
ABSTRACT
Manipulation of glycosylation patterns, i.e., glycoengineering, is incorporated in the therapeutic antibody development workflow to ensure clinical safety, and this approach has also been used to modulate the biological activities, functions, or pharmacological properties of antibody drugs. Whereas most existing glycoengineering strategies focus on the canonical glycans found in the constant domain of immunoglobulin G (IgG) antibodies, we report a new strategy to leverage the untapped potential of atypical glycosylation patterns in the variable domains, which naturally occur in 15% to 25% of IgG antibodies. Glycosylation sites were added to the antigen-binding regions of two functionally divergent, interleukin-2-binding monoclonal antibodies. We used computational tools to rationally install various N-glycosylation consensus sequences into the antibody variable domains, creating “glycovariants” of these molecules. Strikingly, almost all the glycovariants were successfully glycosylated at their newly installed N-glycan sites, without reduction of the antibody’s native function. Importantly, certain glycovariants exhibited modified activities compared to the parent antibody, showing the potential of our glycoengineering strategy to modulate biological function of antibodies involved in multi-component receptor systems. Finally, when coupled with a high-flux sialic acid precursor, a glycovariant with two installed glycosylation sites demonstrated superior in vivo half-life. Collectively, these findings validate a versatile glycoengineering strategy that introduces atypical glycosylation into therapeutic antibodies in order to improve their efficacy and, in certain instances, modulate their activity early in the drug development process.
Introduction
Glycosylation influences the safety, bioactivity, and pharmacokinetic (PK) properties of protein therapeutics, features that are critical for drug efficacy.Citation1–3 Careful control of glycosylation is now a well-recognized, US Food and Drug Administration (FDA)-mandated, aspect of therapeutic protein manufacturing. Because regulatory agencies primarily address the safety of glycosylation, the bulk of the biomanufacturing industry’s efforts to control glycosylation focus on meeting these safety-related regulatory guidelines, rather than on leveraging modern glycoengineering efforts to improve therapeutic protein function. Moreover, most efforts have focused on manipulating glycosylation in the antibody constant domains (specifically the Fc region), with considerably less attention paid to modification of glycosylation in the variable (Fv) domains. However, N-glycan sites within the Fv do occur naturally during somatic hypermutation,Citation4,Citation5 and there is precedent for engineering N-glycans in the Fv domain, as exemplified by strategies to modify the Fv N-glycan of cetuximab.Citation6,Citation7
A common glycoengineering approach designed to improve efficacy is the introduction of new glycosylation sites into a therapeutic protein. This approach was used in the design of darbepoetin, a marketed form of erythropoietin (EPO). Two N-glycan sites were installed into native EPO to yield darbepoetin, which has a serum half-life that is approximately three-fold longer than that of EPO.Citation8 Other examples of therapeutic proteins that have been glycoengineered to incorporate “built-in” N-glycans include an insulin with improved PK properties;Citation9 human immunodeficiency virus (HIV) neutralizing antibodies with improved potency;Citation10 and a therapeutic enzyme ectonucleotide pyrophosphatase phospodiesterase-1 (ENPP-1) with improved bioavailability and PK properties.Citation11
Despite the established benefits of glycoengineering, the pharmaceutical industry has been hesitant to embrace this approach because of the increased product complexity and heterogeneity that result from the diversity of glycan structures, which can complicate regulatory approval.Citation2 Another challenge is that newly added glycans introduced through glycoengineering may increase the number of asialo-, galactose-terminated N-glycan antennae, leading to rapid clearance via the asialoglycoprotein receptor (ASGPR).Citation12,Citation13 To provide context, asialylated EPO has a clearance rate of ~10 min, compared to about eight hours for the naturally sialylated protein,Citation8 dramatically illustrating the efficacy of ASPGR-mediated clearance against glycans extending away from a protein into the solvent, as is the case for virtually all non-Fc region N-glycans.
The pitfall of rapid ASGPR-mediated clearance is avoided by current FDA-approved antibody drugs because they are largely limited to the immunoglobulin G (IgG) class,Citation14 which contains a single glycosylation site (Asn297) in the Fc region, with cetuximab, which has an Fv N-glycan, as a notable exception.Citation6,Citation7 Because Fc region glycans are buried between the two heavy chains of an IgG dimer, they are sterically inaccessible to ASGPR clearance. As a result, antibodies typically persist in circulation for at least one week, and often up to 6 or more weeks,Citation14,Citation15 despite having 2% or less sialylation when produced in Chinese hamster ovary (CHO)-K1 cells. For example, the half-life of the commercial anti-HER2 antibody drug trastuzumab is 28 daysCitation16 even though only ~1.1% of its Fc N-glycans are sialylated.Citation17,Citation18
Antibodies that have atypical, non-Fc region glycans are commonly discontinued from commercial development, which eliminates the potential for new medicines with unique recognition, binding, stability, and pharmacokinetic properties.Citation4 Early intervention in the antibody therapeutic pipeline can have substantial implications for downstream drug development. In particular, introduction of non-canonical glycans has potential to improve the PK properties and enhance the overall efficacy of antibodies. Two promising early-stage therapeutic antibodies with potential for glycoengineering include the IgG class antibodies MAB60219,Citation24 (hereafter denoted 602) and F5111,Citation20 which target the interleukin-2 (IL-2) cytokine and bias its immunoregulatory activities. IL-2 is a pleiotropic cytokine that coordinates immune cell differentiation, activity, proliferation, and survival. IL-2 signals through either an intermediate-affinity (Kd~1 nM) ternary complex with the IL-2 receptor-β (IL-2Rβ) and common γ (γc) chains, or a high-affinity (Kd~10 pM) quaternary complex that also includes the IL-2 receptor-α (IL-2Rα/CD25) chain.Citation21,Citation22 Thus, expression of the non-signaling IL-2Rα subunit regulates cytokine sensitivity, whereas IL-2Rβ and γc mediate signaling.Citation21,Citation23 Because IL-2Rα is abundantly expressed in regulatory T cells (TRegs), but is virtually absent from naïve immune effector cells (Effs, i.e., CD4+/CD8+ effector T and natural killer [NK] cells), TRegs are 100-fold more sensitive to endogenous IL-2 than Effs.Citation21 The 602 and F5111 antibodies function by forming complexes with IL-2 that skew the cytokine’s interactions with its receptor subunits and thereby preferentially stimulate Effs or TRegs, respectively. Briefly, IL-2/602 complexes block the IL-2/IL-2Rα interaction while also enhancing the IL-2/IL-2Rβ interaction through bivalent presentation of the cytokine.Citation19,Citation24 These effects potentiate IL-2 activity in all cells, and eliminate the sensitivity advantage typically conferred to IL-2RαHigh TRegs, thus favoring expansion of IL-2RαLow Effs. Conversely, F5111 stabilizes IL-2 in a structural conformation that preferentially promotes IL-2RαHigh TReg over IL-2RαLow Eff expansion by sterically blocking the IL-2/IL-2Rβ interaction and inducing mild allosteric disruption of the IL-2/IL-2Rα interaction.Citation20 Antibody-mediated immune bias presents an exciting opportunity to develop targeted cytokine therapies to treat cancer and other immune-linked diseases. Indeed, IL-2/602 complexes induce potent antitumor activity in mice without inducing adverse effects typically associated with systemic IL-2 administration,Citation19,Citation24 and IL-2/F5111 complexes reverse type 1 diabetes in non-obese diabetic mice.Citation20
The complementary pair of the 602 and F5111 IL-2 modulating antibodies already show promising preclinical activities, but we wondered whether glycoengineering could provide additional benefits. In particular, we wished to determine whether modification of the native glycosylation patterns would either compromise or enhance the biological activity of these antibodies. To address these possibilities, we built in additional, atypical N-linked glycosylation sites into the Fv regions of these two antibodies as a new paradigm for therapeutic antibody design. As mentioned above, a potential pitfall of building additional N-glycan sites is that, if these sites are not fully sialylated, any benefits from the newly added glycans could be offset by increased clearance by the ASGPR. To ameliorate this potential problem, we used a “high-flux” sialic acid precursor, 1,3,4-O-Bu3ManNAc, to increase flux through the sialic acid pathwayCitation25,Citation26 and improve sialylation of our glycoengineered antibodies.Citation27 We found that, in contrast with concerns of functional disruption, most of our glycoengineered antibodies had identical activity to their respective parent molecules. Moreover, we observed that some glycoengineered antibodies modulated function in potentially productive ways. Our glycoengineering strategy culminated in a double gain-of-glycan variant of the 602 antibody that exhibited both increased sialylation and an improved in vivo half-life without compromising function, illustrating the potential therapeutic benefits of our glycoengineering approach. We anticipate that the glycoengineering strategy we introduce could be extended to other antibodies involved in multi-component systems as a potential approach for improving the efficacy and modulating the activity of candidate therapeutics early in the clinical pipeline.
Results
Design of glycoengineered F5111 variants
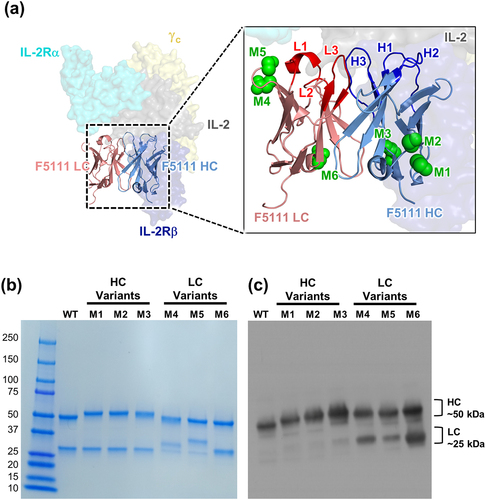
To demonstrate that atypical N-linked glycan sites could be incorporated into the Fv region of antibodies without compromising their activity or PK properties, we adopted a computationally and structurally guided approach to glycoengineer the F5111 antibody. First, the heavy chain (HC) (human IGHV4, 98% identity, IGHD4, IGHJ2, 93% identity) and light chain (LC) (human IGLV6, 100% identity, IGLJ3, 100% identity) Fv sequences were screened for amino acid substitutions that could introduce the Asn-X-Ser/Thr consensus sequence required for N-glycan insertion. We preferentially selected sites that were solvent exposed, located outside of the complementarity-determining regions (CDRs), and likely to be glycosylated (>55% probability based on the NetNGlyc server).Citation28 Six potential insertion sites satisfied these criteria, three located in the HC (denoted M1-M3) and three located in the LC (denoted M4-M6) ( and Supplementary Table S1). To assess whether these substitutions would yield glycovariants that were properly folded and that successfully incorporated N-glycans, the six antibodies were expressed in human embryonic kidney (HEK)-293 F cells. Reducing SDS-PAGE analysis revealed that all six glycovariants were successfully expressed, and molecular weight shifts suggested that the engineered chains were glycosylated as designed (). Successful glycosylation of each glycovariant was confirmed by immunoblotting with Con-A (), a lectin that has broad specificity for N-glycans. All six constructs showed glycosylation of the HC, a reflection of both canonical Fc glycosylation and newly introduced HC variable domain N-glycan. The F5111 glycovariants with N-glycans introduced into the LC (M4, M5, and M6), showed LC glycosylation, as designed.
Figure 1. Engineering novel N-glycan sites into the F5111 antibody. (a) Crystallographic structure of the heavy chain (HC) and light chain (LC) variable domains of the F5111 antibody in complex with the IL-2 cytokine (PDB ID: 5UTZ), overlaid with the heterotrimeric IL-2 receptor (PDB ID: 2B5I). Complementarity-determining loops of the F5111 HC (H1, H2, and H3) and LC (L1, L2, and L3) are indicated (inset). Green spheres indicate the location of the asparagine residue in the engineered N-glycan site. (b) Reducing SDS-PAGE analysis of six glycovariants of the F5111 antibody with inserted N-linked glycosylation sites in the HC (M1, M2, and M3) or LC (M4, M5, and M6). (c) Con-A lectin blot demonstrating glycosylation of F5111 variants. WT, wild type.
As the installed glycans could negatively affect various biochemical and biophysical properties of the antibody,Citation3 we evaluated several additional properties related to developability of the representative glycovariant F5111 M1 in comparison with wild-type F5111. The addition of N-glycan sites minimally affected the expression of the glycovariants in CHO-S cells (Supplementary Figure S1a) and did not lead to any detectable high molecular weight aggregates based on size-exclusion chromatography (SEC) analysis (Supplementary Figure S1b). Dynamic light scattering (DLS) similarly did not reveal any detectable aggregation resulting from the addition of the N-glycan (Supplementary Figure S1c). Furthermore, the isoelectric point was decreased, as expected, due to the addition of a glycan (Supplementary Figure S1d). Importantly, the melting profile and temperatures were similar for the glycovariant (Supplementary Figures 1e, f, Supplementary Table S3), indicating that protein stability was not affected by glycoengineering. Overall, these assays demonstrated that the installation of N-glycans into the Fv domain did not affect key developability criteria for glycovariants of the F5111 antibody.
Glycoengineered F5111 variants recapitulate functional activity of the parent antibody
Having successfully engineered glycovariants of the F5111 antibody and produced these antibodies in human HEK 293 F cells, we next evaluated whether F5111 glycovariants produced in CHO cells commonly used in commercial monoclonal antibody biomanufacturing would retain the binding and functional properties of the parent antibody. Biolayer interferometry (BLI) studies using immobilized IL-2 demonstrated that glycovariants bound the target cytokine with similar affinity and kinetic properties compared to the wild-type antibody, independent of binding assay topology (, Supplementary Figure S2a, Supplementary Table S2).Citation29 The affinity was also observed to be similar between wild-type F5111 and representative glycovariant M1 antibodies when antibodies were immobilized and IL-2 was soluble (Supplementary Figure S2a). The F5111 antibody biases the IL-2 cytokine by sterically blocking interaction with the IL-2Rβ subunit and imposing mild allosteric disruption on the IL-2/IL-2Rα interaction.Citation20 To demonstrate that the competitive binding behaviors of F5111 were retained by glycovariants, BLI studies were conducted in which binding of saturating concentrations of soluble IL-2 to immobilized IL-2 receptor subunits was measured in the presence of F5111 antibody. Similar to the parent F5111 antibody, the six F5111 glycovariants did not affect the IL-2/IL-2Rα interaction (), but fully blocked the IL-2/IL-2Rβ interaction (). Interestingly, glycovariant M5 showed slightly weaker competition with the IL-2/IL-2Rα interaction compared to the parent F5111 antibody and the other glycovariants ().
Figure 2. F5111 glycovariants show similar binding and functional properties to parent antibody. (a) BLI studies depicting the interaction between immobilized human IL-2 and soluble antibody (either F5111 or glycovariants thereof). (b, c) Competitive BLI-based IL-2 binding studies between F5111 or glycovariants thereof and IL-2 receptor subunits. Equilibrium binding of a saturating concentration of IL-2 (600 nM) to immobilized IL-2Rα (b) or IL-2Rβ (c) in the presence of titrated amounts of F5111 antibody. (d, e) IL-2 signaling pathway activation induced by a 1:1 molar ratio of F5111:IL-2 complexes on human YT-1 human NK cells with (d) and without (e) IL-2Rα expression. STAT5 phosphorylation was detected via flow cytometry. Error bars represent standard deviation. (f) Comparison of YT-:YT+ EC50 values for signaling activation by IL-2 or various IL-2/antibody complexes. WT, wild type.
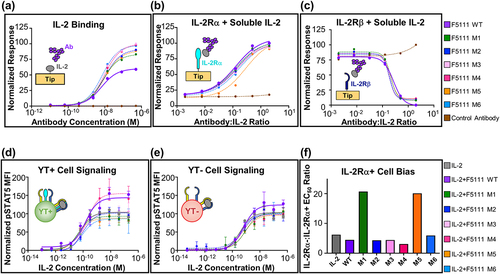
To determine whether the functional activity of F5111 was retained by our engineered glycovariants, we made use of an IL-2 responsive human NK cell line denoted YT-1,Citation30 which inducibly expresses the IL-2Rα subunit.Citation31 In this system, IL-2Rα− YT-1 cells (YT-) and IL-2Rα+ YT-1 cells (YT+) serve as surrogates for IL-2RαLow Effs and IL-2RαHigh TRegs, respectively. Phosphorylation of signal transducer and activator of transcription 5 (STAT5) in YT- and YT+ cells was quantified as a readout for IL-2-induced signaling. The parent F5111 is known to bias signaling effects toward cells that express the IL-2Rα subunit, which results in preferential expansion of TRegs.Citation20 Consistent with this paradigm, we observed more potent activation of YT+ cells compared to YT- cells by complexes of IL-2 and the wild-type F5111 antibody ( and Supplementary Table S2). Encouragingly, all F5111 glycovariants reproduced the parent antibody’s activation of both YT+ and YT- cells. Using the EC50 ratio of IL-2Rα−:IL-2Rα+ YT cells as a metric for bias toward TReg-like cells, we observed that whereas most glycovariants showed a similar extent of bias as the parent F5111 antibody, two glycovariants (M1 and M5) showed improved bias toward IL-2Rα+ cells (). This important finding suggests that glycovariants can potentially augment the functional activities of therapeutic antibodies. This could be due to steric interference of the M1 glycan with IL-2Rβ and the M5 glycan with IL-2Rα (Supplementary Figure S1g), both of which would be expected to increase antibody bias toward IL-2Rα+ versus IL-2Rα− YT cells. Overall, these binding and activation studies demonstrate that our pipeline can be used to design atypical N-linked glycans that do not disrupt, and could possibly enhance, the bioactivity of a therapeutic antibody.
Design of glycoengineered 602 variants
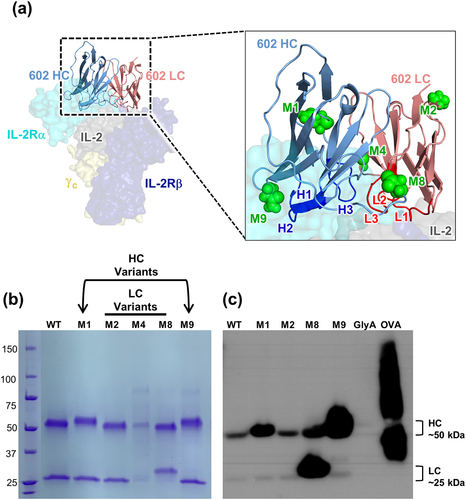
To demonstrate that the glycoengineering strategy we used for F5111 could be generalized to another antibody, we applied this approach to a functionally divergent, anti-IL-2 antibody denoted 602. The 602 antibody biases IL-2 signaling toward Effs over TRegs, and IL-2/602 complexes have promising antitumor activity in mouse models of cancer.Citation19,Citation24 The HC (mouse IGHV2, 95% identity, IGHD6, IGHJ3, 86% identity) and LC (mouse IGKV13, 93% identity, IGKJ2, 100% identity) variable region sequences of 602 were evaluated for amino acid substitutions that could yield novel N-linked glycosylation sites, following the same approach used for the F5111 antibody. In this case, a molecular structure was not available, and thus solvent-exposed sites were determined based on a threaded model structure of the IL-2/602 antigen-binding fragment (Fab) complex.Citation32–34 A total of five potential glycovariants were selected, two of which entailed HC substitutions (denoted M1 and M9) and three of which entailed LC substitutions (denoted M2, M4, and M8) ( and Supplementary Table S4). Of these five potential glycovariants, four were robustly expressed in HEK 293 F cells (), and, of those 4, 3 showed evidence of glycosylation, based on expected molecular weight shifts in the HC (M1 and M9) or LC (M8) in SDS-PAGE analysis (). Con-A lectin immunoblots verified glycosylation of our engineered glycovariants (). The wild-type 602 and all glycovariants displayed canonical Fc glycosylation in the HC, as expected. Glycovariants M1 and M9 showed increased HC glycosylation and M8 showed increased LC glycosylation, as designed. Attenuation of glycosylation upon digestion with PNGase F corroborated that the glycans present in the glycovariants were predominantly N-linked (Supplementary Figure S3a). To demonstrate the potential for incorporation of therapeutically beneficial sialylated glycans, a Sambucus nigra lectin (SNA, B-1305-2) blot was performed (Supplementary Figure S3b). We detected sialylation in all four glycovariants, and variants M1, M8, and M9 showed enhanced sialylation compared to the parent antibody, in their respective glycoengineered chains. Taken together with the results from the F5111 antibody, these data for the 602 antibody illustrate the versatility of our glycoengineering strategy for generating efficiently expressed and successfully glycosylated variants of antibody therapeutic candidates.
Figure 3. Engineering novel N-linked glycosylation sites into the 602 antibody. (a) Threaded computational model of the heavy chain (HC) and light chain (LC) variable domains of the 602 antibody in complex with the IL-2 cytokine (generated using the iTASSER server), overlaid with the heterotrimeric IL-2 receptor (PDB ID: 2B5I). Complementarity-determining loops of the HC (H1, H2, and H3) and LC (L1, L2, and L3) are indicated (inset). Green spheres indicate the location of the asparagine residue in the engineered N-glycan site. (b) Reducing SDS-PAGE analysis of 6 glycovariants of the 602 antibody with inserted N-linked glycosylation sites in the HC (M1 and M9) or LC (M2, M4, M8). (c) Con-A lectin blot demonstrating glycosylation of 602 variants. Glycophorin A (GlyA) and ovalbumin (OVA) were used as controls for O-linked and N-linked glycosylation, respectively. WT, wild type.
Glycoengineered 602 variants recapitulate functional activity of the parent antibody
As with F5111 glycovariants, the binding and signaling properties of the 602 glycovariants prepared in CHO-S cells were compared to those of the parent antibody. BLI studies against immobilized IL-2 demonstrated that all 602 glycovariants bound IL-2 with similar affinity compared to the parent antibody ( and Supplementary Table S5). The 602 antibody orchestrates IL-2 bias by sterically blocking the cytokine’s interaction with the IL-2Rα, but not the IL-2Rβ subunit.Citation24 BLI-based competition studies between the IL-2Rα subunit and 602 antibody variants for cytokine binding demonstrated that all 3 validated glycovariants retain the inhibitory properties of the parent antibody (). Moreover, IL-2/IL-2Rβ interaction was observed in the presence of the wild-type 602 antibody, and this interaction was unaffected by the glycovariants, with the exception of M9, which slightly reduced the cytokine/receptor affinity ().
Figure 4. 602 glycovariants recapitulate binding and functional properties of parent antibody. (a) BLI studies depicting the interaction between immobilized human IL-2 and soluble antibody (either 602 or glycovariants thereof). (b, c) BLI-based competitive IL-2 binding studies between 602 or glycovariants thereof and IL-2 receptor subunits. Equilibrium binding of a saturating concentration of IL-2 (600 nM) to immobilized IL-2Rα (b) or IL-2Rβ (c) in the presence of titrated amounts of 602 antibody is shown. (d, e) IL-2 signaling pathway activation induced by a 1:1 molar ratio of 602:IL-2 complexes on human YT-1 human NK cells with (d) and without (e) IL-2Rα expression. STAT5 phosphorylation was detected via flow cytometry. Error bars represent standard deviation. (f) Ratio of YT-:YT+ EC50 values for signaling activation by IL-2 or various IL-2/antibody complexes. WT, wild type.
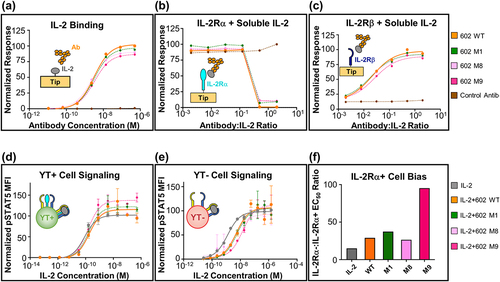
To verify that the activities of 602 glycovariants recapitulate those of the parent antibody, we evaluated the effects of IL-2/glycovariant antibody complexes on IL-2-mediated signaling. STAT5 phosphorylation studies indicated that complexes between IL-2 and the 602 glycovariants behave similarly to the IL-2/602 complex on both IL-2Rα− and IL-2Rα+ YT-1 cells (). However, we observed that M9 diverged from the other glycovariants, skewing IL-2 signaling further toward IL-2Rα+ (TReg-like) cells over IL-2Rα− (Eff-like) cells. The distinctive behavior of M9 may be due to steric interference introduced by the installed glycan proximal to the IL-2 binding site of the 602 antibody (). As was observed for F5111 glycoengineering, these results show that addition of glycans can be achieved without modifying antibody activity while, in some cases (e.g., for M9), glycans can serve as an additional lever to modulate function. Based on their validated glycosylation and unperturbed binding and signaling properties compared to 602, the M1 and M8 variants were chosen as candidates for further design.
A double gain-of-glycan 602 variant shows a further increase in glycosylation
To evaluate whether glycoengineering two atypical glycans into the Fv region of an antibody would provide additional benefits compared to a single engineered N-glycan, we designed a double gain-of-glycan variant of the 602 antibody. The N-linked glycosylation sites associated with the M1 and M8 602 antibody variants were installed into a single construct, denoted M18, that had newly introduced glycosylation sites on both the HC and LC (). The resulting antibody was expressed in CHO-S cells, and glycosylation was observed in both the HC and LC, as evidenced by both SDS-PAGE analysis and lectin immunoblotting (). Further, treatment with the 1,3,4-O-Bu3ManNAc sugar analog, a metabolic precursor of sialic acid,Citation25,Citation26 elicited an apparent increase in glycosylation in both the heavy and light chains for the double M18 mutant, as illustrated by lectin immunoblotting (). To demonstrate that the two introduced glycans did not harm developability criteria, we evaluated additional biochemical properties for wild-type 602 and the 602 M18 glycovariant. Introduction of the two glycans did not lead to detectable high-molecular-weight aggregates by SEC analysis (Supplementary Figure S4b), although minor levels aggregation were observed with DLS for the 602 M18 (Supplementary Figure S4c). As anticipated, the isoelectric point of 602 M18 was significantly shifted compared to the parent antibody due to the introduction of multiple glycans (Supplementary Figure S4d). Importantly, the melting profile and temperature of 602 M18 were similar to those of the wild-type antibody (Supplementary Figures S4e,f and Supplementary Table S6), demonstrating comparable stability between the 602 and 602 M18 antibodies. Taken together, these biophysical analyses revealed no significant adverse consequences with respect to developability for the glycoengineered antibodies. Moreover, consistent with the single gain-of-glycan constructs, the M18 double gain-of-glycan construct maintained the IL-2 binding properties of the parent 602 antibody independent of binding study topology (Supplementary Figures S2b, S5a-c, and Supplementary Table S5). Accordingly, complexes of IL-2 and M18 recapitulated the STAT5-mediated signaling properties of IL-2/602 complexes (Supplementary Figure S5d-Sf and Supplementary Table S5).
Figure 5. Glycosylation and pharmacokinetic properties of the M18 double gain-of-glycan 602 variant. (a) Threaded computational model of the heavy chain (HC) and light chain (LC) variable domains of the 602 antibody in complex with IL-2 (generated using the iTASSER server). The two installed glycosylation sites for the M18 variant are indicated as green spheres. (b,c) Reducing SDS-PAGE analysis (b) and ConA lectin immunoblot (c) showing glycosylation of the wild-type 602 antibody compared to the M18 variant. Proteins were expressed in HEK293 F (HEK) or CHO-S (CHO) cells, as indicated, in the absence (-) or presence (+) of 1,3,4-O-Bu3ManNAc. (d) Fragmentation spectra of wild-type and corresponding engineered N-linked glycovariant peptides from the heavy and light chains of both 602 wild-type and 602 M18, respectively. Oxonium ions from 602 M18 glycopeptides are boxed in red. (e) In vivo pharmacokinetic study of the parent 602 antibody versus the M18 variant, prepared with or without 1,3,4-O-Bu3ManNAc. Serum concentration is plotted at various time points following r.o. injection (n = 3). WT, wild type.
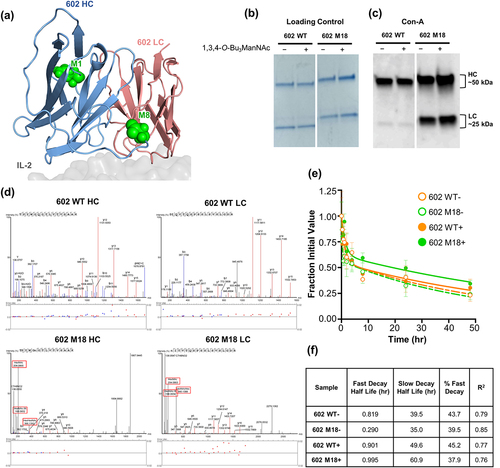
We rigorously characterized the glycosylation of M18 by mass spectrometry (MS) to better probe the composition of the installed N-glycans. In these experiments, wild-type 602 and the M18 glycovariant were produced in translationally relevant CHO-S cells. MS unambiguously detected peptides of the wild-type 602 sequence in both the wild-type 602 HC and LC (). Fragmenting engineered N-linked glycopeptides from these regions in the heavy and light chains of the 602 M18 glycovariant produced glycan-derived oxonium ions. No glycopeptides or oxonium ions were detected from the corresponding wild-type sequences in either chain of the wild-type 602 (). Furthermore, MS analysis of the glycovariant peptides after treating the HC and LC of 602 M18 with Endo-H or PNGase F unambiguously confirmed the engineered N-linked glycan consensus site and produced the predicted HexNAc or deaminated asparagine (Asn)-containing fragment ions, respectively, corroborating that both chains were successfully glycosylated at the engineered sites (Supplementary Tables S7–S12). Taken together, characterization of the M18 double gain-of-glycan variant established that the strategy of combining single gain-of-glycan variants can be exploited to develop higher order glycovariants with enhanced levels of glycosylation.
The M18 double gain-of-glycan 602 variant exhibits enhanced pharmacokinetic properties
Finally, to evaluate whether the doubly glycoengineered M18 antibody exhibited altered PK properties, we measured the serum half-life of the M18 and wild-type 602 antibodies in mice. The animals were administered a bolus dose (10 mg/kg) of fluorescent dye-labeled antibody, and peripheral blood was collected over a 48-hour time period to monitor clearance. In the absence of sialic acid analog treatment, 602 and M18 showed similar slow decay half-lives (39.5 h and 35.0 h, respectively). Addition of the sialic acid precursor 1,3,4-O-Bu3ManNAc extended the slow decay half-lives for both the 602 and M18 antibodies, consistent with the previous observation that sialylation can slow the clearance of proteins from serum.Citation8 Consistent with its increased glycosylation due to the installed N-linked glycan sites, and putatively increased sialylation from 1,3,4-O-Bu3ManNAc treatment, the glycoengineered M18 demonstrated a longer slow decay half-life than analog-treated 602 (60.9 h versus 49.6 h, respectively). The increased half-life was not attributable to changes in neonatal Fc receptor recycling, as the affinities of wild-type 602 and 602 M18 for mouse FcRn were similar (Supplementary Figure S4g and Supplementary Table S6). The prolonged half-life for 602 M18 demonstrated that increasing glycosylation levels, and specifically targeted augmentation of sialic acid levels, can extend the serum half-life of candidate antibody therapeutics.
Discussion
Most commercial antibody design pipelines avoid incorporation of atypical Fab glycans,Citation1 depriving the drug development community of viable, and even potentially superior, therapeutic molecules. In this study we combined multiple glycoengineering strategies for therapeutic proteins that are typically used in isolation, including installation of N-linked glycosylation sitesCitation8–11 and metabolic supplementation with sugar analogues such as 1,3,4-O-Bu3ManNAc.Citation11 Leveraging these approaches, we successfully converted conventional IgG antibodies into glycovariants with sialylated, atypical Fv glycans. Our study had two motivations. The first was to demonstrate that more highly glycosylated IgG antibodies can serve as viable drug candidates by converting a therapeutically promising conventionally glycosylated IgG antibody into an atypically glycosylated antibody. As antibodies with non-canonical glycosylation (15–25% of all IgG antibodies)Citation4 are largely avoided in translational pipelines, with cetuximab as a notable exception,Citation6,Citation7 our intention was to lay a scientific and practical foundation for the clinical translation of these potential drug candidates. Our second motivation was to investigate whether non-canonical glycosylation could actually improve the biological activity of antibodies, as this approach has already showed promise for other therapeutic proteins (e.g., EPO, insulin)Citation8,Citation9 as well as for certain antibodies (e.g., HIV-neutralizing antibodies).Citation10
Here, we have devised a strategy to engineer N-linked sites into the Fv region of IgG antibodies to introduce atypical glycosylation. To do so, we used a sliding window approach that integrated sequence, computational, and structural prediction cues. This strategy proved to be broad-based and versatile, as it successfully yielded several glycovariants based on two functionally distinct IL-2-targeted antibodies. The majority of engineered glycovariants were found to recapitulate the biological activities of the parent antibodies, and in some cases glycovariants introduced subtle modifications with the potential to expand the functional capabilities of the antibody in question. Importantly, the additional glycans did not negatively impact several important developability criteria, including protein expression, aggregation, and thermal stability (Supplementary Figures S1 and S4). Furthermore, we demonstrated the therapeutic benefit of a double gain-of-glycan variant, which exhibited superior serum half-life in mice (). In practice, a glycoengineered antibody with improved PK properties could reduce the dose required for therapeutic efficacy, or equally important, reduce the frequency of dosing. Overall, our glycoengineering approach presents a new strategy for antibody design that can be harnessed to modulate antibody activity and therapeutic efficacy.
A prominent concern for glycoengineering approaches is that the presence of newly installed glycans could disrupt the natural activity of the engineered molecule. For example, if placed in certain positions, glycans installed into IL-2 interacting antibody could interfere with the cytokine’s interactions with its receptor subunits. Accordingly, our design sought to insert the newly introduced Fv glycans at amino acid positions for which interference with binding partners was not anticipated. Consistent with this approach, most glycovariants retained similar affinity and receptor biasing activities compared to the respective parent antibody (). In fact, two glycovariants of the F5111 antibody appear to have superior biasing activity toward Treg-like cells compared to the parent antibody (). Conversely, one glycovariant of the 602 antibody showed similar IL-2 binding properties, but less potent biasing activity toward Effs compared to the parent antibody (). These results demonstrate how even modest sets of semi-rationally designed glycovariants resulted in modulated, bidirectional tuning, which could both be beneficial in different contexts. Future high throughput glycoengineering screens could identify additional candidates that exhibit a range of functional activities. The notion of building in glycans to improve the bioactivity of antibodies is in its infancy, and our findings resonate with other efforts to install glycans in the Fv domain of antibodies in order to modulate interactions with the target antigen.Citation7,Citation10 Further demonstrating the robustness of our design workflow, incorporation of glycosylation sites into both the heavy and light chains in the double gain-of-glycan M18 glycovariant of the 602 antibody did not compromise its biological activity or stability compared to the parent antibody (Supplemental Figures S4 and S5). Overall, studies with both the F5111 and 602 antibodies demonstrated that glycoengineering did not have substantial effects on the biophysical or biochemical properties of the parent antibodies, and that glycan modifications did not negatively impact developability criteria. These data demonstrate that our strategy can generate glycovariants that can either leave antibody/antigen interactions unperturbed or, if desired, selectively tune antibody/antigen interactions.
Another major concern for the introduction of glycans into a therapeutic protein is that this modification could shorten in vivo half-life, thus negating any potential benefits of glycoengineering efforts. In our case, introduction of two glycosylation sites (in the case of M18) did not reduce serum half-life, suggesting that the newly installed N-glycans did not engage ASGPRs, which clear proteins that lack sialic acids at the termini of their glycans.Citation12,Citation13 To further reduce the possibility of ASGPR clearance of our antibodies, we included the “high-flux” sialic acid precursor, 1,3,4-O-Bu3ManNAc, in our work flow to increase sialylation of the antibody. Furthermore, these mutations had no significant effect on FcRn binding (Supplementary Figure S4g), suggesting that the observed changes in PK properties were, in fact, due to introduced glycosylation. Promisingly, use of this metabolic precursor for sialic acid led to an increase in slow decay half-life for the glycoengineered M18 antibody (60.9 h for M18 in the presence of analog versus 39.5 h for the parent 602 antibody in the absence of analog and 49.6 h for 602 in the presence of analog). These results suggest that the analog-treated, double gain-of-glycan variant had slower clearance when more strongly sialylated, in agreement with other reports of atypically glycosylated antibodies that showed prolonged half-life with increased sialic acid content.Citation35 Moreover, our findings demonstrate that introduction of engineered N-glycans can not only avoid reduced serum half-life (as has been demonstrated with other recombinant proteins),Citation8 but it can actually enhance serum half-life. We believe that these results provide precedent for embracing rather than avoiding antibodies with non-canonical Fv glycans in the biopharmaceutical development pipeline.
Overall, this study illustrates the potential advantages that can be conferred by leveraging Fv glycosylation in glycoengineering approaches. The presence of Fv glycans has been known to affect antigen/protein interactions, often disrupting desired activity.Citation4 Here we show that atypical glycans can be installed within various sites on the Fv that can both maintain or potentially toggle, the selective activity of antibodies involved in multi-component systems. The improvement in PK properties also makes this a promising strategy for improving drug efficacy and facilitating downstream development. As our methodology for screening new glycosylation sites was fairly conservative, subsequent designs could explore larger spaces of glycan sites that might confer further benefits to functional activity or PK properties. An exciting implication of our strategy is that this approach can be used to screen any IgG antibody for potential glycosylation sites, regardless of specificity. Our successful glycoengineering of both F5111, representative of an immunosuppressive therapeutic, and 602, as an immunostimulatory therapeutic, demonstrates how this approach can be impartial to the therapeutic context. However, changes in the activity and selectivity of glycoengineered antibodies are expected to be dictated by the size, orientation, and cellular context of the target antigen. Moreover, it remains to be explored if the specific novel N-glycan sites described here can be grafted between antibody backbones or whether further iterations are needed to identify conserved framework mutations that will constitute a modular approach implementable for any IgG antibody.
Materials and methods
Cell culture
HEK 293 F cells (Thermo Fisher Scientific) were cultivated in Freestyle 293 Expression Medium (Thermo Fisher Scientific) supplemented with 2 U/mL penicillin-streptomycin (Gibco). CHO-S cells (Thermo Fisher Scientific) were cultivated in Freestyle CHO Expression Medium (Thermo Fisher Scientific) supplemented with 2 U/mL penicillin streptomycin (Gibco) and 8 mM L-glutamine (Gibco). Unmodified YT-130 and IL-2R α+ YT-131 human NK cells were cultured in RPMI complete medium (RPMI 1640 medium supplemented with 10% fetal bovine serum [FBS], 2 mM L-glutamine, 1x minimum non-essential amino acids, 1 mM sodium pyruvate, 25 mM HEPES, and 100 U/mL penicillin-streptomycin [Gibco]). All cell lines were maintained at 37°C in a humidified atmosphere with 5% CO2.
Engineering N-linked glycosylation sites within loops of interest
To preserve antibody function and maximize likelihood of glycan modification, we sought to install glycosylation sites into solvent-exposed framework regions distal to the antigen-binding domains of the F5111 or 602 antibodies. Structures were visualized in PyMOL using the resolved crystallographic coordinates for the IL-2/F5111 complex (PDB ID: 5UTZ)Citation20 and a homology model generated using the iTASSER serverCitation32–34 (with the IL-2/S4B6 structure PDB ID: 4YUE as a template)Citation36 for the IL-2/602 complex. Using a Python script, we used a sliding window method to identify all possible sites for insertion of an N-linked glycosylation sequence by modifying existing sequences to code for the Asn–X–Ser/Thr consensus sequence, where X is any amino acid except proline (Pro).Citation37 For each loop of interest, every amino acid triplet was enumerated to produce a set of possible N-linked glycosylation positions. Amongst these triplets, all single and double amino acid substitutions were considered as potential sites for N-linked glycosylation sequence insertion. Triple amino acid substitutions were not considered, in order to avoid potential disruption to the antibody structure. The output of this process was a series of modified loops containing the N-X-S and N-X-T motifs. The NetNGlyc Server, an online N-linked glycosylation prediction server,Citation28 was used to estimate the likelihood that each of the possible engineered glycosylation sites would be successfully glycosylated. Sites with low likelihood of glycosylation (glycosylation likelihood of <0.55) were discarded. Using either the Pymol mutagenesis wizard or the Rosetta software package, the structures of each engineered glycosylation site within the F5111 and 602 antibodies were modeled to: 1) avoid steric interference with the antibody itself; 2) avoid steric interference with IL-2 antigen recognition; and 3) avoid steric interference with other proteins that form complexes with the IL-2 antigen (namely IL-2Rα, IL-2Rβ, and γc).
Design of F5111 and 602 antibody variants with engineered N-linked glycosylation sites
The HC sequences for the F5111 human immunoglobulin G1 (IgG1) and the LC sequences for the human lambda light chain were separately cloned into the gWiz vector (Genlantis).Citation20 Similarly, the HC sequences for the 602 mouse immunoglobulin G2a (IgG2a) and the LC sequence for the mouse kappa light chain were separately cloned into the gWiz vector (Genlantis).Citation38 Commercial QuikChange mutagenesis kits (Agilent) were used to create plasmid vectors containing mutated DNA sequences encoding the F5111 and 602 glycovariant HC and LC sequences. The glycoengineered F5111 and 602 antibody variant HCs and LCs were separately cloned into the gWiz vector, and used to prepare the 6 F5111 and 5 602 glycovariants.
Protein purification and expression
Antibodies were expressed recombinantly in either HEK 293 F cells or CHO K1 cells via transient co-transfection of plasmids encoding the respective HC and LC constructs. HC and LC plasmids were titrated in small-scale co-transfection tests to determine optimal ratios for large-scale expression. HEK 293 F or CHO-S cells were grown to 1.2 × 106 cells/mL and diluted to 1.0 × 106 cells/mL. For transient co-transfection of HEK 293 F cells, plasmid DNA (1 mg total of HC and LC plasmids per liter of cells) and polyethyleneimine (PEI, Polysciences; 2 mg per liter of cells) were independently diluted to 0.05 and 0.1 mg/mL, respectively, in OptiPro medium (Thermo Fisher Scientific). Diluted DNA and PEI were then incubated at room temperature for 15 min, mixed together, and subsequently incubated at room temperature for an additional 15 min. For transient co-transfection of CHO-S cells, plasmid DNA (4 mg total of HC and LC plasmids per liter cells) and Poly(β‐amino ester) (PBAE) 4-4-6 (base polymer 4, side chain polymer 4, and end-cap structure 6) (253 mg per liter cells) were independently diluted to 0.05 and 12.7 mg/mL, respectively, in 25 mM MgAc2 buffer, pH 5.0. Diluted DNA and PBAE 4-4-6 were mixed and then incubated at room temperature for 15 min.
The diluted HEK 293 F or CHO-S cells and 40 mL of DNA/PEI mixture or 40 mL DNA/PBAE 4-4-6 per liter of cells were added to a shaking flask and incubated at 37°C and 5% CO2 with rotation at 125 rpm for 5 days. Secreted antibodies were purified from cell supernatants 5 days post-transfection via protein G agarose (Thermo Fisher Scientific) affinity chromatography followed by SEC using a Superdex 200 Increase 10/300 GL column (Cytiva) on a fast protein liquid chromatography (FPLC) instrument, equilibrated in HEPES-buffered saline (HBS, 150 mM NaCl in 10 mM HEPES pH 7.3). Purity was verified by SDS-PAGE analysis. Dynamic light scattering chromatograms of antibodies were collected using a Zetasizer Pro (Malvern). Native isoelectric focusing was performed using Criterion IEF (Bio-Rad) gels according to the manufacturer’s protocol. Antibody melt curves were obtained using a Protein Thermal Shift Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Melting temperatures were determined by maxima of the differential melt-curve.
Human IL-2 (amino acids 1–133), IL-2Rα ectodomain (amino acids 1–217), and IL-2Rβ ectodomain (amino acids 1–214) containing a C-terminal hexahistidine tag were produced and purified from HEK 293 F cells, as described for antibodies, and purified via Ni-NTA affinity chromatography followed by SEC on an FPLC instrument. A C-terminal biotin acceptor peptide (BAP) GLNDIFEAQKIEWHE sequence was included to allow for biotinylation of IL-2, IL-2Rα, and IL-2Rβ. Following Ni-NTA affinity chromatography, the cytokine and receptors were biotinylated with the soluble BirA ligase enzyme in 0.5 mM bicine pH 8.3, 100 mM ATP, 100 mM magnesium acetate, and 500 mM biotin (Sigma). Excess biotin was removed by SEC on an ÄKTATM FPLC instrument using a Superdex 200 column (Cytiva).
Biolayer interferometry binding measurements
Biotinylated human IL-2, IL-2Rα, IL-2Rβ, and mouse FcRn (Sino, Cat CT083-M27H-B) were immobilized to streptavidin (SA)-coated biosensors (Sartorius) in 0.45 μm-filtered phosphate-buffered saline pH 7.2 containing 0.1% (w/v) bovine serum albumin (PBSA). For inverted topology-binding studies, F5111 and 602 wild-type and glycovariant antibodies were immobilized on anti-human Fc capture or anti-mouse Fc capture biosensors (Sartorius). IL-2 and IL-2Rβ were immobilized at a concentration of 50 nM for 120 s and IL-2Rα was immobilized at a concentration of 100 nM for 120 s. Less than 5 signal units (nm) were immobilized to minimize mass transfer effects. Once baseline measurements were collected in PBSA, binding kinetics were measured by submerging the biosensors in wells containing serial dilutions of the appropriate analytes for 300 s (association) followed by submerging the biosensor in wells containing only PBSA for 500 s (dissociation). For Mouse FcRn binding studies, all baseline and samples buffers were PBSA adjusted to pH 5.6, and tips were regenerated in pH 7.4 PBSA. For competitive-binding studies, IL-2/antibody complexes were formed by pre-incubating a saturating concentration (600 nM) IL-2 with antibody for 30 min at room temperature. These complexes were then serially diluted in 600 nM hIL-2 to maintain saturating levels of cytokine. A non-IL-2-binding protein (in house recombinantly expressed monoclonal antibody ipilimumab) was used as a control for nonspecific binding. Tips were regenerated in 0.1 M glycine pH 2.7. Data was processed and kinetic parameters were calculated using the Octet® Data Analysis software version 7.1 (Sartorius), assuming a 1:1 Langmuir binding model. Equilibrium binding was determined by total response measured after 295 s. Equilibrium titration curve fitting and KD value determination was implemented using GraphPad Prism, assuming all binding interactions to be first order. Experiments were reproduced two times with similar results.
YT-1 cell activation studies
Approximately 2 × 105 IL-2Rα- or IL-2Rα+ YT-1 human NK cells (YT- and YT+, respectively,Citation30,Citation31 obtained as a kind gift from Junji Yodoi) were plated in each well of a 96-well plate and resuspended in 20 µL of RPMI-1640 complete medium containing serial dilutions of unbound IL-2 or IL-2/antibody complexes. IL-2/antibody complexes were formed by incubating a 1:1 molar ratio of cytokine to antibody (reflecting co-delivery strategies typically used in therapeutic studiesCitation20,Citation24,Citation36,Citation39) for 30 min at room temperature. Cells were stimulated for 20 min at 37°C and immediately fixed by addition of paraformaldehyde to 1.5% (w/v) and 10 min incubation at room temperature. Permeabilization of cells was achieved by resuspension in 200 µL of ice-cold 100% methanol for 30 min at 4°C. Fixed and permeabilized cells were washed twice with PBSA and incubated with a 1:50 dilution of Alexa Fluor 647-conjugated anti-phospho-STAT5 (pY694, BD Biosciences) diluted in 20 µL PBSA for 2 h at room temperature. Cells were then washed twice in PBSA and analyzed on a CytoFLEX flow cytometer (Beckman Coulter). Dose-response curves were fitted to a logistic model and half-maximal effective concentrations (EC50 values) were calculated using GraphPad Prism data analysis software after subtraction of the mean fluorescence intensity (MFI) of unstimulated cells and normalization to the maximum signal intensity. Experiments were conducted in triplicate and performed at least twice with similar results.
Lectin blots
Each antibody sample was reduced and denatured in 4X Laemmli sample buffer (Bio-Rad) and 2-mercaptoethanol (Sigma-Aldrich) at 90°C for 10 min. The reduced and denatured antibody was loaded at 5 μg per lane on 4–15% Tris-Glycine eXtended (TGX) SDS-PAGE gels (Bio-Rad) that were run in duplicate. One of two gels was stained with Coomassie stain (Bio-Rad) overnight following the manufacturer’s protocols, de-stained in water, and imaged the following day to serve as loading control. The other gel was electroblotted onto nitrocellulose membrane. The membrane was blocked for 1 h in 5% bovine serum albumin in tris-buffered saline with 0.1% tween-20 (TBST) blocking buffer, incubated for 1 h in 2 μg/mL of biotinylated Concanavalin A (Vector Lab) or biotinylated Sambucus nigra lectin (Vector Lab), and washed three times with TBST. Next, the membrane was incubated for 1 h in streptavidin-HRP (Cell Signaling Technology, Cat 3999S) (1:150,000) and imaged.
Mass spectrometry
Each antibody sample was reduced and denatured and then loaded onto an SDS-PAGE gel as described above. The HC and LC bands were excised from the gel, reduced in 10 mM dithiothreitol, 1 h, 56°C followed by alkylation with 55 mM iodoacetamide for 45 min. At room temperature in the dark, and digested overnight at 37C by adding 500 ng of Trypsin/LysC mixture (V5071, Promega) in 20 mM TEAB. Resulting peptides were desalted using Oasis HLB uElution solid phase extraction plates (Waters), resuspended in 2% acetonitrile in 0.1% formic acid and separated on a 75 μm x 150 mm ProntoSIL-120-5-C18 H nanocolumn (5 µm, 120 Å (BISCHOFF), www.bischoff-chrom.com) using 2–90% acetonitrile gradient at 300 nL/min over 90 min on a EasyLC nanoLC 1000 (Thermo Scientific) interfaced with Q-Exactive Plus (QE Plus, Thermo Scientific) mass spectrometer. Survey scans (full ms) were acquired from 350–1800 m/z with data-dependent monitoring of up to 15 peptide masses (precursor ions), each individually isolated in a 1.9 Da window and fragmented using HCD activation collision energy 32 and 15 s dynamic exclusion. Precursor and the fragment ions were analyzed at resolutions 70,000 and 35,000, respectively, with automatic gain control (AGC) target values at 3e6 with 100 ms maximum injection time (IT) and 1e5 with 150 ms maximum IT, respectively. Isotopically resolved masses in precursor (MS) and fragmentation (MS/MS) spectra were extracted from raw MS data in Proteome Discoverer (PD) software (v2.4, Thermo Scientific) searched using Mascot (2.8.0; www.matrixscience.com) against the RefSeq protein database. The following 4 criteria were set for all database searches: 1) sample’s species; 2) trypsin/LysC as the enzyme, allowing three missed cleavages; 3) cysteine carbamidomethylation as fixed modifications; and 4) methionine oxidation, asparagine, and glutamine deamidation as variable modifications. Peptide identifications from Mascot searches were filtered at 1% False Discovery Rate confidence threshold, based on a concatenated decoy database search, using the Proteome Discoverer. PEAKS (v10.6 Proteome Software) and Byonic (v4.1.10 Protein Metrics) was used to annotate the same peptide spectral matches present in PD2.4
Pharmacokinetic studies
All procedures conducted were approved through the Johns Hopkins University Animal Care and Use Committee, in accordance with the NIH Guide for the Care and Use of Laboratory Animals. To compare the PK parameters of the glycoengineered and wild-type antibodies, antibodies were labeled with NHS-Rhodamine according to the manufacturer’s protocol, and administered retro-orbitally to 4-week-old C57BL/6 J mice (n = 3 per cohort). Each mouse received a single dose of 10 mg antibody/kg body weight. 10–15 μL blood samples were collected by tail snip and mixed with 90 μL of PBS-EDTA (150 mM NaCl, 12 mM Na2HPO4, 5 mM EDTA, pH 8). Blood samples were obtained at 5 min, 25 min, 50 min, 70 min, 2 h, 4 h, 8 h, 24 h, and 48 h post injection. Samples were centrifuged at 1000 g for 15 min to remove cells, diluted plasma was transferred to a 96-well plate, and fluorescence was read on a Biotek Synergy 2 Multi-Mode Reader (Excitation/Emission 540/590). Antibody serum concentration was fit to a two-phase exponential implemented using GraphPad Prism.
Author Contributions:
S.L. and J.R. designed and cloned the glycovariant constructs. S.L. and Z.J.B. biophysically characterized the glycovariants. S.L. performed the in vivo PK studies. C.A. and K.D. characterized the glycosylation patterns of the constructs. J.P. and R.N.C. performed mass-spectrometry. J.B.S and K..J.Y. conceptualized the study and supervised all research. S.L., Z.B., J.B.S., and K.J.Y. wrote the manuscript. All authors participated in the review and editing of the manuscript.
Abbreviations
BLI Biolayer interferometry
CDR Complementarity-determining region
CHO cells Chinese hamster ovary cells
ConA Concanavalin A
EC50 Half maximal effective concentration
Fab Fragment antigen-binding
Fc Fragment crystallizable
FcRn Neonatal Fc receptor
Fv Fragment variable
γc Common gamma chain
GlyA Glycophorin A
HC Heavy Chain
HEK cells Human embryonic kidney cells
IL-2 Interleukin-2
KD Equilibrium constant
koff Dissociation rate constant
kon Association rate constant
Ig Immunoglobulin
LCLight Chain
M Molar
MFI Mean fluorescent intensity
mAb Monoclonal antibody
OVAOvalbumin
PK Pharmacokinetic
STAT Signal transducer and activator of transcription
VH Variable domain of
Supplemental Material
Download Zip (3.1 MB)Acknowledgments
The authors are grateful to the Johns Hopkins University Mass Spectrometry Core and Translational Tissue Engineering Center administration for their contributions to this project.
Disclosure statement
The authors have filed intellectual property claims covering the technologies described herein.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2095704
Additional information
Funding
References
- Ghaderi D, Zhang M, Hurtado-Ziola N, Varki A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev. 2012;28(1):147–13. doi:10.5661/bger-28-147.
- Hossler P, Khattak SF, Li ZJ. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology. 2009;19(9):936–49. doi:10.1093/glycob/cwp079.
- Buettner MJ, Shah SR, Saeui CT, Ariss R, Yarema KJ. Improving immunotherapy through glycodesign. Front Immunol. 2018;9:2485. doi:10.3389/fimmu.2018.02485.
- van de BFS, Hafkenscheid L, Rispens T, Rombouts Y. The emerging importance of IgG Fab glycosylation in immunity. J Immunol. 2016;196(4):1435–41. doi:10.4049/jimmunol.1502136.
- van de Bovenkamp Fs, Derksen NIL, Ooijevaar-de Heer P, van Schie Ka, Heer P O-D, Berkowska MA, van der Schoot Ce, IJspeert H, van der Burg M, Gils A, et al. Adaptive antibody diversification through N -linked glycosylation of the immunoglobulin variable region. Proc Natl Acad Sci. 2018;115(8):1901–06. doi:10.1073/pnas.1711720115.
- Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianférani S. Characterization of therapeutic antibodies and related products. Anal Chem. 2013;85(2):715–36. doi:10.1021/ac3032355.
- Giddens JP, Lomino JV, DiLillo DJ, Ravetch JV, Wang L-X. Site-selective chemoenzymatic glycoengineering of Fab and Fc glycans of a therapeutic antibody. Proc Natl Acad Sci. 2018;115(47):12023–27. doi:10.1073/pnas.1812833115.
- Egrie JC, Dwyer E, Browne JK, Hitz A, Lykos MA. Darbepoetin alfa has a longer circulating half-life and greater in vivo potency than recombinant human erythropoietin. Exp Hematol. 2003;31(4):290–99. doi:10.1016/S0301-472X(03)00006-7.
- Guan X, Chaffey PK, Wei X, Gulbranson DR, Ruan Y, Wang X, Li Y, Ouyang Y, Chen L, Zeng C, et al. Chemically precise glycoengineering improves human insulin. ACS Chem Biol. 2018;13(1):73–81. doi:10.1021/acschembio.7b00794.
- Song R, Oren DA, Franco D, Seaman MS, Ho DD. Strategic addition of an N-linked glycan to a monoclonal antibody improves its HIV-1–neutralizing activity. Nat Biotechnol. 2013;31(11):1047–52. doi:10.1038/nbt.2677.
- Stabach PR, Zimmerman K, Adame A, Kavanagh D, Saeui CT, Agatemor C, Gray S, Cao W, De La Cruz EM, Yarema KJ, et al. Improving the pharmacodynamics and in vivo activity of ENPP1‐Fc through protein and glycosylation engineering. Clin Transl Sci. 2021;14(1):362–72. doi:10.1111/cts.12887.
- Schwartz AL, Ashwell G. The hepatic asialoglycoprotein receptor. CRC Crit Rev Biochem. 1984;16(3):207–33. doi:10.3109/10409238409108716.
- Ellies LG, Ditto D, Levy GG, Wahrenbrock M, Ginsburg D, Varki A, Le DT, Marth JD. Sialyltransferase ST3Gal-IV operates as a dominant modifier of hemostasis by concealing asialoglycoprotein receptor ligands. Proc Natl Acad Sci U S A. 2002;99(15):10042–47. doi:10.1073/pnas.142005099.
- Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacomet Syst Pharmacol. 2017;6(9):576–88. doi:10.1002/psp4.12224.
- Liu L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell. 2018;9(1):15–32. doi:10.1007/s13238-017-0408-4.
- Boekhout AH, Beijnen JH, Schellens JHM. Trastuzumab. The Oncologist. 2011;16(6):800–10. doi:10.1634/theoncologist.2010-0035.
- Nakano M, Higo D, Arai E, Nakagawa T, Kakehi K, Taniguchi N, Kondo A. Capillary electrophoresis-electrospray ionization mass spectrometry for rapid and sensitive N-glycan analysis of glycoproteins as 9-fluorenylmethyl derivatives. Glycobiology. 2009;19(2):135–43. doi:10.1093/glycob/cwn115.
- Del Val IJ, Polizzi KM, Kontoravdi C. A theoretical estimate for nucleotide sugar demand towards Chinese hamster ovary cellular glycosylation. Sci Rep. 2016;6(1):28547. doi:10.1038/srep28547.
- Létourneau S, van Leeuwen Emm, Krieg C, Martin C, Pantaleo G, Sprent J, Surh CD, Boyman O, van Leeuwen EMM. IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor α subunit CD25. Proc Natl Acad Sci. 2010;107(5):2171–76. doi:10.1073/pnas.0909384107.
- Trotta E, Bessette PH, Silveria SL, Ely LK, Jude KM, Le DT, Holst CR, Coyle A, Potempa M, Lanier LL, et al. A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat Med. 2018;24(7):1005–14. doi:10.1038/s41591-018-0070-2.
- Liao W, Lin J-X, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38(1):13–25. doi:10.1016/j.immuni.2013.01.004.
- Wang X, Rickert M, Garcia KC. Structure of the Quaternary Complex of Interleukin-2 with Its α, ß, and γ c Receptors. Science. 2005;310(5751):1159–63. doi:10.1126/science.1117893.
- Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26(1):453–79. doi:10.1146/annurev.immunol.26.021607.090357.
- Krieg C, Létourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci U S A. 2010;107(26):11906–11. doi:10.1073/pnas.1002569107.
- Aich U, Campbell CT, Elmouelhi N, Weier CA, Sampathkumar S-G, Choi SS, Yarema KJ. Regioisomeric SCFA attachment to hexosamines separates metabolic flux from cytotoxicity and MUC1 suppression. ACS Chem Biol. 2008;3(4):230–40. doi:10.1021/cb7002708.
- Almaraz RT, Aich U, Khanna HS, Tan E, Bhattacharya R, Shah S, Yarema KJ. Metabolic oligosaccharide engineering with N-Acyl functionalized ManNAc analogs: cytotoxicity, metabolic flux, and glycan-display considerations. Biotechnol Bioeng. 2012;109(4):992–1006. doi:10.1002/bit.24363.
- Yin B, Wang Q, Chung C-Y, Bhattacharya R, Ren X, Tang J, Yarema KJ, Betenbaugh MJ. A novel sugar analog enhances sialic acid production and biotherapeutic sialylation in CHO cells. Biotechnol Bioeng. 2017;114(8):1899–902. doi:10.1002/bit.26291.
- Gupta R, Brunak S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing. 2002;7:310–22.
- Joy C, Krista W, Charles W, Sae C, Danfeng Y, Henrik P, Jing W, Pu L, Bettina H, Weilei M, et al. Label-free detection of biomolecular interactions using BioLayer Interferometry for kinetic characterization. Comb Chem High Throughput Screen. 2009;12(8):791–800. doi:10.2174/138620709789104915.
- Yodoi J, Teshigawara K, Nikaido T, Fukui K, Noma T, Honjo T, Takigawa M, Sasaki M, Minato N, Tsudo M. TCGF (IL 2)-receptor inducing factor(s). I. Regulation of IL 2 receptor on a natural killer-like cell line (YT cells). J Immunol. 1985;134:1623–30.
- Kuziel WA, Ju G, Grdina TA, Greene WC. Unexpected effects of the IL-2 receptor α subunit on high affinity IL-2 receptor assembly and function detected with a mutant IL-2 analog. J Immunol. 1993;150:3357–65.
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–38. doi:10.1038/nprot.2010.5.
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008;9(1):40. doi:10.1186/1471-2105-9-40.
- Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8. doi:10.1038/nmeth.3213.
- Goletz S, Danielczyk A, Stoeckl L. Fab-glycosylated antibodies [Internet]. 2016 [cited 2021 Sep 20]; Available from: https://patents.google.com/patent/US9359439B2/en
- Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, Votavova P, Pepper M, Kovar M, Garcia KC. Antibodies to interleukin-2 elicit selective T cell subset potentiation through distinct conformational mechanisms. Immunity. 2015;42(5):815–25. doi:10.1016/j.immuni.2015.04.015.
- Hart GW, Brew K, Grant GA, Bradshaw RA, Lennarz WJ. Primary structural requirements for the enzymatic formation of the N-glycosidic bond in glycoproteins. Studies with natural and synthetic peptides. J Biol Chem. 1979;254(19):9747–53. doi:10.1016/S0021-9258(19)83579-2.
- Spangler J, Tomala J, Leff MI, Ludwig S, Leonard EK. Methods and materials for targeted expansion of immune effector cells [Internet]. 2020; [accessed 2022 Jun 5]. https://patents.google.com/patent/WO2020264321A1/en?q=Ludwig+leonard&inventor=Spangler&oq=Spangler+Ludwig+leonard
- Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311(5769):1924–27. doi:10.1126/science.1122927.