
ABSTRACT
Affinity maturation, an essential component of antibody engineering, is crucial for developing therapeutic antibodies. Cell display system coupled with somatic hypermutation (SHM) initiated by activation-induced cytidine deaminase (AID) is a commonly used technique for affinity maturation. AID introduces targeted DNA lesions into hotspots of immunoglobulin (Ig) gene loci followed by erroneous DNA repair, leading to biased mutations in the complementary determining regions. However, systems that use an in vivo mimicking mechanism often require several rounds of selection to enrich clones possessing accumulated mutations. We previously described the human ADLib® system, which features autonomous, AID-mediated diversification in Ig gene loci of a chicken B cell line DT40 and streamlines human antibody generation and optimization in one integrated platform. In this study, we further engineered DT40 capable of receiving exogenous antibody genes and examined whether the antibody could be affinity matured. The Ig genes of three representative anti-hVEGF-A antibodies originating from the human ADLib® were introduced; the resulting human IgG1 antibodies had up to 76.4-fold improvement in binding affinities (sub-picomolar KD) within just one round of optimization, owing to efficient accumulation of functional mutations. Moreover, we successfully improved the affinity of a mouse hybridoma-derived anti-hCDCP1 antibody using the engineered DT40, and the observed mutations remained effective in the post-humanized antibody as exhibited by an 8.2-fold increase of in vitro cytotoxicity without compromised physical stability. These results demonstrated the versatility of the novel B cell-based affinity maturation system as an easy-to-use antibody optimization tool regardless of the species of origin.
Abbreviations: ADLib®: Autonomously diversifying library, ADLib® KI-AMP: ADLib® knock-in affinity maturation platform, AID: activation-induced cytidine deaminase, CDRs: complementary-determining regions, DIVAC: diversification activator, ECD: extracellular domain, FACS: fluorescence-activated cell sorting, FCM: flow cytometry, HC: heavy chainIg: immunoglobulin, LC: light chain, NGS: next-generation sequencing, PBD: pyrrolobenzodiazepine, SHM: somatic hypermutation, SPR: surface plasmon resonance
Introduction
Monoclonal antibodies are becoming an indispensable therapeutic modality owing to their remarkable clinical benefits.Citation1,Citation2 Advances in antibody discovery and engineering technologies have allowed for highly rapid and efficient therapeutic antibody generation.Citation3 Affinity maturation is a key step for developing antibodies exhibiting therapeutic properties. Many techniques, including in vitro display-based approaches combined with random- or targeted-mutagenesis and chain shuffling and in silico computational approaches, have been established and widely adopted.Citation4 These well-researched techniques, however, require in-depth knowledge of antibody engineering for artificial mutation library design and involve complicated experimental procedures, including reformatting of antibody fragments into full-length antibodies. Thus, these techniques are usually unsuccessful at processing multiple antibodies simultaneously. The limitations of these approaches can potentially delay further downstream processes of drug development.
An alternative approach for in vitro affinity maturation mimics the in vivo antibody maturation mechanism. In animals, antibody affinity maturation occurs in B cells by somatic hypermutation (SHM). In activated B cells, mutations occur in the immunoglobulin (Ig) loci at a frequency of 10−5–10−3 per base, which is 106-fold more frequent than spontaneous mutations occurring in other non-immunoglobulin genes.Citation5 Activation-induced cytidine deaminase (AID) is an initiator for the SHM process (). AID preferentially deaminates cytosine in DNA to uracil under the context of WRC/GYW DNA motifs (W = A/T, R = A/G, Y = C/T), called “AID hotspots”, whereas SYC (S = G/C) and the third C in the trinucleotides GAC, GGC, CAC and TTC are rarely deaminated and are known as “AID coldspots”.Citation6,Citation7 These hotspots and coldspots are expected to be spread on Ig genes to maximize mutations at complementary-determining regions (CDRs) and minimize mutations at structurally important framework regions (FRs).Citation8,Citation9 Numerous affinity-enhancing mutations artificially introduced using methods such as random mutagenesis and saturation mutagenesis have unexpectedly reduced antigen specificity and/or manufacturability of antibodies.Citation10–12 However, mutations generated via an in vivo mechanism can prevent such drawbacks, and the process can be conducted without a priori knowledge of antibody engineering.Citation13 Although SHM is capable of efficiently introducing functional mutations, including unique mutations with amino acid length changes and/or multiple amino acid replacements that are difficult to obtain using site-directed mutagenesis or in silico approaches, mutations occur stochastically in an AID activity-dependent manner. Multiple rounds of the mutation accumulation process are usually required to obtain the final antibodies.Citation14–21 Therefore, introduction and accumulation of functional mutations in a short amount of time and with subsequent effective enrichment and isolation of the best candidate from the cell pool are critical to performing antibody maturation efficiently using an in vivo mechanism.
Figure 1. Schematic representations of the Ig gene diversification mechanism in DT40 and the ADLib® platforms.
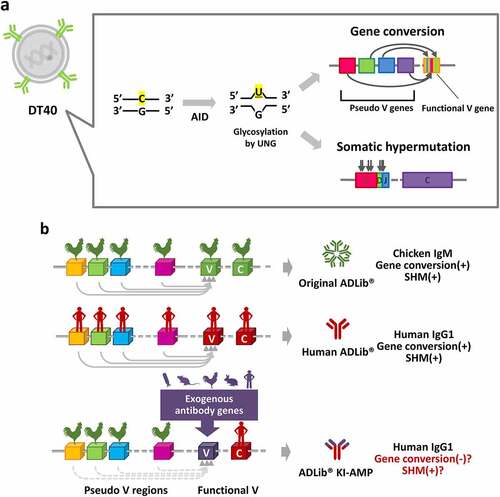
The chicken pre-B cell line DT40 has been used for a wide range of applications in de novo antibody generation,Citation22–27 affinity maturation,Citation15,Citation20,Citation22 and molecular evolution of non-immunoglobulin proteins.Citation28–30 In DT40 cells, AID mediates SHM and gene conversion, a gene shuffling mechanism occurring between the functional antibody genes in Ig loci and the antibody pseudogenes upstream of the functional genes ().Citation31 Gene conversion plays an indispensable role in diversifying B cell repertoires in chickens.Citation31 Autonomously diversifying library (ADLib®) is an antibody discovery and optimization platform fully utilizes the antibody gene diversification machinery of chicken B cell line DT40.Citation22–25,Citation32,Citation33 The original ADLib®, our first platform, expresses chicken IgM (); the human version ADLib® expresses full-length human IgG1 as an in vitro antibody discovery platform.Citation22 Both ADLib® systems are capable of generating target antigen-specific monoclonal antibodies. Notably, the cloned cells expressing the target-specific antibodies retain the ability to further diversify their endogenous Ig genes; therefore, the platform uniquely enables seamless transition from antibody discovery to subsequent antibody maturation. Owing to its short doubling time (7–8 hours),Citation33 this unique in vitro affinity maturation process using DT40 is highly efficient and can generate antibodies exhibiting tens to thousands-fold enhanced antigen-binding capacity within just one or two rounds of maturation cycles (Table S1).Citation22 However, straightforward affinity maturation has only been demonstrated with clones derived from the ADLib® system per se.
Here, we describe the ADLib® knock-in affinity maturation platform (ADLib® KI-AMP), in which DT40 cells are further engineered to be capable of receiving V regions of attempted antibody genes and enhancing their antigen reactivity. The Ig genes subjected to maturation are knocked-in to the endogenous Ig loci of DT40 in this platform. The cells in the ADLib® KI-AMP do not contain synthetic human antibody pseudogenes, unlike those in the human ADLib®;Citation22 instead, these cells possess endogenous chicken pseudogenes upstream of the functional V. It has been reported that the deletion of pseudogenes in DT40 abolishes gene conversion and activates SHM,Citation34 and transgenes inserted into the Ig loci without gene conversion donors readily available nearby can be diversified by SHM induced by AID.Citation29 Therefore, we hypothesize that low homologies between functional V and pseudogenes can impede gene conversion and promote SHM in DT40 without altering the endogenous chicken pseudogenes (). As a result, mutations found in the artificially inserted Ig genes were predominantly introduced by SHM, instead of gene conversion in the ADLib® KI-AMP. Using this novel platform, we demonstrated successful affinity maturation of the desired antibodies regardless of their species of origin while maintaining binding specificity and physical stability.
Results
Design and construction of the ADLib® KI-AMP
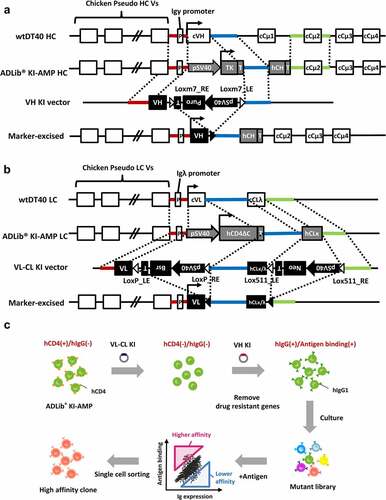
For affinity maturation of antibody candidates using DT40, endogenous chicken Ig genes must be replaced by desired Ig genes. Knockout of the endogenous Ig genes and subsequent knock-in of the desired Ig genes into the cell was performed using gene targeting. To enable the easy construction of cells in which exogenous Ig genes are knocked-in using fluorescence-activated cell sorting (FACS), we generated modified DT40 cells for the ADLib® KI-AMP. In these platform cells, the heavy chain (HC) constant region of the endogenous chicken Cμ1 gene was replaced by the HC constant region of human IgG1 gene as in the human ADLib® ().Citation22 The endogenous light chain (LC) constant region of the chicken LCλ (CLλ) gene was also replaced by the LC constant region of human LCκ (CLκ) (). The V regions of the endogenous chicken HC and LC were replaced by the marker genes. As a result, the knock-in platform cells expressed the marker molecules instead of endogenous antibodies. The Ig genes to be optimized were cloned into the knock-in vectors and the LC gene and the HC gene were sequentially introduced into the platform cells (). Once the exogenous Ig genes were successfully introduced into cells, they expressed full-length antibodies on the cell surface instead of the marker proteins. The cells containing exogenous Ig genes were cultured to introduce mutations to their V regions, and subsequently, the clones exhibiting improved antigen reactivity were enriched and isolated from the mutant library using FACS ().
Figure 2. Schematic representation of the construction of the cells expressing the exogenous antibodies and the standard workflow of affinity maturation using the ADLib® KI-AMP.
ADLib® KI-AMP can introduce mutations in desired antibodies, resulting in improved antigen reactivity
We examined whether mutations occur in the desired Ig gene introduced into the ADLib® KI-AMP cells and whether these mutations could affect antibody affinity. Three anti-human VEGF-A (hVEGF-A) antibody sequences previously obtained from the human ADLib®,Citation22 VEGF_A033 (A033), VEGF_D018 (D018), and VEGF_D058 (D058), were used as the model human antibody sequences and introduced to the platform cells (). A033 was used as a positive control because the binding affinity of this antibody was successfully improved in the previous research.Citation22 The clones expressing hVEGF-A-specific human IgG on the cell surface were isolated using FACS (Figure S1A). We confirmed that the Ig V region sequences of the isolated clones were identical to those of the original clones obtained from human ADLib® using genomic DNA sequencing. We also confirmed that the antibody secreted by the clones showed identical antigen reactivity to that of the original antibodies (Figures S1B and S1C).
Table 1. Summary of the model antibody profiles used in the study.
Each clone expressing the introduced A033, D018, and D058 Ig genes was cultured for 2 weeks, and mutations were accumulated. Flow cytometry (FCM) analysis showed a scattered distribution of cell populations exhibiting variable antigen reactivity (). Up to 96 cells were sorted into independent wells using FACS from a small cell population with increased antigen reactivity. Notably, most of the viable sorted clones showed higher antigen reactivity than the parental clones (). Genomic DNA sequence analysis revealed that all the sampled clones contained mutations resulting in amino acid changes ( and ), and most of these mutations were found in their CDRs. Amino acid mutations occurring in CDRs were likely to affect antigen-binding capacity. Surface plasmon resonance (SPR) analysis confirmed that all of the unique clones, including those with amino acid length changes and/or multiple amino-acid replacement, exhibited improved antigen reactivity up to 76.4-fold ( and Figure S2).
Table 2. Summary of affinity maturation against the model human antibodies, anti-hVEGF-A, using the ADLib® KI-AMP.
Table 3. Summary of binding affinities, kinetics, and amino acid mutations generated using affinity maturation against anti-hVEGF-A antibodies using the ADLib® KI-AMP.
Figure 3. Affinity maturation against the model human antibodies using ADLib® KI-AMP.
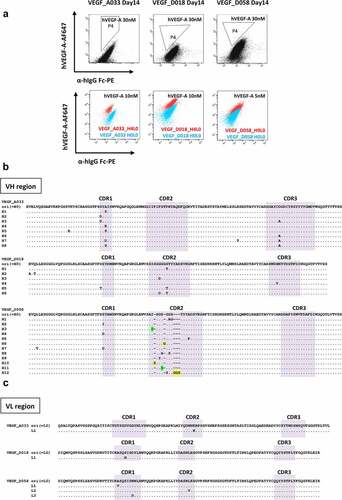
In the case of A033, among all viable clones (26 clones) with improved antigen reactivity, nine unique antibodies were obtained. These affinity-matured clones had different amino acid mutations from those obtained previously.Citation22 In the D018 clones, 37.9% of the viable clones (25 of 66 clones) exhibited improved antigen reactivity, and seven unique antibodies were obtained. The D018_H6L0 clone coincidentally had both mutations that were separately found in the D018_H3L0 and D018_H1L0 clones. Mutations conferring antibody affinity improvement may not be limited to a single position; thus, clones containing functional mutations at multiple sites were successfully obtained within a single round of the affinity maturation process. The SPR analysis showed that the double-mutated D018_H6L0 clone had slightly higher antigen binding affinity than the single-mutation clones D018_H3L0 and D018_H1L0. It is possible that the combination of single mutations synergistically affects the affinity for the antigen.
In the case of D058, 42.5% of the viable clones (31 of 72 clones) exhibited improved antigen reactivity and 15 antibodies with unique V regions were obtained. Amino acid insertions and deletions were frequently observed in VH CDR2 of the clones. A tandem repeat in the corresponding region was identified and the insertion and deletion of the amino acids were found near the repeated sequence (Figure S3). Such tandem repeats promote intermediate loop formation resulting in nucleotide deletions and duplications during repair.Citation35 As multiple AID hotspots are identified in this region, AID deamination is likely to accelerate this deletion and duplication. Indeed, such mutations have been reported in human immune systemCitation35 and other cell-based affinity maturation platforms utilizing AID-mediated SHM.Citation17 Thus, the deletion and insertion observed in VH CDR2 of D058 are in line with the mutational characteristics resulting from AID-mediated SHM.
Collectively, these results show that the ADLib® KI-AMP is capable of inducing SHM onto the exogenous Ig genes introduced into the cell and improving antibody affinities. Of note, decreased binding specificity due to mutations is another concern during the antibody engineering process.Citation10–12 We evaluated antigen-binding specificity of the affinity-matured clones using ELISA and confirmed that all clones maintain target-binding specificity (Figure S4A). In addition, no poly-reactivity against three different types of cell lines was observed by FCM (Figure S4B). These results further verify that improved binding affinity of the affinity-matured antibody derived from the ADLib® KI-AMP is not accompanied by increased nonspecificity or poly-reactivity.
AID-mediated SHM enhances antigen reactivity of desired antibodies in the ADLib® KI-AMP
To gain more insights into sequence diversity and analyze mutation frequencies and preferences, we performed next-generation sequencing (NGS) of each cell population derived from the knocked-in parental clones of anti-VEGF model antibodies. In this experiment, the HC and LC V region sequences of each clone in three cell populations emerged after 2 weeks of cell culture and characterized by different antigen reactivities in the FCM plots (named “high,” “middle,” and “low” in ) were analyzed for comparison. The average number of quality-filtered sequences used in the analysis for each group was 37,135 (Table S2). As the Ig genes of DT40 are constantly mutated, we also analyzed the sequences before starting cell culture (Day 0), and they were used as the control sequences for comparison among those obtained at the end of the culturing. On Day 14, combined sequence diversity for the HC and LC genes of all cells before cell sorting (referred herein as Day 14 whole) increased by 2.5%–9.5% from Day 0 (). The combined mutation rates for the HC and LC genes on Day 14 whole in A033, D018, and D058 were estimated as 9.18 × 10−6, 2.42 × 10−6, and 9.33 × 10−6 mutations per base pair and division, respectively. The mutation patterns identified using the NGS analysis were diverse (). Although the most common type of mutation was a single-base substitution, mutational characteristics of AID-mediated SHM, such as consecutive substitution of multiple bases and amino acid chain length change mutations, were also observed among all three model antibody sequences. The observed base substitutions were biased toward G and C ( and S5A), consistent with the characteristic of AID-induced base substitutions in DT40 reported previously.Citation36,Citation37 These mutation patterns are indicative of an operating AID-mediated mutation mechanism.
Table 4. Summary of NGS analysis against the anti-hVEGF-A clones generated using the ADLib® KI-AMP.
Figure 4. In-depth sequence analysis of the clones obtained from the ADLib® KI-AMP using NGS.
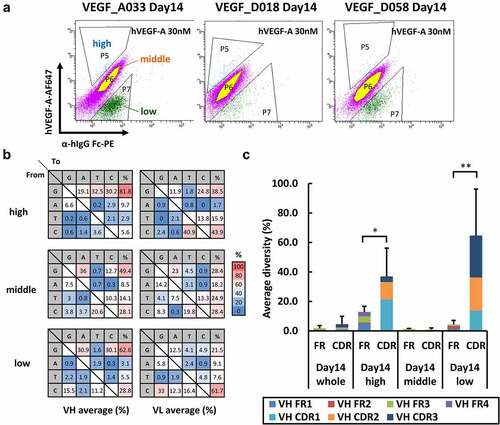
The NGS analysis revealed that more mutations occurred in the HC than in the LC and in CDRs than in FRs (, and S5B). A comparison among all data sets on Day 14 showed that the average sequence diversity of CDRs was markedly higher than that of FRs in the antigen reactivity “high” and “low” populations, whereas the sequence diversities of the CDRs and FRs in the “middle” population were similar. This result suggests that the mutations occurring in CDRs affect antigen-binding capacity more significantly than mutations in FR.
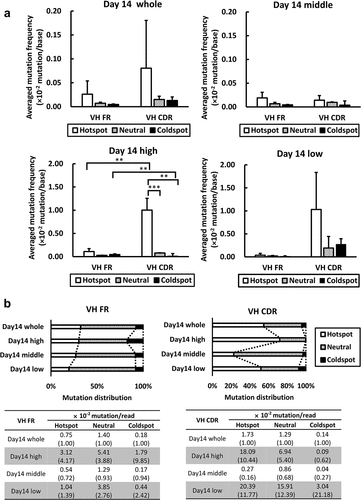
We further investigated the mutation frequencies of all cell populations on Day 14 in both FRs and CDRs grouped by known motifs of AID hotspot, coldspot, and the remaining (hereinafter referred to as “neutral”), respectively. On Day 14, although the averaged mutation frequencies (per base) calculated for each group of Day 14 whole showed large variations, they were higher in the hotspots in CDRs than in FRs ( and S6A). This tendency became apparent in both HC and LC of the antigen reactivity “high” population. These results suggest that the hotspots in CDRs are preferred as a target of AID than those in FRs, and that mutations at the hotspots in CDRs contribute to the alteration of antigen reactivity. Conversely, while more mutations were observed at coldspots in CDRs than in FRs among the whole cells (Day 14 whole), the coldspots in CDRs were less frequently mutated than those in FRs in the antigen reactivity “high” population ( and S6A). Moreover, we also analyzed the frequency of mutations introduced to each group per read ( and S6B) and found that the mutation distribution observed in each group in FRs was similar before and after the antigen reactivity-based cell sorting (, left panel), whereas those observed in CDRs showed larger variations (, right panel). Indeed, the mutation frequencies of the populations with altered antigen reactivities (the “high” and “low” populations) increased on Day 14. In contrast, the “middle” population, with an insignificant change in antigen reactivity, showed decreased mutation frequencies across all groups. Notably, the frequency at the coldspots in CDRs of the antigen reactivity “high” population alone decreased, whereas that of the antigen reactivity “low” population showed the largest increase from Day 14 whole ( and S6B, right panel). These observations clearly indicate that the mutations occurring at the hotspots in CDRs contribute to antigen reactivity change, whereas the mutations occurring at the coldspots in CDRs seem to be unfavorable for enhancing antigen reactivity.
Figure 5. Comparison of AID hotspot mutations in HC among populations exhibiting different antigen reactivities.
We then compared the mutations found in the NGS readings from the Day 14 antigen reactivity “high” population with those observed in the affinity-matured clones exhibiting the highest antigen reactivity, which were sorted into a 96-well plate as described in the previous section (). All mutations that frequently appeared in the NGS readings (>10%) were found in the affinity-matured clones whose affinities were verified using antigen-binding SPR (). The comparison also showed that matured clones containing rare mutations in the NGS readings (e.g., 0.1%–0.4% appearance) can be isolated. Because more stringent FCM gating criteria on the flow cytometer in terms of antigen reactivity were applied during the isolation of potentially affinity-matured clones than the cell populations for NGS analysis, our results demonstrated that an appropriate FCM gating approach can effectively acquire clones containing mutations important for enhanced antigen reactivity, even if such mutations are rare in the diversified cell pool of the ADLib® KI-AMP.
Table 5. Comparison among the isolated affinity-matured clones and their appearances in NGS readings.
Antigen reactivity of a therapeutic antibody lead derived from mouse hybridoma is improved using the ADLib® KI-AMP
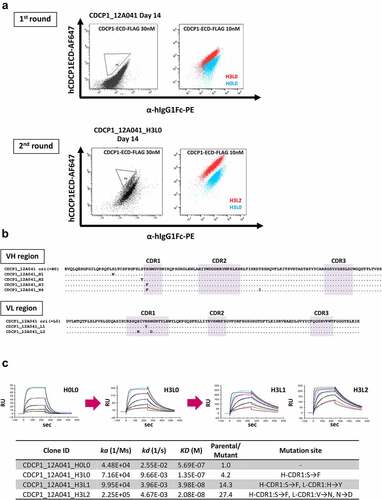
With the successful verification of the ADLib® KI-AMP using three anti-hVEGF-A hIgG1 sequences as a model case, we aimed to extend its applicability to a potential therapeutic antibody candidate obtained using the conventional hybridoma method to further improve its affinity. We focused on in-house established mouse monoclonal antibodies against human CUB domain containing protein 1 (hCDCP1), a novel therapeutic target against cancer.Citation38 One anti-hCDCP1 antibody, 12A041, exhibited moderate antigen-binding activity (KD < 10−7 nM, ). The V gene of 12A041 was cloned into the knock-in vector and introduced into the ADLib® KI-AMP cells (Figure S7A). As 12A041 originated from mouse, the resulting cells express mouse-human chimeric antibodies. The hCDCP1/human IgG Fc-positive DT40 clone was isolated using FACS and specific binding to recombinant hCDCP1 of secreted antibodies was confirmed using ELISA (Figure S7B).
The DT40 clone expressing 12A041 was cultured for 2 weeks to accumulate mutations, and cells exhibiting improved affinity against a soluble hCDCP1-extracellular domain (ECD) were isolated using FACS (). The sequence analysis showed five unique clones. Among mutations contained in these antibody sequences, only the H32Y mutation in the VL CDR1 and S32F mutation in VH CDR1 were thought to confer the affinity improvement ( and S7C). We attempted an additional round of affinity maturation using the clones obtained from the first round as the starting clones and could obtain cells exhibiting increased antigen reactivity only from 12A041_H3L0. The sequence analysis revealed two unique additional mutations, both of which were in the LC (). One was the identical mutation to the one observed in the first maturation cycle (sequence L1), and the other one was newly observed (sequence L2). The best clone generated after two maturation cycles (12A041_H3L2) showed 27.4-fold reduction in the KD value in SPR analysis in comparison with the parental antibody (12A041_H0L0, ).
Figure 6. Affinity maturation against therapeutic lead antibodies generated by mouse hybridoma method using the ADLib® KI-AMP.
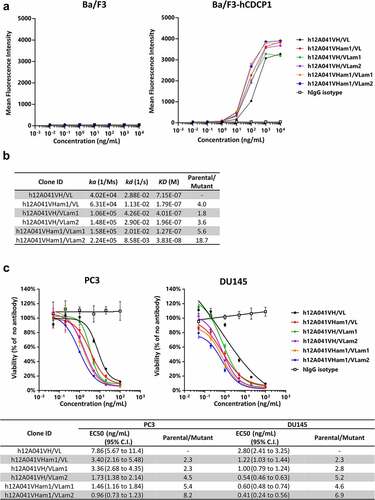
ADLib® KI-AMP derived mutations enhance the potency of post-humanized therapeutic antibody leads
As the affinity-enhancing mutations in CDRs described in the previous section are thought to be involved in stabilizing the interaction between CDR structure and epitope of the antigen, it is plausible that these mutations can enhance the affinity even when the framework regions are humanized. To this end, we humanized the variable domain of 12A041 using a CDR grafting method (h12A041VH/VL), and mutations described in the previous section were introduced to the corresponding residues of h12A041VH/VL. All mutated antibodies exhibited superior binding activity to hCDCP1 stably expressed on Ba/F3 cell surfaces than the parental h12A041VH/VL and maintained low nonspecific binding to non-transfected Ba/F3 cells (). Antigen-binding specificity was also confirmed using ELISA (Figure S8A). The binding kinetics of these antibodies were analyzed using SPR. Introduction of the mutations led to an 18.7-fold reduction in KD values compared with those of the parental antibody ( and S8B). To evaluate the potency of these affinity-matured anti-hCDCP1 antibodies as antibody-drug conjugates, we performed a secondary immunotoxin assay using the human prostate carcinoma cell-line DU145. DU145 cells were cultured with the anti-hCDCP1 antibodies together with pyrrolobenzodiazepine (PBD)-conjugated anti-human IgG-Fc antibodies. The results showed that the matured antibodies exhibited enhanced cytotoxicity compared with that of the parental antibody (). Finally, we analyzed the stability of the affinity-matured antibody because introduction of mutations often deteriorates physicochemical properties of antibodies.Citation10–12 A thermal shift assay showed that the matured antibodies exhibit identical melting temperature to the parental antibody (Table S3). Size exclusion chromatography revealed that all antibodies bearing affinity-enhancing mutations maintain 95% monomeric purity, one of the generally accepted criteria for antibody developability (Table S4).Citation39 Although h12A041VH/VLam1 showed slightly higher level of aggregates compared to the parental antibody, the double mutant h12A041VHam1/VLam1, which possesses an additional mutation in the VH to afford improved affinity, had a similar degree of aggregation as the parental antibody. Furthermore, all matured antibodies showed comparable or negligible increases in aggregates relative to the parental clone after three freeze/thaw cycles (Table S4).
Figure 7. Functional validation of the humanized anti-hCDCP1 antibody involving the mutations generated by the ADLib® KI-AMP.
Discussion
A cell display system coupled with AID-mediated SHM is a commonly used technique for in vitro affinity maturation, and several examples of antibody affinity maturation applied to single-chain variable fragment (scFv), IgM, and IgG using DT40 have been reported.Citation14,Citation15,Citation20,Citation22 These studies took advantage of DT40ʹs Ig gene diversification mechanism and successfully showed the utility of the cells for de novo antibody generation and affinity maturation. However, in previous studies, the antibodies subjected to maturation were obtained from DT40 platform cells; thus, their applicability is limited. Therefore, we developed an antibody maturation platform capable of receiving attempted antibody genes and improving their antigen reactivity. This novel platform, the ADLib® KI-AMP, described herein could enhance antigen-binding affinity of the antibodies discovered from other platforms, such as the human ADLib® and mouse hybridoma. Owing to easier genetic manipulation and rapid growth of DT40 in comparison to other mammalian cells,Citation33 it takes approximately a month from cloning the model antibody genes into the knock-in vectors to isolation of the clones expressing these antibodies by applying a simplified knock-in process with FACS. Using the platform, binding affinities of antibodies sourced from different origins were efficiently enhanced to sub-picomolar KD without compromising target specificity by just a single round of cell culture lasting 2 weeks followed by cell sorting. This is the first report demonstrating successful antibody maturation against the attempted exogenous antibodies exploiting the Ig gene diversification mechanism of DT40, regardless of the species of origin.
As the ADLib® KI-AMP cells do not possess pseudogenes homologous to the functional V region, affinity maturation against the desired antibody genes is expected to be driven by SHM rather than gene conversion. A previous study has shown that the disruption of pseudogene loci resulted in the inhibition of gene conversion and activation of SHM.Citation34 This finding has been applied to successful engineering of non-antibody moleculesCitation29,Citation30 and scFv15 using DT40. In these works, the DT40 cells we used did not harbor target molecule-corresponding pseudogenes but endogenous chicken ones, which show no or low homologies to the introduced non-antibody gene or scFv genes. Here, we similarly observed SHM in human V genes, which were knocked-in to the Ig loci of DT40 harboring chicken pseudogenes. Most importantly, we demonstrated that AID-mediated SHM is capable of improving the antigen reactivity of antibodies introduced into the ADLib® KI-AMP cells as full-length IgGs. Effective introduction and accumulation of functional mutations are critical to efficient antibody affinity maturation. In our platform, affinity-matured clones can be quickly enriched and isolated from the diversified cell pool using FACS. Using the model human antibody sequences, 37.9–100% of the viably sorted clones exhibited improved binding affinity in the SPR analysis. This result illustrates that the platform is capable of introducing mutations to enhance antigen reactivity to the level that enables the discrimination between high-affinity clones and low-affinity ones using FACS within a single round of the maturation process. The overall mutation rate observed in the ADLib® KI-AMP was moderate in comparison to that of other platforms.Citation15,Citation16,Citation29,Citation40 Thus, the estimated frequency of mutagenesis of target genes cannot fully account for our successful antibody maturation.
A comprehensive sequence analysis against the cell populations generated in the diversified cell pool revealed that AID-mediated SHM in DT40 was biased toward G- and C-bases. Mutations in CDRs and AID hotspots were frequently introduced, and the comparison among the cell populations exhibiting different antigen reactivity showed that such mutations became prominent in the clones with altered antigen reactivity. Moreover, the cell population exhibiting increased antigen reactivity contained more mutations at AID hotspots and fewer mutations at coldspots in their CDRs. This observation clearly demonstrates that the biased mutations correlated with improved antigen reactivity. Based on these results, the platform utilizing AID-mediated SHM is a rational option for effective antibody affinity maturation. Besides B cells, non-B cells overexpressing AID can generate mutations if the target sequences are highly transcribed,Citation41 and successful examples of affinity maturation using non-B cells overexpressing AID have been reported.Citation16,Citation17,Citation19
We demonstrated that affinity maturation of antibody can be readily achieved within a single round of the process using the ADLib® KI-AMP, in contrast to the notion that AID-mediated SHM occurs stochastically and thus generally requires multiple maturation cycles. Our platform features naïve Ig gene diversification mechanism of DT40 cells. Not only AID but also other factors such as translesion DNA polymerases,Citation37,Citation42,Citation43 error-prone DNA polymerases,Citation37,Citation42,Citation43 and the diversification activator (DIVAC)Citation44–47 have been implicated in the introduction of hypermutations in DT40. Although the initiation of AID-mediated SHM has not yet been fully understood, the intrinsic gene diversification mechanism itself is expected to be the key element supporting successful antibody maturation. This mechanism might also contribute to antibody maturation without compromising binding specificity and physical stability.Citation13
It is plausible to improve the affinity more effectively by adjusting the culture period or performing iterative process of cell culture and sorting. In this study, we empirically set the culture period as 14 days per round and subsequently verified that a 14-day culture was able to yield high-affinity antibodies with KD values in the picomolar range. As the potential for affinity increase is sequence-dependent, defining an optimal culture period to be applied for all different clones can be challenging. It would be best to determine the timing of isolation of potentially affinity-matured clones from each parental clone by monitoring the distribution of cell population on FCM indicative of effective accumulation of functional mutations over the course of cell culture. Nevertheless, we believe the results of this study serve as a good starting point for setting the culture period for the ADLib® KI-AMP in general.
In-depth sequence analysis using NGS also provides several insights into further understanding of antibody maturation driven by AID-mediated SHM. Our results showed that more mutations occurred at AID hotspots than at either coldspots or neutral, and that the mutation frequencies observed at the hotspots in CDRs were particularly higher than those in FRs. While it has been reported that some hotspot overlapping (WGCW motif) in CDRs exhibit exceptionally high mutation rates in humans,Citation48 this phenomenon was not observed in our study. This suggests that additional factors may have contributed to a biased mutation in CDRs. One potential cis-acting factor other than the conventional AID hotspot/coldspot is the WA/TW motifs preferentially mutated by DNA polymerase η, an error-prone DNA polymerase that is also involved in SHM processing.Citation49,Citation50 The longer motif covering the additional region upstream and downstream of the conventional AID hotspot has also been recently proposed.Citation51 As multiple translesion DNA polymerases, including DNA polymerase η, have been reported to be involved in SHM of DT40,Citation37,Citation42,Citation52 the overlapped unknown “preferences” of these polymerases might result in the biased mutation in CDRs. We also found that the mutation frequency of coldspots in CDRs was significantly decreased in the cell population exhibiting increased antigen reactivity, whereas the mutation frequency was increased in the antigen reactivity-reduced population. This finding suggests that mutations occurring in this region do not contribute to enhanced antigen-binding capacity; rather, such mutations are unfavorable to maintaining antigen reactivity. It is possible that the positions where mutations favoring or not favoring antigen reactivity can occur have been fixed as AID hotspots and coldspots, respectively, during molecular evolution.
While NGS was used to examine the mutational characteristics of AID-mediated SHM in the ADLib® KI-AMP, the possibility of integrating NGS into the workflow of screening affinity-matured clones by retrieving comprehensive VH and VL sequences can also be envisioned. Indeed, affinity-improving mutations described in this study, which were detected by Sanger sequencing following single-cell sorting of DT40, have also been identified in our NGS analysis. Tailoring antibody affinity or stability can therefore be achievable by exploiting NGS data. With the aid of additional VH:VL pairing information, the consequence of each observed mutation can be more precisely interpreted in terms of antibody maturation. This will allow us to further explore the potential of ADLib® KI-AMP in the future.
Materials and methods
Cell culture
DT40 cells were cultured at 39.5°C in 5% CO2 with Iscove’s modified Dulbecco’s medium (Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS; Cytiva), 1% chicken serum (Thermo Fisher Scientific), 1% penicillin/streptomycin (Nacalai Tesque), and 50 µM monothioglycerol (FUJIFILM Wako pure chemical). Ba/F3 cells were cultured at 37°C in 5% CO2 with RPMI1640 (Merck) containing 10% FBS, 1% penicillin/streptomycin, 50 µM monothioglycerol, and 1 ng/mL mouse IL-3 (R&D systems, #403-ML-050). PC3 cells were cultured at 37°C in 5% CO2 with Ham’s F-12 K (FUJIFILM Wako pure chemical) containing 7% FBS and 1% penicillin/streptomycin. DU145 cells were cultured at 37°C in 5% CO2 with RPMI1640 containing 10% FBS and 1% penicillin/streptomycin.
Antigen preparation
The Flag-tagged recombinant proteins used in this study, FLAG-hVEGF-A (UniProt: P15692-4), FLAG-hHer2-ECD (P04626), and hCDCP1-ECD-His-FLAG (9QH5V8), were transiently expressed using FreeStyle 293-F cells (Thermo Fisher Scientific) and purified using anti-FLAG M2 affinity chromatography (Merck, #A2220) followed by gel filtration chromatography (HiLoad 26/600 Superdex 200 pg; Cytiva). To prepare the labeled antigens for affinity maturation, the purified FLAG-hVEGF-A and FLAG-hHer2 were biotinylated using an EZ-Link NHS-PEG4 biotinylation kit (Thermo Fisher Scientific, #A39259).
Plasmid construction for the ADLib® KI-AMP
To construct the ADLib® KI-AMP, the knock-in vectors previously generated for human ADLib® constructionCitation22 were used with modifications for the present purpose. The HC constant region knock-in vector was used for replacing the HC constant region of an endogenous chicken Cμ1 gene of DT40 by the HC constant region of human IgG1 gene. To introduce thymidine kinase gene to the HC V region of DT40, the targeting vector (thymidine kinase knock-in vector) was constructed by assembling a CMV promoter, thymidine kinase (GenBank: KM222725.1, synthesized by Azenta Life Sciences), and SV40 promoter followed by a puromycin resistance gene in reverse orientation flanked by loxPRE (5’-ATAACTTCGTATAATGTATGCTATACGAACGGTA-3’) and loxPLE (5’-ATAACTTCGTATAATGTATGCTATACGAACGGTA-3’) to replace the V region and CAG promoter-blasticidin (reverse orientation) flanked by Vlox sequences (VloxM1; TCAATTTCC- GAGAATGACAGTTCTCGGAAATTGA) of the human HC V region knock-in vector. For introduction of hCD4ΔC marker gene to the LC V region of DT40, the gene of the human CD4 lacking a cytoplasmic domain (hCD4ΔC) was cloned from a pMACS4.1 vector (Miltenyi Biotech, #130-091-886) and fused with an extra sequence to express the N-terminal 2 × FLAG-tag (DYKDDDDK)-fused hCD4ΔC protein. We constructed the hCD4ΔC and kappa LC constant region targeting vector in which the V region of the LC V and constant region knock-in vector with neomycin resistance and blasticidin resistance genes were replaced by a CMV promoter-2 × FLAG-hCD4ΔC followed by a BGH terminator.
Construction of the exogenous Ig gene knock-in vectors
Genomic DNA of VEGF_A033, D018, and D058 was extracted from the DT40 clones previously obtained from human ADLib®.Citation22 The Ig genes of CDCP1_12A0416 were synthesized (by Azenta Life Sciences) based on the sequence determined from the mouse hybridoma cDNA. To introduce the exogenous HC V genes, the synthesized CDCP1_12A041 HC V gene and VEGF_A033, D018, and D058 HC V genes amplified using the primer F1(5’-CCCCACAGGGCTGATGGCGGAGGTG-3’) and R1 (5’- GAACGGTAGGGGATCCATAAAATCG −3’) were assembled with SV40 promoter-puromycin flanked by loxm7LE and loxm7RE and cloned into the EcoRV and BamHI sites of the HC V region knock-in vector. To introduce the exogenous LC V and constant region genes, the synthesized CDCP1_12A041 LC V gene and VEGF_A033, D018, and D058 LC V genes were amplified using the primer F1(5’- TTGCAGCATGAGCCCTTTGTTGTC −3’) and R1 (5’- TTCTATGAAGGGAGCCATAGCCTG −3’) and cloned into the BmgBI – NdeI site of the hCD4ΔC and kappa LC constant region targeting vector. Furthermore, the kappa LC constant region of VEGF_A033 LC V knock-in vector was replaced with human lambda LC constant region amplified from the human lambda LC V and constant region knock-in vector of human ADLib®.
DNA Transfection
All plasmids were linearized before transfection. The restriction enzymes used in this study were purchased from New England Biolabs. The exogenous HC V region knock-in vector and the thymidine kinase knock-in vector were linearized using NotI and KpnI. The exogenous LC V region knock-in vector and the hCD4ΔC knock-in and kappa LC constant region targeting vector were linearized using NotI and AscI. The HC constant region knock-in vector was linearized using SalI. Further, 30 µg of the linearized vectors were transfected with 1.0 × 107 DT40 cells by electroporation (Bio-rad GenePulser equipped with 24-well electroporation plate at 420 V and 125 µF). After 16 h, transfected clones were selected with the medium containing 0.5 μg/mL puromycin (Merck) to obtain the clones knocked-in the HC V region and thymidine kinase, or the medium containing 2 mg/mL geneticin® (Thermo Fisher Scientific) to obtain the clones knocked-in the hCD4ΔC and the LC V and constant region and the HC constant region, respectively. To excise the drug resistance genes, a total of 3 × 106 cells were collected and transfected with 10 µg of a vector expressing an EGFP-Cre recombinase fusion protein by Nucleofector 2b (Lonza) using Cell Line Nucleofector Kit T (Lonza). After 16 h of incubation, transfected cells were sorted using BD FACSAria fusion (BD biosciences) based on green fluorescent protein expression.
Flow cytometry
Cells (up to 5 × 105 cells) were stained with the appropriate antigen and/or antibodies diluted in FCM buffer (phosphate-buffered saline (PBS)/0.5% bovine serum albumin (BSA)/2 mM EDTA) for 30 min on ice at each staining step. The cells were washed using PBS after every staining step and the stained cells were diluted with FCM buffer and analyzed using BD FACSCantoII (BD Biosciences). FCM plots were generated using FlowJo software (BD Biosciences).
Surface human CD4 expression was analyzed by staining with phycoerythrin (PE)-conjugated anti-human CD4 Antibody (Biolegend, #317410). Surface human IgG expression was analyzed by staining with R-PE-conjugated goat anti-human IgG (gamma chain specific; SouthernBiotech, #2040-09). Binding to hVEGF-A was analyzed using the staining with AlexaFluor 647-labeled streptavidin (Thermo Fisher Scientific, #S21374) via biotinylated recombinant FLAG-hVEGF-A. Human CDCP1-ECD-His-FLAG bound by biotinylated anti-FLAG M2 antibody (Merck, #F9291) was stained with AlexaFluor 647-labeled streptavidin.
For the poly-reactivity analysis using FCM, the antibody secreted in the culture supernatant or Human IgG1 Kappa-UNLB (SouthernBiotech, #0151 K-01) were adjusted to 3 μg/mL in the medium and reacted with Ba/F3, HEK293, and HUVEC, respectively. Antibodies that reacted with these cells were detected with R-PE-conjugated goat anti-human IgG antibody (gamma chain specific). As positive control antibodies, PE-conjugated anti-mouse CD45 antibody (Biolegend, #103106) was used for Ba/F3, and mouse anti-human EGFR antibody (Thermo Fisher Scientific, #MS-268-PABX) followed by PE-conjugated goat anti-mouse Ig antibody (BD Biosciences, #550589) was used for both HEK293 and HUVEC.
For the analysis using hCDCP1-expressing Ba/F3 cells, surface hCDCP1 expression was detected by staining with R-PE-conjugated goat anti-human IgG (gamma chain specific) followed by staining with serially diluted humanized hCDCP1 antibodies.
ELISA
ELISA was performed as previously described.Citation22 Briefly, antigens were immobilized onto MaxiSorp 384-well plates (Thermo Fisher Scientific) at 62.5 ng/well overnight at 4°C and then blocked with PBS containing 1% BSA. The cell culture supernatants or the diluted antibodies at 2.5 µg/mL in PBS/1% BSA subjected to analysis were added to the wells and incubated for 1 h at room temperature. The human IgG1 specifically bound to the immobilized antigens were detected by mouse anti-hIgG-Fc HRP-conjugated (SouthernBiotech, #9040-05). The assay was developed with TMB substrate (Dako), and the reaction was stopped with 1 N sulfuric acid (Nacalai Tesque). The absorbance at 450 nm was acquired using Infinite M1000 instrument (TECAN).
Affinity maturation
The ADLib® KI-AMP clones harboring the desired antibody genes were cultured in the medium for 2 weeks. At the end of the culture, the cells exhibiting improved antigen reactivity were isolated using FACS. Subsequently, 1.0 × 107 cells were stained with 30 nM biotinylated FLAG-Her2-ECD, an irrelevant antigen, as negative control and further stained with AlexaFluor 488-labeled streptavidin (Thermo Fisher Scientific, #S11223). The cells were subsequently incubated with 30 nM target antigens and/or biotinylated anti-FLAG M2 antibody followed by staining with AlexaFluor 647-labeled streptavidin and mouse anti-hIgG-Fc PE-conjugated (SouthernBiotech, #9040-09). The cells that did not react to the negative control and exhibited higher reactivity against the target antigens at the given IgG expression levels were sorted using BD FACSAria Fusion (BD Biosciences) onto the 96-well plate. The antigen reactivity of each viable clone was confirmed using FCM, and antibody sequences of the affinity-matured clones were determined.
SPR analysis
SPR analysis was performed as previously described.Citation23 Briefly, anti-hIgG capture antibodies (Cytiva, #BR100839) were immobilized onto the surface of a CM5 sensor chip (Cytiva, #BR100530) via amine coupling. For kinetic analysis, the culture supernatants or the purified antibodies were obtained. The purified antigens serially diluted with HBS-EP+ (hVEGF-A: 100 and 25 nM, hCDCP1-ECD: 300, 100, 33.3, 11.1, 3.7, and 1.23 nM) were injected for 240 s at a flow rate of 30 µL/min followed by 300 s of dissociation. Data were fitted with Biacore T200 evaluation software (Cytiva) using a 1:1 binding model.
Next-generation sequence
Genomic DNA was isolated from ~5,000 to 1,000,000 cells before and after the affinity maturation process. The VL regions were amplified using PCR with the sense primer 5’-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTCCAGGTTCCCTGGTGCAGGC-3’ (where N indicates index sequences) and antisense primer 5’-AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACGCTCTTCCGATCTCATATGAGCGACTCAC-3’. The VH regions were amplified with the sense primer 5’-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTCAGCGCTCTCTGTCCTTCC-3’ and antisense primer 5’-AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACGCTCTTCCGATCT CCAAAATCGCCGCGGC-3’. The PCR products were cleaned with left-side size selectin using SPRIselect (0.8 ×, Beckman Coulter). The final products were pooled with different indexes and sequenced on Illumina MiSeq (by Azenta Life Sciences) to obtain 2 × 300 base pairs read chemistry. The index sequences for the PCR amplifications are listed in Table S5.
Analysis of NGS data
The raw NGS sequence readings were cleaned up using the quality filtration protocol with Trimmomatic.Citation53 Briefly, the leading and trailing positions with quality < 4 (low quality) and the readings with an average quality per base < 15 at the scanning in a 4-base wide sliding window were trimmed, and readings with the lengths less than 36 bp were rejected. The remaining read pairs were merged into a single sequence using FLASHCitation54 and the merged sequences that did not contain PCR primer sequence were filtered out. The primer sequences that did not overlap to the antibody framework regions were trimmed and the remaining sequence was defined as the qualified sequence. The qualified sequences were aligned to the reference sequences of each strain and sectioned into the immunoglobulin framework and CDR according to the Kabat numbering scheme. Sequences of each strain before maturation (Day 0) were used as controls and sequence changes generated by affinity maturation were analyzed as changes from the control sequences.
Antibody humanization and purification
Humanized VH and VL amino acid sequences were designed according to the general procedure previously described.Citation55 The humanized VH and VL sequences were synthesized (by Azenta Life Sciences) and cloned into the antibody expression vectors, pFUSE-CHIg-hG1 and pFUSE2-CLIg-mk, respectively. Antibodies were transiently expressed using Freestyle 293-F cells and purified using Protein A affinity chromatography (Cytiva) followed by buffer exchange to PBS using PD-10 desalting columns (Cytiva).
Protein thermal shift assay
Thermal shift assay was carried out in a 96-well format using a StepOne plus real-time PCR system (Thermo Fisher Scientific). The antibodies were diluted to be 50 µg/mL in 1% methionine, 150 mM NaCl, 20 mM histidine with the fluorescent dye SYPRO Orange (1/5000, Thermo Fisher Scientific, #S6650). Data were collected at 1°C/min intervals from 25°C to 99°C; the Tm for the Fab domain was calculated from the measured melting curve which was analyzed using Protein Thermal Shift Software (Thermo Fisher Scientific).
Freeze-thaw study using size exclusion chromatography
Aliquot of antibody underwent three freeze-thaw cycles between −80 and 25°C and aggregation/fragmentation profiles of the antibodies were analyzed by size exclusion chromatography using an ACQUITY UPLC H-Class PLUS system (Waters) equipped with an ACQUITY UPLC Protein BEH SEC Column (200 Å, 1.7 µm, 4.6 mm×150 mm, Waters). Antibody solutions were diluted to be 0.2 mg/mL in 1% methionine, 150 mM NaCl, 20 mM histidine and the mobile phase consisted of 50 mM Na-phosphate, 300 mM NaCl, pH 7.0. Each sample was injected (8 μL) at a flow rate of 0.35 mL/min. UV absorption was measured at a wavelength of 220 nm.
Immunotoxicity assay
The cytotoxicity was examined using a cell viability assay. PC3 cells and DU145 cells were seeded at 2.0 × 103 cells/well in 96-well plates and incubated overnight at 37°C. Humanized anti-hCDCP1 monoclonal antibodies were added at the concentrations of 100, 20, 5, 1, 0.2 or 0.05 ng/mL, and the cells were incubated 5 min at R.T. Next, anti-Human IgG, PBD-conjugated IgGs with Cleavable Linker (Moradec, #AH-106PB-50) were added at the final concentration of 1 µg/mL. The cells were cultured in the medium for an additional 3 days (DU145) or 7 days (PC3). The viability of the cells was examined with a CellTiter-Glo Luminescent Cell Viability Assay (Promega). Statistical analysis was performed using Graphpad Prism software v. 9.3.0 (Graphpad software Inc.).
Author contributions
Conceptualization: H.I. Project administration: H.I., H.M. Methodology: H.M., A.S. Investigation: Vector design and knock-in cell generation - H.M., A.S. Performing affinity maturation - K.T., H.M., N.H. ELISA and thermal shift assay - H.M. FCM analysis and immunotoxicity assay - H.M., S.H. Sanger sequencing - N.H., H.M. NGS analysis - H.M., H.S., K.K. SPR analysis - H.M., K.-Y.L. Antibody stability analysis - H.M. Writing: Original draft - H.M. Review and editing - H.I., K.-Y.L., S.H., H.S., K.O.
Supplemental Material
Download Zip (5.1 MB)Acknowledgments
The authors gratefully thank Dr. Yoshiharu Sato (DNA Chip Research Inc.) for assistance with NGS data analysis; Kenro Shinagawa for assistance with immunotoxicity assay.
Disclosure statement
H.M., A.S., S.H., K.T., K.-Y.L., N.H., K.K., and H.I. are employees of Chiome Bioscience Inc. S.H., K.K., K.O., and H.S. hold stocks of Chiome Bioscience Inc. The authors have no additional financial interests.
Supplementary Material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2122275
Additional information
Funding
References
- Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, Wu HC. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27:1–19. doi:10.1186/s12929-019-0592-z.
- Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to watch in 2020. MAbs. 2020;12:1–24. doi:10.1080/19420862.2019.1703531.
- Valldorf B, Hinz SC, Russo G, Pekar L, Mohr L, Klemm J, Doerner A, Krah S, Hust M, Zielonka S. Antibody display technologies: selecting the cream of the crop. Biol Chem. 2021:000010151520200377. doi:10.1515/hsz-2020-0377.
- Chan DTY, Groves MAT. Affinity maturation: highlights in the application of in vitro strategies for the directed evolution of antibodies. Emerg Top Life Sci. 2021;5:601–08. doi:10.1042/ETLS20200331.
- Rajewsky K, Förster I, Cumano A. Evolutionary and somatic selection of the antibody repertoire in the mouse. Science. 1987;238:1088–94. PMID: 3317826. doi:10.1126/science.3317826.
- Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424:103–07. doi:10.1038/nature01760.
- Yu K, Huang FT, Lieber MR. DNA substrate length and surrounding sequence affect the activation-induced deaminase activity at cytidine. J Biol Chem. 2004;279:6496–500. doi:10.1074/jbc.M311616200.
- Wagner SD, Milstein C, Neuberger MS. Codon bias targets mutation. Nature. 1995;376:732–732. doi:10.1038/376732a0.
- Kepler TB. Codon bias and plasticity in immunoglobulins. Mol Biol Evol. 1997;14:637–43. doi:10.1093/oxfordjournals.molbev.a025803.
- Julian MC, Li L, Garde S, Wilen R, Tessier PM. Efficient affinity maturation of antibody variable domains requires co-selection of compensatory mutations to maintain thermodynamic stability. Sci Rep. 2017;7:1–13. doi:10.1038/srep45259.
- Julian MC, Lee CC, Tiller KE, Rabia LA, Day EK, Schick AJ, Tessier PM. Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng Des Sel. 2015;28:339–50. doi:10.1093/protein/gzv050.
- Rabia LA, Desai AA, Jhajj HS, Tessier PM. Understanding and overcoming trade-offs between antibody affinity, specificity, stability and solubility. Biochem Eng J. 2018;137:365. doi:10.1016/j.bej.2018.06.003.
- Tiler KE, Chowdhury R, Li T, Ludwig SD, Sen S, Maranas CD, Tessier PM. Facile affinity maturation of antibody variable domains using natural diversity mutagenesis. Front Immunol. 2017:8. doi:10.3389/fimmu.2017.00986.
- Cumbers SJ, Williams GT, Davies SL, Grenfell RL, Takeda S, Batista FD, Sale JE, Neuberger MS. Generation and iterative affinity maturation of antibodies in vitro using hypermutating B-cell lines. Nat Biotechnol. 2002;20:1129–34. doi:10.1038/nbt752.
- Lim AWY, Williams GT, Rada C, Sale JE. Directed evolution of human scFvs in DT40 cells. Protein Eng Des Sel. 2015;29(2):39–48. doi:10.1093/protein/gzv058.
- Chen C, Wang J, Zhao Y, Chen S, Hu Z, Chen L, Hang H. Enhancers improve the AID-induced hypermutation in episomal vector for antibody affinity maturation in mammalian cell display. Antibodies. 2018;7(4):42. doi:10.3390/antib7040042.
- Bowers PM, Horlick RA, Neben TY, Toobian RM, Tomlinson GL, Dalton JL, Jones HA, Chen A, Altobell L III, Zhang X, et al. Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proc Natl Acad Sci USA. 2011;108:20455. doi:10.1073/pnas.1114010108.
- Yin Y, Quinlan BD, Ou T, Guo Y, He W, Farzan M. In vitro affinity maturation of broader and more-potent variants of the HIV-1-neutralizing antibody CAP256-VRC26.25. Proc Natl Acad Sci USA. 2021;118:e2106203118. doi:10.1073/pnas.2106203118.
- Wellner A, McMahon C, Gilman MSA, Clements JR, Clark S, Nguyen KM, Ho MH, Hu VJ, Shin J-E, Feldman J, et al. Rapid generation of potent antibodies by autonomous hypermutation in yeast. Nat Chem Biol. 2021;2021:1–8. doi:10.1101/2020.11.11.378778.
- Kajita M, Okazawa T, Ikeda M, Todo K, Magari M, Kanayama N, Ohmori H. Efficient affinity maturation of antibodies in an engineered chicken B cell line DT40-SW by increasing point mutation. J Biosci Bioeng. 2010;110:351–58. doi:10.1016/j.jbiosc.2010.03.006.
- Chen S, Qiu J, Chen C, Liu C, Liu Y, An L, Jia J, Tang J, Wu L, Hang H. Protein & cell affinity maturation of anti-TNF-alpha scFv with somatic hypermutation in non-B cells. Protein Cell. 2012;460–69. doi:10.1007/s13238-012-2024-7.
- Seo H, Masuda H, Asagoshi K, Uchiki T, Kawata S, Sasaki G, Yabuki T, Miyai S, Takahashi N, Hashimoto S, et al. Streamlined human antibody generation and optimization by exploiting designed immunoglobulin loci in a B cell line. Cell Mol Immunol. 2021;18:1545–61. doi:10.1038/s41423-020-0440-9.
- Seo H, Masuoka M, Murofushi H, Takeda S, Shibata T, Ohta K. Rapid generation of specific antibodies by enhanced homologous recombination. Nat Biotechnol. 2005;23:731–35. doi:10.1038/nbt1092.
- Seo H, Ichi HS, Tsuchiya K, Lin W, Shibata T, Ohta K. An ex vivo method for rapid generation of monoclonal antibodies (ADLib® system). Nat Protoc. 2006;1:1502–06. doi:10.1038/nprot.2006.248.
- Hashimoto K, Kurosawa K, Murayama A, Seo H, Ohta K. B Cell-based seamless engineering of antibody Fc domains. PLoS ONE. 2016:1–22. doi:10.1371/journal.pone.0167232.
- Schusser B, Yi H, Collarini EJ, Izquierdo SM, Harriman WD, Etches RJ, Leighton PA. Harnessing gene conversion in chicken B cells to create a human antibody sequence repertoire. PLoS One. 2013;8:e80108. doi:10.1371/journal.pone.0080108.
- Leighton PA, Schusser B, Yi H, Glanville J, Harriman W. A diverse repertoire of human immunoglobulin variable genes in a chicken B cell line is generated by both gene conversion and somatic hypermutation. Front Immunol. 2015;6:126. doi:10.3389/fimmu.2015.00126.
- Kanayama N, Todo K, Takahashi S, Magari M, Ohmori H. Genetic manipulation of an exogenous non-immunoglobulin protein by gene conversion machinery in a chicken B cell line. Nucleic Acids Res. 2006;34:1–9. doi:10.1093/nar/gnj013.
- Arakawa H, Kudo H, Batrak V, Caldwell RB, Rieger MA, Ellwart JW, Buerstedde J-M. Protein evolution by hypermutation and selection in the B cell line DT40. Nucleic Acids Res. 2008;36:1–11. doi:10.1093/nar/gkm616.
- Brindle NPJ, Sale JE, Arakawa H, Buerstedde JM, Nuamchit T, Sharma S, Steele KH. Directed evolution of an angiopoietin-2 ligand trap by somatic hypermutation and cell surface display. J Biol Chem. 2013;288:33205. doi:10.1074/jbc.M113.510578.
- Buerstedde JM, Reynaud CA, Humphries EH, Olson W, Ewert DL, Weill JC. Light chain gene conversion continues at high rate in an ALV-induced cell line. EMBO J. 1990;9:921–27. doi:10.1002/j.1460-2075.1990.tb08190.x.
- Lin W, Hashimoto SI, Seo H, Shibata T, Ohta K. Modulation of immunoglobulin gene conversion frequency and distribution by the histone deacetylase HDAC2 in chicken DT40. Genes to Cells. 2008;13:255–68. doi:10.1111/j.1365-2443.2008.01166.x.
- Seo H, Yamada T, Hashimoto SI, Lin W, Ohta K. Modulation of immunoglobulin gene conversion incChicken DT40 by enhancing histone acetylation, and its application to antibody engineering. Biotechnol Genet Eng Rev. 2013;24:179–94. PMID: 18059633. doi:10.1080/02648725.2007.10648099.
- Arakawa H, Saribasak H, Buerstedde JM. Activation-induced cytidine deaminase initiates immunoglobulin gene conversion and hypermutation by a common intermediate. PLoS Biol. 2004:2. doi:10.1371/journal.pbio.0020179.
- Wilson PC, De Bouteiller O, Liu Y-J, Potter K, Banchereau J, Capra JD, Pascual V. Somatic hypermutation introduces insertions and deletions into immunoglobulin V genes. J Exp Med. 1998;187:59–70. doi:10.1084/jem.187.1.59.
- Saberi A, Nakahara M, Sale JE, Kikuchi K, Arakawa H, Buerstedde J-M, Yamamoto K, Takeda S, Sonoda E. The 9-1-1 DNA clamp is required for immunoglobulin gene conversion. Mol Cell Biol. 2008;28:6113–22. doi:10.1128/MCB.00156-08.
- Kohzaki M, Nishihara K, Hirota K, Sonoda E, Yoshimura M, Ekino S, Butler JE, Watanabe M, Halazonetis TD, Takeda S. DNA polymerases ν and θ are required for efficient immunoglobulin V gene diversification in chicken. J Cell Biol. 2010;189:1117–27. doi:10.1083/jcb.200912012.
- Wortmann A, He Y, Deryugina EI, Quigley JP, Hooper JD. The cell surface glycoprotein CDCP1 in gancer—Insights, opportunities, and challenges. IUBMB Life. 2009;61:723–30. doi:10.1002/iub.198.
- Bachl J, Carlson C, Gray-Schopfer V, Dessing M, Olsson C. Predicting antibody developability profiles through early stage discovery screening. MAbs. 2020;12:e1743053. doi:10.1080/19420862.2020.1743053.
- Bachl J, Carlson C, Gray-Schopfer V, Dessing M, Olsson C. Increased transcription levels induce higher mutation rates in a hypermutating cell line. J Immunol. 2001;166:5051–57. doi:10.4049/jimmunol.166.8.5051.
- Martin A, Scharff MD. Somatic hypermutation of the AID transgene in B and non-B cells. Proc Natl Acad Sci USA. 2002;99:12304–08. doi:10.1073/pnas.192442899.
- Abe T, Branzei D, Hirota K. DNA damage tolerance mechanisms revealed from the analysis of immunoglobulin V gene diversification in Avian DT40 cells. Genes. 2018;2018(9):614. doi:10.3390/genes9120614.
- Kawamoto T, Araki K, Sonoda E, Yamashita YM, Harada K, Kikuchi K, Masutani C, Hanaoka F, Nozaki K, Hashimoto N, et al. Dual roles for DNA polymerase η in homologous DNA recombination and translesion DNA synthesis. Mol Cell. 2005;20:793–99. doi:10.1016/j.molcel.2005.10.016.
- Blagodatski A, Batrak V, Schmidl S, Schoetz U, Caldwell RB, Arakawa H, Buerstedde JM. A cis-acting diversification activator both necessary and sufficient for AID-mediated hypermutation. PLoS Genet. 2009:5. doi:10.1371/journal.pgen.1000332.
- Kohler KM, McDonald JJ, Duke JL, Arakawa H, Tan S, Kleinstein SH, Buerstedde J-M, Schatz DG. Identification of core DNA elements that target somatic hypermutation. J Immunol. 2012;189:5314. doi:10.4049/jimmunol.1202082.
- Buerstedde JM, Alinikula J, Arakawa H, McDonald JJ, Schatz DG. Targeting of somatic hypermutation by immunoglobulin enhancer And enhancer-like sequences. PLoS Biol. 2014:12. doi:10.1371/journal.pbio.1001831.
- McDonald JJ, Alinikula J, Buerstedde J-M, Schatz DG. A critical context-dependent role for E boxes in the targeting of somatic hypermutation. J Immunol. 2013;191:1556. doi:10.4049/jimmunol.1300969.
- Wei L, Chahwan R, Wang S, Wang X, Pham PT, Goodman MF, Bergman A, Scharff MD, MacCarthy T. Overlapping hotspots in CDRs are critical sites for V region diversification. Proc Natl Acad Sci USA. 2015;112:728–37. doi:10.1073/pnas.1500788112.
- Matsuda T, Bebenek K, Masutani C, Rogozin IB, Hanaoka F, Kunkel TA. Error rate and specificity of human and murine DNA polymerase η. J Mol Biol. 2001;312:335–46. doi:10.1006/jmbi.2001.4937.
- Mayorov R VI, IB ALR, Gearhart PJ. DNA Polymerase η contributes to strand bias of mutations of A versus T in immunoglobulin genes. J Immunol. 2005;174:7781–86. doi:10.4049/jimmunol.174.12.7781.
- Tang C, Krantsevich A, MacCarthy T. Deep learning model of somatic hypermutation reveals importance of sequence context beyond targeting of AID and Polη hotspots. bioRxiv. 2021:1–44. doi:10.1101/2021.08.03.453264.
- Hirota K, Sonoda E, Kawamoto T, Motegi A, Masutani C, Hanaoka F, Szüts D, Iwai S, Sale JE, Lehmann A, et al. Simultaneous disruption of two DNA polymerases, Polη and Polζ, in Avian DT40 cells unmasks the role of Polη in cellular response to various DNA lesions. PLoS Genet. 2010;6:1–13. doi:10.1371/journal.pgen.1001151.
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi:10.1093/bioinformatics/btu170.
- Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63. doi:10.1093/bioinformatics/btr507.
- Tsurushita N, Hinton PR, Kumar S. Design of humanized antibodies: from anti-Tac to Zenapax. Methods. 2005;36:69–83. doi:10.1016/j.ymeth.2005.01.007.