Res. 2020;8(10):1300–10. doi:10.1158/2326-6066.CIR-20-0304.
- Lázár-Molnár E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc Natl Acad Sci. 2008;105:10483–88.
- Zak KM, Grudnik P, Magiera K, Dömling A, Dubin G, Holak TA. Structural biology of the immune checkpoint receptor PD-1 and its ligands PD-L1/PD-L2. Structure. 2017;25(8):1163–74. doi:10.1016/j.str.2017.06.011.
- Philips EA, Garcia-España A, Tocheva AS, Ahearn IM, Adam KR, Pan R, Mor A, Kong X-P. The structural features that distinguish PD-L2 from PD-L1 emerged in placental mammals. J Biol Chem. 2020;295(14):4372–80. doi:10.1074/jbc.AC119.011747.
- Lee CV, Koenig P, Fuh G. A two-in-one antibody engineered from a humanized interleukin 4 antibody through mutation in heavy chain complementarity-determining regions. In: MAbs. Taylor & Francis; 2014. p. 622–27.
- Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67(2):95–106. doi:10.1016/j.molimm.2015.01.003.
- Gong S, Wu C. Generation of Fabs-in-tandem immunoglobulin molecules for dual-specific targeting. Methods. 2019;154:87–92. doi:10.1016/j.ymeth.2018.07.014.
- Sampei Z, Igawa T, Soeda T, Okuyama-Nishida Y, Moriyama C, Wakabayashi T, Tanaka E, Muto A, Kojima T, Kitazawa T, et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PloS one. 2013;8(2):e57479. doi:10.1371/journal.pone.0057479.
- Smith EJ, Olson K, Haber LJ, Varghese B, Duramad P, Tustian AD, Oyejide A, Kirshne JR, Canova L, and Menon J. A novel, native-format bispecific antibody triggering T-cell killing of B-cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci Rep. 2015;5:1–12.
- Klein C, Schaefer W, Regula JT, Dumontet C, Brinkmann U, Bacac M, Umaña P. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods. 2019;154:21–31. doi:10.1016/j.ymeth.2018.11.008.
- Wu X, Sereno AJ, Huang F, Lewis SM, Lieu RL, Weldon C, Torres C, Fine C, Batt MA, and Fitchet JR. Fab-based bispecific antibody formats with robust biophysical properties and biological activity. In: MAbs. Taylor & Francis; 2015. p. 470–82.
- Steinmetz A, Vallée F, Beil C, Lange C, Baurin N, Beninga J, Capdevila C, Corvey C, Dupuy A, and Ferrari P. CODV-Ig, a universal bispecific tetravalent and multifunctional immunoglobulin format for medical applications. In: MAbs. Taylor & Francis; 2016. p. 867–78.
- Jakob CG, Edalji R, Judge RA, DiGiammarino E, Li Y, Gu J, and Ghayur T. Structure reveals function of the dual variable domain immunoglobulin (DVD-Ig™) molecule. In: MAbs. Taylor & Francis; 2013. p. 358–63.