
ABSTRACT
Affinity maturation is often a necessary step for the development of potent therapeutic molecules. Many different diversification strategies have been used for antibody affinity maturation, including error-prone PCR, chain shuffling, and targeted complementary-determining region (CDR) mutation. Although effective, they can negatively impact antibody stability or alter epitope recognition. Moreover, they do not address the presence of sequence liabilities, such as glycosylation, asparagine deamidation, aspartate isomerization, aggregation motifs, and others. Such liabilities, if present or inadvertently introduced, can potentially create the need for new rounds of engineering, or even abolish the value of the antibody as a therapeutic molecule. Here, we demonstrate a sequence agnostic method to improve antibody affinities, while simultaneously eliminating sequence liabilities and retaining the same epitope binding as the parental antibody. This was carried out using a defined collection of natural CDRs as the diversity source, purged of sequence liabilities, and matched to the antibody germline gene family. These CDRs were inserted into the lead molecule in one or two sites at a time (LCDR1-2, LCDR3, HCDR1-2) while retaining the HCDR3 and framework regions unchanged. The final analysis of 92 clones revealed 81 unique variants, with each of 24 tested variants having the same epitope specificity as the parental molecule. Of these, the average affinity improved by over 100 times (to 96 pM), and the best affinity improvement was 231-fold (to 32 pM).
Abbreviations
CDR: complementarity determining region; FACS: fluorescence-activated cell sorting; ka: association rate; kd: dissociation rate; KD: dissociation constant; scFv: single-chain variable fragment; SPR: surface plasmon resonance
Introduction
The quality of a monoclonal antibody is often measured by its affinity to the target antigen. Hence, a common step in the development of a therapeutic antibody is submitting selected leads to affinity maturation campaigns on the assumption that higher affinities will lead to higher drug potency.
Campaigns follow three basic steps: 1) diversification, 2) selection, and 3) validation, and many successful methods have been used. Diversification has exploited error-prone PCR,Citation1,Citation2 chain shuffling,Citation3–5 targeted complementary-determining region (CDR) mutation,Citation6–11 and others,Citation12,Citation13 and will usually create some variants with higher affinities, and many with unchanged or lower affinities. By using display platforms, such as phage,Citation10,Citation11,Citation14,Citation15 yeast,Citation6,Citation12 or ribosome display,Citation16–18 and the application of selective pressure, the new variants with improved affinities can be separated from the rest. Finally, the population obtained can either undergo additional rounds of diversification and selection, or individual clones validated for affinity.
While these diversification techniques are efficient in creating molecules with improved affinities, they often create challenges for downstream development. For example, a biologically validated lead undergoing maturation must retain the same epitope: aggressive randomization strategies such as chain shuffling may lead to epitope driftCitation19 and instead of increased potency result in a loss of biological activity. Random mutations throughout the entire VH or VL domains will inevitably mutate regions unrelated to antigen binding and may generate unstable molecules with poor developability.Citation20 The same is true for the insertion of degenerate sequences within CDRs that can lead to unstable antibodies arising from unnatural sequences causing covariance violations which may affect the overall antibody structure.Citation21,Citation22 Finally, none of these techniques address the issue of sequence liabilities, such as glycosylation, asparagine deamidation, aspartate isomerization, and aggregation-prone motifs, which can negatively impact drug development. Sequence liabilities may be present in the parental molecule or introduced as a result of affinity maturation.
Here, we demonstrate the affinity improvement of an antibody lead to picomolar KD values while retaining identical epitope binding and reducing the number of sequence liabilities. This relies on a new diversification strategy exploiting defined collections of natural CDRs purged of sequence liabilities, which we have previously used to generate highly effective naïve antibody libraries yielding subnanomolar antibodies.Citation23,Citation24 In this approach, diversity is targeted based on the germline gene family of the antibody to be affinity matured, and not to the specific antibody sequence, i.e., two antibodies belonging to the same germline gene family will use the same source of CDR diversity, however different the individual CDR sequences of the parental molecules are.
To generate the CDR diversity, CDR sequences were identified by next-generation sequencing (NGS) of a naïve library,Citation25 oligonucleotides encoding individual CDRs were chemically synthesized without degenerations. CDRs containing sequence liabilities were eliminated, and the resultant CDR diversity was inserted into the lead molecule one or two sites at a time (LCDR1-2; LCDR3; HCDR1-2), as shown in . HCDR3 and the framework regions were kept constant throughout the process. After selecting improved affinity variants using yeast display, the resulting variable regions of each library were combined and further selected. Final analysis revealed >80 unique variants, with all 24 matured antibodies tested having picomolar affinities and competing with the parental for antigen binding.
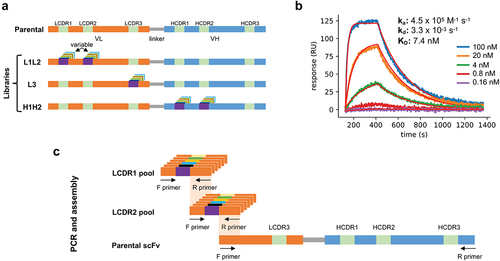
Figure 1. (a) Schematic representation of the parental antibody scFv and the three libraries produced in phase 1: L1L2, L3, and H1H2. Each library has diversity introduced in the indicated CDRs. (b) Surface-plasmon resonance sensorgram of the parental antibody binding to the antigen at increasing concentrations (0.16 nM to 100 nM with a 5-fold stepwise increase). (c) Schematic representation of the L1L2 library assembly.
Results
Phase 1: parental antibody, libraries construction, and selections
An antibody against B7-H4, a monomeric human protein of therapeutic interest, was chosen to be affinity matured using the proposed method. The antibody, comprising a VH3 heavy chain and a Vλ3 light chain, was initially identified by biopanning a naïve human single-chain variable fragment (scFv) phage display library built based on the same principles as Erasmus et al.Citation25 Analysis of the CDRs revealed three sequence liabilities with the potential to affect downstream clinical development: two distinct aspartate isomerization sites, which can lead to chemical degradation and loss of potency,Citation26 in LCDR2 and LCDR3, and a GG nonspecificity motifCitation27 in HCDR2. The scFv affinity (KD) of the parental antibody was determined to be 7.4 nM (ka = 4.5x105 M−1s;−Citation1 kd = 3.3x10−3 s−1) by surface plasmon resonance (SPR) ().
We used deep sequencing data (MiSeq and NovaSeq) from the same antibody library used for panning and its previous iterationCitation25 to identify CDRs that could be shuffled into the parental sequence for affinity maturation. For LCDR1-2 we considered only CDR sequences coming from the Vλ3 germline family and for HCDR1-2 we considered only sequences from the VH3 family, assuming these sequences, since they belonged to the same germline gene families as the parental antibody, would be better tolerated and minimize potential structural disruption. For LCDR3, however, we included all λ light chain LCDR3 sequences, regardless of the specific family or germline since the CDR3 region should be able to support a more diverse set of sequences. To minimize the chance of selecting antibodies with unfavorable developability profiles, sequences containing undesirable liabilities such as glycosylation sites and unpaired cysteines were eliminated (see for the full list of excluded liabilities). Finally, the identified CDRs were generated with flanking framework sequences matching the parental antibody and produced using array-based DNA synthesis. This allowed us to rescue the full diversity at each CDR site by using framework primers.
Table 1. List of sequence liabilities removed from CDRs designed for the affinity maturation libraries. Any CDR containing any of the listed motifs was excluded from the final oligonucleotide pool. Liabilities are not listed in any particular order of impact.
To decrease the initial combinatorial diversity and fully explore the potential at each site, we created three separate libraries: L1L2, H1H2, and L3 (). These allowed us to generate more transformants than the maximum combinatorial diversities for each library (theoretical diversity ranging from 1.7 × 105 to 3.2 × 107). Also, by fixing at least four parental CDRs, including the all-important HCDR3, in each library, search space was decreased, increasing the chances of finding new variants binding to the antigen in the same way, which is essential to retaining biological activity. In addition to the incorporated CDR diversity, each library also included the parental CDR sequences at the same abundance as the other introduced CDRs, even if they contained sequence liabilities. This was to ensure retained activity in the case that specific parental CDR sequences were essential for binding.
Table 2. The number of different sequences introduced in each CDR position of light and heavy chain. Theoretical diversity is calculated by the combinatorial potential of the CDRs and the reported number of transformants corresponds to the yeast display libraries created.
Libraries were assembled by combining the newly produced CDR pools with the remaining fixed regions derived from the scFv parental genes. For example, the L1L2 library was assembled by: 1) amplifying the LCDR1 and LCDR2 with the flanking frameworks from the synthetic oligo pool, 2) amplifying the remaining parts of the scFv from the parental clone, 3) assembling the produced fragments by overlap PCR, and 4) transforming the produced scFv cassettes into S. cerevisiae along with the digested yeast display vector ().
Two different selection strategies were used in this phase: equilibrium selection and kinetic selection ( and ). Citation12,Citation33 Equilibrium selection was performed by incubating the scFv-displaying yeast cells with a defined concentration of biotinylated antigen and sorting labeled cells immediately after reaching equilibrium. Incubations are often performed with decreasing antigen concentrations as the selection rounds progress. However, decreasing the antigen concentration cannot be carried out indefinitely because displayed antibodies on the yeast surface will deplete antigen from the solution before reaching equilibrium.Citation34 Avoiding antigen depletion requires the use of large and impractical incubation volumes with small numbers of cells. An alternative to equilibrium selection is kinetic selection: scFv-displaying yeast cells are incubated with the labeled antigen, washed, and incubated with unlabeled antigen to select only clones with stable binding to the antigen (slow off-rate – kd). The far greater excess of unlabeled antigen prevents rebinding of the displaced labeled antigen. After a defined period, cells still bound to the labeled antigen are sorted.
Figure 2. (a) Schematic representation of yeast display selections using equilibrium and kinetic protocols. (b) Outline of the selection rounds performed in phase 1 with the three-phase 1 libraries (L1L2, L3, and H1H2). (c) The yeast display binding profiles of the parental scFv and the three starting phase 1 libraries at increasing antigen concentration assessed by flow cytometry. Binding to antigen (APC fluorescence) is shown on the Y-axis and scFv display (PE fluorescence) is shown on the X-axis. (d) Yeast display binding profile of the parental scFv and the three-phase 1 libraries after 5 rounds of selection. A reference gate representing the parental population at the corresponding given concentration is shown. In the last two columns, after labeled antigen incubation, cells were washed and incubated with an excess of unlabeled antigen for 2 h and 4 h to evaluate overall improvement in off-rates.

We performed an initial flow cytometric assessment of the libraries using decreasing antigen concentrations (). The light chain libraries, L1L2 and L3, showed a small population binding to the antigen mostly at the highest concentration used (at 100 nM binding populations of 3% and 1.5%, respectively), while for the H1H2 heavy chain library significant binding could be observed from 1.2 nM (2.9% of the population) to 100 nM (8.3% of the population), suggesting higher improvement potential for the heavy chain CDRs as opposed to the light chains for this particular clone.
Given the size of each of these libraries, we performed first and second rounds of selection using magnetic-assisted cell sorting (MACS) at antigen concentrations of 10 nM and 1 nM, respectively. This allowed us to label and sort a larger number of cells than would have been practical using a flow cytometer. For subsequent rounds, we used fluorescence-activated cell sorting (FACS) to enable more precise sorting of the cells of interest. After the first three rounds of equilibrium sorting, we performed two rounds of kinetic sorting with 4 hours of competition for the L3 and H1H2 libraries. Only one 4 h kinetic sort was performed for the L1L2 library, followed by a negative sort, where the population was only incubated with secondary reagents and negative cells were sorted, which was done out of concern that enrichment of polyreactive antibodies may occur in light of the weak positivity observed in the absence of antigen
An assessment of the population obtained for each of these libraries after five rounds show that in all cases a significant improvement of affinity can be observed (): all generated populations show significant binding to the antigen even 4 hours after removal of labeled antigen, a time at which the parental antibody shows minimal residual binding. Even though we had not yet assessed affinities of individual clones, it is not unreasonable to assume, given the yeast display staining profile, that one could already find binders with satisfactory affinities at this stage, depending on the requirements of the project.
Phase 2: combo libraries construction and selections
After selecting CDRs at each position in the three initial libraries that mediated improved binding, we combined the selection outputs to obtain even longer off-rates. This was done by PCR amplifying the regions of interest from each library, i.e., LCDR1 and LCDR2 plus flanking frameworks from the L1L2 final round population, LCDR3 from the L3 and HCDR1 and HCDR2 from the H1H2 library
Two “combo” libraries were assembled by PCR: the first (Combo 1, comprising 2.27 × 108 transformants) was created by combining the output of all three libraries (L1L2, L3, and H1H2), while the second (Combo 2, with 1.04 × 108 transformants) omitted the L1L2 output and used the parental CDRs at LCDR1 and LCDR2 (). Equilibrium and kinetic sorts were again used on these libraries () with two successive rounds of kinetic sorting at 4 h and 16 h with unlabeled antigen, respectively, followed by a final equilibrium round at 0.1 nM antigen concentration.
Figure 3. (a) Schematic representation of parental antibody scFv and the two Combo libraries produced: Combo 1 has diversity in LCDR1-3 and HCDR1-2 and Combo 2 has diversity in LCDR3 and HCDR1-2. (b) Outline of the selection rounds performed in phase 2 with the two Combo libraries. (c) Yeast display binding profile of the combo libraries before any rounds of selection were performed. Binding to antigen (APC fluorescence) is shown on the Y-axis and scFv display (PE fluorescence) is shown on the X-axis. (d) Yeast display binding profile of the parental scFv and the combo libraries after 3 rounds of selection.
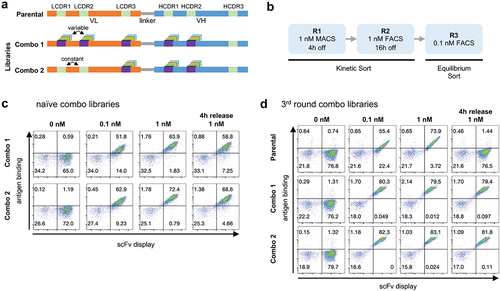
Immediately after transformation, the libraries showed binding to the antigen even at 0.1 nM – 51.8% and 62.9% of the population for Combo 1 and 2, respectively (). After three rounds of sorting, the difference between the combo libraries and the parental antibody is striking (): at 0.1 nM >80% of the yeast population bound to the antigen for both libraries as opposed to 55% for the parental antibody. When stained with 1 nM of antigen, and subsequently incubated with an excess of unlabeled Ag for 4 h, both combo libraries show very little signal loss compared to the staining at 1 nM of antigen with no competition period, whereas the parental completely lost binding under these conditions.
Sequence of the Affinity-Matured Clones
To evaluate the clones obtained we converted the populations obtained after the third round of selection of the Combo libraries to an scFv-Fc format to facilitate affinity screening. A total of 92 clones (45 from Combo 1 and 47 from Combo 2) were sequenced and 81 unique sequences were identified: 38 from Combo 1, 39 from Combo 2, and 4 in both libraries (, Supplementary Table 1). These unique sequences were often formed by different combinations of the same CDRs () found in LCDR1-3 and HCDR1-2 (, Supplementary Table 1). Interestingly, the heavy chain CDRs were more diverse than the light CDRs. This relates to the observed binding pattern of the first naïve libraries that showed a higher binding signal for the H1H2 library () suggesting that the heavy CDRs indeed were more tolerant to sequence changes.
Table 3. The number of unique CDRs found in the 92 sequenced clones (81 unique found).
Figure 4. (a) Venn diagram representing the number of unique clones identified by Sanger sequencing coming from Combo libraries 1 and 2. (b) Chord diagram showing the connections between each of the CDRs identified in the 81 unique affinity-matured clones. (c) Sequence logosCitation35 comparing the CDRs from the parental scFv, designed for the library, and observed in the sequenced clones. Letter heights indicate the frequency of the given amino acid and letter width represents the frequency of non-gap at the position. Dashed squares show regions that converged for an amino acid different from the parental. (d) Heat map showing the number of amino acid changes from parental in each CDR and the total number per clone. Each row represents an identified clone. Clones are ordered from least to most total changes. (e) Histograms represent the number of amino acid changes in each CDR identified in the starting library and the total number of changes per clone after final selection (Levenshtein distance).
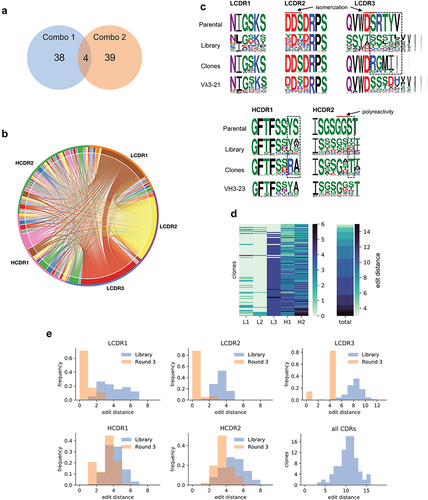
Sequence comparison of the CDRs present in the parental antibody, the maturation libraries, and the identified clones (, Supplementary Figure 1) shows that in many cases the amino acid in the parental antibody was already optimal since after several selection rounds the clones converge back to the parental sequence. Strikingly, LCDR1 and LCDR2 seemed to tolerate far fewer changes than the other CDRs, and for LCDR2, almost all clones reverted to the parental sequence, justifying the inclusion of the parental CDR sequences in the libraries. In LCDR3, despite introducing a diversity of 166,196 different CDRs from all λ germlines, we only identified 5 different sequences, all from the same family as the parental, with the most abundant present in 76 of the 92 clones sequenced. As for HCDR1 and HCDR2, much of the diversity found is concentrated in a few positions, suggesting that these may be less relevant for binding (e.g., position 6 at both HCDR1 at HCDR2), while some other positions showed clear convergence to amino acids different to the parental (, dashed boxes). Also of note, only two HCDR2 sequences did not have a substitution comprising a positively charged amino acid (K or R) at positions 6 or 7, suggesting a strong role for electrostatic interactions in this specific region.
The number of CDR modifications in the antibodies ranged from only 3 total to 15 amino acid changes (, Supplementary Figure 1). However, approximately half the clones came from the Combo 2 library in which LCDR1 and LCDR2 were kept constant. That said, only one LCDR1 had 4 modifications from the parental with all others having 2 or fewer (). For LCDR3, the dominant sequence had 4 modifications from parental, and for HCDR1 and HCDR2 the most frequently observed change was 3 modifications. It is important to notice that modifications were calculated using the Levenshtein distance which encompasses substitutions, insertions, and deletions, and the final number will be calculated as the minimum number of changes needed to transform one sequence into the other.
None of the affinity-matured antibodies had sequence liabilities in LCDR1, HCDR1, HCDR2, and HCDR3, which remained unchanged from the parental. The GG sequence liabilityCitation27 present in the parental HCDR2 was most often replaced by GS, GA, GT, and GD (present in 21, 19, 13, and 11 unique clones, respectively). For LCDR3, 67 of the 81 clones did not have any liabilities, 13 had the same sequence as the parental and therefore retained a DS aspartate isomerization site, and one had the same isomerization site plus a VV polyreactivity motif, probably arising from a synthesis or PCR error since this CDR was not included in the initial design. For LCDR2, only 1 of the 81 unique clones, G05, did not have the DDS aspartate isomerization motif, replaced by an EDV, while 72 had the exact sequence of the parental LCDR2 harboring the liability. Eight others were similar to the parental sequence and still harbored the same motif (again, likely coming from synthesis/PCR errors) (). In summary, 68 affinity-matured clones had a single CDR liability (Asp isomerization), 12 clones had 2, and 1 clone had 3, showing that the method can be effectively used to reduce developability issues simultaneously with affinity maturation without compromising affinity improvement – shows that the affinity of G05, which lacks the aspartate isomerization motifs (DD and DS in DDS) in LCDR1, is comparable to the best affinity-matured clones.
Table 4. LCDR2 sequences identified in the 81 affinity-matured clones. The first sequence corresponds to the parental clone and the last sequence corresponds to clone G05, not having the Asp isomerization motifs.
Figure 5. (a) Antigen binding to yeast displaying the parental scFv when competing with 24 affinity-matured clones (A01-A02, A04-A12, B01-B12, G05) and an unrelated scFv-Fc (control). Binding values are given by the median APC fluorescence of the yeast displaying population. There is no binding to the target by the parental clone whenever the antigen is pre-blocked with one of the selected clones, but binding is retained when the antigen is pre-incubated with a control clone. (b) Observed on-rates (ka) and off-rates (kd) for the parental and 24 affinity-matured clones (clone G05, which does not have the LCDR2 liabilities is highlighted in Orange). Reported values are the mean of at least 4 measurements. Isoaffinity curves (KD) are shown as dashed diagonal lines. (c) SPR sensorgrams for 24 affinity-matured clones. The name of the clones and calculated KD are shown in each plot.
Epitope, affinity, and polyreactivity screening
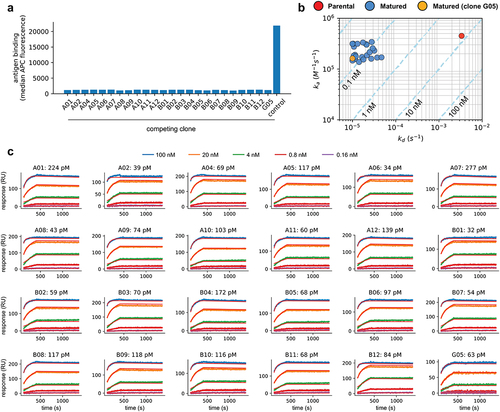
Of the identified clones, 24 were expressed as scFv-Fc in yeast: 12 from the Combo 1 library (A01-A02, A03-A06, B01-B06, G05) and 12 from the Combo 2 library (A07-A12, B07-B12). These had 3 to 13 amino acid changes from the parental. To determine whether they retained the same epitope as the initial lead, we tested if these were able to inhibit the parental scFv from binding to the antigen. We incubated the scFv supernatants with the labeled antigen (10 nM) for 15 minutes, added the yeast cells displaying the parental molecule to the mixture, and stained to detect binding. All 24 affinity-matured clones showed significantly decreased binding to the antigen by the parental, while a control scFv supernatant directed against an unrelated target did not (). Although the precise determination of the epitope recognized by all clones would require structural analysis, the fact that all these retain the same HCDR3 and framework regions of the parental molecule and compete for antigen binding suggests they indeed recognize the same epitope in the target molecule.
We determined the affinity of the 24 clones using high-throughput SPR. The supernatants were arrayed by non-covalent capture in a medium-density chip previously coupled with an anti-human polyclonal antibody (at least 4 replicates per clone). Non-biotinylated antigen was used for all affinity measurements and sensorgrams were fitted using a 1:1 Langmuir kinetic model.Citation36 The evaluated clones had affinities (KD) on average 103x better than the parental molecule (average 96 pM), dissociation rates (kd) 71–334 fold slower, and association rates (ka) within the same order of magnitude (, – capture, curve fitting, and statistics for all replicates shown on Supplementary Table 2). In fact, it is notable that the off-rates for some of the tested antibodies are probably better than 10–Citation5 ( and , ), as indicated by the bunching of matured antibodies at that value (, ) – such slow off-rates are challenging to accurately measure using SPR under the specific experimental settings used. No affinity difference was observed between clones coming from one combo library versus the other. The improvement of off-rates, but not on-rates, is a reflection of the protocols and selective pressure used during library panning: extended periods of antigen release favored stable binders with long off-rates, while no effort was made to rescue clones that had a faster association. Dissociation constants (KD) for the replicates showed good agreement (Supplementary Table 1).
Table 5. Kinetic measurements for affinity-matured clones and parental. Reported values are the mean for at least 4 replicates and standard errors are reported for the KD. Asterisks indicate clones that reached the minimum value allowed for the dissociation rate in our measurements (10−5 s−1).
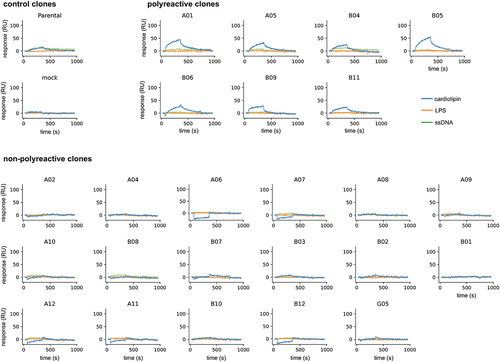
Finally, we also used SPR to assess the polyreactivity of the same clones. After the scFv-Fc fusions were captured, unrelated protein antigens were injected (interferon α, ubiquitin carboxyl-terminal hydrolase 11 [USP11], and IL17α-IL17ß heterodimer), and also polyreactivity detection reagents we routinely use in our laboratory (cardiolipin, lipopolysaccharide (LPS), and ssDNA). None of the clones reacted against the unrelated protein antigens (Supplementary Figure 2–5). Interestingly, the parental molecule showed a detectable signal against cardiolipin and ssDNA, suggesting that it had some degree of polyreactivity to start with (). Seven of the tested clones also showed discernible binding to cardiolipin and one of them (B04) reacted with ssDNA as well. The remaining 17 clones did not react with any of the tested reagents (), suggesting that the method can also be effective in removing this biophysical liability even though we have not screened for it beforehand or implemented steps during selection to mitigate it. No obvious pattern was observed among the polyreactive clones in terms of CDR usage.
Figure 6. Sensorgrams obtained by surface plasmon resonance for the polyreactivity test using cardiolipin (blue), ssDNA (green), and LPS (Orange). Clones were classified into three categories: controls – parental and mock supernatant, polyreactive – when discernible association and dissociation binding signal was observed against one or more analytes, and non-polyreactive – when no discernible association and dissociation signal was observed.
Discussion
Technology for introducing diversity into proteins has long existed and has been extensively used to evolve molecules with desired characteristics, including affinity maturation of antibodies in vitro.Citation5–8, 10−13,Citation15,Citation17,Citation18,Citation37–53 Our approach of mimicking natural CDR diversification to improve affinity shares similarities with previously published works,Citation6,Citation54 with the major difference that instead of using degenerations to simulate the frequency of naturally occurring amino acids at a given position, we rescued complete natural CDR sequences from the human repertoire and had them synthesized. This takes advantage of recent advances in synthetic biology that enable large-scale and low-cost production of oligonucleotide pools.Citation55,Citation56 In this way, only true natural CDRs are incorporated, rather than diversity that merely resembles natural CDRs. We have not tested other traditional techniques side-by-side using the same clone to directly compare the results, but one can infer that the proposed method has potential advantages: 1) It may avoid covariance violations,Citation21,Citation22 since the CDRs are more likely to fold correctly as they have been derived from natural antibodies, and are matched to the VH and VL families of the parental; 2) It allows the elimination of sequence liabilities; 3) A “generic” set of oligonucleotides can be used for each germline gene family, rather than designing and synthesizing an oligo set specific for the sequence of each antibody to be affinity matured; and 4) The method can be seamlessly combined with other ones: for example, during phase 1, one could also have created a saturation mutagenesis library of the HCDR3 and later combined its output during phase 2 to add another layer to the affinity maturation process. Of note, the libraries sequenced for our source diversity were derived from naïve CD19+ B-cells, with most V regions having fewer mutations when compared to fully mature IgG repertoire. Also, we used the IMGT CDR definition for all CDRs except LCDR2 (Kabat), but the method could easily accommodate different CDR schemes.
In this work, we were able to improve the affinity of our lead while eliminating sequence liabilities present in the CDRs. Most LCDR2s still had the original isomerization motifs (DD and DS), but one liability-free LCDR2 sequence (clone G05, and ) was identified that showed affinity similar to the other improved clones (KD = 63 pM). In theory, one could omit original parental CDRs containing sequence liabilities in the affinity maturation libraries, but at the risk of being unable to mature the clone if the eliminated motif is essential for binding. Removing liabilities from a therapeutic monoclonal antibody has a series of benefits: de-risking the lead upfront can improve developability and minimize chances of clinical failure due to poor molecule properties.Citation57 In addition, increased immunogenicity risk (e.g., glycosylation) and loss of potency (e.g., asparagine deamidation) are critical quality attributes that require tracking and risk management strategies to be put in place, increasing the resource burden on the development process and on the overall drug development life cycle. Carrying out separate engineering campaigns to independently improve affinity and developability is not only time and resource-consuming, but can also lead to an endless developmental loop since affinity improvements can lead to developability problems and vice-versa.Citation31,Citation58,Citation59 By using defined collections of natural CDR sequences lacking sequence liabilities, the motifs present in the final antibody can be easily modulated, and affinity maturation is carried out simultaneously with the elimination of sequence liabilities. It is important to note that the list of liabilities used here is not exhaustive and one has the flexibility to add or remove sequence motifs depending on the requirements of the project or the perceived importance of a given liability to the final product.
In addition to removing sequencing liabilities, we were also able to reduce/remove detectable polyreactivity from the parental clone against ssDNA and cardiolipin. At the onset of the project, we had not yet screened the parental lead for polyreactivity, therefore no deselection steps or other strategies were implemented to mitigate it during the affinity maturation. It is obvious now that one should always perform this type of test beforehand to appropriately guide the selection strategy. In this case, however, we were able to identify high-affinity variants that showed no binding to these polyspecificity reagents.
After the initial identification of leads binding to a target of interest, clones are often tested for their ability to achieve the desired biological activity. At this stage, the epitope recognized by the antibody will dictate if the antibody will be an agonist, antagonist, or have no activity whatsoever. After leads with desired activity are identified, it is generally assumed that increasing affinity will also increase potency,Citation60,Citation61 and retaining the recognition site is one of the basic requirements of this assumption. More aggressive maturation techniques such as chain shuffling (especially heavy chain) have a higher potential to cause epitope drift and loss of activity, making a previously valuable molecule useless. Here we have shown that a stepwise approach where at least 4 CDRs remain constant, and very importantly, the HCDR3 remains unchanged, was effective in obtaining very high affinities while retaining recognition of the same epitope. Whereas every antibody-antigen pair will have its own peculiarities, we believe the centrality of the HCDR3 in epitope recognitionCitation62,Citation63 justifies the retention of the parental HCDR3 to avoid epitope drift.
It is noteworthy that in the affinity maturation of this antibody we observed that HCDR1 and HCDR2 were much more amenable to substitutions than LCDR1 and LCDR2. This was reflected in the naïve L1L2 libraries showing significantly less binding than the H1H2 at the beginning of the process, as well as in the final number of different CDRs identified for each position at the end. Hypothesizing that the lack of significant improvement with the L1L2 library may reflect the presence of (close to) optimal sequences for these CDRs, we built a second combo library where parental sequences were used for LCDR1-2. The performances of both combo libraries were equivalent during sorting, and affinity measurements also showed similar results. In fact, 17 of the 45 unique clones identified from the first combo library had the unmutated parental LCDR1-2 as well, confirming the importance of these CDRs in binding. This suggests that for some antibodies, affinity maturation of all CDRs may not be necessary, or even possible. Given that the importance of individual CDRs in binding varies significantly between different antibodiesCitation62,Citation63 and, in the absence of structural information, it is difficult to predict this in advance, we believe creating combination libraries where all improved CDRs are shuffled is probably the safest approach. However, an approach in which phase 1 libraries showing limited improvement (e.g., L1L2 in this case) are omitted from the final combination library appears likely to be a valid approach, and allows for the construction of smaller combinatorial libraries, reducing overall effort. Given the flow cytometry results, we expect even the phase 1 libraries to include clones with significantly higher affinities than the parental. Thus, depending on the goal of the affinity maturation campaign, one can evaluate each position in phase 1 and decide which libraries should move to phase 2, or even stop there.
Library size is often a concern when performing in vitro evolution of any sort since one is limited by the number of transformants that can be conveniently obtained during library generation: 109–1010 for phage and E. coli display and 108–109 for yeast display in S. cerevisiae. We opted for a step-wise approachCitation64 since it would allow us to explore the sequence space more effectively: in phase 1 we combined LCDR1 with LCDR2, HCDR1 with HCDR2, and used LCDR3 by itself, generating libraries that were larger than the corresponding theoretical combinatorial diversity. We have no reason to believe this is the optimal approach and did so because it was the most convenient set of combinations for library construction. Alternatives such as splitting HCDR1 and HCDR2 or combining them with LCDR1 or LCDR2 may provide advantages, although, given the affinity improvements observed here, this is not immediately obvious. Alternatively, one could use a purely molecular technique such as ribosome display, which is not limited by transformation efficiencies but presents its own challenges.
We used scFv yeast display because of the high precision in retrieving the desired population when combined with flow cytometry. However, we expect this diversification approach to be equally effective in the phage or ribosome display context and could even expand possibilities due to larger library sizes. However, experimental design during selection would have to be adapted to these platforms since they are expected to show different behavior due to their monovalent nature as opposed to the multivalent nature of yeast: whereas a polyvalent yeast cell exists in a continuum from antigen saturation to no antigen-binding varying over time in proportion to the antibody off-rate, monovalent systems can only exist in the binary bound or non-bound states. This difference can be overcome, for example, by using a larger number of displaying particles and relying on the population binding decay over time as opposed to the single-cell decay. We routinely use the scFv format for antibody discovery and engineering and have found conversion to the IgG format occurs with 70–95% success, depending upon scaffold and target. However, if conversion is a concern, the techniques described here would be easily applied to a Fab display system. In addition, one could insert thermal stability selection rounds into the process to potentially improve this characteristic in the matured antibodies.Citation65
This work demonstrates the possibility of performing affinity maturation of a low-nanomolar affinity antibody to the low-picomolar range by replacing all CDRs, except HCDR3, with a collection of known, defined human CDRs. Replacing no more than two CDRs in phase 1 with compatible CDRs from similar, naturally occurring antibodies, along with retention of the HCDR3, is expected to maintain epitope binding, given the importance of HCDR3 in antibody binding.Citation63 Affinity gain came mostly from improved off-rate (kd), which has been suggested to better correlate with the biological activity than the affinity itself (KD).Citation60,Citation61 Off-rates for some of the antibodies were measured as <10,−Citation5 the measurement limit for SPR under our experimental settings. It is possible that off-rates are actually longer, and corresponding affinities higher, if measured using alternative methods, such as kinetic exclusion assays.Citation66 Although we have used a set of CDRs in this work that comes from an internal library, one could easily assemble a similar set of sequences from publicly available datasets, such as Observed Antibody Space.Citation67 The tolerance for mutations in each of the specific CDRs observed here is not meant to be generalized for different antibodies and it is expected that each antibody-antigen pair will have its own tolerance for mutations at distinct positions. Nonetheless, the concepts and overall experimental design we propose are expected to be equally useful for other antibodies as well.
Materials and methods
CDR design
Two internal naïve human natural phage display librariesCitation25 were sequenced using MiSeq and NovaSeq. Light chain and heavy chain sequences were annotated using IgBlastCitation68 and the IMGT schemeCitation69 was used for CDRs except for LCDR2 where Kabat was used.Citation70 For LCDR1-2 all IGLV3 family CDRs were considered (family-specific); for LCDR3 all IGLV CDRs (not family-specific); and for HCDR1-2, all IGHV3 CDRs were considered (family-specific). CDRs containing any liabilities () were discarded. The remaining CDRs were synthesized as an oligo pool (Agilent Technologies, USA). Flanking framework regions were added to each CDR sequence to enable amplification and assembly.
Phase 1 library construction
The five different collections of CDR sequences (LCDR1-3, HCDR1-2) were amplified by PCR from the oligo pool using specific primers and Q5 polymerase (NEB #M0491L). Primers were designed to match the flanking regions of the CDRs and to have a Tm of ~60°C. The PCR reactions were carried out following the manufacturer’s instructions. The remaining regions were amplified by PCR from the parental scFv. The amplified fragments were gel extracted using a standard column purification kit (Zymo Research Corporation, #D4001). The fragments were then assembled in a PCR-like reaction without primers and using Q5 polymerase and cycled 15 times. After the initial assembly, external primers were added to the reaction and the cycle was repeated 8 more times to amplify the assembled scFv. The assembled scFv was gel purified as before and reamplified in a large volume PCR (800 μl divided in 8 × 100 μl reactions), and then cleaned up by standard column purification (Zymo Research Corporation, #D4033. The purified scFv amplicons from each library were transformed into yeast along with 4 μg of the yeast display vector pSYD previously digested with the enzymes BssHII and NheI (NEB #R0199S and #R0131S) by electroporation using the method described previously.Citation71
Phase 2 library combo construction
For the Combo 1 library, the regions of interest were amplified from the round 5 populations of each library (L1L2, L3, H1H2) and assembled by PCR (same method described above). For the Combo 2 library, LCDR1 and LCDR2 were amplified from the parental scFv. Assembled scFv libraries were transformed into yeast as before.
Antigen
The B7-H4 purified monomeric recombinant human protein was ordered from ACROBiosystems (#B74-H5222) and handled according to manufacturer protocols. For use in the yeast display experiments, the protein was chemically biotinylated using EZ-Link NHS-LC-Biotin following the manufacturer’s instructions (Thermo Scientific).
Yeast display screening
Yeast display selections were performed as in Ferrara et al (2012).Citation72 Briefly, cells were induced in selective media containing 2% galactose overnight at 20°C. 105 induced cells were washed twice with cold washing buffer (PBS pH 7.4 0.5% BSA) and incubated at room temperature with the biotinylated antigen diluted in PBS. For the equilibrium sort protocol, after the biotinylated antigen incubation step the cells were washed and stained promptly with the anti-SV5 labeled with phycoerythrin (PE; labels cells displaying scFv) and streptavidin labeled with Alexa Fluor 633 (Thermo Scientific; labels cells bound to biotinylated antigen) and cells binding the antigen were sorted either by FACS (fluorescence-activated cell sorting) or MACS (magnetic-activated cell sorting). For the MACS procedure, paramagnetic beads coated with streptavidin were used (Miltenyi Biotec). For the kinetic sort protocol, after cells were incubated with the biotinylated antigen, they were washed and incubated with unlabeled antigen (10x more concentrated than the biotinylated antigen) to allow for discrimination of biotinylated antigen release according to the off-rate of the displayed scFv. The excess unlabeled antigen was added to prevent the rebinding of biotinylated antigen. After a defined period of time cells were stained and sorted as described before.
Screening and protein expression
The scFv from the final population (round 3, combo libraries) was bulk cloned into a yeast expression vector containing a human IgG1 Fc region to be expressed in the scFv-Fc format. For this, the scFv region was amplified by PCR, digested with BssHII and NheI restriction enzyme (New England Biolabs), and cloned into the pDNL9 vector. 92 clones were analyzed by Sanger sequencing and 24 of these plus the parental were expressed using S. cerevisiae strain YVH10 (ATCC MYA-4940). scFv-Fc fusions were expressed for 72 h at 20°C in the presence of galactose.
Affinity measurement
The Carterra LSA SPR system was used for the affinity measurements. Briefly, anti-Human IgG Fc (Southern Biotech, #2048-01) was covalently coupled to an HC30M chip following the manufacturer’s protocols. Crude yeast supernatants containing the scFv-Fc fusions were arrayed on the chip. Five antigen antigens injections were performed, each having a 5-fold increase in concentration (0.16 nM to 100 nM) to determine association and dissociation rates (5 minutes association and 15 minutes dissociation). Running buffer was 10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.05% Tween 20 with 0.5 mg/mL BSA. Binding data were double referenced by subtracting the responses from an interspot (local reference) surface and the responses from the leading blank injection and globally fit to a 1:1 Langmuir binding model for estimation of ka (association rate constant), kd (dissociation rate constant), and KD (affinity) using the Carterra Kinetics software. The Rmax parameter was kept constant for all analyte concentrations. Data analysis and kinetic parameters calculations were performed using Carterra software. For the affinity-matured clones, the leading baseline was cropped to enable fitting.
Qualitative polyreactivity assay
The Carterra LSA SPR system was used for the polyreactivity measurements. The clones were captured as described above and a series of unrelated analytes were injected: cardiolipin (50 μg/ml, Sigma-Aldrich #C0563-10 MG, sodium salt from bovine heart), ssDNA (4 μg/ml, Sigma-Aldrich #D8899-1 MG), LPS-EB (10 μg/ml, Invivogen #tlrl-eblps), USP11 (100 nM, supplied by the Structural Genomic Consortium [SGC-Toronto] as AviTag-His purified protein), IL17α-IL17ß heterodimer (100 nM, Sino Biologicals #CT047-HNAE), IFNα (100 nM, Genscript #Z03002), and also the target protein. Data analysis was performed using Carterra software. The samples were double-referenced using the reference ROI and the leading buffer injection, the baseline was aligned for all samples. Sensorgrams showing discernible association and dissociation phases (not due to bulk shift) were considered binding events.
Binding inhibition of the parental scFv by the affinity-matured clones
The scFv-Fc supernatants of the same 24 clones used for affinity analysis (plus an unrelated control) were incubated with the labeled antigen (10 nM) for 15 minutes. Approximately 2 × 105 yeast cells displaying the parental molecule were added to the mixture and incubated for 30 min at room temperature. Cells were washed twice and stained to detect binding using anti-SV5 labeled with PE (labels cells displaying scFv) and streptavidin labeled with Alexa Fluor 633. Populations were analyzed by flow cytometry for binding (Intellicyt iQue3 machine). The yeast population displaying the scFv (PE fluorescence) was gated and the median allophycocyanin (APC) fluorescence was used to quantify antigen binding.
Supplemental Material
Download Zip (1.4 MB)Disclosure statement
AART and CLL received financing from Specifica Inc.
SD, MFE, FF, LPS, LN, EM, TM, ADA, KP, and ARMB are employed by Specifica Inc.
RAB, and HGN are employed by Incyte Research Institute. SS was employed by Incyte Research Institute at the time this work was carried out.
SD, MFE, FF, LN, and ARMB are shareholders at Specifica Inc.
AART, SD, and ARMB are inventors on a patent application describing the technology
The antibodies and methods in this work are part of patent applications US 63/323,632 and US 63/218,919
RAB and HGN are shareholders at Incyte Research Institute
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2115200
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Gram H, Marconi LA, Barbas CFs3rd, Collet TA, Lerner RA, Kang AS. In vitro selection and affinity maturation of antibodies from a naive combinatorial immunoglobulin library. Proc Natl Acad Sci U S A. 1992;89(8):1–14. doi:10.1073/pnas.89.8.3576.
- Daugherty PS, Chen G, Iverson BL, Georgiou G. Quantitative analysis of the effect of the mutation frequency on the affinity maturation of single chain Fv antibodies. Proc Natl Acad Sci U S A. 2000;97:2029–34. doi:10.1073/pnas.030527597.
- Lou J, Geren I, Garcia-Rodriguez C, Forsyth CM, Wen W, Knopp K, Brown J, Smith T, Smith LA, Marks JD, et al. Affinity maturation of human botulinum neurotoxin antibodies by light chain shuffling via yeast mating. Protein Eng Des Sel. 2010;23:311–19. doi:10.1093/protein/gzq001.
- Park SG, Lee JS, Je EY, Kim IJ, Chung JH, Choi IH. Affinity maturation of natural antibody using a chain shuffling technique and the expression of recombinant antibodies in Escherichia coli. Biochem Biophys Res Commun. 2000;275:553–57. doi:10.1006/bbrc.2000.3336.
- Marks JD, Griffiths AD, Malmqvist M, Clackson TP, Bye JM, Winter G. By-passing immunization: building high affinity human antibodies by chain shuffling. Biotechnology (N Y). 1992;10:779–83. doi:10.1038/nbt0792-779.
- Tiller KE, Chowdhury R, Li T, Ludwig SD, Sen S, Maranas CD, Tessier PM. Facile affinity maturation of antibody variable domains using natural diversity mutagenesis. Front Immunol. 2017;8:986. doi:10.3389/fimmu.2017.00986.
- Barderas R, Desmet J, Timmerman P, Meloen R, Casal JI. Affinity maturation of antibodies assisted by in silico modeling. Proc Natl Acad Sci U S A. 2008;105:9029–34. doi:10.1073/pnas.0801221105.
- Rajpal A, Beyaz N, Haber L, Cappuccilli G, Yee H, Bhatt RR, et al. A general method for greatly improving the affinity of antibodies by using combinatorial libraries. Proc Natl Acad Sci U S A. 2005;102:8466–71. doi:10.1073/pnas.0503543102.
- Schier R, Bye J, Apell G, McCall A, Adams GP, Malmqvist M, Weiner LM, Marks JD. Isolation of high-affinity monomeric human anti-c-erbB-2 single chain Fv using affinity-driven selection. J Mol Biol. 1996;255:28–43. doi:10.1006/jmbi.1996.0004.
- Schier R, Marks JD. Efficient in vitro affinity maturation of phage antibodies using BIAcore guided selections. Hum Antibodies Hybridomas. 1996;7:97–105. doi:10.3233/HAB-1996-7302.
- Yang WP, Green K, Pinz-Sweeney S, Briones AT, Burton DR, Barbas CF. 3rd. CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into the picomolar range. J Mol Biol. 1995;254:392–403. doi:10.1006/jmbi.1995.0626.
- Boder ET, Midelfort KS, Wittrup KD. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc Natl Acad Sci USA. 2000;97:10701–05. doi:10.1073/pnas.170297297.
- Low NM, Holliger PH, Winter G. Mimicking somatic hypermutation: affinity maturation of antibodies displayed on bacteriophage using a bacterial mutator strain. J Mol Biol. 1996;260:359–68. doi:10.1006/jmbi.1996.0406.
- Lamdan H, Gavilondo JV, Munoz Y, Pupo A, Huerta V, Musacchio A, Pérez L, Ayala M, Rojas G, Balint RF, et al. Affinity maturation and fine functional mapping of an antibody fragment against a novel neutralizing epitope on human vascular endothelial growth factor. Mol Biosyst. 2013;9:2097–106. doi:10.1039/c3mb70136k.
- Schier R, McCall A, Adams GP, Marshall KW, Merritt H, Yim M, Crawford RS, Weiner LM, Marks C, Marks JD, et al. Isolation of picomolar affinity anti-c-erbB-2 single-chain Fv by molecular evolution of the complementarity determining regions in the center of the antibody binding site. J Mol Biol. 1996;263:551–67. doi:10.1006/jmbi.1996.0598.
- Chan DTY, Jenkinson L, Haynes SW, Austin M, Diamandakis A, Burschowsky D, Seewooruthun C, Addyman A, Fiedler S, Ryman S, et al. Extensive sequence and structural evolution of Arginase 2 inhibitory antibodies enabled by an unbiased approach to affinity maturation. Proc Natl Acad Sci U S A. 2020;117:16949–60. doi:10.1073/pnas.1919565117.
- Hanes J, Jermutus L, Weber-Bornhauser S, Bosshard HR, Pluckthun A. Ribosome display efficiently selects and evolves high-affinity antibodies in vitro from immune libraries. Proc Natl Acad Sci U S A. 1998;95:14130–35. doi:10.1073/pnas.95.24.14130.
- Hanes J, Schaffitzel C, Knappik A, Pluckthun A. Picomolar affinity antibodies from a fully synthetic naive library selected and evolved by ribosome display. Nat Biotechnol. 2000;18:1287–92. doi:10.1038/82407.
- Ohlin M, Owman H, Mach M, Borrebaeck CAK. Light chain shuffling of a high affinity antibody results in a drift in epitope recognition. Mol Immunol. 1996;33:47–56. doi:10.1016/0161-5890(95)00123-9.
- Julian MC, Lee CC, Tiller KE, Rabia LA, Day EK, Schick AJ, et al. 3rd Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng Des Sel. 2015;28:339–50. doi:10.1093/protein/gzv050.
- Chou K-C, Némethy G, Pottle M, Scheraga HA. Energy of stabilization of the right-handed βαβ crossover in proteins. J Mol Biol. 1989;205:241–49. doi:10.1016/0022-2836(89)90378-1.
- Du Q, Wei D, Chou K-C. Correlations of amino acids in proteins. Peptides. 2003;24:1863–69. doi:10.1016/j.peptides.2003.10.012.
- Teixeira AAR, Erasmus MF, D’Angelo S, Naranjo L, Ferrara F, Leal-Lopes C, Durrant O, Galmiche C, Morelli A, Scott-Tucker A, et al. Drug-like antibodies with high affinity, diversity and developability directly from next-generation antibody libraries. MAbs. 2021;13:1980942. doi:10.1080/19420862.2021.1980942.
- Ferrara F, Erasmus MF, D’Angelo S, Leal-Lopes C, Teixeira AA, Choudhary A, et al. A pandemic-enabled comparison of discovery platforms demonstrates a naïve antibody library can match the best immune-sourced antibodies. Nat Commun. pp.13. 2022. doi:10.1038/s41467-021-27594-w
- Erasmus MF, D’Angelo S, Ferrara F, Naranjo L, Teixeira AA, Buonpane R, et al. A single donor is sufficient to produce a highly functional in vitro antibody library. Commun Biol. pp.4. 2021. doi:10.1038/s42003-020-01536-6
- Sydow JF, Lipsmeier F, Larraillet V, Hilger M, Mautz B, Molhoj M, et al. Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. PLoS One. 2014;9:e100736. doi:10.1371/journal.pone.0100736.
- Kelly RL, Le D, Zhao J, Wittrup KD. Reduction of nonspecificity motifs in synthetic antibody libraries. J Mol Biol. 2018;430:119–30. doi:10.1016/j.jmb.2017.11.008.
- Kumar S, Singh SK. Developability of Biotherapeutics: computational approaches. Florida (US): CRC Press; 2016.
- Gavel Y, von Heijne G. Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng. 1990;3:433–42. doi:10.1093/protein/3.5.433.
- Bethea D, Wu SJ, Luo J, Hyun L, Lacy ER, Teplyakov A, Jacobs SA, O’Neil KT, Gilliland GL, Feng Y, et al. Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng Des Sel. 2012;25:531–37. doi:10.1093/protein/gzs047.
- Wu SJ, Luo J, O’Neil KT, Kang J, Lacy ER, Canziani G, Baker A, Huang M, Tang QM, Raju TS, et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng Des Sel. 2010;23:643–51. doi:10.1093/protein/gzq037.
- Vlasak J, Ionescu R. Fragmentation of monoclonal antibodies. MAbs. 2011;3:253–63. doi:10.4161/mabs.3.3.15608.
- Boder ET, Wittrup KD. Optimal screening of surface-displayed polypeptide libraries. Biotechnol Prog. 1998;14:55–62. doi:10.1021/bp970144q.
- VanAntwerp JJ, Wittrup KD. Fine affinity discrimination by yeast surface display and flow cytometry. Biotechnol Prog. 2000;16:31–37. doi:10.1021/bp990133s.
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–90. doi:10.1101/gr.849004.
- Lundström I. Real-time biospecific interaction analysis. Biosens Bioelectron. 1994;9:725–36. doi:10.1016/0956-5663(94)80071-5.
- Kang AS, Jones TM, Burton DR. Antibody redesign by chain shuffling from random combinatorial immunoglobulin libraries. Proc Natl Acad Sci U S A. 1991;88:11120–23. doi:10.1073/pnas.88.24.11120.
- Hawkins RE, Russell SJ, Winter G. Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J Mol Biol. 1992;226:889–96. doi:10.1016/0022-2836(92)90639-2.
- Cannon DA, Shan L, Du Q, Shirinian L, Rickert KW, Rosenthal KL, Korade M, van Vlerken-Ysla LE, Buchanan A, Vaughan TJ, et al. Experimentally guided computational antibody affinity maturation with de novo docking, modelling and rational design. PLoS Comput Biol. 2019;15:e1006980. doi:10.1371/journal.pcbi.1006980.
- Hu D, Hu S, Wan W, Xu M, Du R, Zhao W, Gao X, Liu J, Liu H, Hong J, et al. Effective optimization of antibody affinity by phage display integrated with high-throughput DNA synthesis and sequencing technologies. PLoS One. 2015;10:e0129125. doi:10.1371/journal.pone.0129125.
- Frigotto L, Smith M, Brankin C, Sedani A, Cooper S, Kanwar N, Evans D, Svobodova S, Baar C, Glanville J, et al. Codon-Precise, synthetic, antibody fragment libraries built using automated hexamer codon additions and validated through next generation sequencing. Antibodies. 2015;4:88–102. doi:10.3390/antib4020088.
- Thakkar S, Nanaware-Kharade N, Celikel R, Peterson EC, Varughese KI. Affinity improvement of a therapeutic antibody to methamphetamine and amphetamine through structure-based antibody engineering. Sci Rep. 2014;4:3673. doi:10.1038/srep03673.
- Votsmeier C, Plittersdorf H, Hesse O, Scheidig A, Strerath M, Gritzan U, Pellengahr K, Scholz P, Eicker A, Myszka D, et al. Femtomolar Fab binding affinities to a protein target by alternative CDR residue co-optimization strategies without phage or cell surface display. MAbs. 2012;4:341–48. doi:10.4161/mabs.19981.
- Gonzalez-Munoz A, Bokma E, O’Shea D, Minton K, Strain M, Vousden K, et al. Tailored amino acid diversity for the evolution of antibody affinity. MAbs. 2012;4:4. doi:10.4161/mabs.4.1.18821.
- Yang GH, Yoon SO, Jang MH, Hong HJ. Affinity maturation of an anti-hepatitis B virus PreS1 humanized antibody by phage display. J Microbiol. 2007;45:528–33.
- Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25:1171–76. doi:10.1038/nbt1336.
- Garcia-Rodriguez C, Levy R, Arndt JW, Forsyth CM, Razai A, Lou J, Geren I, Stevens RC, Marks JD. Molecular evolution of antibody cross-reactivity for two subtypes of type A botulinum neurotoxin. Nat Biotechnol. 2007;25:107–16. doi:10.1038/nbt1269.
- Lu D, Shen J, Vil MD, Zhang H, Jimenez X, Bohlen P, Witte L, Zhu Z. Tailoring in vitro selection for a picomolar affinity human antibody directed against vascular endothelial growth factor receptor 2 for enhanced neutralizing activity. J Biol Chem. 2003;278:43496–507. doi:10.1074/jbc.M307742200.
- Rader C, Popkov M, Neves JA, Barbas CF. 3rd. Integrin alpha(v)beta3 targeted therapy for Kaposi’s sarcoma with an in vitro evolved antibody. FASEB J. 2002;16:2000–02. doi:10.1096/fj.02-0281fje.
- Wu H, Beuerlein G, Nie Y, Smith H, Lee BA, Hensler M, Huse WD, Watkins JD. Stepwise in vitro affinity maturation of Vitaxin, an α v β 3 -specific humanized mAb. Proc Natl Acad Sci U S A. 1998;95:6037–42. doi:10.1073/pnas.95.11.6037.
- Hemminki A, Niemi S, Hoffren AM, Hakalahti L, Soderlund H, Takkinen K. Specificity improvement of a recombinant anti-testosterone Fab fragment by CDRIII mutagenesis and phage display selection. Protein Eng. 1998;11:311–19. doi:10.1093/protein/11.4.311.
- Thompson J, Pope T, Tung JS, Chan C, Hollis G, Mark G, et al. Affinity maturation of a high-affinity human monoclonal antibody against the third hypervariable loop of human immunodeficiency virus: use of phage display to improve affinity and broaden strain reactivity. J Mol Biol. 1996;256:77–88. doi:10.1006/jmbi.1996.0069.
- Jackson JR, Sathe G, Rosenberg M, Sweet R. In vitro antibody maturation. Improvement of a high affinity, neutralizing antibody against IL-1 beta. J Immunol. 1995;154:3310–19.
- Julian MC, Li L, Garde S, Wilen R, Tessier PM. Efficient affinity maturation of antibody variable domains requires co-selection of compensatory mutations to maintain thermodynamic stability. Sci Rep. 2017;7:45259. doi:10.1038/srep45259.
- Kosuri S, Church GM. Large-scale de novo DNA synthesis: technologies and applications. Nat Methods. 2014;11:499–507. doi:10.1038/nmeth.2918.
- Kosuri S, Eroshenko N, Leproust EM, Super M, Way J, Li JB, Church GM. Scalable gene synthesis by selective amplification of DNA pools from high-fidelity microchips. Nat Biotechnol. 2010;28:1295–99. doi:10.1038/nbt.1716.
- Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A. 2017;114:944–49. doi:10.1073/pnas.1616408114.
- Tiller KE, Li L, Kumar S, Julian MC, Garde S, Tessier PM. Arginine mutations in antibody complementarity-determining regions display context-dependent affinity/specificity trade-offs. J Biol Chem. 2017;292:16638–52. doi:10.1074/jbc.M117.783837.
- Pepinsky RB, Silvian L, Berkowitz SA, Farrington G, Lugovskoy A, Walus L, Eldredge J, Capili A, Mi S, Graff C, et al. Improving the solubility of anti-LINGO-1 monoclonal antibody Li33 by isotype switching and targeted mutagenesis. Protein Sci. 2010;19:954–66. doi:10.1002/pro.372.
- Rosenfeld R, Alcalay R, Mechaly A, Lapidoth G, Epstein E, Kronman C, J Fleishman S, Mazor O. Improved antibody-based ricin neutralization by affinity maturation is correlated with slower off-rate values. Protein Eng Des Sel. 2017;30:611–17. doi:10.1093/protein/gzx028.
- Hurlburt NK, Seydoux E, Wan YH, Edara SsVV, AB, Feng J, Feng J, Suthar MS, McGuire AT, Stamatatos L, Pancera M, et al. Structural basis for potent neutralization of SARS-CoV-2 and role of antibody affinity maturation. Nat Commun. 2020;11:5413. doi:10.1038/s41467-020-19231-9.
- Akbar R, Robert PA, Pavlovic M, Jeliazkov JR, Snapkov I, Slabodkin A, Weber CR, Scheffer L, Miho E, Haff IH, et al. A compact vocabulary of paratope-epitope interactions enables predictability of antibody-antigen binding. Cell Rep. 2021;34:108856. doi:10.1016/j.celrep.2021.108856.
- Xu JL, Davis MM. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity. 2000;13:37–45. doi:10.1016/S1074-7613(00)00006-6.
- Hemminki A, Niemi S, Hautoniemi L, Söderlund H, Takkinen K. Fine tuning of an anti-testosterone antibody binding site by stepwise optimisation of the CDRs. Immunotechnology. 1998;4:59–69. doi:10.1016/S1380-2933(98)00002-5.
- Traxlmayr MW, Shusta EV. Directed evolution of protein thermal stability using yeast surface display. Methods Mol Biol. 2017;1575:45–65.
- Darling RJ, Brault PA. Kinetic exclusion assay technology: characterization of molecular interactions. Assay Drug Dev Technol. 2004;2:647–57. doi:10.1089/adt.2004.2.647.
- Kovaltsuk A, Leem J, Kelm S, Snowden J, Deane CM, Krawczyk K. Observed antibody space: a resource for data mining next-generation sequencing of antibody repertoires. J Immunol. 2018;201:2502–09. doi:10.4049/jimmunol.1800708.
- Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41:W34–40. doi:10.1093/nar/gkt382.
- Giudicelli V, Brochet X, Lefranc MP. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb Protoc. 2011;2011:695–715. doi:10.1101/pdb.prot5633.
- Kabat EA, Wu TT, Bilofsky H. Evidence supporting somatic assembly of the DNA segments (minigenes), coding for the framework, and complementarity-determining segments of immunoglobulin variable regions. J Exp Med. 1979;149:1299–313. doi:10.1084/jem.149.6.1299.
- Benatuil L, Perez JM, Belk J, Hsieh CM. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng Des Sel. 2010;23:155–59. doi:10.1093/protein/gzq002.
- Ferrara F, Naranjo LA, Kumar S, Gaiotto T, Mukundan H, Swanson B, et al. Using phage and yeast display to select hundreds of monoclonal antibodies: application to antigen 85, a tuberculosis biomarker. PLoS One. 2012;7:e49535. doi:10.1371/journal.pone.0049535.