
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Computational prediction of the behavior of concentrated protein solutions is particularly advantageous in early development stages of biotherapeutics when material availability is limited and a large set of formulation conditions needs to be explored. This review provides an overview of the different computational paradigms that have been successfully used in modeling undesirable physical behaviors of protein solutions with a particular emphasis on high-concentration drug formulations. This includes models ranging from all-atom simulations, coarse-grained representations to macro-scale mathematical descriptions used to study physical instability phenomena of protein solutions such as aggregation, elevated viscosity, and phase separation. These models are compared and summarized in the context of the physical processes and their underlying assumptions and limitations. A detailed analysis is also given for identifying protein interaction processes that are explicitly or implicitly considered in the different modeling approaches and particularly their relations to various formulation parameters. Lastly, many of the shortcomings of existing computational models are discussed, providing perspectives and possible directions toward an efficient computational framework for designing effective protein formulations.
Introduction
Protein-based therapeutics are at the frontier of development in pharmaceutical industry with a fast-growing global market, where monoclonal antibodies (mAbs) represent the largest class of biotherapeutics with more than 80 mAb drugs approved to date in the United States alone.Citation1, Citation2 In addition to the challenges associated with their structural and functional design, protein solutions often exhibit physical instabilities such as aggregation and phase separation that arise from a complex interaction network among protein molecules with solution components. As current trends in biologics pipeline shift toward high concentration formulations, controlling protein instabilities is becoming more challenging. At elevated protein concentrations (>100 mg/mL), phenomena such as multi-body interactions and crowding exacerbate physical instabilities and might lead to other undesirable behaviors such as elevated viscosity and thermodynamic instabilities.Citation3 While challenging, achieving stable high concentration protein formulations is necessary for both moving toward a patient-centric drug product and expanding the biologics drug market. As such, there is a need for rapidly advancing our understanding of the behavior of biotherapeutics at elevated protein concentrations.
Indeed, mitigating protein instabilities during the development of commercially viable biotherapeutics requires identifying optimal but phase-appropriate formulations. This entails exploring the space that governs the relations between formulation conditions and solution behavior. However, this formulation space is vast, where many parameters describing the solution conditions (e.g., protein concentration, pH, buffer, and excipients) are closely related to many protein properties such as hydrophobicity, charge distribution, morphology, and size. In fact, high concentration protein formulations constitute complex solutions, where formulation parameters are strongly interconnected to protein behavior such that a change in one parameter could cause contradictory effects on the relation between formulation and protein stability.Citation3,Citation4 Moreover, due to limitations in material, time and resources availability during early-stage development (e.g., drug-candidate selection and preclinical development), a thorough experimental exploration of the formulation space becomes significantly challenging. In this regard, the implementation of fundamentally and statistically based computational models provides complementary tools for in-depth elucidation of the protein behavior, as well as for the subsequent identification of potentially relevant formulations. Specifically, these models can help design biologic drug formulations by: (1) constraining the formulation space to be experimentally investigated; (2) providing understanding of the underlying mechanisms for the different instability processes; and/or (3) identifying the mechanisms by which different solution components (or excipients) modulate protein behavior in the formulation.
Over the past two decades, an increasing number of studies focusing on the development and implementation of a variety of computational modeling tools have been reported. These studies have focused on understanding and/or predicting the behavior of protein solutions from either a biological or a biopharmaceutical standpoint.Citation5–12 As such, this review aims to provide a survey of the state-of-art of the in-silico application of a wide-range of computational models for effectively studying physical instabilities in protein solutions within the context of concentrated conditions. The models summarized here span various techniques and length-scales, ranging from atomistic simulations, coarse-grain representations, kinetic models, as well as novel approaches that combine resolutions from different molecular representations with other types of statistical and mathematical implementations. The review starts with an overview of the diverse classes of computational approaches that one commonly finds for evaluating the physical processes involved in destabilizing protein solutions. A particular emphasis is given on highlighting the range of length- and timescales that they can cover, as well as the underlying assumptions of each type of model. This overview aims to provide a summary of key physical considerations, practical and conceptual advantages, and missing components in the different classes of models. Thereafter, the different adverse thermodynamic and transport phenomena commonly affecting high-concentration protein formulations are revisited, exploring protein instabilities such as protein aggregation, phase separation and elevated solution viscosity. From a formulation perspective, protein–protein and protein–excipient interactions are the controlling knobs to modulate protein instabilities. Considering the landscape of mathematical models presented above, the manuscript continues with an overview of the proposed approaches used for evaluating protein interactions in both diluted and concentrated conditions. The review closes with a discussion about the perspectives and possible directions toward an efficient computational framework for designing effective protein formulations. This includes an analysis of the shortcomings of existing computational models in terms of computational cost, accessibility and inherent modeling limitations in capturing relevant experimental outcomes, as well as an examination of emerging multi-scale modeling approaches such as combining atomistic or coarse-grained models with machine learning or continuum models.
Types of protein models
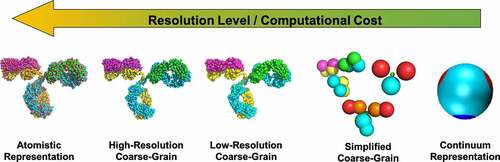
Several computational models and tools have been developed in recent years, addressing many of the pharmaceutics-related protein stability problems such as protein self-association, protein aggregation, phase separation and elevated viscosity. These models and tools are very broad in terms of how proteins are represented and what “key” protein features are incorporated to study the different instability processes. Due to the multi-scale nature of protein stability, it is computationally prohibitive to use a single model to study both nanometer-scale problems (e.g., conformational changes, protein–protein interactions) and macroscopic issues (e.g., aggregation, phase-separation). As a result, the computational study of protein stability lends itself to a hierarchy of models (), which can be classified as: 1) atomistic; 2) coarse-grain; and 3) continuum models. Although other types of modeling such as quantum mechanical representations and statistical approaches have been also used, this review primarily focuses on classical modeling studies applied to understanding and predicting protein stability phenomena, and problems particularly related to high-concentration protein formulations. provides a summary of the different classes of models and their applicability to physical instability phenomena in protein solutions.
Table 1. Application of different model resolutions to protein instability processes
Figure 1. Representation of the hierarchy of computational protein models based on their level of resolution, using an IgG2 mAb (PDB: 1IGT) as an example. These types of models include: atomistic; high-resolution coarse-grain (based on the model from Bereau and DesernoCitation13); low resolution coarse-grain from Blanco et al.;Citation14 simplified coarse-grain using the 12-bead model from Calero-Rubio et al.Citation15 and the 4- and 7-bead models from Blanco et al.;Citation16 and a continuum model based on Wertheim’s theory adapted by Skar-Gislinge et al.Citation17 The arrow indicates the direction in which the resolution-level increases for each model.
Atomistic models
Atomistic representations of both protein and all other species in the formulation (e.g., water, excipients) provide the most fundamental modeling approach to evaluate the behavior of protein solutions. In all-atom simulations, each atom is explicitly modeled as a single bead, and the protein solution behavior is characterized through the potential energy function. This function accounts for the bonded interactions (e.g., bond stretching, angle bending, torsions and improper angles), as well as intra- and inter-molecular non-bonded interactions (e.g., electrostatics, van der Waals, hydrogen bonding) for all molecular species. Consequently, the large number of different atomic forces yield an even larger number of parameters to capture the strength, range, and equilibrium energy for each type of interaction. The set of parameters or force fields for basic biomolecular systems (namely, proteins, nucleic acids, lipids, and biologically relevant ions) have been determined by several groups, where CHARMMCitation18 and AMBERCitation19 are among the most commonly used force fields. This class of models provide valuable insight into the early stages of different stability processes beyond experimental resolution capabilities. As such, they have been successfully applied to study various protein phenomena in crowded environments such as the kinetics and thermodynamics of protein conformational changes,Citation20 early stages of protein aggregation in peptides and small proteins,Citation6 protein–protein/excipient interactions,Citation21 among other processes.Citation22,Citation23
However, there are some limitations of atomistic simulations in protein systems. Due to the significantly large number of particles or atoms that needs to be considered, in-silico experiments of protein solutions are generally constrained to processes occurring in timescales smaller than microseconds. While advances in enhanced sampling algorithms in tandem with the use of supercomputers have allowed for modeling large systems involving crowded environments and high protein concentrations,Citation24,Citation25explicit simulations of phenomena of biopharmaceutical interest such as particle formation remains unreachable due to the long time- and length-scale for these stability processes.Citation3 Similarly, due to typical parameterization strategies of atomistic models against experimental data, the inherent complexity of force fields leads to difficulties regarding transferability and relevance of these models between different types of biomolecular systems. Such issues become more evident when studying concentrated protein solutions, as the balance between protein–protein and protein-solvent/excipient interactions is not adequately captured.Citation26–28 Moreover, parametrization of force fields have been largely based on systems of biological interest rather than biopharmaceutical relevance, which poses a challenge when studying the effects of various formulation components such as polysorbatesCitation29 and cryoprotectants.Citation30 Despite the substantial advances in recent years toward the improvement of force fields,Citation26–28 further efforts may be needed to fine-tune the different terms in the non-bonded interactions when simulating relevant protein formulations.
Coarse-grain models
Coarse-grain (CG) protein models have emerged as an alternative approach for studying protein stability problems, as they offer the potential for overcoming some of the inherent limitations of all-atom simulations.Citation12,Citation13,Citation31 In this class of models, the complexity of protein systems is simplified by grouping two or more atoms into a single particle (CG site), reducing the degrees of freedom in the system and expanding the range of time- and length-scales achievable by atomistic models.Citation7,Citation9 Interactions between CG sites is however independent of the set of atoms used for mapping these sites, as they are developed to capture key physicochemical factors to successfully studying the phenomenon of interest. As such, the resulting mapping of a given set of atoms onto a CG site is neither unique nor arbitrary. This flexibility in coarse-graining proteins has yielded a wide range of CG models, from simplified models to highly detailed CG representations (). highlights selected models and their applications based on the hierarchy of coarse-graining resolutions.
Table 2. Selected examples of coarse-grain models and their applications
In high-resolution CG models, residues are generally represented by four to seven CG sites. Most of these CG sites are assigned to the peptide backbone to preserve the dynamics of protein secondary structure, while the remaining sites represent the residue’s side chain to incorporate the identity of the protein sequence through amino acid-specific interactions. Examples of these models include the MARTINI model,Citation35 the OPEP model,Citation39 and the PRIMO model.Citation46 Because of the high level of structural details that these CG models provide, they have been successfully implemented for studying protein–protein interactions,Citation74 the mechanisms for protein folding and aggregation in small to medium size proteins and in crowded environments,Citation13,Citation44 the self-assembly pathways of virus proteins,Citation75 as well as to facilitate the refinement of NMR and crystallographic structures.Citation76 High-resolution models represent a significant improvement over atomistic models in terms of computational cost, enabling the evaluation of protein process with characteristic timescales on the order of milliseconds.Citation9,Citation77
By contrast, low-resolution CG models take the representation of proteins one step further by using only 1–3 CG sites per residue, where the specificity of different residue–residue interactions is maintained by explicitly incorporating the type and nature of each amino acid.Citation12,Citation78 By reducing most of the degrees of freedom from the backbone, these models enable faster sampling of systems with multiple proteins or with characteristic length-scales on the order of ~100 nm.Citation79 In spite of the limited amount of structural details of the protein backbone, some of these CG models still account for the flexibility of the peptide bond (i.e., the distribution of distances, planar and torsional angles) from statistical analysis of the structural properties of known peptides and proteins.Citation59,Citation77,Citation78 For instance, the UNRES model,Citation59 a 2-CG site per residue representation, has been extensively used for studies of protein folding, structure prediction and the mechanism of protein fibrillation.Citation60–62 Other CG models in this category have been used to evaluate weak protein–protein interactions,Citation14,Citation50 the effect of mutations on protein self-association,Citation48 the self-assembly mechanism of large protein complexes,Citation55,Citation58 and the role of post-translational modifications on protein micro-phase separation.Citation80
On the other end of the spectrum of protein coarse-graining, there are simplified CG models, where fragments, or even entire proteins, are modeled by a single CG site. These models sacrifice sequence-level resolution to facilitate studying systems with as many as 105 molecules.Citation71 Nonetheless, elaborated force fields that include orientational-dependent interactions are developed for some of these type of models in order to capture key properties of the sequence heterogeneity. Moreover, their simplicity has enabled simulating systems of large proteins like mAbs without using state-of-the-art supercomputers. Indeed, a number of different simplified CG models for mAbs have been developed in recent years, where mAb representations span from 3 to 26 beads with different levels of intramolecular flexibility.Citation15,Citation16,Citation66,Citation81,Citation82 These mAb models, as well as other simplified CG models for globular proteins, have been used for explicitly simulating macroscopic behavior of concentrated protein solutions such as crystallization, liquid–liquid phase separation, fibril formation, and their transport properties.Citation70,Citation73,Citation83–85
Similar to atomistic models, there are a number of different challenges in the development and implementation of coarse-grain models. These models are simplified representations of their all-atom counterparts; however, scaling between CG and atomistic models is not symmetric, as different CG sites within a given model might correspond to a different number of atoms with different chemical properties. Such asymmetry can result in both a non-uniform scaling of the system dynamics and a bias of the interactions between different CG sites, which may lead to difficulties in appropriately capturing the kinetics and thermodynamics of a given protein process.Citation7,Citation86 Furthermore, CG models are generally “custom-made” toward studying a particular system of interest, where their parameterization is usually based on reproducing the system behavior at a given thermodynamic condition. As a consequence, it is often observed that these models cannot be transferred between different protein systems, or they are even unable to predict the behavior of the same system at different thermodynamic states.Citation82,Citation87,Citation88 Finally, most CG models are developed in an implicit-solvent framework, where the behavior of the solvent and any excipient in solution is averaged out and absorbed in the potential energy function for protein–protein interactions. If one were interested in studying the effects of excipients on the stability of protein solutions (e.g., during formulation screening studies), a different set of CG force-fields for each combination of excipients would be required to carry out such in-silico studies.
Continuum models
Continuum models have emerged as alternative tools for explicitly simulating protein processes with very large characteristic time- and length-scales (e.g., timescales larger than seconds and length-scales larger than micrometers), which are out of reach for atomistic and CG models.Citation89–91 These models aim to solve mechanistic thermodynamic, hydrodynamic and/or kinetic equations of a process of interest. However, unlike the other types of models discussed above, continuum models incorporate minimal molecular or structural details of the system to be studied. Instead, they generally rely on physicochemical properties of both the protein and solution such as diffusivities, surface tension, and various free-energies and kinetic rate constants, while molecular aspects such as protein–protein and protein-solvent/excipient interactions are often treated in a mean-field approximation.Citation92–94 This relative simplicity of the continuum models facilitates studying the behavior of protein solutions with significantly less computational resources than molecular models.
Among the different types of continuum models, those based in computational fluid dynamics (CFD) are arguably the most broadly used in the pharmaceutical industry, with various applications to different stages of upstream and downstream process development.Citation95–97 The success of CFD to study problems involving fluid flows has led to the development of a number of different models for studying the behavior of concentrated protein solutions under mechanical stresses such as shear forces in pre-filled syringesCitation92,Citation98 and dense environments like subcutaneous tissue.Citation99 Similarly, several mechanistic kinetic models for protein aggregation have been developed to assess and predict the effect of different formulation parameters (e.g., pH, ionic strength, temperature) on the nucleation and growth of high molecular weight species. These aggregation kinetic models include mass-action kinetic approaches,Citation100 fixed-point approaches,Citation101 and stochastic approaches.Citation102 Other examples of continuum models include the application of theories based on statistical mechanics such as the Self-Consistent Ornstein-Zernike Approximation, Kirkwood-Buff solution theory, and Wertheim’s perturbation theory for evaluating high-concentration phenomena such as protein self-assembly,Citation103 liquid–liquid phase separation,Citation104 protein interactions,Citation105,Citation106 and solution rheology.Citation107,Citation108
Overall, continuum models are advantageous in connecting in-silico analysis of the behavior of protein solution to experimental development of biologic drugs. These models focus on evaluating stability phenomena on a scale comparable to that of most experimental techniques. In fact, they are often used for fitting experimental data to augment the information obtained from different assays, as well as to predict the behavior of protein formulations at different solution conditions.Citation85,Citation107,Citation109 However, special considerations are needed when implementing these types of modeling. While the underlying theories that constitutes most continuum models are rigorous, multiple assumptions are required to adapt these theories for modeling complex systems such as protein solutions. Due to these assumptions, the accuracy and relevance of the models may only hold for a small subset of solution conditions, which might result in misinterpreting the behavior of the solution beyond the conditions directly compared against experiments. For instance, a common simplification in statistical mechanics theories is to neglect high-order dependence of protein concentration.Citation104 Although such assumption is valid at diluted conditions, it fails to correctly describe the behavior of concentrated solutions due to factors like multi-body interactions and long-range spatial correlations.Citation110 Likewise, because of the large simplification in the molecular resolution of the system, the individual effects from different physical factors (e.g., different types of interactions, molecular anisotropy and heterogeneity) are reduced to a few parameters in the continuum models, leading to potential challenges in analyzing the results from the models.Citation17 The balance between these physical factors is a function of the local protein environment, and thus one should expect that the reduced model parameters are not constant but change with the solution conditions.Citation82 Additionally, the roles that the different physical factors play on the behavior of the solution are not independent from each other, which might lead to multiple combinations of the model parameters to describe the behavior of the protein system at a given solution condition.Citation100 Because of these challenges, the degree of predictability and transferability of the continuum models to different protein systems and formulations is very limited.
High-concentration physical instabilities
Protein self-association and aggregation
Among the different physical stability issues affecting proteins, aggregation is indisputably the most prevalent problem during the development of biotherapeutics. The formation of high molecular weight species and particulate matter can reduce the efficacy and affect the appearance of the drug product, in addition to potentially lead to unwanted immunogenicity.Citation3 Predicting and minimizing protein aggregation is therefore an important drug development goal. However, this remains a challenging task, as aggregation is an ubiquitous process to any protein, where the rate for aggregate formation is very sensitive to both protein structure and solution conditions.Citation111,Citation112 Moreover, aggregation is a complex multistep process governed by intra- and intermolecular interactions, with characteristic time and length scales that span many orders of magnitude.Citation100,Citation111
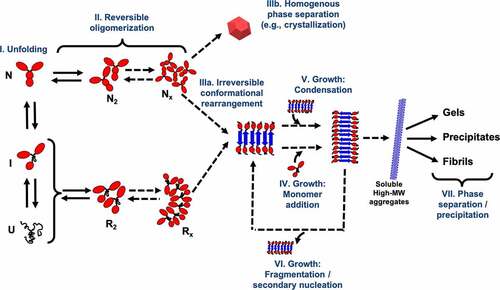
Mechanistically, protein aggregation occurs through a series of both reversible and irreversible stagesCitation100,Citation111 (), which include: conformational change of the protein monomer to form an aggregation-prone or reactive species (stage I); nucleation via protein self-association (stage II); the formation of the smallest irreversible aggregate species (stage IIIa) or a homogeneous phase separation when native self-association occurs (stage IIIb); aggregate growth via monomer addition (stage IV), aggregate–aggregate coalescence (stage V), and aggregate fragmentation (stage VI); and the phase separation or precipitation of the high molecular weight species (stage VII). Depending on the specific protein system, the relevance and rates of these stages can differ, leading to a variety of aggregation mechanisms. In this regard, in-silico models provide invaluable tools to gain insight into the critical factors affecting the different aggregation stages. Although most of the existing computational models cannot capture the complete range of relevant time and length scales, different types of modeling approaches have been developed and implemented to independently assess these stages, making predictions of the aggregation propensity and long-term stability of a given protein formulation.Citation86,Citation113,Citation114
Figure 2. Schematic representation of the generalized protein aggregation mechanism for multidomain proteins such as mAbs. The stages shown in the diagram correspond to either effectively reversible steps (double arrows) or irreversible steps (single arrows). Protein oligomerization can occur through self-association of the native monomer (N) or a (partially) unfolded reactive species R. The mechanism also considers the case that N self-associates to a critical size (NX) to nucleate a homogeneous phase separation (e.g., liquid-liquid separation or crystallization).
Based on the generalized aggregation reaction in , different (continuum) aggregation kinetic models have been established.Citation93,Citation100,Citation102,Citation109,Citation115,Citation116 All these models express the extent of aggregation via mass balance equations for the elementary reactions, but they differ by how the aggregate size distribution is treated. One of the most comprehensive aggregation models is that developed by the Roberts’ group, which follows a Lumry-Eyring Nucleated-Polymerization approach (LENP).Citation100 This model incorporates most of the aggregation stages outlined above, while it explicitly captures a broad range of aggregate species to yield a discrete aggregate size distribution. The LENP model has been successfully applied to characterize the effect of solution conditions on the aggregation mechanism of globular proteinsCitation117 and antibodies.Citation118 However, implementation of the LENP model is somewhat limited to diluted conditions, as relevant factors to the mass transport in concentrated protein solutions (e.g., crowding, viscosity effects, ion binding) are not incorporated in the model. Alternative approaches combine the aggregation mass balance equations with the Smoluchowski coagulation equations, which allows for coupling the elementary rate constants to the diffusion and fractal dimension of the aggregated species.Citation114,Citation116,Citation119,Citation120 This type of kinetic model has been shown to fit experimental data for mAbs reasonably well over a wide range of solution conditions and protein concentrations as high as 60 mg/mL.Citation116 Nonetheless, in a modified Smoluchowski-based model that incorporates concentration-dependent viscosity, Nicoud et al.Citation109 found for a concentrated mAb solution that large discrepancies between the aggregation model and experiments are obtained when strong anisotropic protein interactions and/or native aggregation are suspected. On the other hand, other aggregation kinetic models have focused on solving the mass balance equations in terms of probability density functionsCitation121 and generating functionsCitation102,Citation115,Citation122 to recover the aggregate size distribution. These models have successfully captured the time-evolution at various protein concentrationsCitation101,Citation102 and at crowded conditions.Citation121 Moreover, some of these models, as those developed by Knowles and collaborators,Citation101,Citation122,Citation123 yield a simplified analytical solution to the set of reaction equations, which reduces the computational burden and facilitates their implementation for fitting experimental data. However, most, if not all, of these latter kinetic models have been exclusively implemented to study fibrillation in peptides and small proteins. As such, they generally simplify aggregate growth to occur only via monomer addition, while other stages commonly observed during amyloid formation (e.g., secondary nucleation, auto-catalysis, and stochastic kinetics) are incorporated.
An alternative computational approach is also commonly used to evaluate and predict aggregation propensity and kinetics, which is based on information derived from single-molecule features. Different experimental mutagenesis studies on amyloidogenic proteins have shown that aggregation is correlated to physicochemical features in protein structure such as hydrophobicity, charge, β-strand propensity, and surface arrangement of amino acids.Citation124,Citation125 These observations initially spurred the development of a first-generation of aggregation predictors based solely on protein sequence, which identify aggregation-prone regions (APRs) from pattern matching and heuristic equations validated against experimental databases of hundreds of amyloid-forming short peptides (). Among these predictors, there are methods such AGGRESCAN,Citation126 TANGO,Citation130 PASTA,Citation129 and WALTZ.Citation131 The interested reader is referred to recent reviews on this specific topic.Citation86,Citation139 These sequence-based algorithms provide fast and computationally inexpensive tools to identify APRs and rank proteins based in their intrinsic aggregation propensity, and they have been used for guiding modification of mAb sequences to improve their stability.Citation140 However, these predictors present several limitations for a broader applicability in the biopharmaceutical industry. Firstly, most of these sequence-based tools predict APRs based on the aggregation mechanism of peptides and amyloidogenic proteins, which may not be representative of the aggregation pathway of larger proteins. Secondly, available experimental data for building and validating these algorithms is limited to a few solution conditions (mainly, physiological conditions), and therefore more data is required to expand their usefulness for predicting APRs in biotherapeutics over relevant formulations (e.g., larger pH range, different buffers and excipients). Lastly, the inherent assumptions of these sequence-based algorithms prevent them from capturing the role that the three-dimensional structure plays on aggregation. In fact, when applied to large proteins such as mAbs, these predictors often lead to a larger number of false positive and negative results, identifying regions that are not solvent exposed and failing to capture APRs involving residues from non-continuous sequence fragments.Citation8,Citation141
Table 3. Selected examples of aggregation-propensity predictors
To overcome some of the limitations of the sequence-based tools, a second generation of algorithms for predicting APRs have emerged, which explicitly accounts for the folded structure of proteins. That is, rather than relying solely on the protein sequence, the protein structure (e.g., from crystallographic or homology models) is leveraged to assess the likelihood that a given APR might be involved at the interface for aggregate formation. Some of these structure-based predictors, such as SolubiS,Citation138 CamSol,Citation134 and AGGRESCAN-3D,Citation52 combine the predictions from sequence-based tools with calculations of conformational stability and residue energy to weight APRs based on their tendency to be solvent-exposed or to interact with other fragments. Other predictors like SAP,Citation136 Developability Index,Citation135 and AggScore,Citation133 predominantly use the 3D protein structure to assess the residue solvent accessibility and/or partial charges (e.g., via short Molecular Dynamics simulations), which are then correlated to aggregation propensity. As a result, these structure-based predictors do not only outperform their sequence-based counterparts, but they have also been found useful for screening and re-engineering biotherapeutics candidates to reduce their aggregation propensity.Citation8 Moreover, Wolf et al.Citation142 found that the results obtained from some of these in-silico algorithms for a series of mAbs are well correlated with several experimental techniques used to evaluate their early-stage developability. In other studies, Trout and collaborators have combined the calculation from SAP with Molecular Dynamics simulations of protein-excipient interactions to gain insight into how aggregation propensity and viscosity of mAb formulations are affected by excipients such as carbohydratesCitation30 and ionic species.Citation143 Despite the significant improvements of the structure-based predictors, there are a few downsides that need to be considered for their implementation. Calculations of the different structural properties are carried out from fluctuations around the native protein structure, and thus they inherently bias the results toward APRs involved in a native-aggregation pathway. Likewise, these algorithms are based on single-molecule simulations, where the general assumption is that each APR is independent of each other in terms of their contribution to protein aggregation. As a consequence, they neglect synergistic effects that may arise from the proximity of two or more APRs.Citation8 The single-molecule calculations also make impossible to distinguish protein concentration effects on aggregation such as the role of multibody interactions and excluded volume.Citation111 Nevertheless, as more experimental data on aggregation of biotherapeutics becomes available, these limitations might be overcome by improvements in the underlying heuristics to correlate structural properties to aggregation propensity. Promising efforts in this direction have been recently seen by Lai et al.,Citation144 where different structural properties, including those used in SAP, were fed into a machine learning algorithm to predict the aggregation rate of 21 mAbs at high-concentration.
The use of molecular models has not been limited to investigating the molecular properties related to aggregation. Both atomistic and CG models have been widely used to understand the dynamics and thermodynamics of various aspects of the aggregation mechanism.Citation6,Citation78,Citation113 The development of enhanced sampling methods such as Replica-Exchange Molecular Dynamics, Metadynamics, Umbrella Sampling, and Markov modeling, among others, have facilitated expanding the use of these different types of protein models to study critical steps from the generalized aggregation mechanism (). Reviews that provide a comprehensive summary of advanced computational methods used for studying protein aggregation and other processes are available.Citation10,Citation24 In the case of all-atom simulations, studies on protein aggregation are often focused on small protein fragments from known APRs. In-silico studies of homopeptides (e.g., poly-alanine, poly-valine, poly-glycine) and several fragments from amyloidogenic proteins have enabled our understanding, at a molecular level, of the early-stage mechanism of fibril formation.Citation145–147 These studies have shown that peptides initially collapse into a partially ordered oligomeric state up to a critical size nucleus of 6–8 strands, to then evolve into ordered β-sheet structures. Luiken and BolhuisCitation148 showed that this nucleation process can change from a one-step to a two-step nucleation mechanism as the peptide hydrophobicity increases. Atomistic models have also provided insight into the growth stages of fibrillation.Citation6,Citation31,Citation113 These models have highlighted the role that conformational fluctuations play during elongationCitation149,Citation150 and secondary nucleation,Citation151 as well as enabled the characterization of key residue–residue and residue–water interactions that govern the stability and fragmentation of fibrils from different amyloid-prone peptides.Citation152,Citation153 Note that the implementation of atomistic models have mainly focused on amyloid formation, whereas their application to the phenomenon of aggregation in biotherapeutics have been directed to characterize the local dynamics of antibody fragments to help explain experimental observations of different aggregation behaviors.Citation154
On the other hand, computational studies of the aggregation mechanism of larger proteins and/or longer length-scales have been possible via coarse-grain models from all different resolution-levels.Citation7,Citation31,Citation155 Different simplified CG representations have been developed to elucidate how the interplay between soluble and aggregation-reactive conformations affect the nucleation and elongation stages of fibrillation.Citation71,Citation155,Citation156 In an interesting report by Ŝarić et al.Citation73 using one of these simplified CG models, the authors found that monomers can spontaneously aggregate without a nucleation step at high protein concentrations, while the formation of a small oligomeric nucleus is a prerequisite for aggregation at low protein concentrations. Likewise, low-resolution CG models have led to new insights into protein self-association and aggregate growth.Citation78 Phenomenological models such as those from Shea’s and Caflisch’s groups have been used to describe the effect of hydrophobic and charge residues on the nucleation stage,Citation157 as well as the dependence of the different pathways for fibril growth on protein conformation.Citation158,Citation159 Other low-resolution CG models based on rigid representations of proteins have been used to understand the role of surface residues and solution conditions of protein self-association.Citation49,Citation50,Citation62 For instance, Blanco et al.Citation14 used a 1-bead per residue protein model for γD-crystallin to evaluate the effect of ionic strength on modulating preferential protein orientations during self-association, enabling the identification of key mutations to reduce the aggregation rate.Citation48 Notably, protein aggregation studies using different high-resolution CG models have arrived at similar conclusions regarding the mechanisms of nucleation and aggregate growth (albeit using smaller fragments of the same proteins).Citation40,Citation160,Citation161
Liquid-liquid phase equilibrium
Another concerning physical stability issue in high-concentration protein formulations is their potential to become opalescent and undergo a liquid–liquid phase separation (LLPS) process during refrigerated conditions, where the solution separates into protein-rich and protein-poor phases.Citation11,Citation162,Citation163 This phenomenon can both affect the esthetics of the drug product and trigger other stability issues.Citation163 While LLPS is typically a reversible process (e.g., increasing temperature brings the solution back to a homogeneous, single phaseCitation3) the partitioning of the different solution components (particularly, ionic species) between both phases might trigger other phenomena. The imbalance of the buffer/excipient species between the equilibrium phases might shift the pH and ionic strength toward unfavorable conditions and result in protein unfolding, irreversible aggregation or protein precipitation.Citation163 This problem is not exclusive to the biopharmaceutical industry, as LLPS also occurs in living cells and is related to mechanisms of intracellular organization and various diseases.Citation164,Citation165 As such, understanding the phase separation of protein solutions remains an active research area in many disciplines from both experimental and computational standpoints.
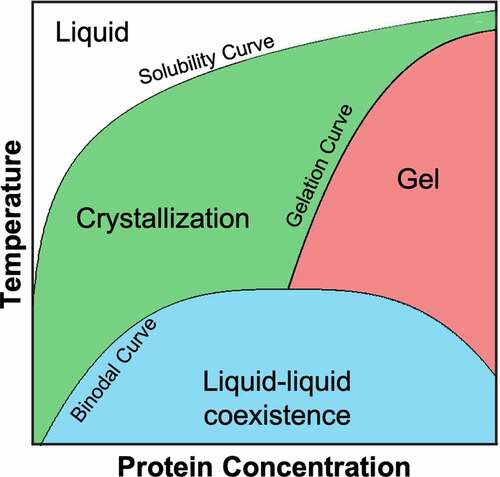
Experimental studies on several globular proteins such as lysozymeCitation166 and γ-crystallinsCitation167 have provided a comprehensive picture of the phase behavior of these proteins, with prominent common features such as a metastable LLPS with respect to crystallization and the formation of an arrested or gel-like state above the critical concentration (). The resemblance of the phase diagram of globular proteins with that of short-ranged attractive colloidal particles spurred most of the earliest computational studies in LLPS for proteins.Citation168 In fact, simplified CG models consisting of hard-spheres with an isotropic short-range attraction are able to qualitatively reproduce both the metastability of the liquid–liquid transition and the high-concentration arrested state provided that the range of the intermolecular interactions is sufficiently small (e.g., ~1/8 of the protein diameter).Citation169 Moreover, these simplified colloidal protein models have led to the discovery of an “extended law of corresponding states” for LLPS in globular proteins.Citation169,Citation170 That is, the LLPS of globular proteins collapses into a master curve when representing the binodal curve in terms of the strength of the attractive interactions (via the osmotic second viral coefficient, ) and an effective protein volume that accounts for the screened protein charge. However, these isotropic models are unable to quantitatively capture the shape of the phase boundaries and the concentration for crystallization, and instead CG models with patchy or directional interactions are required for better quantitative agreement with experimental data.Citation11,Citation162 Numerous groups have not only demonstrated that these patchy models yield broader binodal curves like those of globular proteins,Citation171–173 but they also provide insight into the variety of space groups on protein crystals.Citation84 Moreover, it has been shown through these patchy models that LLPS can be either suppressed or triggered by protein oligomerization based on the extent of aggregation.Citation83,Citation171 The computational study of LLPS has not been limited to simplified CG models, as other CG models with different levels of resolution, as well as continuum models, have also been developed for evaluating LLPS in globular and disordered proteins.Citation104,Citation165,Citation174 In a very interesting approach, Wertheim’s thermodynamic perturbation theory was adapted to capture directional interactions of globular proteins,Citation104 and it was even extended to study protein-phase separation in the presence of different buffersCitation175 and salts.Citation176
Figure 3. Generic phase diagram for globular proteins adapted from Muschol and Rosenberger.Citation166 The regions below the solubility curve (i.e., the gel and liquid-liquid coexistence regions) are metastable with respect to crystallization. The liquid-liquid coexistence region, bounded by the binodal curve, corresponds to the thermodynamic state where the solution separates into protein-rich and protein-poor phases. The gelation curve indicates the boundary for the formation of an arrested state. For any protein, the relative position between the solubility, binodal and gelation curves depends on both the protein sequence and solution conditions. Redrawn from Ref. 166.
In-silico studies of LLPS have also been applied to proteins of pharmaceutical interest such as monoclonal antibodies.Citation177,Citation178 The liquid–liquid binodal curve of mAbs significantly differs from that of globular proteins, as the critical point of antibody solutions typically occurs at lower temperatures and concentrations, while the binodal curve is broader.Citation11,Citation163 Sun et al.Citation177 used simplified CG models representing mAbs as flexible molecules composed of 3 to 7 CG-sites to demonstrate that both the ‘Y’ shape and the flexibility of the hinge region contribute to the asymmetrical shape of the binodal, but they have minimal effect on determining both the critical concentration and elevated density of the protein-rich phase. Instead, it was found that the inner subdomains (i.e., CH1, CH2 and CL) need to be net repulsive or less attractive in comparison to the other subdomains in order to obtain a phase coexistence curve quantitatively comparable to experiments. Hinge flexibility, on the other hand, still plays an important role in facilitating quaternary structural rearrangement to achieve a more compact but stable solution structure at very high mAb concentrations.Citation16 Recently, Vlachy and collaboratorsCitation178 arrived at similar conclusions by extending Wertheim’s theory for a 7-CG-site mAb model. These authors found that the critical temperature and concentration are sensitive to the imbalance of the interactions involving the CH3 and variable fragment (FV), but they are marginally affected by the actual strength of the intermolecular interactions. This latter continuum model has been implemented to semi-quantitatively reproduce experimental LLPS data for two different mAbs,Citation178 as well as to evaluate the effect of polymersCitation179 and bulky agentsCitation180 on the phase separation of antibodies.
The development and implementation of both simplified CG and continuum models has driven much of our understanding of the phase behavior of proteins. These models have not only provided insight about the relevance of anisotropic or patchy interactions on the LLPS of proteins, but they have also allowed us to identify simple guidelines to qualitatively identify conditions that lead to phase separation (e.g., based on for globular proteins or the imbalance of interactions between mAb fragments). Nonetheless, further research is still needed to streamline the use of modeling for robustly screening protein drug candidates or drug product candidates against phase behavior. Firstly, while patchy models represent the state-of-the-art for quantifying LLPS, the definition of a protein “patch” remains loose. There is no comprehensive rationale to select the degree of anisotropy to connect the CG patchy model to the protein structure or sequence. Commonly, patches are placed in either a random or symmetric fashion, and experimental data is fit based on the number, size and interacting strength of these patches.Citation106 Although this approach can be effective, it does not give insight about specific molecular features related to LLPS for a given protein or class of proteins. Different efforts have been made to relate surface features or sequence fragments to interacting patches,Citation181,Citation182 but it is unclear what the relevant characteristics are for these patches to influence LLPS and other instability phenomena. In this regard, the use of higher resolution CG models might provide an alternative approach to overcome these issues, as they can provide information about residue-level interactions related to LLPS. In fact, such models have been recently used to assess the LLPS of intrinsically disordered proteins.Citation164,Citation165 Another outstanding challenge comes from the inherent modeling limitations for evaluating phase separation processes. From a computational perspective, identifying and characterizing phase coexistence curves are some of the most expensive and intensive modeling tasks, as they require sampling over millions of configurations and/or very large system volumes to overcome the generally large thermodynamic barriers between the phases at equilibrium. Moreover, these simulations typically need to be carried out over several thermodynamic states (e.g., multiple sets of temperature and pressure) in order to reconstruct the binodal curves. As such, novel and clever approaches need to be developed or adapted to reduce the computational burden for assessing LLPS. Some recent methodologies based on multi-scaling,Citation183 Widom insertion,Citation184 and thermodynamic extrapolationCitation185 might provide a path forward in this regard. Last but not least, there is a lack of experimental data for phase behavior of proteins, and in particular for pharmaceutically relevant biologics. LLPS data have only been reported for a few proteins, which represents a challenge for identify generalized guidelines to predict potential problems for a given drug candidate with respect to phase behavior.
Transport properties of high-concentration solution
Solution viscosity is a critical attribute for the development of high-concentration protein formulations, as an elevated viscosity (>30 cP) can significantly impact the pressure and flow in various unit operations such as filtration, ultrafiltration-diafiltration, and filling,Citation95,Citation96 as well as limit the development of devices for drug administration (e.g., auto-injectors and pre-filled syringes).Citation186 From a physicochemical standpoint, it has been proposed that the presence of transient protein clusters is the root cause of high viscosity in protein solutions, which is in turn driven by protein–protein interactions.Citation187,Citation188 As such, understanding the relationships between interactions, solution structure and solution rheology is key for establishing appropriate formulation strategies to achieve a suitable viscosity in high-concentration biotherapeutics. In this regard, computational protein models have greatly contributed to our current knowledge of the molecular origins of viscosity behavior in protein solutions.
Early computational work focused on investigating the viscosity problem using globular proteins as model systems, where well-established (continuum) colloidal models for spherical particles such as the mode-coupling theoryCitation189 (MCT) could provide insight into the relation between protein interactions and solution viscosity. Several groups have studied the rheological behavior of solutions of bovine serum albumin (BSA) up to 300 mg/mL at different buffer conditions and salt concentrations.Citation190–192 Interestingly, colloidal models based on particles with hard-sphere repulsion and long-range electrostatic interactions were able to fit reasonably well the concentration-dependent viscosity of BSA, indicating that long-range repulsive interactions govern the behavior of viscosity.Citation191 A similar conclusion was also found by Foffi et al.Citation193 for α-crystallin, where screening of electrostatic repulsions allows the solution viscosity to be captured by a simple polydisperse hard-sphere model and by MCT. Despite the success of these repulsive colloidal models, many proteins interact through a combination of short-range attractions and long-range repulsions. Different studies with lysozyme have demonstrated that the competition between these types of interactions facilitates the formation of protein clusters through an intermediate-range order, where screening electrostatics yields a higher solution viscosity.Citation107,Citation110,Citation194 In such cases, previously used colloidal models such as MCT fail to capture the viscosity of concentrated lysozyme solutions.Citation110 In an interesting recent report, von Bülow et al.Citation195 used all-atom Molecular Dynamics simulations to evaluate the concentration dependence of viscosity for four small globular proteins (≤ 14 kDa) for protein concentrations as high as 200 mg/mL. The authors found that protein crowding strongly affects both translational and rotational diffusions, but the slowdown on the rotational diffusivity is mainly related to the formation of weak, dynamic protein clusters with dissociation constants of ~20 mM. Indeed, they derived a heuristic model that well reproduces experimental diffusivities based on the mean cluster size and viscosity calculated from the simulations.
When modeling the viscosity of pharmaceutically relevant proteins such as mAbs, the situation is more dire than in the case of globular proteins. The multi-domain nature and anisotropic shape of mAbs make the implementation of existing colloidal spherical models difficult for predicting solution viscosity. Moreover, under identical changes in formulation conditions, some antibodies show opposite trends in viscosity behavior.Citation4,Citation188 These observations have suggested that the elevated viscosity of mAb formulations is driven by local sequence and structural features rather than net colloidal effects. As such, a large portion of the computational research carried out in this area has focused on the implementation of all-atom models to correlate local molecular descriptors (e.g., protein charge, solvent accessible area, dipole moment) with experimental viscosity data.Citation8 In one of the earliest works with mAbs, Li et al.Citation196 evaluated a series of molecular descriptors for 11 homology models of mAbs based on electrostatic and solvophobic properties. The authors correlated these descriptors with viscosity measurements of the antibodies under equivalent solution conditions, finding that the viscosity behavior for a given mAb isotype is correlated to descriptors associated with the FV domain, such as charge, pI, zeta potential, and aggregation propensity. Likewise, Tomar et al.Citation197 used experimental measurements of 16 different mAbs to develop a computational scheme to predict their concentration-dependent viscosity based on the electrostatic and solvophobic properties of both the FV and the full mAb structure. In addition to the electrostatic properties of the FV, this latter work identified that both the hydrophobic surface area of the mAb and the charge of hinge region are important to the predictability of viscosity. In agreement with these observations, Sharma et al.Citation141 used 14 mAbs to correlate experimental viscosity values with in-silico molecular descriptors, including properties calculated from Molecular Dynamics simulations. The authors concluded that the solution viscosity was correlated with the hydrophobicity and charge dipole of the FV region. More recently, Lai et al.Citation144 used 27 mAbs approved by the Food and Drug Administration to expand this type of modeling analysis, combining molecular descriptors obtained from atomistic models with machine learning feature selection. Both the net charge of the full mAb and the number of solvophobic residues in the FV region were highlighted by the machine learning algorithm as key features for the viscosity behavior.
An alternative computational approach has focused on using simplified CG models for mAbs to study the relation between protein interactions and solution structure, seeking to correlate changes in the spatial correlations of mAbs with experimental measurements of viscosity. Chaudhri et al.Citation66,Citation67 used two different simplified CG resolution levels to study the solution structure for two antibodies, which differ by only a few mutations in the complementarity-determining regions (CDRs) but exhibit different concentration-dependent viscosity. One of the CG models represents mAbs with one bead per subdomain (i.e., a 12-CG-site mAb representation), while the other model adds further resolution at the CDR and hinge regions by using 26 CG-sites to represent mAbs. Based on calculations of the potential of mean force at different protein concentrations via Molecular Dynamics simulations, it was found that complementary electrostatic interactions involving both the antigen-binding fragment (Fab) and FC leads to the formation of antibody clusters, networks, and higher-order structures, which correlates with the viscosity behavior of both mAbs. Interestingly, the use of a higher level of coarse-graining did not provide much further insight into the solution structure of these mAb systems, as the 12-CG-site model was sufficient to determine the underlying cause of the concentration-dependent viscosity. A similar conclusion was also obtained when further implementing the same modeling approach on 4 additional mAbs.Citation68 Wang et al.Citation69 combined the 12-CG-site representation with Brownian dynamics simulations to semi-quantitatively reproduce the experimental transport properties (e.g., self-diffusivity, structure factor and viscosity) of the same mAbs used by Chaudhri et al.Citation66 The authors identified as the cause of changes in transport properties the formation of weakly interacting protein clusters rather than dense or strong networks. Nonetheless, it was required to impose nonphysical constraints to maintain rigidity in the protein cluster in order to reproduce the high-concentration viscosity behavior of one of the mAbs. This model was recently improved by Lai et al.Citation70 by incorporating anisotropy into the short-range interactions between the constant and variable regions of the mAbs instead of an uniform van der Waals interaction term for all the CG-sites. The improved 12-CG-site model was evaluated against experimental viscosity measurements of 27 antibodies, where model parameterization was directly coupled to a previous machine learning approach.Citation144
In a different set of works, Dear et al.Citation85 and Chowdhury et al.Citation88 used a similar 12-CG-site mAb model in combination with small-angle X-ray scattering (SAXS) experiments to predict the concentration-dependent viscosity of two mAbs over a broad range of formulations. SAXS data was used to determine whether anisotropic protein interactions were relevant at a given solution condition, as well as to parameterize the CG force field. The resulting CG model was used for calculating the cluster size distribution of the solution, which in turn was used to reproduce the solution viscosity via an empirical equation.Citation88 This approach predicted reasonably well the changes in viscosity with respect to protein concentrations for both mAbs in most of the tested formulations, as well as for a polyclonal IgG;Citation198 however, it failed to capture these changes when protein clustering is driven by strong anisotropic interactions. More recently, Izadi et al.Citation81 developed a 10-CG-site mAb model, where the FC domain is represented by only 2 CG-sites. Unlike all previous CG models, parameterization of the CG force field was based solely on data from atomistic simulations to incorporate the multipole moments of the charge distribution. The resulting model was compared against experimental data from 16 antibodies in two different formulations (low and high ionic strength), and it was qualitatively correlated with transport properties such as the diffusion interaction parameter and viscosity. Other recent models based on Wertheim’s theory have arrived at similar conclusions, showing promising results for semi-quantitatively capturing the relation between anisotropic interactions (between FV and CH3 domains) and solution viscosity.Citation17,Citation199
Although both atomistic and CG models have been fundamental to expand our understanding of the role that electrostatic and anisotropic interactions play in the concentration-dependent viscosity of mAbs and other proteins, these models have some limitations, and as such, present opportunities for improvement. Benchmarking and validation of the different models have been limited to no more than 27 different biotherapeutics and only a few different solution conditions. A larger and more diversified set of molecules and formulations is needed to extend the validity, accuracy, and robustness of any predictive scheme. Additionally, the connection between protein interactions and viscosity behavior across the majority of the models have relied on either heuristic equations or statistical analysis. While these approaches allow us to establish correlations between the molecular and macroscopic behavior of the protein solution, they do not identify the underlying cause. Development of further phenomenological theories and models is still needed to elucidate the molecular origins for the concentration-dependent viscosity of a given biologic formulation. Moreover, most of existing models for assessing the viscosity behavior of protein solutions uses implicit solvent approximations. This approach facilitates reducing computational cost, but it significantly limits our ability to rationally identify the type of viscosity-modifier excipients that can be used for a given drug product formulation. While the work by von Bülow et al.Citation195 has demonstrated that it is possible to use all-atom modeling for explicitly assessing protein solution rheology (albeit for small proteins), further advancements in computational hardware are still required to permit the application of that type of models outside of supercomputers, as well as to extend their application to biologics of pharmaceutical interest. Finally, the empirical nature and reduced training data sets of most of these computational schemes limits their transferability to different molecules or formulation conditions. Generally, a new set of statistical correlations or CG force field parameters needs to be derived for each new protein system, which can be inefficient and unsuitable for screening over hundreds of drug candidates and/or formulations. Lai et al.Citation70 suggested, as a potential solution to this challenge, the use of databases to relate sets of model parameters to viscosity values for linear interpolation.
Protein-protein and protein-excipient interactions
As highlighted in the previous section, protein interactions are the keystone to understanding and predicting the different physical instability phenomena in protein solutions. The balance between different intermolecular forces (from both protein–protein and protein-excipient interactions) and solution conditions defines the likelihood that proteins self-associate, which in turn triggers the different stability issues discussed above. While protein–protein interactions account for the total contribution from attractive (e.g., hydrophobic, van der Waals, dipoles, hydrogen bonding) and repulsive forces (e.g., electrostatic, sterics), the protein environment plays a critical role in constraining the relevant ensemble of configurations that determines the net interactions and protein behavior.Citation106 As such, at diluted conditions, long-range interactions and the distribution of interacting sites at the protein surface govern net protein interactions due to the large average separation distances between proteins. On the other hand, at elevated concentrations, factors such as multibody interactions, crowding effects, spatial correlations and ion binding become important in controlling the distribution of proteins in solution, and thus they are equally important to net protein interactions.Citation3,Citation111,Citation200 From an experimental standpoint, protein interactions at diluted conditions are captured through parameters such as the second virial coefficient and the dynamic interaction parameter
, while other parameters like the Kirkwood-Buff integral
and structure factor
are used for assessing high-concentration protein interactions.Citation201,Citation202 Likewise, the preferential interaction parameter
is also used as a metric for protein-excipient interactions.Citation203 In fact, this assortment of parameters is central to many theories, models and heuristics for predicting the high-concentration behavior of protein solutions.Citation201,Citation204 This section focuses on summarizing the computational approaches used to capture these parameters and their application to evaluate and predict protein stability problems.
Among the different parameters used to describe protein–protein interactions in diluted conditions, is arguably the most broadly used. This parameter provides a measurement of the orientational- and solvent-averaged protein–protein interactions, and it has been correlated to phenomena such as protein aggregationCitation112 and protein-phase behavior.Citation169 One of the earliest, yet widely used computational approaches to calculate
is derived from the Derjaguin-Landau and Verwey-Overbeek (DLVO) theory for colloidal systems.Citation205 This theory represents proteins as spherical particles interacting through a combination of van der Waals and screened electrostatic forces. An extended version of DLVO (xDLVO) has also been developed to incorporate other types of interactions such as dispersion forces and osmotic potential.Citation206,Citation207 Different reports have shown that both DLVO and xDLVO can predict the overall trends of
for protein solutions as a function of ionic excipient concentrationCitation208 and in the presence of polymers.Citation206 Nonetheless, these theories provide only qualitative representations for protein interactions, as they are unable to accurately capture how
is affected by anisotropy in terms of both protein shape and surface heterogeneity.Citation106,Citation209
Pusara et al.Citation210 recently developed a coarse-grained xDLVO model to correct for the spherical approximation on the DLVO theory, though the results only show a modest improvement for predicting values as a function of ionic strength on both globular proteins and immunoglobulins. These results are not surprising, as Grünberger et al.Citation211 previously demonstrated that a CG representation of at least one bead per residue is the minimum resolution-level required to reasonably reproduce steric effects on
on different classes of proteins. In this regard, the low-resolution CG models from Kim and HummerCitation55 and Blanco et al.Citation14 offer suitable options to predict
, as they provide a resolution of one CG-site per residue that interacts through amino acid-specific short-range attractions and screened electrostatics (akin to a classical DLVO theory). These models differ by the type of protein interactions they were validated to capture. The former model was designed to reproduce “lock-and-key” and moderately strong protein interactions in order to evaluate the formation and behavior of protein complexes.Citation58 On the other hand, the latter model can represent the effects of pH and ionic strength on
, and it has been implemented to evaluate weak protein–protein interactions on globular proteinsCitation48 and mAb formulations.Citation50 Notably, atomistic and high-resolution CG models such as MARTINI have also been implemented for reproducing experimental
values of globular proteins in different solution conditions, though they require significant re-parameterization of the corresponding force fields.Citation212,Citation213
Although low-resolution CG models constitute simple but accurate protein representations for evaluating and identifying the effect of specific residues in the colloidal stability, they present practical limitations for their implementation in formulation development applications. When considering the use of these models for screening a broad range of formulation conditions, the computational burden of simulating hundreds of conditions makes them unsuitable for this type of application due to typically constrained timelines for drug development. As such, both continuum and simplified CG models provide alternative approaches for efficiently evaluating protein interactions. Wertheim’s perturbation theory has been applied to evaluate
, showing the ability of this type of continuum model to semi-quantitatively capture the relationship between anisotropic protein interactions and the nature of the solution buffer and other ionic excipients.Citation104,Citation176 Likewise, Roberts and collaborators have extensively evaluated the use of simplified CG models for capturing the behavior of
on globular proteinsCitation87 and mAbs.Citation15,Citation65 In an initial report, Calero-Rubio et al.Citation15 used a series of mAb model representations (ranging from 1 to 12 CG-sites) to compare the effect of a number of physical factors and model parameters (e.g., hinge flexibility, charge distribution, and the strength of attractions) on protein–protein interactions at diluted and concentrated conditions. The authors found that highly anisotropic charge distributions lead to nonphysically realistic
values, while hinge flexibility has minimal impact on protein interactions. More importantly, they identified that mAb models of 6 or 12 CG-sites provided a reasonable balance between computational cost and numerical uncertainty when comparing
results against low-resolution CG models. The same group later fit these simplified CG models against experimental data of
versus ionic strength for two different mAbs in order to evaluate their ability to predict the behavior of mAb solutions at high-concentrations.Citation63,Citation64 Interestingly, it was shown that both types of CG models are able to reproduce the osmotic compressibility over a wide range of mAb concentrations as long as net protein interactions are repulsive or mildly attractive. More recently, Shahfar et al.Citation65 also compared different simplified CG models against
values for five different mAbs and arrived at a similar conclusion, where domain-resolution models (e.g., a 6- or 12-bead mAb representation) are only able to reasonably predict
for repulsive and mildly attractive conditions. The authors also found that a 12-bead mAb model with explicit incorporation of charged amino acids on the protein surface can reproduce
for conditions dominated by attractive electrostatic interactions.
Computational models are also often used to evaluate the static structure factor and the osmotic compressibility as metrics for protein–protein interactions at elevated concentrations.Citation201
is related to the Fourier Transform of the protein radial distribution function and provides a measurement of spatial correlations at all length-scales.Citation16 The osmotic compressibility is instead related to the zero-limit value of the structure factor (i.e.,
), and therefore it provides information regarding how protein molecules are correlated with each other in the bulk solution.Citation202 From the standpoint of continuum models, there exists a number of different approaches for calculating
based on integral equation theory, where all of these models represent molecules as spherical particles interacting via various continuous or discontinuous potentials.Citation82,Citation105,Citation107 As discussed in the previous section, the high-concentration behavior of protein solutions is driven by anisotropic interactions, and thus these isotropic models present limited applicability to capture
for protein solutions.Citation82 Nonetheless, akin to the case of
, Wertheim’s theory has also been extended to evaluate the role of specific anisotropic attractions on
for globular proteinsCitation104 and mAbs.Citation199 Alternatively, MintonCitation214,Citation215 developed a continuum model for evaluating
(defined by the author as
) from light scattering experiments, which incorporates molecular anisotropy by representing proteins as hard convex particles based on the approximation than molecular crowding is the dominant force at elevated protein concentrations. Note that this model only considers excluded volume effects as direct protein interactions, while it implicitly captures the effects of longer-range attractions and repulsions by: (1) accounting for a thermodynamic equilibrium between monomer and protein clusters; and (2) allowing the protein diameter to differ from the actual molecular size. As a result, Minton’s model has been successfully applied to evaluate protein self-association at high concentrations,Citation216,Citation217 and to study how weak protein clusters are related to solution viscosityCitation201,Citation218,Citation219 and protein-excipient interactions.Citation220,Citation221 However, due to the inherent assumptions regarding protein morphology and the nature of the intermolecular interactions, this model might lead to overestimation of net protein interactions and cluster formation when the solution behavior is dominated by strong protein-protein/excipient interactions.Citation201,Citation221,Citation222
Similarly, simplified CG models have been commonly used for fitting experimental profiles from small-angle scattering experiments on mAb systems.Citation69,Citation82,Citation85 For instance, Corbett et al.Citation82 evaluated
for mAb solutions at different pH and protein concentrations as high as 160 mg/mL, using a 3 CG-site mAb model that interacts only via short-range attractions. The authors found that such a simplified CG model is able to capture generic features of
related to the quaternary protein structure and the nearest-neighbor interaction shell of the proteins. However, when the 3 CG-site mAb model is parameterized using solely data from diluted conditions, it is unable to reproduce high-concentration protein interactions as measured by both
and
. Likewise, Dear et al.Citation85 and Chowdhury et al.Citation88,Citation198 used a 12 CG-site model to evaluate experimental
from two different mAbs and a bovine immunoglobulin in various formulation conditions, where the CG-sites interact through a weak short-range attraction and an electrostatic repulsion. Although these reports show that the 12 CG-site model is able to reasonably reproduce the regions of
related to the bulk behavior and nearest-neighbor shell (i.e., the low- and intermediate-
regions), the addition of a strong attractive potential to the outer CG-sites of the model is required for fitting the data from net attractive formulations.
Most of the above in-silico models mainly focus on evaluating protein–protein interactions, while the effects of excipient are implicitly treated and absorbed in the resulting model parameters. That is, a different set of parameters for the corresponding CG force field might be required for representing different solution conditions of the same protein. As a result, these models efficiently screen protein–protein interactions for different formulation conditions, but they are unable to provide information about the mechanism of action for a given excipient. In that regard, atomistic models have been used to explore how different excipients interact with the protein surface and/or disrupt protein-protein/solvent interactions.Citation21,Citation204,Citation223,Citation224 Generally, these studies of protein-excipient interactions follow similar docking methodologies than those used for identifying binding free-energies for the formation of protein-ligand complexes.Citation10 However, unlike protein–ligand interactions, pharmaceutically relevant excipients such as carbohydrates, nonionic surfactants and free amino acids interact weakly with proteins (). As a result, these studies have shown that protein-excipient interactions occur through multiple interacting regions rather than specific binding pockets. Trout group proposed the use of the preferential interaction parameter as a metric for comparing protein-excipient interactions, which quantifies the excess of excipient molecules in the vicinity of the protein as compared to the bulk solution.Citation204 Cloutier et al.Citation30,Citation143 have investigated
for the interaction of three different mAbs with sugars (sorbitol, sucrose and trehalose) and ionic excipients (NaCl, arginine and proline) in the context of protein aggregation and solution viscosity. The authors found that carbohydrates often interact with aromatic residues, whereas free amino acids interact through both charge–charge and cation–π interactions. Nonetheless, the effects of these excipients on protein stability are not generalized, as they lead to different aggregation and viscosity behaviors across the different tested mAbs. The same authors further extended this approach by developing a machine learning algorithm that combines the calculation of
with in-silico molecular descriptors (e.g., protein charge, surface area, hydrophobicity) in order to screen biotherapeutic formulations.Citation225
Figure 4. Illustrative example of protein-excipient interactions for the variable region of a mAb as calculated by the preferential interaction parameter (). Panels show the interactions of the antibody with different excipients: (a) Proline; (b) arginine-HCl; and (c) NaCl. Coloring indicates local values of
, where red indicates preferential inclusion (i.e., attractive interactions). Notably, for all excipients, multiple regions of preferential inclusion are identified along the protein surface. Figure adapted from Cloutier et al.Citation143

Using a different computational approach, Jo et al.Citation224 and Somani et al.Citation226 recently studied the relation between protein-excipient interactions and mAb solution viscosity by evaluating the binding free-energies of a number of excipients (including free amino acids and sugars) to three different Fabs, for which they used the Site-Identification by Ligand Competitive Saturation (SILCS) technology.Citation227 SILCS consists of simulating the protein in an aqueous environment containing a number of different probe, organic molecules, which provides information regarding the affinity of the protein toward specific functional groups. This information is thereafter used to estimate the binding free-energy of different protein regions to the excipients of interest via a perturbation approach, and thus reducing the overall computational cost of individually simulating each of the protein-excipient systems. By applying SILCS, the authors were able to map the regions of the proteins with higher affinity toward interactions with the different excipients, where some of these regions exclusively interact with specific excipients. Comparison of the patterns for protein-excipient interactions against similar patterns for protein–protein interactions and experimental data of high-concentration viscosity yielded an interesting negative correlation between the number of binding sites for a given excipient and viscosity.Citation224 The analysis led the authors to hypothesize the mechanism of action by which lysine increased viscosity for one of the studied mAbs.Citation226 However, the computational results by SILCS could not explain how some of the excipients such as arginine modulate the behavior of the studied mAbs. While SILCS presents a promising methodology for efficiently performing excipient selection during formulation screening, further efforts are still needed to adapt this new technology to the type of problems faced in the biotherapeutic industry, as it was also acknowledged by the same authors.
Future directions
This article reviewed computational protein models of different resolutions, as well as a selection of in-silico studies applied to elucidate some of the biopharmaceutically relevant physical stability issues in high-concentration protein solutions. Models developed to assess protein aggregation,Citation73,Citation100,Citation144 protein-phase separation,Citation83,Citation177,Citation178 elevated solution viscosity,Citation81,Citation195 and protein interactions were included.Citation85,Citation225 highlights several of the computational models used for studying physical instabilities in proteins, which are described throughout the review. Modulating these problems constitutes one of the main goals when developing phase-appropriate drug product formulations in order to gain understanding about their underlying mechanisms and the role that protein environment plays on these phenomena. Besides the inherent challenges in studying these complex issues, experimental assessment of concentrated protein solutions is often restricted by limitations in instrument capabilities, material availability and accelerated timelines for development. In this regard, computational models like those reviewed here could facilitate the identification of suitable drug formulation by helping analyze and augment the information drawn from experiments, providing molecular insight about the stability of the protein in different formulation conditions, and reducing the number of formulation parameters to be assessed in-vitro. Clearly, efforts toward that direction have already begun in earnest, as significant advancements have been achieved in terms of developing novel models and methodologies for efficiently studying the behavior of pharmaceutical proteins such as mAbs, as well as for predicting how protein stability is affected by formulation parameters (e.g., pH, ionic strength, excipients). However, this review also highlights many of the challenges to fully integrate modeling approaches in formulation development workflows.
Table 4. Summary of computational models for studying physical instabilities in protein solutions
The most pressing limitation is without a doubt the lack of benchmarking of existing computational models against diverse experimental data sets, which should contain information about solution behavior over multiple proteins for a wide range of formulation parameters. Many of the current protein models are borrowed or adapted from other fields (e.g., amyloidosis, crystallization, colloids), where similar stability issues are also of interest. However, such models and their corresponding force fields are unable to accurately capture the behavior of proteins in pharamaceutically relevant conditions (e.g., non-biological pH or buffers, interactions of proteins with surfactants). Likewise, for models with a high level of atomistic detail, explicit simulation of concentrated protein solution remains computationally prohibitive, and thus they generally rely on statistical correlations for linking the simulation outcomes to experimental behavior. Due to the lack of comprehensive data, the conclusions of those models present limited transferability and extrapolability to proteins/formulations outside the range of experimental data used to build such correlations. This latter point is also by itself another challenge.
Notably, many in-silico methodologies highlighted here have led to a seemingly evident conclusion regarding the unique nature of protein behavior with respect to formulation conditions. That is, even for proteins sharing a high level of homology between each other (e.g., mAbs of the same isotype), the effects of an excipient, for instance, might drastically vary from one protein to another. As such, there is a need to develop and implement advanced statistical or mathematical methodologies that can evaluate complex data sets and contextualize the broad, but apparently contradictory effects of a given formulation across different proteins. In this regard, Trout’s group has already taken some steps in this direction by combining, via machine learning algorithms, the results from different atomistic models for predicting various instability behaviors with experimental data.Citation144,Citation225 Lastly, even with the use of simplified protein models, the computational time for simulating concentrated protein systems remains unfeasible for efficiently screening over hundreds of formulation conditions and/or drug candidates. Consequently, further efforts are needed regarding the development or implementation of complementary theories or numerical approximations that facilitate reducing either the computational time or the number of simulations. Methodologies such as those proposed by Mahynski et al.Citation185 and Hatch et al.Citation234 for extrapolating modeling results for predicting phase separation and , or the theories developed by Carmichael and ShellCitation235 and Jin et al.Citation183 for multi-scaling in aggregation and phase separation problems might provide a solution to this latter challenge. The complexity of the phenomena related to protein stability and high-concentration formulations is likely to continue fueling the advancement of many more computational models and methodologies, facilitating the continued significance of in-silico workflows to the development of biologic drug products.
Abbreviations
Table
Acknowledgments
The author thanks numerous MSD colleagues for their input at various stages of this work. Dr. Hanmi Xi and Dr. Timothy Rhodes are gratefully thanked for comments to the manuscript. Dr. Pouria Mistani is also thanked for many helpful and stimulating discussions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Kaplon H, Reichert JM. Antibodies to watch in 2021. MAbs. 2021;13(1):1860476. doi:10.1080/19420862.2020.1860476.
- Strickley RG, Lambert WJ. A review of formulations of commercially available antibodies. J Pharm Sci. 2021;110(7):2590–2608 e2556. doi:10.1016/j.xphs.2021.03.017.
- Warne NW, Mahler H-C. Challenges in protein product development. Cham, Switzerland: Springer International Publishing; 2018.
- Inoue N, Takai E, Arakawa T, Shiraki K. Specific decrease in solution viscosity of antibodies by arginine for therapeutic formulations. Mol Pharm. 2014;11(6):1889–24. doi:10.1021/mp5000218.
- Agrawal NJ, Helk B, Kumar S, Mody N, Sathish HA, Samra HS, Buck PM, Li L, Trout BL. Computational tool for the early screening of monoclonal antibodies for their viscosities. MAbs. 2016;8(1):43–48. doi:10.1080/19420862.2015.1099773.
- Carballo-Pacheco M, Strodel B. Advances in the simulation of protein aggregation at the atomistic scale. J Phys Chem B. 2016;120(12):2991–99. doi:10.1021/acs.jpcb.6b00059.
- Kmiecik S, Gront D, Kolinski M, Wieteska L, Dawid AE, Kolinski A. Coarse-grained protein models and their applications. Chem Rev. 2016;116(14):7898–936. doi:10.1021/acs.chemrev.6b00163.
- Kuroda D, Tsumoto K. Engineering stability, viscosity, and immunogenicity of antibodies by computational design. J Pharm Sci. 2020;109(5):1631–51. doi:10.1016/j.xphs.2020.01.011.
- Joshi SY, Deshmukh SA. A review of advancements in coarse-grained molecular dynamics simulations. Mol Simul. 2021;47(10–11):786–803. doi:10.1080/08927022.2020.1828583.
- Lazim R, Suh D, Choi S. Advances in molecular dynamics simulations and enhanced sampling methods for the study of protein systems. Int J Mol Sci. 2020;21(17):6339. doi:10.3390/ijms21176339.
- Stradner A, Schurtenberger P. Potential and limits of a colloid approach to protein solutions. Soft Matter. 2020;16(2):307–23. doi:10.1039/c9sm01953g.
- Giulini M, Rigoli M, Mattiotti G, Menichetti R, Tarenzi T, Fiorentini R, Potestio R. From system modeling to system analysis: the impact of resolution level and resolution distribution in the computer-aided investigation of biomolecules. Front Mol Biosci. 2021;8:676976. doi:10.3389/fmolb.2021.676976.
- Bereau T, Deserno M. Generic coarse-grained model for protein folding and aggregation. J Chem Phys. 2009;130(23):235106. doi:10.1063/1.3152842.
- Blanco MA, Sahin E, Robinson AS, Roberts CJ. Coarse-grained model for colloidal protein interactions, b22, and protein cluster formation. J Phys Chem B. 2013;117(50):16013–28. doi:10.1021/jp409300j.
- Calero-Rubio C, Saluja A, Roberts CJ. Coarse-grained antibody models for “weak” protein-protein interactions from low to high concentrations. J Phys Chem B. 2016;120(27):6592–605. doi:10.1021/acs.jpcb.6b04907.
- Blanco MA, Hatch HW, Curtis JE, Shen VK. Evaluating the effects of hinge flexibility on the solution structure of antibodies at concentrated conditions. J Pharm Sci. 2019;108(5):1663–74. doi:10.1016/j.xphs.2018.12.013.
- Skar-Gislinge N, Ronti M, Garting T, Rischel C, Schurtenberger P, Zaccarelli E, Stradner A. A colloid approach to self-assembling antibodies. Mol Pharm. 2019;16(6):2394–404. doi:10.1021/acs.molpharmaceut.9b00019.
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmuller H, MacKerell AD Jr. Charmm36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods. 2017;14(1):71–73. doi:10.1038/nmeth.4067.
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. Ff14sb: improving the accuracy of protein side chain and backbone parameters from ff99sb. J Chem Theory Comput. 2015;11(8):3696–713. doi:10.1021/acs.jctc.5b00255.
- Yu I, Mori T, Ando T, Harada R, Jung J, Sugita Y, Feig M. Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. Elife. 2016:5. doi:10.7554/eLife.19274.
- Kalayan J, Curtis RA, Warwicker J, Henchman RH. Thermodynamic origin of differential excipient-lysozyme interactions. Front Mol Biosci. 2021;8:689400. doi:10.3389/fmolb.2021.689400.
- Mereghetti P, Wade RC. Atomic detail brownian dynamics simulations of concentrated protein solutions with a mean field treatment of hydrodynamic interactions. J Phys Chem B. 2012;116(29):8523–33. doi:10.1021/jp212532h.
- Duran T, Minatovicz B, Bai J, Shin D, Mohammadiarani H, Chaudhuri B. Molecular dynamics simulation to uncover the mechanisms of protein instability during freezing. J Pharm Sci. 2021;110(6):2457–71. doi:10.1016/j.xphs.2021.01.002.
- Sugita Y, Feig M. Chapter 14. All-atom molecular dynamics simulation of proteins in crowded environments. In: Ito Y, Dötsch V, Shirakawa M, eds. In-cell NMR Spectroscopy. London, UK: The Royal Society of Chemistry, 2020:228–248.
- Musiani F, Giorgetti A. Protein aggregation and molecular crowding: perspectives from multiscale simulations. Int Rev Cell Mol Biol. 2017;329:49–77. doi:10.1016/bs.ircmb.2016.08.009.
- Riniker S. Fixed-charge atomistic force fields for molecular dynamics simulations in the condensed phase: an overview. J Chem Inf Model. 2018;58(3):565–78. doi:10.1021/acs.jcim.8b00042.
- Lindorff-Larsen K, Maragakis P, Piana S, Eastwood MP, Dror RO, Shaw DE. Systematic validation of protein force fields against experimental data. PLoS One. 2012;7(2):e32131. doi:10.1371/journal.pone.0032131.
- Petrov D, Zagrovic B. Are current atomistic force fields accurate enough to study proteins in crowded environments? PLoS Comput Biol. 2014;10(5):e1003638. doi:10.1371/journal.pcbi.1003638.
- Arsiccio A, Pisano R. Surfactants as stabilizers for biopharmaceuticals: an insight into the molecular mechanisms for inhibition of protein aggregation. Eur J Pharm Biopharm. 2018;128:98–106. doi:10.1016/j.ejpb.2018.04.005.
- Cloutier T, Sudrik C, Mody N, Sathish HA, Trout BL. Molecular computations of preferential interaction coefficients of igg1 monoclonal antibodies with sorbitol, sucrose, and trehalose and the impact of these excipients on aggregation and viscosity. Mol Pharm. 2019;16(8):3657–64. doi:10.1021/acs.molpharmaceut.9b00545.
- Morriss-Andrews A, Shea JE. Computational studies of protein aggregation: methods and applications. Annu Rev Phys Chem. 2015;66(1):643–66. doi:10.1146/annurev-physchem-040513-103738.
- Bereau T, Bachmann M, Deserno M. Interplay between secondary and tertiary structure formation in protein folding cooperativity. J Am Chem Soc. 2010;132(38):13129–31. doi:10.1021/ja105206w.
- Paik BA, Blanco MA, Jia X, Roberts CJ, Kiick KL. Aggregation of poly(acrylic acid)-containing elastin-mimetic copolymers. Soft Matter. 2015;11(9):1839–50. doi:10.1039/c4sm02525c.
- Calero-Rubio C, Paik B, Jia X, Kiick KL, Roberts CJ. Predicting unfolding thermodynamics and stable intermediates for alanine-rich helical peptides with the aid of coarse-grained molecular simulation. Biophys Chem. 2016;217:8–19. doi:10.1016/j.bpc.2016.07.002.
- Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink SJ. The martini coarse-grained force field: extension to proteins. J Chem Theory Comput. 2008;4(5):819–34. doi:10.1021/ct700324x.
- Yu H, Han W, Ma W, Schulten K. Transient β-hairpin formation in α-synuclein monomer revealed by coarse-grained molecular dynamics simulation. J Chem Phys. 2015;143(24):243142. doi:10.1063/1.4936910.
- Han W, Schulten K. Fibril elongation by aβ17–42: kinetic network analysis of hybrid-resolution molecular dynamics simulations. J Am Chem Soc. 2014;136(35):12450–60. doi:10.1021/ja507002p.
- Freeman R, Han M, Alvarez Z, Lewis JA, Wester JR, Stephanopoulos N, McClendon MT, Lynsky C, Godbe JM, Sangji H, et al. Reversible self-assembly of superstructured networks. Science. 2018;362(6416):808–13. doi:10.1126/science.aat6141.
- Sterpone F, Derreumaux P, Melchionna S. Protein simulations in fluids: coupling the opep coarse-grained force field with hydrodynamics. J Chem Theory Comput. 2015;11(4):1843–53. doi:10.1021/ct501015h.
- Chiricotto M, Melchionna S, Derreumaux P, Sterpone F. Hydrodynamic effects on β-amyloid (16-22) peptide aggregation. J Chem Phys. 2016;145(3):035102. doi:10.1063/1.4958323.
- Kynast P, Derreumaux P, Strodel B. Evaluation of the coarse-grained opep force field for protein-protein docking. BMC Biophysics. 2016;9(1):4. doi:10.1186/s13628-016-0029-y.
- Chiricotto M, Melchionna S, Derreumaux P, Sterpone F. Multiscale aggregation of the amyloid aβ16–22 peptide: from disordered coagulation and lateral branching to amorphous prefibrils. J Phys Chem Lett. 2019;10(7):1594–99. doi:10.1021/acs.jpclett.9b00423.
- Cheon M, Chang I, Hall CK. Extending the prime model for protein aggregation to all 20 amino acids. Proteins. 2010;78(14):2950–60. doi:10.1002/prot.22817.
- Latshaw DC, Cheon M, Hall CK. Effects of macromolecular crowding on amyloid beta (16-22) aggregation using coarse-grained simulations. J Phys Chem B. 2014;118(47):13513–26. doi:10.1021/jp508970q.
- Cheon M, Kang M, Chang I. Polymorphism of fibrillar structures depending on the size of assembled abeta17-42 peptides. Sci Rep. 2016;6(1):38196. doi:10.1038/srep38196.
- Kar P, Gopal SM, Cheng YM, Panahi A, Feig M. Transferring the primo coarse-grained force field to the membrane environment: simulations of membrane proteins and helix-helix association. J Chem Theory Comput. 2014;10(8):3459–72. doi:10.1021/ct500443v.
- Hatherley R, Brown DK, Glenister M, Tastan Bishop O. Primo: an interactive homology modeling pipeline. PLoS One. 2016;11(11):e0166698. doi:10.1371/journal.pone.0166698.
- O’Brien CJ, Blanco MA, Costanzo JA, Enterline M, Fernandez EJ, Robinson AS, Roberts CJ. Modulating non-native aggregation and electrostatic protein-protein interactions with computationally designed single-point mutations. Protein Eng Des Sel. 2016;29(6):231–43. doi:10.1093/protein/gzw010.
- Ferreira GM, Shahfar H, Sathish HA, Remmele RL Jr., Roberts CJ. Identifying key residues that drive strong electrostatic attractions between therapeutic antibodies. J Phys Chem B. 2019;123(50):10642–53. doi:10.1021/acs.jpcb.9b08355.
- Ferreira GM, Calero-Rubio C, Sathish HA, Remmele RL Jr., Roberts CJ. Electrostatically mediated protein-protein interactions for monoclonal antibodies: a combined experimental and coarse-grained molecular modeling approach. J Pharm Sci. 2019;108(1):120–32. doi:10.1016/j.xphs.2018.11.004.
- Kolinski A. Protein modeling and structure prediction with a reduced representation. Acta Biochim Pol. 2004;51(2):349–71. doi:10.18388/abp.2004_3575.
- Kuriata A, Iglesias V, Pujols J, Kurcinski M, Kmiecik S, Ventura S. Aggrescan3d (a3d) 2.0: prediction and engineering of protein solubility. Nucleic Acids Res. 2019;47(W1):W300–W307. doi:10.1093/nar/gkz321.
- Kurcinski M, Pawel Ciemny M, Oleniecki T, Kuriata A, Badaczewska-Dawid AE, Kolinski A, Kmiecik S. Cabs-dock standalone: a toolbox for flexible protein-peptide docking. Bioinformatics. 2019;35(20):4170–72. doi:10.1093/bioinformatics/btz185.
- Pulawski W, Jamroz M, Kolinski M, Kolinski A, Kmiecik S. Coarse-grained simulations of membrane insertion and folding of small helical proteins using the cabs model. J Chem Inf Model. 2016;56(11):2207–15. doi:10.1021/acs.jcim.6b00350.
- Kim YC, Hummer G. Coarse-grained models for simulations of multiprotein complexes: application to ubiquitin binding. J Mol Biol. 2008;375(5):1416–33. doi:10.1016/j.jmb.2007.11.063.
- Rozycki B, Kim YC, Hummer G. Saxs ensemble refinement of escrt-iii chmp3 conformational transitions. Structure. 2011;19(1):109–16. doi:10.1016/j.str.2010.10.006.
- Dignon GL, Zheng W, Kim YC, Best RB, Mittal J. Sequence determinants of protein phase behavior from a coarse-grained model. PLoS Comput Biol. 2018;14(1):e1005941. doi:10.1371/journal.pcbi.1005941.
- Jost Lopez A, Quoika PK, Linke M, Hummer G, Kofinger J. Quantifying protein-protein interactions in molecular simulations. J Phys Chem B. 2020;124(23):4673–85. doi:10.1021/acs.jpcb.9b11802.
- Liwo A, Baranowski M, Czaplewski C, Golas E, He Y, Jagiela D, Krupa P, Maciejczyk M, Makowski M, Mozolewska MA, et al. A unified coarse-grained model of biological macromolecules based on mean-field multipole-multipole interactions. J Mol Model. 2014;20(8):2306. doi:10.1007/s00894-014-2306-5.
- He Y, Mozolewska MA, Krupa P, Sieradzan AK, Wirecki TK, Liwo A, Kachlishvili K, Rackovsky S, Jagiela D, Slusarz R, et al. Lessons from application of the unres force field to predictions of structures of casp10 targets. Proc Natl Acad Sci U S A. 2013;110(37):14936–41. doi:10.1073/pnas.1313316110.
- Kachlishvili K, Maisuradze GG, Martin OA, Liwo A, Vila JA, Scheraga HA. Accounting for a mirror-image conformation as a subtle effect in protein folding. Proc Natl Acad Sci U S A. 2014;111(23):8458–63. doi:10.1073/pnas.1407837111.
- Rojas A, Liwo A, Browne D, Scheraga HA. Mechanism of fiber assembly: treatment of abeta peptide aggregation with a coarse-grained united-residue force field. J Mol Biol. 2010;404(3):537–52. doi:10.1016/j.jmb.2010.09.057.
- Calero-Rubio C, Saluja A, Sahin E, Roberts CJ. Predicting high-concentration interactions of monoclonal antibody solutions: comparison of theoretical approaches for strongly attractive versus repulsive conditions. J Phys Chem B. 2019;123(27):5709–20. doi:10.1021/acs.jpcb.9b03779.
- Calero-Rubio C, Ghosh R, Saluja A, Roberts CJ. Predicting protein-protein interactions of concentrated antibody solutions using dilute solution data and coarse-grained molecular models. J Pharm Sci. 2018;107(5):1269–81. doi:10.1016/j.xphs.2017.12.015.
- Shahfar H, Forder JK, Roberts CJ. Toward a suite of coarse-grained models for molecular simulation of monoclonal antibodies and therapeutic proteins. J Phys Chem B. 2021;125(14):3574–88. doi:10.1021/acs.jpcb.1c01903.
- Chaudhri A, Zarraga IE, Kamerzell TJ, Brandt JP, Patapoff TW, Shire SJ, Voth GA. Coarse-grained modeling of the self-association of therapeutic monoclonal antibodies. J Phys Chem B. 2012;116(28):8045–57. doi:10.1021/jp301140u.
- Chaudhri A, Zarraga IE, Yadav S, Patapoff TW, Shire SJ, Voth GA. The role of amino acid sequence in the self-association of therapeutic monoclonal antibodies: insights from coarse-grained modeling. J Phys Chem B. 2013;117(5):1269–79. doi:10.1021/jp3108396.
- Buck PM, Chaudhri A, Kumar S, Singh SK. Highly viscous antibody solutions are a consequence of network formation caused by domain-domain electrostatic complementarities: insights from coarse-grained simulations. Mol Pharm. 2015;12(1):127–39. doi:10.1021/mp500485w.
- Wang G, Varga Z, Hofmann J, Zarraga IE, Swan JW. Structure and relaxation in solutions of monoclonal antibodies. J Phys Chem B. 2018;122(11):2867–80. doi:10.1021/acs.jpcb.7b11053.
- Lai PK, Swan JW, Trout BL. Calculation of therapeutic antibody viscosity with coarse-grained models, hydrodynamic calculations and machine learning-based parameters. MAbs. 2021;13(1):1907882. doi:10.1080/19420862.2021.1907882.
- Vacha R, Frenkel D. Relation between molecular shape and the morphology of self-assembling aggregates: a simulation study. Biophys J. 2011;101(6):1432–39. doi:10.1016/j.bpj.2011.07.046.
- Vacha R, Linse S, Lund M. Surface effects on aggregation kinetics of amyloidogenic peptides. J Am Chem Soc. 2014;136(33):11776–82. doi:10.1021/ja505502e.
- Saric A, Buell AK, Meisl G, Michaels TCT, Dobson CM, Linse S, Knowles TPJ, Frenkel D. Physical determinants of the self-replication of protein fibrils. Nat Phys. 2016;12(9):874–80. doi:10.1038/nphys3828.
- Baaden M, Marrink SJ. Coarse-grain modelling of protein-protein interactions. Curr Opin Struct Biol. 2013;23(6):878–86. doi:10.1016/j.sbi.2013.09.004.
- Zhang L, Lua LH, Middelberg AP, Sun Y, Connors NK. Biomolecular engineering of virus-like particles aided by computational chemistry methods. Chem Soc Rev. 2015;44(23):8608–18. doi:10.1039/c5cs00526d.
- Badaczewska-Dawid AE, Kolinski A, Kmiecik S. Computational reconstruction of atomistic protein structures from coarse-grained models. Comput Struct Biotechnol J. 2020;18:162–76. doi:10.1016/j.csbj.2019.12.007.
- Singh N, Li W. Recent advances in coarse-grained models for biomolecules and their applications. Int J Mol Sci. 2019;20(15):3774. doi:10.3390/ijms20153774.
- Morriss-Andrews A, Shea JE. Simulations of protein aggregation: insights from atomistic and coarse-grained models. J Phys Chem Lett. 2014;5(11):1899–908. doi:10.1021/jz5006847.
- Pak AJ, Voth GA. Advances in coarse-grained modeling of macromolecular complexes. Curr Opin Struct Biol. 2018;52:119–26. doi:10.1016/j.sbi.2018.11.005.
- Perdikari TM, Jovic N, Dignon GL, Kim YC, Fawzi NL, Mittal J. A predictive coarse-grained model for position-specific effects of post-translational modifications. Biophys J. 2021;120(7):1187–97. doi:10.1016/j.bpj.2021.01.034.
- Izadi S, Patapoff TW, Walters BT. Multiscale coarse-grained approach to investigate self-association of antibodies. Biophys J. 2020;118(11):2741–54. doi:10.1016/j.bpj.2020.04.022.
- Corbett D, Hebditch M, Keeling R, Ke P, Ekizoglou S, Sarangapani P, Pathak J, Van Der Walle CF, Uddin S, Baldock C, et al. Coarse-grained modeling of antibodies from small-angle scattering profiles. J Phys Chem B. 2017;121(35):8276–90. doi:10.1021/acs.jpcb.7b04621.
- Blanco MA, Shen VK. Effect of the surface charge distribution on the fluid phase behavior of charged colloids and proteins. J Chem Phys. 2016;145(15):155102. doi:10.1063/1.4964613.
- Staneva I, Frenkel D. The role of non-specific interactions in a patchy model of protein crystallization. J Chem Phys. 2015;143(19):194511. doi:10.1063/1.4935369.
- Dear BJ, Bollinger JA, Chowdhury A, Hung JJ, Wilks LR, Karouta CA, Ramachandran K, Shay TY, Nieto MP, Sharma A, et al. X-ray scattering and coarse-grained simulations for clustering and interactions of monoclonal antibodies at high concentrations. J Phys Chem B. 2019;123(25):5274–90. doi:10.1021/acs.jpcb.9b04478.
- Prabakaran R, Rawat P, Thangakani AM, Kumar S, Gromiha MM. Protein aggregation: in silico algorithms and applications. Biophys Rev. 2021;13(1):71–89. doi:10.1007/s12551-021-00778-w.
- Woldeyes MA, Calero-Rubio C, Furst EM, Roberts CJ. Predicting protein interactions of concentrated globular protein solutions using colloidal models. J Phys Chem B. 2017;121(18):4756–67. doi:10.1021/acs.jpcb.7b02183.
- Chowdhury A, Bollinger JA, Dear BJ, Cheung JK, Johnston KP, Truskett TM. Coarse-grained molecular dynamics simulations for understanding the impact of short-range anisotropic attractions on structure and viscosity of concentrated monoclonal antibody solutions. Mol Pharm. 2020;17(5):1748–56. doi:10.1021/acs.molpharmaceut.9b00960.
- Antosiewicz JM, Shugar D. Poisson-boltzmann continuum-solvation models: applications to ph-dependent properties of biomolecules. Mol Biosyst. 2011;7(11):2923–49. doi:10.1039/c1mb05170a.
- Hanson B, Richardson R, Oliver R, Read DJ, Harlen O, Harris S. Modelling biomacromolecular assemblies with continuum mechanics. Biochem Soc Trans. 2015;43(2):186–92. doi:10.1042/BST20140294.
- Mills ZG, Mao W, Alexeev A. Mesoscale modeling: solving complex flows in biology and biotechnology. Trends Biotechnol. 2013;31(7):426–34. doi:10.1016/j.tibtech.2013.05.001.
- Zhang Y, Han D, Dou Z, Veilleux JC, Shi GH, Collins DS, Vlachos PP, Ardekani AM. The interface motion and hydrodynamic shear of the liquid slosh in syringes. Pharm Res. 2021;38(2):257–75. doi:10.1007/s11095-021-02992-3.
- Schreck JS, Bridstrup J, Yuan JM. Investigating the effects of molecular crowding on the kinetics of protein aggregation. J Phys Chem B. 2020;124(44):9829–39. doi:10.1021/acs.jpcb.0c07175.
- Radhakrishnan R, Yu HY, Eckmann DM, Ayyaswamy PS. Computational models for nanoscale fluid dynamics and transport inspired by nonequilibrium thermodynamics. J Heat Transfer. 2017;139(3):0330011–19. doi:10.1115/1.4035006.
- Wutz J, Waterkotte B, Heitmann K, Wucherpfennig T. Computational fluid dynamics (cfd) as a tool for industrial uf/df tank optimization. Biochem Eng J. 2020;160:107617. doi:10.1016/j.bej.2020.107617.
- Pohar A. A review of computational fluid dynamics (cfd) simulations of mixing in the pharmaceutical industry. Biomed J Sci Tech Res. 2020:27. doi:10.26717/bjstr.2020.27.004494.
- Ladner T, Odenwald S, Kerls K, Zieres G, Boillon A, Boeuf J. Cfd supported investigation of shear induced by bottom-mounted magnetic stirrer in monoclonal antibody formulation. Pharm Res. 2018;35(11):215. doi:10.1007/s11095-018-2492-4.
- Hanslip S, Desai KG, Palmer M, Kemp I, Bell S, Schofield P, Varma P, Roche F, Colandene JD, Nesta DP. Syringe filling of a high-concentration mab formulation: experimental, theoretical, and computational evaluation of filling process parameters that influence the propensity for filling needle clogging. J Pharm Sci. 2019;108(3):1130–38. doi:10.1016/j.xphs.2018.10.031.
- Kuttler A, Dimke T, Kern S, Helmlinger G, Stanski D, Finelli LA. Understanding pharmacokinetics using realistic computational models of fluid dynamics: biosimulation of drug distribution within the csf space for intrathecal drugs. J Pharmacokinet Pharmacodyn. 2010;37(6):629–44. doi:10.1007/s10928-010-9184-y.
- Li Y, Roberts CJ. Lumry-eyring nucleated-polymerization model of protein aggregation kinetics. 2. competing growth via condensation and chain polymerization. J Phys Chem B. 2009;113(19):7020–32. doi:10.1021/jp8083088.
- Dear AJ, Meisl G, Michaels TCT, Zimmermann MR, Linse S, Knowles TPJ. The catalytic nature of protein aggregation. J Chem Phys. 2020;152(4):045101. doi:10.1063/1.5133635.
- Shen JL, Tsai MY, Schafer NP, Wolynes PG. Modeling protein aggregation kinetics: the method of second stochasticization. J Phys Chem B. 2021;125(4):1118–33. doi:10.1021/acs.jpcb.0c10331.
- Ben-Naim A. Theoretical aspects of self-assembly of proteins: a kirkwood-buff-theory approach. J Chem Phys. 2013;138(22):224906. doi:10.1063/1.4810806.
- Kastelic M, Kalyuzhnyi YV, Hribar-Lee B, Dill KA, Vlachy V. Protein aggregation in salt solutions. Proc Natl Acad Sci U S A. 2015;112(21):6766–70. doi:10.1073/pnas.1507303112.
- Gazzillo D, Pini D. Self-consistent ornstein-zernike approximation (scoza) and exact second virial coefficients and their relationship with critical temperature for colloidal or protein suspensions with short-ranged attractive interactions. J Chem Phys. 2013;139(16):164501. doi:10.1063/1.4825174.
- Roberts CJ, Blanco MA. Role of anisotropic interactions for proteins and patchy nanoparticles. J Phys Chem B. 2014;118(44):12599–611. doi:10.1021/jp507886r.
- Liu Y, Porcar L, Chen J, Chen WR, Falus P, Faraone A, Fratini E, Hong K, Baglioni P. Lysozyme protein solution with an intermediate range order structure. J Phys Chem B. 2011;115(22):7238–47. doi:10.1021/jp109333c.
- Sapir L, Harries D. Macromolecular stabilization by excluded cosolutes: mean field theory of crowded solutions. J Chem Theory Comput. 2015;11(7):3478–90. doi:10.1021/acs.jctc.5b00258.
- Nicoud L, Jagielski J, Pfister D, Lazzari S, Massant J, Lattuada M, Morbidelli M. Kinetics of monoclonal antibody aggregation from dilute toward concentrated conditions. J Phys Chem B. 2016;120(13):3267–80. doi:10.1021/acs.jpcb.5b11791.
- Godfrin PD, Hudson SD, Hong K, Porcar L, Falus P, Wagner NJ, Liu Y. Short-time glassy dynamics in viscous protein solutions with competing interactions. Phys Rev Lett. 2015;115(22):228302. doi:10.1103/PhysRevLett.115.228302.
- Nicoud L, Owczarz M, Arosio P, Morbidelli M. A multiscale view of therapeutic protein aggregation: a colloid science perspective. Biotechnol J. 2015;10(3):367–78. doi:10.1002/biot.201400858.
- Roberts CJ. Therapeutic protein aggregation: mechanisms, design, and control. Trends Biotechnol. 2014;32(7):372–80. doi:10.1016/j.tibtech.2014.05.005.
- Ilie IM, Caflisch A. Simulation studies of amyloidogenic polypeptides and their aggregates. Chem Rev. 2019;119(12):6956–93. doi:10.1021/acs.chemrev.8b00731.
- Hirota N, Edskes H, Hall D. Unified theoretical description of the kinetics of protein aggregation. Biophys Rev. 2019;11(2):191–208. doi:10.1007/s12551-019-00506-5.
- Michaels TC, Lazell HW, Arosio P, Knowles TP. Dynamics of protein aggregation and oligomer formation governed by secondary nucleation. J Chem Phys. 2015;143(5):054901. doi:10.1063/1.4927655.
- Zidar M, Kuzman D, Ravnik M. Characterisation of protein aggregation with the smoluchowski coagulation approach for use in biopharmaceuticals. Soft Matter. 2018;14(29):6001–12. doi:10.1039/c8sm00919h.
- Li Y, WFt W, Roberts CJ. Characterization of high-molecular-weight nonnative aggregates and aggregation kinetics by size exclusion chromatography with inline multi-angle laser light scattering. J Pharm Sci. 2009;98(11):3997–4016. doi:10.1002/jps.21726.
- Brummitt RK, Nesta DP, Chang L, Kroetsch AM, Roberts CJ. Nonnative aggregation of an igg1 antibody in acidic conditions, part 2: nucleation and growth kinetics with competing growth mechanisms. J Pharm Sci. 2011;100(6):2104–19. doi:10.1002/jps.22447.
- Imamura H, Honda S. Kinetics of antibody aggregation at neutral ph and ambient temperatures triggered by temporal exposure to acid. J Phys Chem B. 2016;120(36):9581–89. doi:10.1021/acs.jpcb.6b05473.
- Arosio P, Rima S, Lattuada M, Morbidelli M. Population balance modeling of antibodies aggregation kinetics. J Phys Chem B. 2012;116(24):7066–75. doi:10.1021/jp301091n.
- Bridstrup J, Schreck JS, Jorgenson JL, Yuan JM. Stochastic kinetic treatment of protein aggregation and the effects of macromolecular crowding. J Phys Chem B. 2021;125(23):6068–79. doi:10.1021/acs.jpcb.1c00959.
- Meisl G, Kirkegaard JB, Arosio P, Michaels TC, Vendruscolo M, Dobson CM, Linse S, Knowles TP. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat Protoc. 2016;11(2):252–72. doi:10.1038/nprot.2016.010.
- Michaels TCT, Saric A, Curk S, Bernfur K, Arosio P, Meisl G, Dear AJ, Cohen SIA, Dobson CM, Vendruscolo M, et al. Dynamics of oligomer populations formed during the aggregation of Alzheimer’s abeta42 peptide. Nat Chem. 2020;12(5):445–51. doi:10.1038/s41557-020-0452-1.
- Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424(6950):805–08. doi:10.1038/nature01891.
- Lopez de La Paz M, Serrano L. Sequence determinants of amyloid fibril formation. Proc Natl Acad Sci U S A. 2004;101(1):87–92. doi:10.1073/pnas.2634884100.
- Conchillo-Sole O, de Groot NS, Aviles FX, Vendrell J, Daura X, Ventura S. Aggrescan: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform. 2007;8(1):65. doi:10.1186/1471-2105-8-65.
- Burdukiewicz M, Sobczyk P, Rodiger S, Duda-Madej A, Mackiewicz P, Kotulska M. Amyloidogenic motifs revealed by n-gram analysis. Sci Rep. 2017;7(1):12961. doi:10.1038/s41598-017-13210-9.
- Tartaglia GG, Cavalli A, Pellarin R, Caflisch A. Prediction of aggregation rate and aggregation-prone segments in polypeptide sequences. Protein Sci. 2005;14(10):2723–34. doi:10.1110/ps.051471205.
- Walsh I, Seno F, Tosatto SC, Trovato A. Pasta 2.0: an improved server for protein aggregation prediction. Nucleic Acids Res. 2014;42(W1):W301–307. doi:10.1093/nar/gku399.
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22(10):1302–06. doi:10.1038/nbt1012.
- Maurer-Stroh S, Debulpaep M, Kuemmerer N, Lopez de La Paz M, Martins IC, Reumers J, Morris KL, Copland A, Serpell L, Serrano L, et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat Methods. 2010;7(3):237–42. doi:10.1038/nmeth.1432.
- Tartaglia GG, Vendruscolo M. The zyggregator method for predicting protein aggregation propensities. Chem Soc Rev. 2008;37(7):1395–401. doi:10.1039/b706784b.
- Sankar K, Krystek SR Jr., Carl SM, Day T, Maier JKX. Aggscore: prediction of aggregation-prone regions in proteins based on the distribution of surface patches. Proteins. 2018;86(11):1147–56. doi:10.1002/prot.25594.
- Sormanni P, Aprile FA, Vendruscolo M. The camsol method of rational design of protein mutants with enhanced solubility. J Mol Biol. 2015;427(2):478–90. doi:10.1016/j.jmb.2014.09.026.
- Lauer TM, Agrawal NJ, Chennamsetty N, Egodage K, Helk B, Trout BL. Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J Pharm Sci. 2012;101(1):102–15. doi:10.1002/jps.22758.
- Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci U S A. 2009;106(29):11937–42. doi:10.1073/pnas.0904191106.
- Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The foldx web server: an online force field. Nucleic Acids Res. 2005;33:W382–388. doi:10.1093/nar/gki387.
- Van Durme J, De Baets G, Van Der Kant R, Ramakers M, Ganesan A, Wilkinson H, Gallardo R, Rousseau F, Schymkowitz J. Solubis: a webserver to reduce protein aggregation through mutation. Protein Eng Des Sel. 2016;29(8):285–89. doi:10.1093/protein/gzw019.
- Santos J, Pujols J, Pallares I, Iglesias V, Ventura S. Computational prediction of protein aggregation: advances in proteomics, conformation-specific algorithms and biotechnological applications. Comput Struct Biotechnol J. 2020;18:1403–13. doi:10.1016/j.csbj.2020.05.026.
- Wang X, Singh SK, Kumar S. Potential aggregation-prone regions in complementarity-determining regions of antibodies and their contribution towards antigen recognition: a computational analysis. Pharm Res. 2010;27(8):1512–29. doi:10.1007/s11095-010-0143-5.
- Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A. 2014;111(52):18601–06. doi:10.1073/pnas.1421779112.
- Wolf Perez AM, Sormanni P, Andersen JS, Sakhnini LI, Rodriguez-Leon I, Bjelke JR, Gajhede AJ, De Maria L, Otzen DE, Vendruscolo M, et al. In vitro and in silico assessment of the developability of a designed monoclonal antibody library. MAbs. 2019;11(2):388–400. doi:10.1080/19420862.2018.1556082.
- Cloutier TK, Sudrik C, Mody N, Hasige SA, Trout BL. Molecular computations of preferential interactions of proline, arginine.Hcl, and nacl with igg1 antibodies and their impact on aggregation and viscosity. MAbs. 2020;12(1):1816312. doi:10.1080/19420862.2020.1816312.
- Lai PK, Fernando A, Cloutier TK, Kingsbury JS, Gokarn Y, Halloran KT, Calero-Rubio C, Trout BL. Machine learning feature selection for predicting high concentration therapeutic antibody aggregation. J Pharm Sci. 2021;110(4):1583–91. doi:10.1016/j.xphs.2020.12.014.
- Baftizadeh F, Biarnes X, Pietrucci F, Affinito F, Laio A. Multidimensional view of amyloid fibril nucleation in atomistic detail. J Am Chem Soc. 2012;134(8):3886–94. doi:10.1021/ja210826a.
- Karandur D, Wong KY, Pettitt BM. Solubility and aggregation of gly 5 in water. J Phys Chem B. 2014;118(32):9565–72. doi:10.1021/jp503358n.
- Ma B, Nussinov R. Molecular dynamics simulations of alanine rich beta-sheet oligomers: insight into amyloid formation. Protein Sci. 2002;11(10):2335–50. doi:10.1110/ps.4270102.
- Luiken JA, Bolhuis PG. Primary nucleation kinetics of short fibril-forming amyloidogenic peptides. J Phys Chem B. 2015;119(39):12568–79. doi:10.1021/acs.jpcb.5b05799.
- Tofoleanu F, Yuan Y, Pickard F, Tywoniuk B, Brooks BR, Buchete NV. Structural modulation of human amylin protofilaments by naturally occurring mutations. J Phys Chem B. 2018;122(21):5657–65. doi:10.1021/acs.jpcb.7b12083.
- Collu F, Spiga E, Chakroun N, Rezaei H, Fraternali F. Probing the early stages of prion protein (prp) aggregation with atomistic molecular dynamics simulations. Chem Commun (Camb). 2018;54(57):8007–10. doi:10.1039/c8cc04089c.
- Schwierz N, Frost CV, Geissler PL, Zacharias M. From abeta filament to fibril: molecular mechanism of surface-activated secondary nucleation from all-atom md simulations. J Phys Chem B. 2017;121(4):671–82. doi:10.1021/acs.jpcb.6b10189.
- Schwierz N, Frost CV, Geissler PL, Zacharias M. Dynamics of seeded aβ40-fibril growth from atomistic molecular dynamics simulations: kinetic trapping and reduced water mobility in the locking step. J Am Chem Soc. 2016;138(2):527–39. doi:10.1021/jacs.5b08717.
- Ndlovu H, Ashcroft AE, Radford SE, Harris SA. Effect of sequence variation on the mechanical response of amyloid fibrils probed by steered molecular dynamics simulation. Biophys J. 2012;102(3):587–96. doi:10.1016/j.bpj.2011.12.047.
- Nichols P, Li L, Kumar S, Buck PM, Singh SK, Goswami S, Balthazor B, Conley TR, Sek D, Allen MJ. Rational design of viscosity reducing mutants of a monoclonal antibody: hydrophobic versus electrostatic inter-molecular interactions. MAbs. 2015;7(1):212–30. doi:10.4161/19420862.2014.985504.
- Ilie IM, den Otter WK, Briels WJ. A coarse grained protein model with internal degrees of freedom. application to α-synuclein aggregation. J Chem Phys. 2016;144(8):085103. doi:10.1063/1.4942115.
- Barz B, Urbanc B. Minimal model of self-assembly: emergence of diversity and complexity. J Phys Chem B. 2014;118(14):3761–70. doi:10.1021/jp412819j.
- Bellesia G, Shea JE. Self-assembly of beta-sheet forming peptides into chiral fibrillar aggregates. J Chem Phys. 2007;126(24):245104. doi:10.1063/1.2739547.
- Bellesia G, Shea J-E. Diversity of kinetic pathways in amyloid fibril formation. J Chem Phys. 2009;131(11):111102. doi:10.1063/1.3216103.
- Pellarin R, Schuetz P, Guarnera E, Caflisch A. Amyloid fibril polymorphism is under kinetic control. J Am Chem Soc. 2010;132(42):14960–70. doi:10.1021/ja106044u.
- Cao Y, Jiang X, Han W. Self-assembly pathways of beta-sheet-rich amyloid-beta(1-40) dimers: markov state model analysis on millisecond hybrid-resolution simulations. J Chem Theory Comput. 2017;13(11):5731–44. doi:10.1021/acs.jctc.7b00803.
- Sørensen J, Periole X, Skeby KK, Marrink S-J, Schiøtt B. Protofibrillar assembly toward the formation of amyloid fibrils. J Phys Chem Lett. 2011;2(19):2385–90. doi:10.1021/jz2010094.
- McManus JJ, Charbonneau P, Zaccarelli E, Asherie N. The physics of protein self-assembly. Curr Opin Colloid In. 2016;22:73–79. doi:10.1016/j.cocis.2016.02.011.
- Raut AS, Kalonia DS. Pharmaceutical perspective on opalescence and liquid-liquid phase separation in protein solutions. Mol Pharm. 2016;13(5):1431–44. doi:10.1021/acs.molpharmaceut.5b00937.
- Dignon GL, Zheng W, Kim YC, Mittal J. Temperature-controlled liquid-liquid phase separation of disordered proteins. ACS Cent Sci. 2019;5(5):821–30. doi:10.1021/acscentsci.9b00102.
- McCarty J, Delaney KT, Danielsen SPO, Fredrickson GH, Shea JE. Complete phase diagram for liquid-liquid phase separation of intrinsically disordered proteins. J Phys Chem Lett. 2019;10(8):1644–52. doi:10.1021/acs.jpclett.9b00099.
- Muschol M, Rosenberger F. Liquid-liquid phase separation in supersaturated lysozyme solutions and associated precipitate formation/crystallization. J Chem Phys. 1997 Doi: 10.1063/1.474547;107(6):1953–62. doi:10.1063/1.474547.
- Siezen RJ, Fisch MR, Slingsby C, Benedek GB. Opacification of gamma-crystallin solutions from calf lens in relation to cold cataract formation. Proc Natl Acad Sci U S A. 1985;82(6):1701–05. doi:10.1073/pnas.82.6.1701.
- Asherie N, Lomakin A, Benedek GB. Phase diagram of colloidal solutions. Phys Rev Lett. 1996;77(23):4832–35. doi:10.1103/PhysRevLett.77.4832.
- Platten F, Valadez-Perez NE, Castaneda-Priego R, Egelhaaf SU. Extended law of corresponding states for protein solutions. J Chem Phys. 2015;142(17):174905. doi:10.1063/1.4919127.
- Noro MG, Frenkel D. Extended corresponding-states behavior for particles with variable range attractions. The Journal of Chemical Physics. 2000;113(8):2941–44. doi:10.1063/1.1288684.
- Dorsaz N, Filion L, Smallenburg F, Frenkel D. Spiers memorial lecture: effect of interaction specificity on the phase behaviour of patchy particles. Faraday Discuss. 2012;159:9–21. doi:10.1039/c2fd20070h.
- Gnan N, Sciortino F, Zaccarelli E. Patchy particle models to understand protein phase behavior. Methods Mol Biol. 2019;2039:187–208. doi:10.1007/978-1-4939-9678-0_14.
- Liu H, Kumar SK, Sciortino F. Vapor-liquid coexistence of patchy models: relevance to protein phase behavior. J Chem Phys. 2007;127(8):084902. doi:10.1063/1.2768056.
- Benayad Z, von Bülow S, Stelzl LS, Hummer G. Simulation of fus protein condensates with an adapted coarse-grained model. J Chem Theory Comput. 2020;17(1):525–37. doi:10.1021/acs.jctc.0c01064.
- Brudar S, Hribar-Lee B. Effect of buffer on protein stability in aqueous solutions: a simple protein aggregation model. J Phys Chem B. 2021;125(10):2504–12. doi:10.1021/acs.jpcb.0c10339.
- Kalyuzhnyi YV, Vlachy V. Explicit-water theory for the salt-specific effects and hofmeister series in protein solutions. J Chem Phys. 2016;144(21):215101. doi:10.1063/1.4953067.
- Sun G, Wang Y, Lomakin A, Benedek GB, Stanley HE, Xu L, Buldyrev SV. The phase behavior study of human antibody solution using multi-scale modeling. J Chem Phys. 2016;145(19):194901. doi:10.1063/1.4966972.
- Kastelic M, Vlachy V. Theory for the liquid-liquid phase separation in aqueous antibody solutions. J Phys Chem B. 2018;122(21):5400–08. doi:10.1021/acs.jpcb.7b11458.
- Kalyuzhnyi YV, Vlachy V. Modeling the depletion effect caused by an addition of polymer to monoclonal antibody solutions. J Phys Condens Matter. 2018;30(48):485101. doi:10.1088/1361-648X/aae914.
- Hvozd T, Kalyuzhnyi YV, Vlachy V. Aggregation, liquid-liquid phase separation, and percolation behaviour of a model antibody fluid constrained by hard-sphere obstacles. Soft Matter. 2020;16(36):8432–43. doi:10.1039/d0sm01014f.
- Rego NB, Xi E, Patel AJ. Identifying hydrophobic protein patches to inform protein interaction interfaces. Proc Natl Acad Sci U S A. 2021;118(6):e2018234118. doi:10.1073/pnas.2018234118.
- Brunsteiner M, Flock M, Nidetzky B. Structure based descriptors for the estimation of colloidal interactions and protein aggregation propensities. PLoS One. 2013;8(4):e59797. doi:10.1371/journal.pone.0059797.
- Jin J, Yu A, Voth GA. Temperature and phase transferable bottom-up coarse-grained models. J Chem Theory Comput. 2020;16(11):6823–42. doi:10.1021/acs.jctc.0c00832.
- Qin S, Zhou HX. Fast method for computing chemical potentials and liquid-liquid phase equilibria of macromolecular solutions. J Phys Chem B. 2016;120(33):8164–74. doi:10.1021/acs.jpcb.6b01607.
- Mahynski NA, Blanco MA, Errington JR, Shen VK. Predicting low-temperature free energy landscapes with flat-histogram monte carlo methods. J Chem Phys. 2017;146(7):074101. doi:10.1063/1.4975331.
- Jameel F, Skoug JW, Nesbitt RR. Development of biopharmaceutical drug-device products. Cham, Switzerland: Springer International Publishing, 2020.
- Zhang ZH, Liu Y. Recent progresses of understanding the viscosity of concentrated protein solutions. Curr Opin Chem Eng. 2017;16:48–55. doi:10.1016/j.coche.2017.04.001.
- Tomar DS, Kumar S, Singh SK, Goswami S, Li L. Molecular basis of high viscosity in concentrated antibody solutions: strategies for high concentration drug product development. MAbs. 2016;8(2):216–28. doi:10.1080/19420862.2015.1128606.
- Götze W. Complex dynamics of glass-forming liquids. New York, USA: Oxford University Press, 2008.
- Yadav S, Shire SJ, Kalonia DS. Viscosity analysis of high concentration bovine serum albumin aqueous solutions. Pharm Res. 2011;28(8):1973–83. doi:10.1007/s11095-011-0424-7.
- Heinen M, Zanini F, Roosen-Runge F, Fedunova D, Zhang FJ, Hennig M, Seydel T, Schweins R, Sztucki M, Antalik M, et al. Viscosity and diffusion: crowding and salt effects in protein solutions. Soft Matter. 2012;8(5):1404–19. doi:10.1039/c1sm06242e.
- Sharma V, Jaishankar A, Wang YC, McKinley GH. Rheology of globular proteins: apparent yield stress, high shear rate viscosity and interfacial viscoelasticity of bovine serum albumin solutions. Soft Matter. 2011;7(11):5150–60. doi:10.1039/c0sm01312a.
- Foffi G, Savin G, Bucciarelli S, Dorsaz N, Thurston GM, Stradner A, Schurtenberger P. Hard sphere-like glass transition in eye lens alpha-crystallin solutions. Proc Natl Acad Sci U S A. 2014;111(47):16748–53. doi:10.1073/pnas.1406990111.
- Riest J, Nagele G, Liu Y, Wagner NJ, Godfrin PD. Short-time dynamics of lysozyme solutions with competing short-range attraction and long-range repulsion: experiment and theory. J Chem Phys. 2018;148(6):065101. doi:10.1063/1.5016517.
- von Bulow S, Siggel M, Linke M, Hummer G. Dynamic cluster formation determines viscosity and diffusion in dense protein solutions. Proc Natl Acad Sci U S A. 2019;116(20):9843–52. doi:10.1073/pnas.1817564116.
- Li L, Kumar S, Buck PM, Burns C, Lavoie J, Singh SK, Warne NW, Nichols P, Luksha N, Boardman D. Concentration dependent viscosity of monoclonal antibody solutions: explaining experimental behavior in terms of molecular properties. Pharm Res. 2014;31(11):3161–78. doi:10.1007/s11095-014-1409-0.
- Tomar DS, Li L, Broulidakis MP, Luksha NG, Burns CT, Singh SK, Kumar S. In-silico prediction of concentration-dependent viscosity curves for monoclonal antibody solutions. MAbs. 2017;9(3):476–89. doi:10.1080/19420862.2017.1285479.
- Chowdhury A, Guruprasad G, Chen AT, Karouta CA, Blanco MA, Truskett TM, Johnston KP. Protein-protein interactions, clustering, and rheology for bovine igg up to high concentrations characterized by small angle x-ray scattering and molecular dynamics simulations. J Pharm Sci. 2020;109(1):696–708. doi:10.1016/j.xphs.2019.11.001.
- Kastelic M, Dill KA, Kalyuzhnyi YV, Vlachy V. Controlling the viscosities of antibody solutions through control of their binding sites. J Mol Liq. 2018;270:234–42. doi:10.1016/j.molliq.2017.11.106.
- Garidel P, Blume A, Wagner M. Prediction of colloidal stability of high concentration protein formulations. Pharm Dev Technol. 2015;20(3):367–74. doi:10.3109/10837450.2013.871032.
- Hung JJ, Dear BJ, Karouta CA, Chowdhury AA, Godfrin PD, Bollinger JA, Nieto MP, Wilks LR, Shay TY, Ramachandran K, et al. Protein-protein interactions of highly concentrated monoclonal antibody solutions via static light scattering and influence on the viscosity. J Phys Chem B. 2019;123(4):739–55. doi:10.1021/acs.jpcb.8b09527.
- Blanco MA, Perevozchikova T, Martorana V, Manno M, Roberts CJ. Protein-protein interactions in dilute to concentrated solutions: alpha-chymotrypsinogen in acidic conditions. J Phys Chem B. 2014;118(22):5817–31. doi:10.1021/jp412301h.
- Timasheff SN. The control of protein stability and association by weak interactions with water: how do solvents affect these processes? Annu Rev Biophys Biomol Struct. 1993;22(1):67–97. doi:10.1146/annurev.bb.22.060193.000435.
- Shukla D, Shinde C, Trout BL. Molecular computations of preferential interaction coefficients of proteins. J Phys Chem B. 2009;113(37):12546–54. doi:10.1021/jp810949t.
- Verwey EJ. Theory of the stability of lyophobic colloids. J Phys Colloid Chem. 1947;51(3):631–36. doi:10.1021/j150453a001.
- Herhut M, Brandenbusch C, Sadowski G. Inclusion of mprism potential for polymer-induced protein interactions enables modeling of second osmotic virial coefficients in aqueous polymer-salt solutions. Biotechnol J. 2016;11(1):146–54. doi:10.1002/biot.201500086.
- Herhut M, Brandenbusch C, Sadowski G. Modeling and prediction of protein solubility using the second osmotic virial coefficient. Fluid Phase Equilib. 2016;422:32–42. doi:10.1016/j.fluid.2016.01.020.
- Schleinitz M, Sadowski G, Brandenbusch C. Protein-protein interactions and water activity coefficients can be used to aid a first excipient choice in protein formulations. Int J Pharm. 2019;569:118608. doi:10.1016/j.ijpharm.2019.118608.
- Quang LJ, Sandler SI, Lenhoff AM. Anisotropic contributions to protein-protein interactions. J Chem Theory Comput. 2014;10(2):835–45. doi:10.1021/ct4006695.
- Pusara S, Yamin P, Wenzel W, Krstic M, Kozlowska M. A coarse-grained xdlvo model for colloidal protein-protein interactions. Phys Chem Chem Phys. 2021;23(22):12780–94. doi:10.1039/d1cp01573g.
- Grunberger A, Lai PK, Blanco MA, Roberts CJ. Coarse-grained modeling of protein second osmotic virial coefficients: sterics and short-ranged attractions. J Phys Chem B. 2013;117(3):763–70. doi:10.1021/jp308234j.
- Stark AC, Andrews CT, Elcock AH. Toward optimized potential functions for protein-protein interactions in aqueous solutions: osmotic second virial coefficient calculations using the martini coarse-grained force field. J Chem Theory Comput. 2013;9(9):4176–85. doi:10.1021/ct400008p.
- Qin S, Zhou HX. Calculation of second virial coefficients of atomistic proteins using fast fourier transform. J Phys Chem B. 2019;123(39):8203–15. doi:10.1021/acs.jpcb.9b06808.
- Minton AP. Effective hard particle model for the osmotic pressure of highly concentrated binary protein solutions. Biophys J. 2008;94(7):L57–59. doi:10.1529/biophysj.107.128033.
- Minton AP. Static light scattering from concentrated protein solutions, i: general theory for protein mixtures and application to self-associating proteins. Biophys J. 2007;93(4):1321–28. doi:10.1529/biophysj.107.103895.
- Fernandez C, Minton AP. Static light scattering from concentrated protein solutions ii: experimental test of theory for protein mixtures and weakly self-associating proteins. Biophys J. 2009;96(5):1992–98. doi:10.1016/j.bpj.2008.11.054.
- Scherer TM, Liu J, Shire SJ, Minton AP. Intermolecular interactions of igg1 monoclonal antibodies at high concentrations characterized by light scattering. J Phys Chem B. 2010;114(40):12948–57. doi:10.1021/jp1028646.
- Lilyestrom WG, Yadav S, Shire SJ, Scherer TM. Monoclonal antibody self-association, cluster formation, and rheology at high concentrations. J Phys Chem B. 2013;117(21):6373–84. doi:10.1021/jp4008152.
- Wang W, Lilyestrom WG, Hu ZY, Scherer TM. Cluster size and quinary structure determine the rheological effects of antibody self-association at high concentrations. J Phys Chem B. 2018;122(7):2138–54. doi:10.1021/acs.jpcb.7b10728.
- Scherer TM. Role of cosolute-protein interactions in the dissociation of monoclonal antibody clusters. J Phys Chem B. 2015;119(41):13027–38. doi:10.1021/acs.jpcb.5b07568.
- Fernandez C, Minton AP. Effect of nonadditive repulsive intermolecular interactions on the light scattering of concentrated protein-osmolyte mixtures. J Phys Chem B. 2011;115(5):1289–93. doi:10.1021/jp110285b.
- Wills PR, Winzor DJ. Rigorous analysis of static light scattering measurements on buffered protein solutions. Biophys Chem. 2017;228:108–13. doi:10.1016/j.bpc.2017.07.007.
- Barata TS, Zhang C, Dalby PA, Brocchini S, Zloh M. Identification of protein-excipient interaction hotspots using computational approaches. Int J Mol Sci. 2016;17(6):853. doi:10.3390/ijms17060853.
- Jo S, Xu A, Curtis JE, Somani S, MacKerell AD Jr. Computational characterization of antibody-excipient interactions for rational excipient selection using the site identification by ligand competitive saturation-biologics approach. Mol Pharm. 2020;17(11):4323–33. doi:10.1021/acs.molpharmaceut.0c00775.
- Cloutier TK, Sudrik C, Mody N, Sathish HA, Trout BL. Machine learning models of antibody-excipient preferential interactions for use in computational formulation design. Mol Pharm. 2020;17(9):3589–99. doi:10.1021/acs.molpharmaceut.0c00629.
- Somani S, Jo S, Thirumangalathu R, Rodrigues D, Tanenbaum LM, Amin K, MacKerell AD Jr., Thakkar SV. Toward biotherapeutics formulation composition engineering using site-identification by ligand competitive saturation (silcs). J Pharm Sci. 2021;110(3):1103–10. doi:10.1016/j.xphs.2020.10.051.
- Raman EP, Lakkaraju SK, Denny RA, MacKerell AD Jr. Estimation of relative free energies of binding using pre-computed ensembles based on the single-step free energy perturbation and the site-identification by ligand competitive saturation approaches. J Comput Chem. 2017;38(15):1238–51. doi:10.1002/jcc.24522.
- Nicoud L, Sozo M, Arosio P, Yates A, Norrant E, Morbidelli M. Role of cosolutes in the aggregation kinetics of monoclonal antibodies. J Phys Chem B. 2014;118(41):11921–30. doi:10.1021/jp508000w.
- Baftizadeh F, Pietrucci F, Biarnes X, Laio A. Nucleation process of a fibril precursor in the c-terminal segment of Amyloid-β. Phys Rev Lett. 2013;110(16):168103. doi:10.1103/PhysRevLett.110.168103.
- Rojas AV, Liwo A, Scheraga HA. A study of the α-helical intermediate preceding the aggregation of the amino-terminal fragment of the β amyloid peptide (Aβ1–28). J Phys Chem B. 2011;115(44):12978–83. doi:10.1021/jp2050993.
- Rojas A, Maisuradze N, Kachlishvili K, Scheraga HA, Maisuradze GG. Elucidating important sites and the mechanism for amyloid fibril formation by coarse-grained molecular dynamics. ACS Chem Neurosci. 2017;8(1):201–09. doi:10.1021/acschemneuro.6b00331.
- Kern N, Frenkel D. Fluid-fluid coexistence in colloidal systems with short-ranged strongly directional attraction. J Chem Phys. 2003;118(21):9882–89. doi:10.1063/1.1569473.
- Kastelic M, Kalyuzhnyi YV, Vlachy V. Fluid of fused spheres as a model for protein solution. Condens. Matter Phys 2016;19(2):23801. doi:10.5488/Cmp.19.23801. Artn 23801
- Hatch HW, Jiao S, Mahynski NA, Blanco MA, Shen VK. Communication: predicting virial coefficients and alchemical transformations by extrapolating mayer-sampling monte carlo simulations. J Chem Phys. 2017;147(23):231102. doi:10.1063/1.5016165.
- Carmichael SP, Shell MS. A new multiscale algorithm and its application to coarse-grained peptide models for self-assembly. J Phys Chem B. 2012;116(29):8383–93. doi:10.1021/jp2114994.