
ABSTRACT
T cell-engaging bispecific antibodies (TCEs) are clinically effective treatments for hematological cancers. While the utility of TCEs in solid malignancies is being explored, toxicities arising from antigen expression on normal tissues have slowed or halted several clinical trials. Here, we describe the development of TCEs that preferentially drive T cell-mediated death against target cells co-expressing two tumor-associated antigens. We show that Ly6E and B7-H4 are simultaneously expressed on approximately 50% of breast cancers, whereas normal tissue expression is limited and mostly orthogonal. Traditional bispecific TCEs targeting a singular antigen, either Ly6E or B7-H4, are active when paired with high-affinity CD3-engagers, but normal tissue expression presents a toxicity risk. Treatment with a murine cross-reactive B7-H4-TCE results in rapid and severe weight loss in mice along with damage to B7-H4-expressing tissues. To overcome on-target toxicity, we designed trispecific antibodies co-targeting Ly6E, B7-H4, and CD3 and characterized the impact of dual-antigen binding and the relative placement of each binding domain on tumor killing in vitro and in vivo. In vitro killing of tumor cells co-expressing both antigens correlates to the placement of the higher affinity B7-H4 binding domain, with only modest enhancements seen upon addition of Ly6E binding. In xenograft models, avid binding of appropriately designed trispecific TCEs enables tumor growth inhibition while evading the poor tolerability seen with active bispecific TCEs. Collectively these data highlight the potential for dual-antigen targeting to improve safety and efficacy, and expand the scope of tumors that may effectively be treated by TCEs.
Abbreviations: Chimeric antigen receptor T cells (CAR-Ts), dual-antigen targeted T cell engagers (DAT-TCE), Fragment antigen-binding (Fab), Hematoxylin and eosin (H&E), Institutional Animal Care and Use Committee (IACUC), Immunoglobulin G (IgG), immunohistochemistry (IHC), NOD SCID gamma (NSG), peripheral blood mononuclear cells (PBMCs), surface plasmon resonance (SPR), T cell-engagers (TCEs)
Introduction
The clinical success of blinatumomab sparked a revolution in T-cell redirecting therapies, resulting in dozens of CD3-bispecific antibodies in clinical trials.Citation1 Multiple therapeutics targeting B-cell or myeloid cell antigens, including CD19, CD20, CD33, CD123 and BCMA, have advanced rapidly, with several now in late-stage clinical trials.Citation2 While depletion of immune cell sub-types can be tolerated, most solid tissues do not rapidly regenerate, and solid tumor-targeted T-cell engagers (TCEs), as well as chimeric antigen receptor T-cells (CAR-Ts), cannot inherently distinguish healthy tissues from tumors. Few antigens exist with specific expression on solid tumors and little to no expression elsewhere, and several trials exploring more broadly expressed antigens have been paused or halted due to toxicity and patient deaths.Citation3–5 Based on these limitations multiple groups have explored approaches to expand the scope of tumors addressable with TCEs. Recent reports have described trivalent, bispecific antibodies that exploit avidity as a means to preferentially target tumor cells with elevated expression of tumor-associated antigen (e.g., HER2, CEA, ENPP3).Citation6–8 This approach substantially improves safety margins preclinically, but is limited to antigens with large expression differentials (typically greater than 10-fold) between target cells and cellular models of ‘normal’ tissue. Additional approaches, including antibody masking,Citation9 conditional (pH-dependent) binding,Citation10 split anti-CD3s,Citation11 and dual targeting of CD3 (P07766) and CD28 agonists,Citation12 have been explored as ways to increase the selectivity of T-cell bispecifics, but to date they have not been clinically validated. In light of the substantial unmet need for effective solid tumor treatments, we investigated whether requiring simultaneous binding to two tumor-associated antigens could be used to drive selective tumor killing by TCEs.
Use of a dual-antigen strategy requires: 1) co-expression of two antigens in proximity to each other on tumor cells to drive antibody binding and subsequent cell killing, and 2) limited and non-overlapping expression of the antigens in normal tissues to avoid off-tumor toxicity. While this approach exploits avidity in a manner analogous to trivalent bispecific antibodies, it potentially enables application of avidity-driven selectivity to antigens present at comparable levels in normal and cancerous tissues. These opportunities come at the expense of additional complexity both in the form of multi-dimensional optimization of the trispecific antibodies and biological complexity due to varying antigen densities across tissues.
Here, we describe the identification of Ly6E (Q16553) and B7-H4 (Q7Z7D3) as a pair of antigens that are suitable for this strategy, with substantial co-expression on human breast tumors and orthogonal normal tissue expression. Ly6E and B7-H4 antigens are both highly expressed in tumors with restricted normal tissue expression.Citation13,Citation14 Little is known about their native biological functions. Both Ly6E and B7-H4 have been pursued as antibody-drug conjugate targets based on this preferential tumor expression, but the potency of T cell-based therapies enhances the risk of toxicity for tissues with low levels of antigen expression. CAR-Ts targeting B7-H4 have shown strong preclinical toxicity.Citation15 In an attempt to further expand the number of tumors potentially addressable with TCEs, we explored whether trispecific antibodies targeting both B7-H4 and Ly6E could drive cell killing while sparing tissues that express a singular antigen. We describe the rationale for selection of B7-H4 and Ly6E, validation that incorporation of Fabs against each target result in functional TCEs, development of a murine model reflecting on-target, off-tumor toxicity of the B7-H4 TCE, and TCE optimization resulting in a dual-antigen targeted TCE (DAT-TCE) that drives tumor growth inhibition without corresponding B7-H4-targeted toxicity.
Results
Ly6E and B7-H4 are preferentially co-expressed in tumors
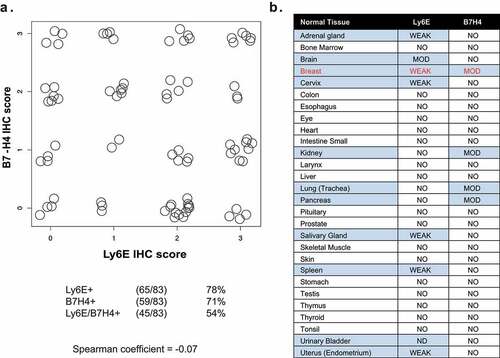
To identify a pair of antigens with properties suitable for avidity-driven tumor selectivity, we re-analyzed historical immunohistochemical (IHC) data characterizing antigen expression in tumors and normal tissues. Comparison of 83 breast cancer biopsies previously evaluated for both Ly6ECitation14 and B7-H4Citation13 found 65/83 (78%) expressed Ly6E while 59/83 (71%) expressed B7-H4 (). Co-expression of the two antigens was observed in 45/83 biopsies (54%), with 19/83 biopsies (23%) showing at least 2+ staining for both antigens. In parallel, we examined published Ly6E and B7-H4 IHC staining of normal human tissue microarrays to determine expression patterns and identify potential tissue specific toxicity risks associated with avid binding of Ly6E and B7-H4 ().Citation13,Citation14 Assessment of 28 duplicate normal tissue cores revealed weak to moderate expression of either Ly6E or B7-H4 across a small number of tissues, while co-expression was only observed in normal breast tissue. Breast tissue is often surgically resected prior to systemic therapy for metastatic breast cancer. As a consequence, we hypothesized that therapeutics requiring simultaneous expression of Ly6E and B7-H4 for activity would show limited toxicity in normal tissues.
Figure 1. Immunohistochemical analysis of Ly6E and B7-H4 in breast tumor and normal tissue biopsies. A. Correlation of IHC staining for Ly6E and B7-H4 across 83 breast cancer biopsies evaluated for both antigens. B. Correlation of IHC staining for Ly6E and B7-H4 across tissue microarrays of normal human tissue.
Ly6E and B7-H4 TCEs are active in vitro and in vivo
We initiated development of dual-targeting trispecific TCEs by characterizing the activities of the parent bispecifics: anti-Ly6E/CD3 and anti-B7-H4/CD3. We used two antibodies previously determined to have activity as bivalent antibody-drug conjugates, anti-Ly6E 9B12Citation14 and anti-B7-H4 1D11.Citation13 Bivalent 1D11 binds the N-terminal IgV domain of human and murine B7-H4 with nearly equivalent affinity (2.9 vs. 5 nM at 25°C).Citation13 In monovalent formats, 1D11 shows ~10x preferential binding for murine B7-H4 (vide infra). 9B12 binds avidly to human Ly6E expressing cells with an affinity of 3.7 nM, but shows very limited monovalent binding (KD>200 nM) and is not cross-reactive to murine Ly6E.Citation14,Citation16
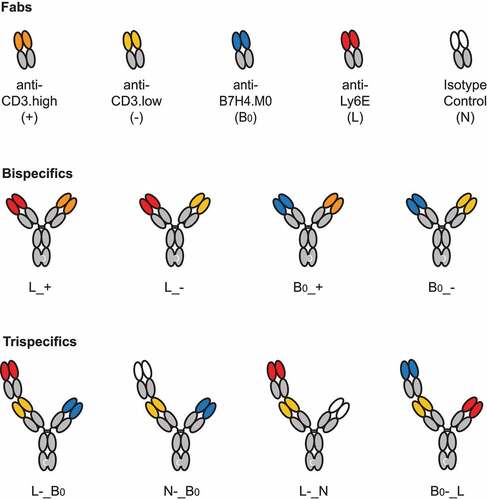
For the anti-CD3 component, we evaluated two closely related antibodies that target the N-terminus of CD3ε, anti-CD3.high and anti-CD3.low, previously reported to have affinities of 0.5 and 50 nM, respectively.Citation17,Citation18 Higher affinity binding to CD3 has been shown to drive TCE activity against low abundance antigens.Citation19 Previous TCEs exploiting avidity to selectively kill cells expressing high levels of a single antigen used a low affinity anti-CD3 arm.Citation6 For simplicity, TCEs containing the anti-CD3.high or anti-CD3.low affinity antibody arm will be denoted by a (+) or (-), respectively ().
Figure 2. A brief guide to key molecules. Visual descriptions and molecule codes are provided for TCEs used in key in vivo experiments.
Tumor antigen- and CD3-targeted half-antibodies were expressed using the knobs-into-holes technology,Citation20,Citation21 assembled into bispecific TCEs (), and evaluated for antigen-dependent killing (). In vitro killing was assessed by co-culture of purified CD8 + T-cells with HCC1569, a human breast tumor cell line that expresses high levels of Ly6E and B7-H4 at both the RNA and cell-surface protein levels (S1 Fig). Consistent with the low affinity of the anti-Ly6E antibody, anti-Ly6E/CD3 TCEs with either the high (L_+) or low (L_-) affinity anti-CD3 arm resulted in minimal in vitro cell killing whereas the anti-B7-H4/CD3 bispecific antibodies (B0_+ and B0_-) resulted in substantial killing that correlated with affinity for CD3 (). An isotype control antibody (N) paired with both the high (N_+) and low (N_-) affinity anti-CD3 arm had no activity, confirming that tumor antigen engagement is necessary for TCE function. In alternative cell lines with high Ly6E expression, partial activity of L_+ could sometimes be detected, while L_- remained inert (S2 Fig), suggesting that a high affinity anti-CD3 arm could partially compensate for low affinity Ly6E binding.
Figure 3. Activities of Ly6E and B7-H4 TCEs. A. Format of knob-into-hole bispecific and Fab-IgG trivalent bispecific antibodies. B. HCC1569 cells were cocultured with purified human CD8+ T-cells at an E:T ratio of 4:1 in the presence of indicated antibodies for three days. Following incubation, cell number was assessed using Cell Titer Glo. Points represent the average of n = 2. C-D. NSG mice bearing HCC1569x2 xenografts and engrafted with human PBMCs were treated once as indicated and body weight (top) and tumor growth (bottom) were monitored over time. Graph legends: thin lines: responses of individual mice; thick black lines: group average; dashed blue line: average response of mice treated with PBMCs alone; red lines: mice that reached the humane endpoints before the study end. Note, in D.i., both B0_+ and vehicle arms were halted prematurely due to the poor tolerability of the B0_+.
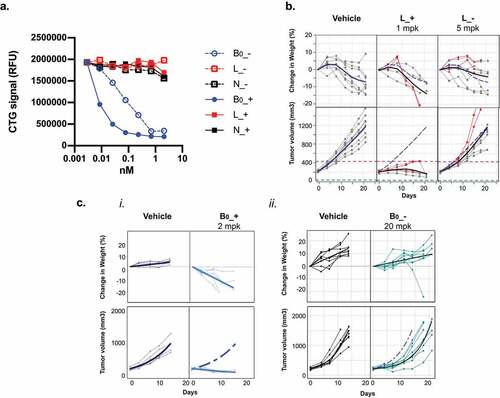
We also evaluated the in vivo activity of the TCEs against HCC1569x2, a cell line adapted from HCC1569 for in vivo growth, in NOD SCID gamma (NSG) mice engrafted with human peripheral blood mononuclear cells (PBMCs). HCC1569x2 cells were previously reported to have substantial expression of both Ly6E and B7-H4 by flow cytometry and maintain IHC 3+ staining in xenografts;Citation13,Citation16 however, extremely poor in vitro growth makes them suboptimal for cell-based studies. Interestingly, while both L_+ and L_- were inactive in vitro, L_+ was able to substantially inhibit HCC1569x2 growth in vivo at 1 mg/kg (). When paired with the low-affinity anti-CD3 binder, anti-Ly6E remained ineffectual at doses up to 20 mg/kg (S3 Fig). Late-onset weight loss was often observed for mice treated with PBMCs alone or combined with L_+. Given the lack of murine cross-reactivity of anti-Ly6E, and similarity these responses, we attributed the poor tolerability to graft-versus-host disease commonly observed in human PBMC-engrafted mice and not to the therapeutic ().Citation22 The somewhat variable toxicity and tumor growth inhibition observed in negative control mice treated with PBMCs between experiments (vide infra) were attributed to the variable activities of the PBMC donors, but the activities of the bispecifics compared to the control remained well-conserved.
Treatment with murine cross-reactive anti-B7-H4 paired with the low-affinity CD3 arm (B0_-) up 20 mg/kg was well tolerated, but failed to substantially inhibit tumor growth (). Conversely, anti-B7-H4 paired with a high affinity CD3 (B0_+) resulted in tumor regression, but also substantial weight loss, jaundice, and lethargy distinct from PBMCs alone (). These findings suggest treatment with murine cross-reactive B0_+ drives on-target, off-tumor toxicities.
B7-H4-targeted TCEs drive pathology in B7-H4-expressing tissues
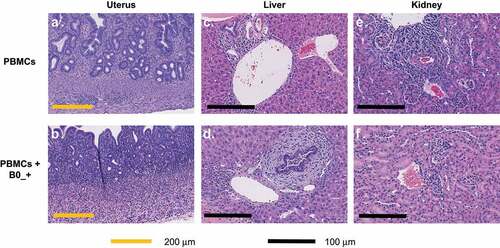
Given the poor tolerability of B7-H4-targeted TCEs, we chose to isolate tissues previously reported to express B7-H4 in mice to investigate antigen-specific toxicity.Citation13,Citation15 Hematoxylin and eosin (H&E) staining of liver, female reproductive tracts, pancreas and kidney from mice treated with 2 mg/kg of B0_+ for 14 d revealed varying degrees of chronic inflammatory cell infiltration composed of lymphocytes, plasma cells and histiocytes (). The staining is perivascular and associated with structures reported to express B7-H4 (e.g., bile ducts, islets endometrium, fallopian tubes). Perivenous infiltrate, necrosis, and obliterative/sclerosing cholangitis were uniformly observed in TCE-treated mice, but rarely or never observed in mice treated with vehicle control. Minimal inflammatory changes were observed in the kidney, with rare samples showing predominantly mild to moderate perivascular and tubular-associated chronic inflammation. Lower grade or no lesions were found in mice treated with PBMCs alone. Collectively, these data suggest that PBMC treatment alone may induce a graft-versus-host-like response evidenced by low grade immune lesions in endometrium, liver, and kidney, but that the observed inflammation is exacerbated by B7-H4-TCE treatment represented by more severe lesions in B7-H4+ tissues from treated mice. Given the low to moderate expression of B7-H4 in normal tissues, the requirement for high affinity anti-CD3 to drive robust anti-tumor activity, and the poor tolerability of the murine cross-reactive anti-B7-H4 TCEs, we concluded B7-H4 TCEs were unlikely to be well tolerated at clinically active doses. While not directly tested due to the lack of murine cross-reactive antibodies, pharmacologically active Ly6E-TCEs were also considered high risk due to low-to-moderate normal tissue expression and the requirement for high potency CD3 binders. Thus, we explored whether dual-antigen targeting might enable more tumor-specific cell killing.
Figure 4. Histology of B7-H4 TCE treated mice. Representative areas of uterus show mild chronic inflammation of endometrium and myometrium in PBMC-treated mice (A, top left), but more pronounced, full-thickness chronic inflammation in the uteri of PBMC with B0/+-treated mice (B, bottom left). Similarly, livers of mice receiving PBMC only show only mild, focal perivascular chronic inflammatory infiltrates (C, top middle) while livers from treated mice show more pronounced portal inflammation with evidence of sclerosing cholangitis (D, bottom middle). Kidneys of both PBMC and treated mice (E, F, top right, bottom right respectively) show only small, rare, areas of mild chronic inflammation.
Generation and characterization of trispecific antibodies
We next investigated whether trispecific antibodies targeting Ly6E, B7-H4, and CD3 could maintain activity on tumors co-expressing both antigens, while reducing the toxicity driven by monovalent (B7-H4-targeted) binding to mouse tissue. Trispecific antibodies were assembled from Fab-IgG and IgG half-Abs (). On the Fab-IgG half-Ab, the N-terminal Fab (Position 1) was linked to the second Fab (Position 2) using the upper hinge as a linker (Fab1-DKTHT-Fab2-Fc)Citation6 and selectively paired with the IgG half-Ab (Position 3) using the knobs-into-holes mutations (). For ease of communication, the trispecifics are abbreviated analogously to our bispecifics with the “_” used to indicate separate half-antibodies. For example, anti-Ly6E(position 1)-CD3.high(position 2)_B7-H4(position 3) is abbreviated to L+_B ().
Figure 5. Cell killing of breast cancer lines using trispecific anti-Ly6E/B7-H4/CD3 antibodies. A. Format of trispecific antibodies. B-D. Breast cancer cell lines were cocultured with purified human T-cells at an E:T ratio of 4:1 in the presence of the indicated trispecific antibodies for three days. Following incubation, cell number was assessed using Cell Titer Glo. RNA levels for each cell line as determined by the cancer genome atlas (TCGA) and reported as normalized reads per kilobase per million total reads (nRPKM) are indicated underneath the respective names.
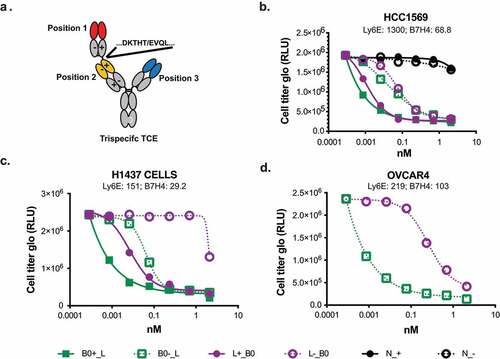
We chose to prioritize constructs with the low-affinity anti-CD3 Fab in Position 2. Lower affinity binders have been previously shown to drive selective avidity-based killing with TCEs, and placement of anti-CD3 binders in this format has validated activity.Citation6,Citation7 This arrangement results in the long distance (position 1-position 3) interaction taking place on the tumor cell surface, while the intercellular tumor-to-T-cell distances (positions 1–2 and 2–3) are minimized. The tighter synapse is thought to improve T cell-mediated killing.Citation23 Surface plasmon resonance (SPR) analysis confirmed anti-B7-H4 affinity was not affected by placement at either of the external positions (S4 Fig), and fluorescence-activated cell sorting analysis confirmed Ly6E drove equivalent levels of avid binding regardless of position (S5 Fig). Cell killing of the trispecific antibodies was assessed in coculture assays with CD8 + T-cells and HCC1569 cells expressing both Ly6E and B7-H4 (). All trispecific antibodies shown were able to induce T cell-mediated death. Interestingly, the relative placement of the anti-Ly6E and anti-B7-H4 appeared to modestly affect the activity, with placement of the higher affinity anti-B7-H4 Fab at position 1 leading to enhanced killing. In additional cell lines with more balanced expression of Ly6E and B7-H4, the impact of the specific Fab placement showed a substantially enhanced effect (, and d).
In vivo analysis of trispecific antibodies
With trispecific molecules in hand, we investigated whether avid binding could drive tumor killing, without recapitulating the observed toxicities. Notably, anti-B7-H4 has higher affinity for murine vs. human antigen (0.7 vs. 13 nM), thus providing a high bar for selective killing. Based on the observation that the relative placement of the tumor antigen-binding arms could influence T-cell engager activity in vitro (), we compared in vivo activities of both B0-_L and L-_B0 (). In contrast to the limited activity seen with bispecific antibodies containing a low affinity CD3 (), both trispecific antibodies were able to suppress tumor growth. Interestingly, B0-_L, which showed more activity in vitro (), drove rapid and profound weight loss and jaundice, while L-_B0 was tolerated similarly to PBMCs alone.
Figure 6. In vivo activities of trispecific antibodies. A-B. The HCC1569x2 model was implanted into NSG mice engrafted with human PBMCs. Once tumors reached ~200 mm3, tumor growth (top) and body weight (bottom.) were assessed following a single administration of the indicated treatments. Graph legends: thin lines: responses of individual mice; thick black lines: group average; dashed blue line: average response of mice treated with PBMCs alone.
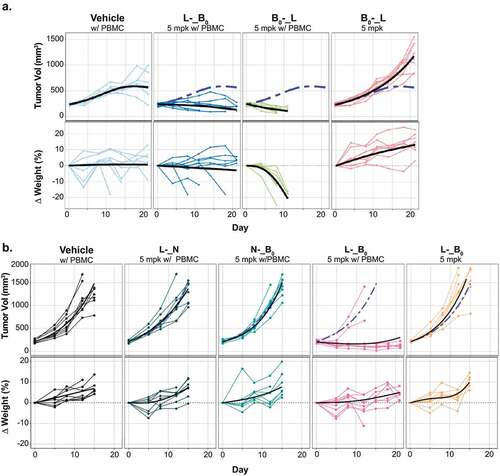
These data provided support for the concept that avid binding could enhance activity in vivo. To model the impact of trispecific binding to tissues expressing only one of the two targeted antigens we generated format-matched trispecific antibodies in which one of the tumor-targeted Fabs was replaced with an isotype control (N). Consistent with the experiment in , treatment with 5 mg/kg of L-_B0 resulted in near complete tumor growth inhibition in 6/8 mice (), without evident weight loss or jaundice. By contrast, both antibodies targeting just a single tumor antigen (N-_B0, and L-_N) failed to affect tumor growth. To concretely distinguish between the effects of simultaneous binding from additive impacts of two antigens, we also compared the activity of the trispecific antibody to the combination of N-_B0 and L-_N (S6 Fig). Combining trispecific antibodies that each bind only Ly6E or B7-H4 failed to inhibit tumor growth, confirming the essential role of avidity in driving the anti-tumor activities of the TCEs. Collectively these data suggest that dual-antigen targeting can enhance therapeutic efficacy while minimizing substantial on-target, off-tumor toxicity.
Discussion
Avidity as a means to enhance binding has been appreciated for more than a century. For antibodies, the high avidity of IgMs is thought to allow widespread surveillance for antigens that have not previously been encountered,Citation24 while bivalent engagement with affinity matured IgGs enables selective but long-lived interactions with their cell-surface targets. Recently, several groups have exploited avidity by combining high affinity ‘targeting’ Fabs with low affinity ‘active’ antibody or ligand.Citation25–28 These approaches allow selective activation of cytokine receptors or checkpoint inhibitors both in vitro and in vivo, but the applicability of this approach is limited to therapeutic strategies where the target antigen expression is highly specific (typically immune cells) or where partial activity of the active moiety in normal tissues is tolerated. Several reports by Mazor and colleagues explored dual-antigen selectivity in the context of EGFR/HER2Citation29 and CD4/CD70Citation30 bispecific antibodies. Avidity-driven EGFR/HER2 inhibition reduces cell growth in vitro and in xenograft models co-expressing both antigens while not impacting the growth of cells expressing only EGFR. With CD4/CD70, Mazor et al. exploited hinge binding to FcγRs to drive selective antibody-dependent cell-mediated killing of T cells co-expressing the antigens in vitro, but extension of this work to in vivo systems was not reported.
Here we describe a process for the selection of optimal target antigens and optimization of trispecific antibodies for targeted T cell-dependent killing of cells co-expressing two tumor-associated antigens. We show that Ly6E and B7-H4 are simultaneously expressed in ~50% of human breast cancer biopsies, including high levels of co-expression (IHC 3+ for both) in 6/83 (7%) of tumors. For comparison, HER2, also explored as an avidity driven target, is expressed on ~15% of breast tumors.Citation6,Citation31 Both Ly6E and B7-H4-targeted TCEs can inhibit xenograft growth with appropriate CD3-binding arms, but, consistent with previous studies with CAR-Ts,Citation15 murine cross-reactive anti-B7-H4 antibodies drive tissue damage in B7-H4-expressing tissues (). The murine cross reactivity of our more active anti-B7-H4 binder enabled simultaneous evaluation of toxicity and efficacy, facilitating selection of advantaged therapeutics. It is worth noting that TCEs drive toxicity both through direct T cell-mediated killing of tissues expressing the target antigen and via cytokine release initiating with on-target activity of T-cells and propagating through the myeloid compartment.Citation32 While step-fractionated dosing can substantially limit uncontrolled cytokine release,Citation33 approaches to reduce on-target toxicity in normal tissues are more limited. In principle, either cytokine release or direct killing could be responsible for the observed weight loss in our studies, but the observation of direct toxicity in B7-H4-expressing tissues, along with jaundice in the affected mice, are supportive of direct T-cell toxicity. The enhanced affinity of our reagents for murine B7-H4 over human B7-H4 presents a high bar for demonstrating activity in the absence of on-target toxicity. Anti-Ly6E is not murine cross-reactive, therefore the approach taken in these studies cannot directly identify Ly6E-associated toxicities, or characterize an increase in therapeutic window for the dual-targeting approach. On human cells, in culture and in xenograft models, Ly6E TCEs paired with low affinity CD3 binders bound weakly to Ly6E-expressing cells and failed to induce any detectable cell death without B7H4 co-engagement.
Our data highlights that engineered avidity, resulting from simultaneously targeting two tumor-associated antigens, is able to enhance TCE-mediated killing (), and that appropriate placement of binding domains can profoundly influence the biological activity of the trispecifics (). We propose that the observed differences in activity are driven by synapse distance and maximal activity is achieved when the more potent Fab is placed at position 1 in proximity to the anti-CD3 Fab.Citation34 These data are consistent with the hypothesis that avid binding to Ly6E can restore activity to a weakly potent (but better tolerated) anti-B7-H4 TCE.
Collectively, this work demonstrates dual-antigen targeting can expand the scope of tumors addressable with TCEs. More generally, the approach of requiring two antigens for cell binding may enable refined cell targeting and/or therapeutic delivery. In addition to more precise targeting of a specific tissue, avidity-driven dual-antigen targeting may also enable therapeutic differentiation between immune (or other highly plastic) cells in different states of activation or maturation, further expanding the effective target space that may be amenable to intervention with targeted therapeutics.
Materials and methods
DNA and protein design and production
Expression vectors for IgG and Fab-IgG half-antibodies were generated as previously described.Citation35 For 1Fab-IgG constructs, the coding region for Ly6E (9B12) or B7-H4 (1D11) targeted Fab directly preceded the coding region of the anti-CD3.knob sequence. The Fc was in hIgG1 format, which includes the N297G mutation to avoid glycosylation, reduces binding to Fc-gamma-receptors,Citation36 and prevents undesired nonspecific cross-linking of CD3. Charge pairing mutations were installed within the Fab-IgG HC and the corresponding light chains to drive appropriate chain-pairing.Citation35 Half-antibodies were produced by transient transfection of Expi293 or Chinese hamster ovary cells at 30 mL (for in vitro assays) or at ≥1 L scale, respectively, and were purified by MAbSelectSure (GE Healthcare).Citation34 Knob and hole half-antibodies were assembled into the desired bi- or tri-specific format by applying procedures previously established for bispecific antibodies.Citation37 Following assembly and purification, all trispecific antibodies were of the appropriate format with >99% monomer by size exclusion chromatography and with the expected mass for the correctly assembled antibody as the major peak by liquid chromatography-mass spectrometry.Citation38 For in vitro screening, trispecific expression and assembly were performed without optimizing each construct’s DNA ratios or fractionating the assembled species, resulting in chain-pairing efficiencies of at least 76% correct, with many constructs in the 85–95% range (S7 Fig). The major impurity contained two copies of the tumor-targeted light chain in the Fab-IgG half-Ab resulting in an inactive anti-CD3 binding site (S7 Fig). For in vivo studies, DNA ratios were optimized to minimize mispairing, and residual mispaired species were separated chromatographically, resulting in >95% correctly paired products.
Histology
Tissues were fixed for up to 24 h in 10% neutral buffered formalin then transferred to 70% ethanol prior to embedding. Samples were paraffin-embedded and 4 μm-thick sections were cut onto Superfrost Plus glass slides. H&E staining was performed according to a routine standard operating procedure using a Leica Autostainer XL (Leica, Buffalo Grove, IL). Tissues were dewaxed and rehydrated before staining. Hematoxylin (American Mastertech, Lodi, CA) was applied for 8 minutes followed by a 30 second differentiation in 0.5% acid alcohol and bluing agent (Richard-Allan, Kalamazoo, MI) for 1 minute after which eosin (Eosin Y, American Mastertech, Lodi, CA) was applied for 30 seconds.
SPR analysis
Binding of trispecific Fab-IgG antibodies to recombinant B7-H4 extracellular domain was evaluated by SPR analysis using a Biacore T200 system. Antibodies in 10 mM HEPES, 150 mM NaCl, 0.05% Tween 20, 1 mM EDTA, pH 7.4, were captured non-covalently on a CM5-based human antibody capture chip and binding of recombinant, soluble B7-H4 ECD-His was monitored in real time using single-cycle kinetics and an assay temperature of 37°C. Data were fit to a 1:1 binding model using Biacore Evaluation Software.
Flow cytometry and cell killing assays
Cells were cultured as recommended by the ATCC. Prior to staining adherent lines were released by Accutase treatment, all cells were filtered and equilibrated in BD Stain buffer. Cells were stained with primary antibodies at indicated concentrations for 1 h at 4°C, washed x 3, stained with secondary Ab (Jackson ImmunoResearch 109–606-003) (1:100) for 1 h at 4°C, washed, stained with Fixable Viability Dye eFluor 780 (1:1000) for 1 h at 4°C, and washed x 2. Samples were fixed in 1% paraformaldehyde in phosphate-buffered saline (PBS) prior to analysis. Samples were assessed on a BD LSRFortessa. Following exclusion of dead cells, singlets were assessed for antibody binding using FlowJo software. Methods for PBMCs and CD8+ separation and cell quantitation by CellTiter-Glo (Promega) were described previously.Citation39 CD8+ cells were used as effectors in a 4:1 effector:target ratio for three days.
Xenograft studies
The efficacy of T cell-directing bispecific or trispecific antibodies targeting B7-H4 and/or Ly6E was investigated in a human breast cancer HCC1569X2 xenograft model in mice. The HCC1569X2 cell line was derived at Genentech from parental HCC1569 cells (ATCC) to provide optimal tumor growth in mice and was authenticated by short tandem repeat profiling using the Promega PowerPlex 16 System. Animal studies using this cell line were carried out at Genentech in compliance with National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the Genentech Institutional Animal Care and Use Committee (IACUC) under study number 18–0468. To establish the xenograft model, three million tumor cells (suspended in 0.2 mL of Hanks’ Balanced Salt Solution with Matrigel) were inoculated into the thoracic mammary fat pad of female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (common name NOD/scid gamma; NSG) mice (Jackson Laboratory, Sacramanto, CA).
When tumors reached the desired volume (~200 mm3), animals were randomized into groups of n = 5–8 with similar distribution of tumor volumes, and received intravenous dose(s) of vehicle (20 mM histidine acetate, 240 mM sucrose, 0.02% polysorbate-20, pH 5.5) or antibodies through the tail vein (Day 0). At 7–10 days prior to randomization, a subset of animals received an intraperitoneal injection of 10 million human PBMCs suspended in 0.1 mL of sterile PBS. PBMCs were purified from the blood of healthy donors to Genentech’s onsite donation program. All donors signed forms indicating their informed consent. PBMCs were isolated from whole blood using the Lymphocyte Separation Medium (MP Biomedical, LLC) and cryopreserved at – 80C, and cultured overnight in non-activating condition before harvest for injection. The treatment information was not blinded during measurement. Tumors were measured in two dimensions (length and width) using calipers and tumor volume was calculated using the formula: Tumor size (mm3) = 0.5 x (length x width x width). Changes in body weights were reported as a percentage relative to the starting weight. Tumor sizes and mouse body weights were recorded twice weekly over the course of the study. Mice whose tumor volume exceeded 2000 mm3 or whose body weight loss was >20% of their starting weight were promptly euthanized per IACUC guidelines.
Supplemental Material
Download MS Word (3 MB)Acknowledgments
The authors would like to thank Ingrid Kim, Kelly Loyet, Kathy Barrett, Mike Dillon, Wilson Phung, Geoff Del Rosario, and the APAT group for technical support in generating and characterizing the antibodies.
Disclosure statement
All authors are former or current employees of Genentech Inc., a member of the Roche group and may have financial interests including salary, equity, and/or existing patent applications.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2115213
Additional information
Funding
References
- Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nature Reviews, Drug Discovery. 2019;18:1–10.
- Mirzaei HR, Rodriguez A, Shepphird J, Brown CE, Badie B. Chimeric antigen receptors T cell therapy in solid tumor: challenges and clinical applications. Front Immunol. 2017;8:1850. doi:10.3389/fimmu.2017.01850.
- Argilés G, Saro J, Segal NH, Melero I, Ros W, Marabelle A, Rodriguez ME, Albanell J, Calvo E, Moreno V, et al. LBA-004 Novel carcinoembryonic antigen T-cell bispecific (CEA-TCB) antibody: preliminary clinical data as a single agent and in combination with atezolizumab in patients with metastatic colorectal cancer (mCRC). Ann Oncol. 2017;28:iii151. doi:10.1093/annonc/mdx302.003.
- Wermke M, Alt J, Kauh J, Back J, Salhi Y, Reddy V, Barve M, Ochsenreither S. 1147PPreliminary results from a phase I study of GBR 1302, a bispecific antibody T-cell engager, in HER2 positive cancers. Ann Oncol. 2018;29:viii408–9. doi:10.1093/annonc/mdy288.020.
- Kebenko M, Goebeler M-E, Wolf M, Hasenburg A, Seggewiss-Bernhardt R, Ritter B, Rautenberg B, Atanackovic D, Kratzer A, Rottman JB, Friedrich M, et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE®) antibody construct, in patients with refractory solid tumors. OncoImmunology. 2018;7(8):e1450710.
- Slaga D, Ellerman D, Lombana TN, Vij R, Li J, Hristopoulos M, Clark R, Johnston J, Shelton A, Mai E, et al. Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3. Sci Transl Med. 2018;10(463):eaat5775. doi:10.1126/scitranslmed.aat5775.
- Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, Bodmer W, Lehmann S, Hofer T, Hosse RJ, et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res. 2016;22(13):3286–97. doi:10.1158/1078-0432.CCR-15-1696.
- Nisthal A, Lee S-H, Kim YK, Bonzon C, Rashid R, Avery KN, Bogaert L, Ardila C, Leung IW, Rodriguez N, et al. Abstract 2286: xmAb30819, an XmAb ® 2+1 ENPP3 x CD3 bispecific antibody for RCC, demonstrates safety and efficacy in in vivo preclinical studies. Immunology. 2020;80:2286–2286.
- Boustany LM, Wong L, White CW, Diep L, Huang Y, Liu S, Richardson JH, Kavanaugh WM, Irving BA. Abstract A164: EGFR-CD3 bispecific ProbodyTM therapeutic induces tumor regressions and increases maximum tolerated dose >60-fold in preclinical studies. Molecular Cancer Therapeutics. 2018:17:A164–A164.
- Cugnetti APG, Liu H, Wang J, Xing C, Wheeler C, Lucas M, Chang C, Frey G, Boyle WJ, Short JM. Abstract 5698: novel conditionally active bispecific T cell engagers targeting solid tumors. Cancer Chem. 2018;2020:5698–5698.
- Banaszek A, Bumm TGP, Nowotny B, Geis M, Jacob K, Wölfl M, Trebing J, Kucka K, Kouhestani D, Gogishvili T, et al. On-target restoration of a split T cell-engaging antibody for precision immunotherapy. Nat Commun. pp.1–10. 2019. doi:10.1038/s41467-018-07882-8
- Skokos D, Waite JC, Haber L, Crawford A, Hermann A, Ullman E, Slim R, Godin S, Ajithdoss D, Ye X, et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci Transl Med. 2020;12(525):eaaw7888. doi:10.1126/scitranslmed.aaw7888.
- Leong SR, Liang W-C, Wu Y, Crocker L, Cheng E, Sampath D, Ohri R, Raab H, Hass PE, Pham T, et al. An anti-B7-H4 antibody-drug conjugate for the treatment of breast cancer. Mol Pharm. 2015;12(6):1717–29. doi:10.1021/mp5007745.
- Asundi J, Crocker L, Tremayne J, Chang P, Sakanaka C, Tanguay J, Spencer S, Chalasani S, Luis E, Gascoigne K, et al. An antibody-drug conjugate directed against lymphocyte antigen 6 complex, locus E (LY6E) provides robust tumor killing in a wide range of solid tumor malignancies. Clin Cancer Res. 2015;21(14):3252–62. doi:10.1158/1078-0432.CCR-15-0156.
- Smith JB, Lanitis E, Dangaj D, Buza E, Poussin M, Stashwick C, Scholler NsJr., Powell, Djp DJ. Tumor regression and delayed onset toxicity following B7-H4 CAR T cell therapy. Mol Ther. 2016;24(11):1987–99. doi:10.1038/mt.2016.149.
- Chuh JDC, Go M, Chen Y, Guo J, Rafidi H, Mandikian D, Sun Y, Lin Z, Schneider K, Zhang P, et al. Preclinical optimization of Ly6E-targeted ADCs for increased durability and efficacy of anti-tumor response. Mabs. 2020;13(1):1862452. doi:10.1080/19420862.2020.1862452.
- Mandikian D, Takahashi N, Lo AA, Li J, Eastham-Anderson J, Slaga D, Ho J, Hristopoulos M, Clark R, Totpal K, et al. Relative target affinities of T-cell–dependent bispecific antibodies determine biodistribution in a solid tumor mouse model. Mol Cancer Ther. 2018;17(4):776–85. doi:10.1158/1535-7163.MCT-17-0657.
- Leong SR, Sukumaran S, Hristopoulos M, Totpal K, Stainton S, Lu E, Wong A, Tam L, Newman R, Vuillemenot BR, et al. An anti-CD3/anti–CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood. 2017;129(5):609–18. doi:10.1182/blood-2016-08-735365.
- Andersen PS, Geisler C, Buus S, Mariuzza RA, Karjalainen K. Role of the T cell receptor ligand affinity in T cell activation by bacterial superantigens*. J Biol Chem. 2001;276(36):33452–57. doi:10.1074/jbc.M103750200.
- Ridgway JBB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody C H 3 domains for heavy chain heterodimerization. Protein Eng Des Sel. 1996;9(7):617–21. doi:10.1093/protein/9.7.617.
- Atwell S, Ridgway JBB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library11Edited by P.E.wright. J Mol Biol. 1997;270(1):26–35. doi:10.1006/jmbi.1997.1116.
- Rijn RSV, Simonetti ER, Hagenbeek A, Hogenes MCH, Weger RAD, Dijk MRC, Weijer K, Spits H, Storm G, Bloois LV, et al. A new xenograft model for graft-versus-host disease by intravenous transfer of human peripheral blood mononuclear cells in RAG2-/- γc-/- double-mutant mice. Blood. 2003;102(7):2522–31. doi:10.1182/blood-2002-10-3241.
- Ellerman D. Bispecific T-cell engagers: towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods [Internet] [accessed 2022 Aug 31]. 2019;154:102–17. Available from https://reader.elsevier.com/reader/sd/pii/S104620231830121X?token=951F0C894A5665B52048BCFFA711B119048F561AA95ACDD3CC157A4264B05D01A472B9B5F0340EA1446F651080257A5F.
- Boes M, Prodeus AP, Schmidt T, Carroll MC, Chen J. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J Exp Med. 1998;188(12):2381–86. doi:10.1084/jem.188.12.2381.
- Dheilly E, Moine V, Broyer L, Salgado-Pires S, Johnson Z, Papaioannou A, Cons L, Calloud S, Majocchi S, Nelson R, et al. Selective blockade of the ubiquitous checkpoint receptor CD47 is enabled by dual-targeting bispecific antibodies. Mol Ther. 2017;25(2):523–33. doi:10.1016/j.ymthe.2016.11.006.
- Cauwels A, Lint SV, Garcin G, Bultinck J, Paul F, Gerlo S, Heyden JVD, Bordat Y, Catteeuw D, Cauwer LD, et al. A safe and highly efficient tumor-targeted type I interferon immunotherapy depends on the tumor microenvironment. Oncoimmunology. 2018;7:e1398876.
- Cauwels A, Lint SV, Paul F, Garcin G, Koker SD, Parys AV, Wueest T, Gerlo S, Heyden JVD, Bordat Y, et al. Delivering Type I interferon to dendritic cells empowers tumor eradication and immune combination treatments. Cancer Res. 2018;78(2):463–74. doi:10.1158/0008-5472.CAN-17-1980.
- Mock J, Stringhini M, Villa A, Weller M, Weiss T, Neri D. An engineered 4-1BBL fusion protein with “activity on demand. Proc National Acad. 2020;117:e1398876.
- Mazor Y, Sachsenmeier KF, Yang C, Hansen A, Filderman J, Mulgrew K, Wu H, Dall’Acqua WF. Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci Rep. 2017;7(1):40098. doi:10.1038/srep40098.
- Mazor Y, Hansen A, Yang C, Chowdhury PS, Wang J, Stephens G, Wu H, Dall’Acqua WF. Insights into the molecular basis of a bispecific antibody’s target selectivity. mAbs. 2015;7(3):461–69. doi:10.1080/19420862.2015.1022695.
- Ahn S, Woo JW, Lee K, Park SY. HER2 status in breast cancer: changes in guidelines and complicating factors for interpretation. J Pathol Transl Med. 2020;54(1):34–44. doi:10.4132/jptm.2019.11.03.
- Godbersen-Palmer C, Coupet TA, Grada Z, Zhang SC, Sentman CL. Toxicity induced by a bispecific T cell–redirecting protein is mediated by both T cells and myeloid cells in immunocompetent mice. J Immunol. 2020;204(11):2973–83. doi:10.4049/jimmunol.1901401.
- Hosseini I, Gadkar K, Stefanich E, Li -C-C, Sun LL, Chu Y-W, Ramanujan S. Mitigating the risk of cytokine release syndrome in a Phase I trial of CD20/CD3 bispecific antibody mosunetuzumab in NHL: impact of translational system modeling. Npj Syst Biology Appl. 2020;6(1):28. doi:10.1038/s41540-020-00145-7.
- Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, DiCara D, Clark V, Hristopoulos M, Cook R, Slaga D, et al. Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell. 2017;31(3):383–95. doi:10.1016/j.ccell.2017.02.001.
- Dillon M, Yin Y, Zhou J, McCarty L, Ellerman D, Slaga D, Junttila TT, Han G, Sandoval W, Ovacik MA, et al. Efficient production of bispecific IgG of different isotypes and species of origin in single mammalian cells. mAbs. 2016;9(2):213–30. doi:10.1080/19420862.2016.1267089.
- Leabman MK, Meng YG, Kelley RF, DeForge LE, Cowan KJ, Iyer S. Effects of altered FcγR binding on antibody pharmacokinetics in cynomolgus monkeys. mAbs. 2014;5(1):896–903. doi:10.4161/mabs.26436.
- Spiess C, Merchant M, Huang A, Zheng Z, Yang N-Y, Peng J, Ellerman D, Shatz W, Reilly D, Yansura DG, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat Biotechnol. 2013;31(8):753–58. doi:10.1038/nbt.2621.
- Yin Y, Han G, Zhou J, Dillon M, McCarty L, Gavino L, Ellerman D, Spiess C, Sandoval W, Carter PJ. Precise quantification of mixtures of bispecific IgG produced in single host cells by liquid chromatography-Orbitrap high-resolution mass spectrometry. Mabs. 2016;8(8):00–00. doi:10.1080/19420862.2016.1232217.
- Junttila TT, Li J, Johnston J, Hristopoulos M, Clark R, Ellerman D, Wang B-E, Li Y, Mathieu M, Li G, et al. Antitumor efficacy of a bispecific antibody that targets HER2 and activates T cells. Cancer Res. 2014;74(19):5561–71. doi:10.1158/0008-5472.CAN-13-3622-T.