
ABSTRACT
Immunoglobulin G (IgG) has served as a traditional framework for antibody-based biology, and engineering approaches for creating multispecific therapeutics have greatly expanded its applicability. Despite these developments, there are limits to the functionality of IgG with respect to effector cells that can be activated and paratope valency that can be obtained. Other Ig isotypes have distinct functions that can engage and activate different effector cells, and some can be found naturally in higher-order assemblies. In an effort to expand the repertoire of multispecific designs for other antibody isotypes, we present here engineering of the first heterodimeric IgA Fc that can be produced at high purity with native IgA-like thermal stability. The crystal structure confirmed the accuracy of the in silico model used for engineering and that the mutations introduced at the CH3 interface do not perturb the overall IgA Fc structure. Affinity measurements and on-cell neutrophil binding demonstrated that the heterodimeric IgA Fc retains the ability to bind FcαRI, an important prerequisite for IgA-based therapeutics designed to interact with effector cells, such as neutrophils. Given the ability of IgA antibodies to multimerize via interaction with the J-chain, the designs presented here could also be used to generate multispecific, multimeric scaffolds that leverage valency to increase clustering and specificity via avidity. Taken together, the newly developed heterodimeric IgA Fc platform allows for the development of novel, multifunctional, and multimeric molecules that have the potential to transform the next generation of antibody therapeutics.
Abbreviations
CE-SDS: capillary electrophoresis sodium dodecyl sulfate; DSC: differential scanning calorimetry; FACS: fluorescence-activated cell sorting; FSA: full-sized antibody; Her2: human epidermal growth factor receptor 2; MFI: mean fluorescent intensity; OAA: one-armed antibody; PBS: phosphate-buffered saline; PDB: Protein Data Bank; SEC: size-exclusion chromatography; prepSEC (preparative SEC); RMSD: root-mean-square deviation; RU: resonance units; SPR: surface plasmon resonance; TAA: tumor-associated antigen; WT: wild-type.
Introduction
To date, all approved antibody-based clinical biologics contain an immunoglobulin G (IgG)-derived framework. Widespread development of IgG-based therapeutics has been made possible due to extensive efforts to optimize methods for IgG manufacturing and to characterize and optimize IgG stability, target binding affinity, effector function, and half-life. IgG-derived monoclonal antibodies have also been used as scaffolds to engineer next-generation multifunctional, multispecific biologics, a number of which are used in the clinic.Citation1 Examples of these include immune cell-redirecting bispecifics, multispecifics targeting multiple tumor-associated antigens, multiple checkpoints or a combination of the two, and biparatopic antibodies that target different epitopes of the same target.Citation1–3
For many of these, a heterodimeric Fc is the key prerequisite to manufacture a stable and pure multispecific therapeutic. Several heterodimeric IgG Fc platforms have been developed using a variety of rational design approaches. For example, electrostaticCitation4–6 and stericCitation6–8 design strategies based on the introduction of point mutations in the CH3 domain of the IgG Fc can drive heterodimer formation and exclude homodimer formation. Other methods are based on engineering chimeric CH3 domains to incorporate portions of IgG and other antibody isotypes (such as IgM, IgA, and IgD) or T-cell receptors that favor heterodimeric over homodimeric Fc pairs.Citation9,Citation10
Although IgG is the most understood and therapeutically utilized antibody isotype, there are limitations to its utility in terms of effector cell types that can be engaged and activated and maximum paratope valency that can be achieved. There is increasing evidence that IgA could represent an avenue toward novel therapeutic strategies due to its ability to form multimers and interact with FcαRI (CD89).Citation11–15 IgA consists of two subclasses, IgA1 and IgA2, and a set of distinct allotypesCitation16 and can form dimeric, tetrameric, and pentameric multimers via the interaction of its tailpiece with the J-chain.Citation17 IgA Fc can bind to FcαRI expressed on myeloid cells, and via this receptor can potently activate neutrophil cellular cytotoxicity. Neutrophils are an integral part of the innate immune system and are the most abundant leukocytes found in the human blood.Citation18 Activation of neutrophils can lead to a variety of potent pro-inflammatory effects, including the recruitment of additional effector cells via chemokine secretion, degranulation, production of reactive oxidative species, and killing of target cells or pathogens via antibody-dependent cellular cytotoxicity and antibody-dependent cellular phagocytosis.Citation18 A recently discovered mechanism for neutrophil-mediated tumor cell clearing by trogocytosis of IgA-opsonized tumor cells has also been described.Citation19–25 Some of these neutrophil-mediated effector functions have recently been exploited in the development of IgA-based therapeutics aimed at solid and liquid tumor cell killing or clearance of infectious disease-causing pathogens.Citation13 Furthermore, it has been shown that the IgA-mediated killing of tumor cells by neutrophils can be enhanced by concurrently eliciting CD47-SIRPα checkpoint blockade.Citation26
Although IgA has broad therapeutic potential and is the focus of a growing body of work,Citation11,Citation27,Citation28 current technical hurdles around IgA half-life (6 days) and manufacturability have stalled translation of IgA-based molecules into clinical candidates. As a result, several antibody engineering strategies aimed at circumventing these limitations while maintaining FcαRI-engagement have been pursued. These range from designing a bispecific IgG antibody, with one binding arm targeting FcαRI,Citation29 to engineering IgG/A hybrid and chimeric molecules aimed at combining long half-life and proven developability of IgG with the effector functionality of IgA.Citation25,Citation30
Taken together, IgA shows potential for a wide array of therapeutic applications ranging from immuno-oncology and infectious diseases to autoimmunity. Similar to how a heterodimeric Fc expanded the accessible formats for IgG, a solution for a heterodimeric IgA Fc could open a new chapter in antibody-based therapeutics. Here, we present engineering of a heterodimeric IgA Fc scaffold with high purity and native IgA-like stability and retained ability to bind FcαRI. This newly designed scaffold provides a first-in-class stable and manufacturable multispecific IgA platform capable of activation of neutrophils via the FcαRI. A multispecific IgA platform in turn provides access to new biological pathways for next-generation, multifunctional, IgA-based immunotherapies and thus functionalities that are untapped by IgG.
Results
A heterodimeric IgA Fc was designed using a rational structure-based in silico approach
To design a heterodimeric IgA Fc, we used a rational structure-guided in silico design approach. As described in the Methods, mutations in the IgA CH3:CH3 interface were structurally modeled and energetically evaluated using physics-based and knowledge-based in silico metrics. Mutations were selected according to their predicted ability to disrupt homodimeric Fc formation (negative design) while stabilizing a heterodimeric Fc (positive design) (). The in silico design metrics used to determine the combinations of mutations to be tested in vitro for their ability to favor formation of heterodimeric IgA Fc are shown in Table S1.
Figure 1. Steric and electrostatic in silico design strategies to drive IgA Fc heterodimer formation and OAA format used to test designs in vitro. (a) Cartoon depictions of steric and electrostatic negative and positive design concepts for in silico modeling of mutations to drive heterodimerization of an IgA Fc (left). Examples of structural models of steric and electrostatic designs in an IgA CH3 predicted to favor heterodimer formation of the Fc (right). Cartoon depiction of concepts are overlayed on the structural models for clarity. (b) Example of in silico knowledge-based and physics-based metrics used to rank favorable steric and electrostatic designs (single design shown). (c) Antibody formats used to test heterodimeric designs in vitro. Mutations favoring formation of a heterodimeric IgA Fc assemble into a OAA species that can be resolved by size from homodimeric Fc-only and full-sized antibody species. The OAA used is composed of anti-Her2 IgG1 Fab, IgG1/IgA2 hybrid hinge and IgA2m1 Fc.
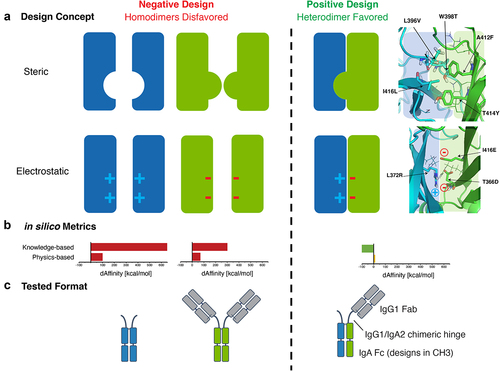
The mutations tested to drive heterodimeric IgA Fc formation and exclude IgA Fc homodimerization can largely be grouped into either steric or electrostatic designs, as illustrated in . Designs were ranked based on the predicted energetic differences between homo- and hetero-dimers. Top steric designs contained mutations that introduced large hydrophobic side chains, such as phenylalanine, tyrosine, methionine, and tryptophan at positions A412 and T414 on one Fc chain (referred to throughout as chain A) and substitution of W398 on the opposite chain (referred to throughout as chain B) for a residue with a small side chain, such as threonine or leucine. Mutations at these three amino acid positions were ultimately selected as the core steric design predicted to favor heterodimer formation and were part of all steric designs tested in vitro ().
Table 1. Summary of purification results and CH3 thermal stability of heterodimeric IgA Fc steric designs.
The electrostatic designs tested here involved the introduction of salt bridges to promote heterodimerization, whereas homodimerization was disfavored by electrostatic repulsion of same charge residues across the CH3:CH3 interface. All electrostatic designs included the replacement of the R372-R418 pi–pi interaction in the CH3:CH3 interface with a salt bridge (Table S2). This newly constructed salt bridge was accompanied by the introduction of additional salt bridges between buried residues in the hydrophobic core of the interface (e.g., T366, L372, I416, ). Replacement of an important arginine pi–pi stacking interaction found in the wild-type IgA Fc, as well as introduction of charged residues in the environment of the buried core as described here, had the potential to significantly skew the energetics of the CH3 interface to favor heterodimerization, but also carried significant risk of destabilization of the protein.
High purity of heterodimeric IgA Fc was observed for steric designs
To assess whether the heterodimeric IgA Fc designs identified in silico resulted in increased heterodimeric purity in vitro, mutations were introduced in a one-armed antibody (OAA) format of IgA where chain A consisted of a modified IgA2m1 FcCitation21 with an IgG Fab and chain B consisting of only the modified IgA2m1 Fc to allow for separation of homodimers and heterodimers by molecular weight (). Using this size-based strategy, mutations that drive the formation of heterodimeric IgA Fc (chain A + chain B OAA, 95 kDa) could be resolved from chain A homodimers (chain A + chain A full-sized antibody (FSA) species, 142 kDa) and chain B homodimers (chain B + chain B dimeric Fc species, 47 kDa). Expression of a wild-type IgA OAA, in the absence of heterodimeric Fc-driving mutations, yielded a mix of species. Specifically, homodimeric FSA (~25%), homodimeric Fc-only (~25%), and heterodimeric OAA species (~50%) were detected in capillary electrophoresis sodium dodecyl sulfate (CE-SDS) and analytical size-exclusion chromatography (SEC) (). This result was consistent with the expected theoretical distribution of antibody species following the equimolar expression of both wild-type IgA Fc chains.Citation7
Figure 2. Lead steric designs 6, 10, and 11 drive formation of a heterodimeric IgA Fc as determined by formation of OAA species in vitro and possess wild-type IgA-like thermal stability. (a) Non-reducing CE-SDS profiles of IgA Fc OAA variants after affinity purification. Data are shown for variants comprising a wild-type IgA CH3 and those containing heterodimer-driving mutations from steric designs 1–4 and 6–11. (b) Analysis of IgA OAA variants by analytical SEC after affinity purification. Data is shown for a wild-type IgA Fc (left) as well as steric design 6 (right). (c) Overlay of DSC thermograms measured for SEC-purified wild-type IgA Fc (black) and heterodimeric IgA Fc containing mutations from steric designs 2 (red), 3 (orange), 6 (yellow), 10 (light green), and 11 (green) OAA variants. The first and second transitions are attributed to CH3 and CH2 + Fab unfolding, respectively.
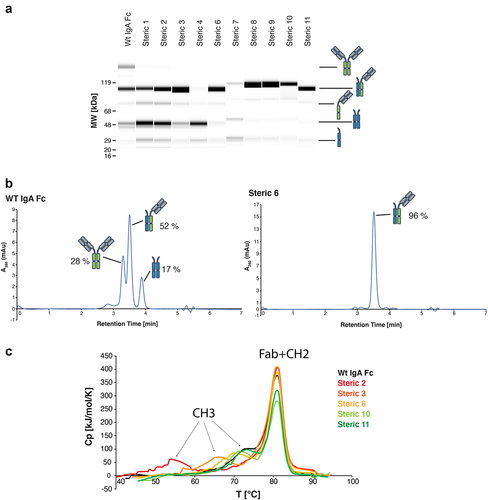
Variants containing mutations that were predicted to promote heterodimer formation showed notably different expressions and distributions of antibody species when compared to wild-type IgA OAA. Variants containing electrostatic design mutations (Table S2) did not result in detectable expression in culture supernatant as determined by SDS-PAGE (data not shown). This result supports our hypothesis regarding the importance of the arginine pi–pi interactions for the stability of the IgA CH3:CH3 interface. Conversely, variants containing mutations to drive heterodimerization of the Fc via steric designs 1–11 () had detectable expression as determined by SDS-PAGE (data not shown). However, steric 5 did not yield sufficient material to support further characterization. All other designs (steric 1–4, steric 6–11) were purified by affinity chromatography for further analysis by CE-SDS and analytical SEC. In addition to the chain A/chain B OAA heterodimers, varying levels of Fc-only chain B homodimers and half antibody species were observed, while FSA chain A homodimers were not present for any of the steric designs (). Steric designs 3, 6, 10, and 11 showed the highest purity of OAA heterodimeric species, with steric 6 reaching heterodimeric purity of >95% by both CE-SDS and analytical SEC (, ). Conversely, steric 4 contained no OAA heterodimer or FSA homodimer species, but only Fc-only homodimer and the corresponding chain B by itself, indicating a problem in the expression of chain A in this design.
Heterodimeric IgA OAA species from steric 1–3 and steric 6–11 designs as well as the wild-type IgA OAA were further purified by prepSEC to remove any FSA (chain A homodimer) or Fc-only species (chain B homodimer). Analysis by CE-SDS and HPLC-SEC was used to confirm purification of highly homogeneous samples containing 90–100% of OAA heterodimeric IgA species and final yields ranged from 30 to 200 mg/L of expression culture ().
Lead steric designs showed thermal stability similar to wild-type IgA by differential scanning calorimetry
Differential scanning calorimetry (DSC) was performed on the purified IgA OAAs to assess how the introduced mutations driving heterodimer formation in the lead steric designs 2, 3, 6, 10, and 11 affected the thermal stability of the heterodimeric IgA Fc compared to wild-type IgA. The DSC thermogram of wild-type IgA OAA with an unmodified IgA CH3:CH3 interface showed two transitions, the first one at 74.2°C and a second higher enthalpy transition at 81.0°C (). The second transition at 81.0°C was detected for all investigated IgA OAAs and can be attributed to the overlapped unfolding of both the Fab and the CH2 domain, neither of which was mutated in the tested designs. The Tm of the first transition varied depending on the mutations introduced in the steric designs and was assigned to the unfolding of the CH3 domain. Of note, this first transition in IgA thermal unfolding has previously been attributed to the unfolding of CH2.Citation25 However, data presented herein suggest that this first, lower temperature transition in IgA stems from CH3 unfolding and that the order of thermal stability of CH2 versus CH3 in IgA is reversed when compared to IgG.
The three designs that showed the highest CH3 thermal stability were steric 6, 10, and 11 with CH3 Tm values observed at 71.9°C, 72.0°C, and 73.6°C, respectively (, ). The modified CH3 in steric designs 3 and 2 were destabilized compared to wild-type IgA CH3 with Tm values of 65.9°C and 55.0°C, respectively (, ). The thermal stability of the CH3 domain of heterodimeric variants bearing steric 6, 10, or 11 design mutations was within ~2°C of the wild-type IgA CH3, and these mutants were identified as lead variants ().
A crystal structure of the heterodimeric IgA Fc revealed a heterodimeric interface consistent with in silico models and a retained overall wild-type-like IgA Fc structure
As efforts to crystallize heterodimeric IgA Fc on its own were not successful, a crystallization strategy using heterodimeric IgA Fc (steric 6) in complex with SSL7 at 1:2 stoichiometric ratio was pursued. shows the heterodimeric IgA Fc structure obtained in complex with SSL7 (Protein Data Bank (PDB) ID 7TRZ). The structure was solved to 2.35 Å resolution by molecular replacement using the previously solved 3.2 Å resolution homodimeric IgA Fc:SSL7 structure (PDB ID 2QEJ) as a search model. Data collection and refinement statistics are summarized in Table S3. The heterodimeric IgA Fc:SSL7 protein complex crystallized with one SSL7 bound to each IgA Fc protomer in the asymmetric unit, analogous to the 2QEJ structure (). Overall, the heterodimeric IgA Fc has a high structural homology with the wild-type homodimeric IgA Fc from both previously reported IgA Fc structures in PDB IDs 2QEJ and 1OW0 with RMSD (root-mean-square deviation) of 0.94 Å and 0.88 Å across Cα atoms in the Fc, respectively. Analysis of the electron density surrounding individual heterodimerizing mutations in IgA Fc steric 6 (chain A – A412F and T414Y; chain B – L369V, W398T, and I416L) revealed a mixture of the two possible orientations of the asymmetrical CH3:CH3 domain interface in the asymmetric unit. Based on the observation that one orientation nonetheless predominated and the inability to accurately deconvolute the two forms, the mutated residues were modeled at 100% occupancy.
Figure 3. The crystal structure of heterodimeric IgA Fc steric design 6 was solved in complex with S. aureus SSL7 (PDB ID: 7TTZ). The heterodimeric IgA Fc has high structural homology with the predicted in silico model and differs from wild-type IgA Fc only at the CH3:CH3 interface. (a) Cartoon representation of the heterodimeric IgA Fc (steric 6) in pale cyan and pale green in complex with two S. aureus SSL7 monomers in pale orange and pale yellow (PDB ID: 7TTZ). (b) Superimposition of the heterodimeric IgA Fc (steric 6) crystal structure (pale blue and pale green) and wild-type IgA Fc (PDB ID: 2QEJ; gray) illustrates the structural homology between heterodimeric and homodimeric IgA CH2 and CH3 domain structure. The peptide backbone is drawn as ribbon and mutated residues and equivalent wild-type residues in the CH3:CH3 interface are drawn as sticks. (c) Cartoon representation of heterodimeric IgA (steric 6) CH3:CH3 interface with mutated residues drawn as sticks (right) compared to wild-type homodimeric IgA Fc (PDB ID: 2QEJ) interface (left) and the predicted in silico model (middle).
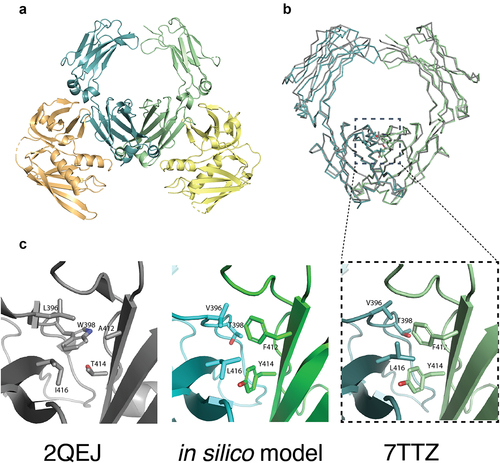
The mutated amino acid side chains in the heterodimeric IgA Fc were found to be fully buried at the CH3:CH3 interface in the crystal structure, except for L396V, which had 18% solvent-exposed surface area (vs. 4% for wild-type L396). In addition to targeting buried residues that are part of direct interactions across the interface and therefore have high energetic contributions to dimerization, this design strategy was intentionally followed to preserve the surface and two-fold rotational symmetry of the native IgA Fc. Consistent with the in silico prediction and modeling, the mutations introduced regions of steric complementarity and promoted assembly of a heterodimeric IgA Fc. The RMSD of the heterodimeric IgA Fc CH3 structure relative to the computationally modeled structure was 1.2 Å across Cα atoms for residues 345–450. When comparing the side-chain orientations, several residues showed different rotamers in the in silico model when compared to the crystal structure, but overall, key interchain interactions were observed as predicted (). For example, the introduction of a T414Y mutation created a new interchain hydrogen bond between the T414Y hydroxyl and the previously unsaturated T368 hydroxyl, as predicted by the in silico model (Figure S1).
The crystal structure of the heterodimeric IgA Fc also provides insight into the disulfide connectivity at the hinge of IgA. The data collected here support a model with intrachain disulfide bonds between C242 and C301 in each chain and an interchain disulfide bond between C299 and C299. This is consistent with the disulfide pattern modeled in PDB ID 2QEJ but differs from PDB ID 1OW0, which models interchain disulfides between Cys 242 and Cys 299 (Figure S2a, c). Furthermore, the analysis of the electron density suggests that a population of molecules in the heterodimeric IgA Fc crystal may also contain an interchain disulfide bond between C241 and C241, though it is not modeled by the final structure (Figure S2b, c). It is possible that there is a mix of reduced cysteines, oxidized cysteines, and interchain disulfide bonded cysteines present at position C241.
Heterodimeric IgA Fc designs show preserved binding to FcαRI by surface plasmon resonance
To investigate the possibility that mutations introduced at the CH3 interface in the steric designs could alter FcαRI-dependent IgA effector function, the binding affinity of lead IgA variants (steric 6, 10, 11) for FcαRI was measured by surface plasmon resonance (SPR). For wild-type IgA Fc, an apparent KD value of 11 nM was observed (Table S4). As the IgA Fc contains two binding sites for the FcαRI, but the data were fitted to a 1:1 Langmuir binding model, a poor fit was obtained (, top). Kinetic binding analysis of heterodimeric IgA Fc (steric 6, 10, and 11) OAAs and wild-type IgA Fc OAA to FcαRI yielded an apparent KD of 23, 22, and 15 nM, respectively (, bottom, , Table S4). The measured 1.5 – 2-fold difference in apparent affinity measured between wild-type and heterodimeric IgA Fc OAA variants suggests that the CH3 interface mutations in steric designs 6, 10, and 11 do not strongly affect binding to FcαRI and are unlikely to compromise downstream cellular effector functions. This finding is further supported by the high structural homology measured between the crystal structure of heterodimeric IgA Fc (steric 6) compared to wild-type IgA Fc ().
Figure 4. IgA heterodimeric Fc variants retain binding to the FcαRI. (a) Surface plasmon resonance sensorgrams for wild-type IgA OAA (WT) (top, apparent KD = 11 nM), IgA OAA FcαRI-KO 1x (middle, KD = 273 nM), and heterodimeric IgA Fc (steric 6) OAA (bottom, apparent KD = 23 nM) measured at FcαRI ligand density of 14 RU, fit using a 1:1 Langmuir model. (b) Dependence of apparent KD of binding of IgA OAA variants to FcαRI on ligand density of FcαRI immobilized on the SPR sensor chip. Data for a wild-type IgA OAA, steric heterodimeric designs 6, 10, 11 as well as a single sided FcαRI-binding-deficient IgA Fc based on heterodimeric design 6 (IgA OAA FcαRI-KO 1x) is shown.
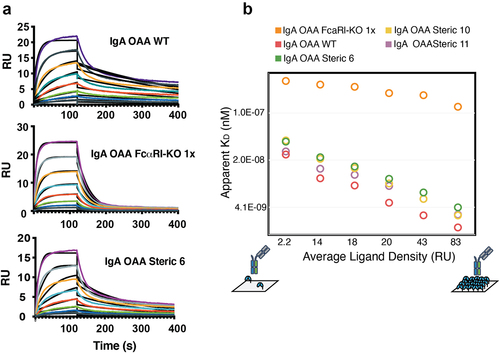
To interrogate the contribution of avidity to the bivalent binding of FcαRI to the IgA Fc, we generated two heterodimeric IgA OAA variants using the steric 6 design that contained either a one-sided (IgA OAA FcαRI-KO 1x) or two-sided FcαRI binding site knockout (IgA OAA FcαRI-KO 2x). Introducing the Fc mutation F443A to both IgA chains in IgA OAA FcαRI-KO 2x was found to abrogate binding to FcαRI (Table S4), consistent with the data reported previously.Citation31 However, when F443A was introduced into a single IgA Fc chain in FcαRI-KO 1x, a 1:1 Langmuir kinetic model yielded a good fit with a KD of 273 nM (, middle, Table S4), one order of magnitude lower affinity than the apparent KD from wild-type IgA OAA. This finding supports that the poor fit of the 1:1 Langmuir model to the data collected for the IgA wild-type and heterodimeric OAAs is due to the bivalent binding of FcαRI to the IgA Fc and demonstrates the gain in apparent affinity due to avidity. The previously reported binding of FcαRI to IgA found apparent KD values ranging from 122 to 455 nM.Citation21,Citation25,Citation30,Citation32,Citation33
We further investigated the effect that different immobilized FcαRI ligand densities had on the measured apparent KD for the IgA OAA variants with SPR. Consistent with the model for bivalent IgA Fc binding to FcαRI, increasing the density of FcαRI resulted in higher apparent affinities for wild-type and heterodimeric IgA Fc variants containing two FcαRI binding sites due to avidity (, Table S4). Similar KD values were measured for the IgA OAA FcαRI-KO 1x across the varying FcαRI ligand densities as expected as the 1:1 binding would not be affected by receptor density on the SPR sensor chip surface (). These data further confirm that both FcαRI binding sites are functional in the IgA heterodimeric Fc steric designs containing mutations at the CH3 interface.
Heterodimeric IgA Fc designs bind FcαRI expressed on neutrophils comparable to wild-type IgA Fc
To translate the in vitro SPR binding results to a more physiologically relevant ex vivo setting, the binding of heterodimeric IgA Fc designs to commercially available, freshly thawed neutrophils was assessed by flow cytometry. Neutrophils were characterized post-freeze-thaw to confirm the expression of FcαRI and the absence of surface human epidermal growth factor receptor 2 (Her2) by flow cytometry, as the IgA variants are Her2 targeted (Figure S4a, b). Heterodimeric IgA Fc designs (steric 6, 10, and 11) were all found to bind neutrophils to similar levels, within 1.6-fold relative to wild-type IgA Fc, when incubated with neutrophils at 1 µM (Figure S4c). As it is known that neutrophil viability decreases quickly post-thaw, a short incubation on ice was used during which it is possible that equilibrium was not reached. Nevertheless, it allowed for the qualitative assessment of the rank order of IgA variant binding. As expected, FcαRI-binding-deficient heterodimeric IgA OAA FcαRI-KO 2x did not bind neutrophils and heterodimeric IgA OAA FcαRI-KO 1x bound to approximately 30% mean fluorescent intensity (MFI) relative to the parental steric 6 heterodimeric IgA Fc design (Figure S4c). Steric design 11 showed a similar binding profile to wild-type IgA. Steric designs 6 and 10 bound neutrophils to higher MFI than wild-type IgA (Figure S4c). This was unexpected and will require further experimentation. Taken together, these on-cell binding measurements of natively expressed FcαRI on human neutrophils support that the heterodimeric IgA Fc designs retain wild-type IgA-like ability to engage neutrophils.
Discussion
Successful design of a stable IgA Fc with high heterodimer specificity was achieved using a structure-guided rational design approach that we had previously used to engineer a heterodimeric IgG Fc.Citation8 Combining in-depth structural and connectivity analysis and extensive engineering experience with rotamer-packing-based modeling and physics- and knowledge-based in silico metrics allowed the identification of a small number of lead designs in relatively few engineering steps, yielding a top design, with >95% pure heterodimeric IgA Fc following a single purification step. As high purity and thermal stability are key biophysical characteristics important for the developability and manufacturability of a biologic,Citation34 obtaining a pure heterodimeric IgA Fc was prioritized in the engineering strategy. Minimizing the propensity of undesired homodimeric side products to form is favorable as they are potentially hard to separate, lower the yield, and increase production cost. Furthermore, as low thermal stability can be a liability for the developability of therapeutics,Citation34 we showed that IgA Fc heterodimer-driving mutations introduced in the lead designs only marginally affect the thermal stability of the heterodimeric IgA Fc compared to the wild-type IgA Fc. We also obtained the crystal structure of one of the lead designs and demonstrated that our modeling approach accurately predicted the immediate structural environment of mutations introduced. Our results confirmed that the heterodimeric IgA Fc structure is conserved when compared to the wild-type IgA Fc and showed comparable binding to FcαRI by SPR and on-cell binding to human neutrophils, indicating that the functionality of the molecules is not affected by the mutations in the designs. These heterodimeric IgA Fc designs allow for the assembly of multispecific IgA-based scaffolds that can facilitate the creation of the next generation of IgA-based therapeutics, a budding field in biological research.Citation11
A clear application of the heterodimeric IgA Fc platform is the development of bispecific IgA therapeutics. Just as platforms for heterodimeric IgG Fc scaffolds have enabled the development of bispecific and biparatopic IgG molecules, IgG-based T-cell engagers, and other IgG-based multispecific scaffolds, engineering of heterodimeric IgA Fc bolsters the therapeutic potential for IgA as well. For example, there is evidence that targeting immunomodulatory receptors found in innate myeloid immune cells (e.g., CD47) at the same time as targeting tumor-associated antigens (TAAs) in cancer cells can increase IgA-mediated neutrophil anti-tumor activity in vitro and in vivo.Citation26 Combining immunomodulatory and TAA targeting in a single, bispecific IgA antibody has the potential to increase the additional activity seen through geometry and avidity not accessible by the combination of two monospecific IgA antibodies ().
Figure 5. Applications of a heterodimeric IgA Fc in the development of IgA multispecifics and interrogation of IgA biology. The heterodimeric IgA Fc designs described herein (center) can be used for a variety of applications. These include engineering of an asymmetric IgA Fc, for example to introduce a FcRn-binding motif to increase the half-life of IgA while preserving binding to FcαRI (top left). A heterodimeric IgA can also be used to create multispecific therapeutics with a variety of formats, ranging from OAAs, bispecifics and biparatopics to multispecific multimers (top right) which can be used to target the mucosa when bound to the secretory compartment (bottom left). Heterodimeric IgAs can furthermore be used as a tool to interrogate fundamental questions surrounding IgA biology such as the effect of FcαRI binding valency on the activation of effector cells such as neutrophils (bottom right).
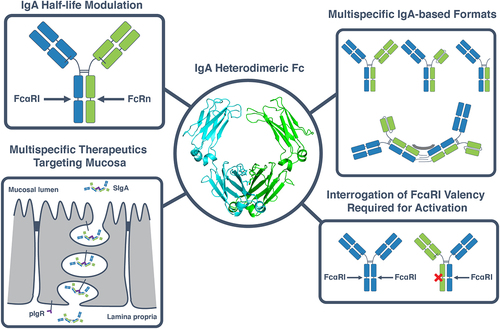
One major hurdle to the translation of IgA-based therapeutics to the clinic is its relatively short half-life of ~6 days compared to ~23 days for IgG.Citation35,Citation36 One of the mechanisms that equips IgG with its relatively long half-life is its ability to bind FcRn and get recycled to the cell surface from the endosomal pathway.Citation37 IgA does not natively bind to FcRn and, therefore, chimeric engineering strategies have been employed to combine FcαRI binding native to IgA and FcRn binding native to IgG.Citation36 One of the challenges is that if IgA and IgG Fc structures are overlayed, FcαRI and FcRn binding sites are located in the same region close to the CH2:CH3 interface and are therefore considered mutually exclusive structurally (Figure S3). Specifically, grafting of the FcαRI binding site from IgA to IgG results in the loss of FcRn bindingCitation30 and the same would likely be the case if the FcRn binding interface would be grafted from IgG to IgA. A heterodimeric Fc enables future engineering of both receptor-binding sites in a single Fc (). For a hybrid molecule with one FcαRI and one FcRn binding site to be viable, both interactions need to be functional when each of the Fc receptors is engaged in a monovalent fashion. While it is known that monovalent engagement of FcRn conveys the ability of IgG to be recycled, thereby increasing half-life,Citation38 there is still debate over the valency dependence of IgA Fc receptor engagement for FcαRI-mediated effector cell activity. It is hypothesized that the 2:1 engagement of FcαRI by IgA Fc is responsible for potent activation of neutrophils by IgA compared to IgG, which binds FcyR on neutrophils at 1:1 stoichiometry.Citation39 Further work is required to test whether 1:1 binding of IgA to FcαRI is sufficient to activate neutrophils productively. An IgA FcαRI-KO 1x accessible via the IgA heterodimeric Fc platform described here () will help answer this question.
Beyond greatly expanding the accessible formats and geometries of a monomeric antibody as has been seen for IgG,Citation40 a heterodimeric IgA Fc also enables a new category of multispecific, multimeric monoclonal antibodies. One feature of IgA is that a higher valency is naturally accessible via J-chain-based multimers.Citation17,Citation32 These multivalent formats can provide avidity effects that increase apparent affinity of low-affinity paratopes and increase clustering and specificity for target cells with high receptor density.Citation17,Citation41 Furthermore, the presence of the J-chain enables covalent interaction of dimeric IgA with pIgR for transcytosis of the mucosa, leaving multimeric IgA bound to the secretory component.Citation42 This feature of IgA could be used to direct a multispecific, multimeric antibody to mucosal environments following intravenous administration.Citation43 Combining this higher valency with a heterodimeric IgA Fc gives rise to geometries and valencies of formats that were previously inaccessible and an opportunity to deliver multispecific IgA to mucosal environments ().
In contrast to IgG Fc, the IgA Fc is not readily crystallizable, and we were unable to produce diffraction quality crystals of an IgA Fc by itself. This is consistent with the fact that only two IgA Fc crystal structures are deposited in the PDB, both in complex with either human FcαRI (PDB ID 1OW0)Citation44 or staphylococcal SSL7 (PDB ID 2QEJ).Citation45 A noteworthy observation is that the heterodimeric IgA Fc co-crystal structure reported herein (PDB ID 7TTZ) has disulfide bonds in the hinge region that are similar to those in 2QEJ but differ from 1OW0 (Figure S2). In addition, the analysis of the electron density obtained for the heterodimeric IgA Fc suggests that an additional interchain hinge disulfide might be present at position C241. In the case of IgG2, for example, it has been shown that different disulfide connectivity between the hinge and the light chain (known as A/B forms) results in molecules with different biological properties.Citation46 Although the biological effect of different hinge disulfide connectivity in IgA crystal structures remains unclear, it is conceivable that interconversion of different IgA forms through different disulfide bond connectivities could result in different biological properties, although further investigation is required.
Using a rational and systematic approach to concurrently address purity and stability design requirements, we engineered a heterodimeric IgA Fc that also retains native IgA Fc-like biophysical properties. This strategy prioritized retaining quality attributes like purity and stability that can positively affect the developability of the next generation of bispecifics and other complex therapeutic proteins. The success of these efforts underlines the impact of protein engineering on the development of a protein scaffold to be used subsequently as a platform for the design of multispecific IgA therapeutics.
Materials and methods
In silico modeling and design of IgA heterodimeric Fc
An extensive structural analysis of the IgA Fc CH3:CH3 interface (PDB ID: 2QEJ) was performed to characterize residues at the interface according to their energetic contribution to dimerization.Citation45 For this analysis, the ZymeCADTM suite of proprietary tools was used to determine the connectivity and energetics derived from knowledge-based and physics-based potentials. This was done on a static structure, as well as a 50 ns explicit molecular dynamics trajectory as described previously.Citation47 Guided by results from this initial analysis and in a first “negative design” round, residues were selected for the introduction of mutations predicted to disrupt dimerization. These mutations were chosen based on two main design concepts illustrated in . Negative electrostatic designs relied on the introduction of same-charge pairs and the associated repulsion across the interface, while negative steric designs were based on the introduction of cavities or steric clashes at the interface. These negative designs were modeled and evaluated energetically using proprietary in silico tools. In a second “positive design” step, additional mutations were introduced with the goal of rescuing heterodimerization. The stabilization of the heterodimeric complex was either based on the introduction of salt bridges via opposing charges across the interface or on the accommodation of residues with large side chains by cavities introduced on the opposite side of the interface. Designs with the largest energetic differences between homodimers and heterodimers were selected to be expressed and evaluated (Table S1).
In vitro assessment of lead heterodimer mutations in IgA one-armed antibody format
To assess the ability of asymmetric mutations introduced into the IgA CH3 domains to drive heterodimeric pairing of an IgA Fc, an IgA OAA format was used (). The sequence of IgA Fc was based on the CH2 and CH3 domains of the IgA2m1 allotype.Citation16 In the OAA format, one-half antibody consisted of an IgG1-based Fab with anti-Her2 paratopeCitation48 that was fused in the heavy chain to an IgA2m1 Fc, referred to as chain A. A chimeric hinge comprising the upper IgG1 hinge (EPKSC) followed by an IgA2 hinge (RVPPPPP) was used to connect the IgG1 Fab to the IgA2m1 Fc. The other Fc-containing chain of the OAA consisted of an IgA2 hinge (RVPPPPP) fused to an IgA2m1 Fc, referred to as chain B. Mutations predicted to drive heterodimeric pairing listed in were introduced in the CH3 domains of the Fc for the OAA constructs. In some constructs, the F443A mutation, which has been reported to abrogate binding to the FcαRI,Citation31 was introduced either on one chain (IgA OAA FcαRI-KO 1x) or on both chains (IgA OAA FcαRI-KO 2x). In all IgA constructs, mutations were made to both heavy chains to remove the cysteine responsible for disulfide bonding to pIgR in wild-type IgA (C311S) and to remove and stabilize a glycosylation site (N337T, I338L, and T339S).Citation21 Furthermore, the C-terminus of the IgA Fc was truncated to residue G453 to remove the tailpiece.
Cloning, expression, and purification of samples
The genes encoding the antibody heavy and light chains were synthesized and subcloned into mammalian expression vector pTT5 containing a signal peptide (EFATMRPTWAWWLFLVLLLALWAPARGCitation49). Mutations were introduced into the IgA CH3 by site-directed mutagenesis. A construct for the expression of human FcαRI (residues 22–227, GenBank: CAA38089.1) was created using the same mammalian expression vector pTT5 and signal peptide with a C-terminal His6 tag preceded by a TEV cleavage site. A construct for recombinant expression of Staphylococcus aureus protein SSL7 (residues 31–231, GenBank accession code WP_000769814) was generated in a pET32a vector containing an N-terminal thioredoxin and His6 tag followed by a thrombin cleavage site.
Transient co-expression of one light chain and two heterodimeric heavy chains A and B using three individual vectors was conducted in Expi293FTM cells (Thermo Fisher, Waltham, MA) for expression of the IgA OAA variants. Clarified culture supernatants were applied to a CaptureSelectTM IgA affinity matrix (ThermoFisher, Waltham, MA, USA) column equilibrated with phosphate-buffered saline (PBS), pH 7.2 for purification. The column was washed with PBS, pH 7.2 and protein eluted with 0.1 M glycine, pH 2.5. The eluted samples were neutralized by adding 10% (v/v) 1 M Tris, pH 9 to yield a final pH of 6–7. Following CaptureSelectTM IgA affinity purification, samples were assessed for heterodimeric purity using CE-SDS (SDS LabChip® GXII, Perkin Elmer, Waltham, MA, USA) and analytical SEC (HPLC-SEC).
HPLC-SEC was performed on an Agilent Technologies 1260 Infinity LC system using an Agilent Technologies AdvanceBio SEC 300A column at 25°C. 5 µL of sample at approximately 1 mg/ml was injected into the column at a flow rate of 1 mL/min using PBS, pH 7.4 as mobile phase. Chromatograms were extracted at 280 nm, and peak integration was performed using the OpenLAB CDS ChemStation software.
IgA samples were further purified by preparative SEC (prepSEC) to separate heterodimeric IgA Fc OAA species from homodimeric Fc species using a Superdex 200 Increase 10/300 GL column (GE Life Sciences) equilibrated with PBS pH 7.4. The eluted fractions corresponding to heterodimeric IgA Fc OAA were pooled and analyzed by CE-SDS and HPLC-SEC to confirm purity.
Recombinant FcαRI-His was expressed in Expi293FTM cells and purified from clarified culture supernatant by Ni2+ affinity chromatography. Eluted protein was buffer exchanged into PBS pH 7.4 and analyzed by CE-SDS and analytical SEC to confirm purity.
Recombinant SSL7 was expressed in Escherichia coli BL21 (DE3) cells as a thioredoxin fusion protein and purified by Ni2+ affinity chromatography. The eluted protein was cleaved using thrombin protease and purified again by Ni2+ affinity chromatography to separate SSL7 from the thioredoxin-His6 tag. SSL7 was further purified by prepSEC as described above using 20 mM HEPES (pH 7.5), 150 mM NaCl as a mobile phase. The heterodimeric IgA Fc:SSL7 complex was prepared by mixing heterodimeric IgA Fc (steric 6) and SSL7 at molar ratio of 1:2.1 and then re-purified by prepSEC using 20 mM HEPES (pH 7.5), 150 mM NaCl as a mobile phase.
Crystallization, data collection, and structure determination of heterodimeric IgA Fc:SSL7 complex
Cocrystals of heterodimeric IgA Fc (steric 6) and SSL7 were generated by vapor diffusion against 30% (v/v) PEG MME 550, 0.1 M HEPES (pH 7.5), and 0.2 M MgCl2. Crystals were briefly soaked in reservoir condition supplemented with ethylene glycol/glycerol mixture for cryoprotection and flash cooled in liquid nitrogen. Diffraction data were collected at the Stanford Synchrotron Radiation Light source and integrated and scaled using autoXDS to 2.35 Å resolution.Citation50
The structure was determined by molecular replacement using PHASER in Phenix.Citation51 The search model consisted of the previously reported IgA Fc:SSL7 structure (PDB ID 2QEJ). We adopted the numbering of residues from that structure herein (see Table S5 for the different IgA numbering systems). The model was built in COOTCitation52 and refinements were performed to a resolution of 2.35 Å using phenix.refine.Citation53 Structural validation was performed with Molprobity,Citation54 including analysis of the Ramachandran plots, with greater than 96% of the residues in the most favored conformations and no non-Gly residues in the disallowed regions. Five percent of reflections were set aside for calculation of Rfree. Data collection and crystallographic refinement statistics are presented in Table S3. Atomic coordinates and structure factors were deposited in the PDB with ID 7TTZ.
Differential scanning calorimetry
Thermal stability of purified heterodimeric IgA Fc OAA variants was measured using DSC. Samples were diluted in PBS, pH 7.4 to 0.5–1 mg/ml for analysis using NanoDSC (TA Instruments, Newcastle, DE, USA). Each sample was injected and heated from 25°C to 95°C with a scan rate of 1°C/min under 60 psi nitrogen pressure. Thermograms were analyzed using the NanoAnalyze software (TA Instruments). The matching PBS, pH 7.4 buffer thermogram was subtracted from the sample thermogram and the baseline fit using a sigmoidal curve. Data were deconvoluted using a two-state scaled DSC model to extract transition temperatures.
Surface plasmon resonance
Binding affinity of IgA variants to FcαRI was measured using a Biacore T200 instrument (Cytiva). Experiments were conducted at a temperature of 25°C using HBS EP+ (10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA, and 0.05% v/v surfactant P20) as running buffer. IgA variant binding affinity was measured using different FcαRI-His ligand densities on a CM5 Series S sensor chip. The surface was prepared by first covalently immobilizing anti-histidine antibodies using standard amine coupling method according to manufacturer’s instruction (Cytiva). Using multi-cycle kinetics, FcαRI-His was then captured onto the anti-His surface to reach a range of ligand densities between 2.6 and 83 resonance units (RU). Eight concentrations of each IgA variant were prepared using a two-fold dilution series. The concentration range was 2.3–300 nM for wild-type and heterodimeric IgA OAA variants and 7.8–1000 nM for IgA FcαRI-binding site knockout variants (FcαRI-KO 1x and FcαRI-KO 2x). Diluted IgA samples and double blank buffers were injected at a flow rate of 30 µl/min for 120 s. The dissociation time used was 500 s and 10 mM glycine-HCl (pH 1.5) was used to regenerate the surface between each cycle for 30 s at 30 µl/min. The binding kinetics and affinity were analyzed using the 1:1 Langmuir binding model in Biacore T200 Evaluation Software v3.0. Kinetic values are summarized in Table S4.
On-cell binding of IgA variants to neutrophils measured by flow cytometry
Frozen vials of purified human peripheral blood neutrophils were purchased from STEMCELL Technologies Inc. (catalog # 200–0384) for measurement of IgA binding to native FcαRI. Surface expression of FcαRI and Her2 was determined by flow cytometry using mouse anti-human FcαRI (CD89) mAb conjugated to PE (catalog # MA5-28547, Invitrogen) and FITC conjugated mouse anti-human Her2 antibody (catalog # 324404, BioLegend). Briefly, neutrophils were thawed and incubated with fluorophore-labeled mAb diluted in fluorescence-activated cell sorting (FACS) buffer (2% fetal bovine serum in PBS) containing 1:1000 eBioscience fixable viability dye eFlour 450 (catalog # 65–0863-14, Invitrogen) for 30 min on ice. Following incubation, cells were washed twice and analyzed on a BD LSR FortessaTM X-20 Cell Analyzer.
To measure binding to neutrophils by wild-type and heterodimeric IgA Fc variants, IgA variants were diluted in a 4 point, 4-fold titration series starting from 2000 nM. Freshly thawed neutrophils were diluted to 1.5 million cells/mL and incubated with IgA variant at 1:1 (v/v) ratio for 30 minutes on ice. Cells were washed and then resuspended with RPE-labeled goat anti-human kappa light chain secondary antibody (cat #206009, Bio-Rad Laboratories Inc.) diluted in FACS buffer containing 1:1000 eBioscience fixable viability dye eFlour 450 (catalog # 65–0863-14, Invitrogen). Samples were incubated with secondary antibody for 30 minutes on ice. Plates were then washed twice and analyzed on a BD LSR FortessaTM X-20 Cell Analyzer.
Supplemental Material
Download Zip (3.5 MB)Acknowledgments
We thank Charles Stevens, Sifa Arrafi, Daniel Patton, Rhonda Wideman, Stephanie Masterman, David Douda, and Brandon Clavette for insightful discussions of in silico tools, purification strategies, business opportunities, project management, and the biology of IgA.
Disclosure statement
FH, MMV, SC, JL, PF, ES, BSM, SD, TSVK, and EEC are current or former employees of Zymeworks Inc., and may hold shares or stock options in Zymeworks Inc. Zymeworks Inc. provided financial support for the study. The work described here is the subject of a patent application assigned to Zymeworks Inc. MJB and FP have no conflicts of interest to declare.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2141637
Additional information
Funding
References
- Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18(8):585–13. doi:10.1038/s41573-019-0028-1.
- Ellerman D. Bispecific T-cell engagers: towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods. 2019;154:102–17. doi:10.1016/j.ymeth.2018.10.026.
- Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, Trichard S, Cesari C, Sapet M, Bosco F, et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell. 2019;177(7):1701–13 e16. doi:10.1016/j.cell.2019.04.041.
- Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, Ng SB, Born T, Retter M, Manchulenko K, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285(25):19637–46. doi:10.1074/jbc.M110.117382.
- De Nardis C, Hendriks LJA, Poirier E, Arvinte T, Gros P, Bakker ABH, de Kruif J. A new approach for generating bispecific antibodies based on a common light chain format and the stable architecture of human immunoglobulin G1. J Biol Chem. 2017;292:14706–17. doi:10.1074/jbc.M117.793497.
- Leaver-Fay A, Froning KJ, Atwell S, Aldaz H, Pustilnik A, Lu F, Huang F, Yuan R, Hassanali S, Chamberlain A, et al. Computationally designed bispecific antibodies using negative state repertoires. Structure. 2016;24(4):641–51. doi:10.1016/j.str.2016.02.013.
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody C H 3 domains for heavy chain heterodimerization. Protein Eng. 1996;9(7):617–21. doi:10.1093/protein/9.7.617.
- Von Kreudenstein TS, Escobar-Carbrera E, Lario PI, D’Angelo I, Brault K, Kelly J, Durocher Y, Baardsnes J, Woods RJ, Xie MH, et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: quality by molecular design. MAbs. 2013;5(5):646–54. doi:10.4161/mabs.25632.
- Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo KM. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel. 2010;23:195–202.
- Skegro D, Stutz C, Ollier R, Svensson E, Wassmann P, Bourquin F, Monney T, Gn S, Blein S. Immunoglobulin domain interface exchange as a platform technology for the generation of Fc heterodimers and bispecific antibodies. J Biol Chem. 2017;292(23):9745–59. doi:10.1074/jbc.M117.782433.
- van Tetering G, Evers M, Chan C, Stip M, Leusen J. Fc engineering strategies to advance IgA antibodies as therapeutic agents. Antibodies. 2020;9. doi:10.3390/antib9040070.
- Gruijs M, Sewnath CAN, Egmond MV. Therapeutic exploitation of neutrophils to fight cancer. Semin Immunol. 2021;57:101581. doi:10.1016/j.smim.2021.101581.
- Davis SK, Selva KJ, Kent SJ, Chung AW. Serum IgA Fc effector functions in infectious disease and cancer. Immunol Cell Biol. 2020;98:276–86. doi:10.1111/imcb.12306.
- de Sousa-Pereira P, Woof JM. IgA: structure, function, and developability. Antibodies. 2019;8(4):57. doi:10.3390/antib8040057.
- Biswas S, Mandal G, Payne KK, Anadon CM, Gatenbee CD, Chaurio RA, Costich TL, Moran C, Harro CM, Rigolizzo KE, et al. IgA transcytosis and antigen recognition govern ovarian cancer immunity. Nature. 2021;591(7850):464–70. doi:10.1038/s41586-020-03144-0.
- Chintalacharuvu KR, Raines M, Morrison SL. Divergence of human alpha-chain constant region gene sequences. A novel recombinant alpha 2 gene. J Immunol. 1994;152:5299–304.
- Kumar N, Arthur CP, Ciferri C, Matsumoto ML. Structure of the secretory immunoglobulin A core. Science. 2020;367(6481):1008–14. doi:10.1126/science.aaz5807.
- Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. The balancing act of neutrophils. Cell Host Microbe. 2014;15(5):526–36. doi:10.1016/j.chom.2014.04.011.
- Horner H, Frank C, Dechant C, Repp R, Glennie M, Herrmann M, Stockmeyer B. Intimate cell conjugate formation and exchange of membrane lipids precede apoptosis induction in target cells during antibody-dependent, granulocyte-mediated cytotoxicity. J Immunol. 2007;179(1):337–45. doi:10.4049/jimmunol.179.1.337.
- Boross P, Lohse S, Nederend M, Jansen JH, van Tetering G, Dechant M, Peipp M, Royle L, Liew LP, Boon L, et al. Ig A EGFR antibodies mediate tumour killing in vivo. EMBO Mol Med. 2013;5(8):1213–26. doi:10.1002/emmm.201201929.
- Lohse S, Meyer S, Meulenbroek LA, Jansen JH, Nederend M, Kretschmer A, Klausz K, Möginger U, Derer S, Rösner T, et al. An anti-EGFR IgA that displays improved pharmacokinetics and myeloid effector cell engagement in vivo. Cancer Res. 2016;76(2):403–17. doi:10.1158/0008-5472.CAN-15-1232.
- Lohse S, Loew S, Kretschmer A, Jansen JHM, Meyer S, Ten Broeke T, Rösner T, Dechant M, Derer S, Klausz K, et al. Effector mechanisms of IgA antibodies against CD20 include recruitment of myeloid cells for antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity. Br J Haematol. 2018;181(3):413–17. doi:10.1111/bjh.14624.
- Dechant M, Vidarsson G, Stockmeyer B, Repp R, Glennie MJ, Gramatzki M, van de Winkel JGJ, Valerius T. Chimeric IgA antibodies against HLA class II effectively trigger lymphoma cell killing. Blood. 2002;100(13):4574–80. doi:10.1182/blood-2002-03-0687.
- Brandsma AM, Ten Broeke T, Nederend M, Meulenbroek LA, van Tetering G, Meyer S, Jansen JHM, Beltrán Buitrago MA, Nagelkerke SQ, Németh I, et al. Simultaneous targeting of fcgammaRs and fcalphari enhances tumor cell killing. Cancer Immunol Res. 2015;3(12):1316–24. doi:10.1158/2326-6066.CIR-15-0099-T.
- Borrok MJ, Luheshi NM, Beyaz N, Davies GC, Legg JW, Wu H, Dall’Acqua WF, Tsui P. Enhancement of antibody-dependent cell-mediated cytotoxicity by endowing IgG with FcalphaRI (CD89) binding. MAbs. 2015;7(4):743–51. doi:10.1080/19420862.2015.1047570.
- Treffers LW, Ten Broeke T, Rosner T, Jansen JHM, van Houdt M, Kahle S, Schornagel K, Verkuijlen PJJH, Prins JM, Franke K, et al. IgA-mediated killing of tumor cells by neutrophils is enhanced by CD47-sirpalpha checkpoint inhibition. Cancer Immunol Res. 2020;8(1):120–30. doi:10.1158/2326-6066.CIR-19-0144.
- Sterlin D, Gorochov G. When Therapeutic IgA Antibodies Might Come of Age. Pharmacology. 2021;106(1–2):9–19. doi:10.1159/000510251.
- Breedveld A, van Egmond M. IgA and FcalphaRI: pathological roles and therapeutic opportunities. Front Immunol. 2019;10:553. doi:10.3389/fimmu.2019.00553.
- Heemskerk N, Gruijs M, Temming AR, Heineke MH, Gout DY, Hellingman T, Tuk CW, Winter PJ., Lissenberg-Thunnissen S, Bentlage AE, de Donatis M. Augmented antibody-based anticancer therapeutics boost neutrophil cytotoxicity. J Clin Invest. 2021;131. doi:10.1172/JCI134680.
- Kelton W, Mehta N, Charab W, Lee J, Lee CH, Kojima T, et al. IgGA: a “cross-isotype” engineered human Fc antibody domain that displays both IgG-like and IgA-like effector functions. Chem Biol. 2014;21(12):1603–09. doi:10.1016/j.chembiol.2014.10.017.
- Posgai MT, Tonddast-Navaei S, Jayasinghe M, Ibrahim GM, Stan G, Herr AB. FcαRI binding at the IgA1 C H 2–C H 3 interface induces long-range conformational changes that are transmitted to the hinge region. Proc Natl Acad Sci U S A. 2018;115(38):E8882–E91. doi:10.1073/pnas.1807478115.
- Lombana TN, Rajan S, Zorn JA, Mandikian D, Chen EC, Estevez A, Yip V, Bravo DD, Phung W, Farahi F, et al. Production, characterization, and in vivo half-life extension of polymeric IgA molecules in mice. MAbs. 2019;11(6):1122–38. doi:10.1080/19420862.2019.1622940.
- Rouwendal GJ, van der Lee MM, Meyer S, Reiding KR, Schouten J, de Roo G, Egging DF, Leusen JH, Boross P, Wuhrer M, et al. A comparison of anti-HER2 IgA and IgG1 in vivo efficacy is facilitated by high N-glycan sialylation of the IgA. MAbs. 2016;8(1):74–86. doi:10.1080/19420862.2015.1102812.
- Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, Cao Y. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A. 2017;114(5):944–49. doi:10.1073/pnas.1616408114.
- Delacroix DL, Elkom KB, Geubel AP, Hodgson HF, Dive C, Vaerman JP. Changes in size, subclass, and metabolic properties of serum immunoglobulin A in liver diseases and in other diseases with high serum immunoglobulin A. J Clin Invest. 1983;71(2):358–67. doi:10.1172/JCI110777.
- Mester S, Evers M, Meyer S, Nilsen J, Greiff V, Sandlie I, Leusen J, Andersen JT. Extended plasma half-life of albumin-binding domain fused human IgA upon pH-dependent albumin engagement of human FcRn in vitro and in vivo. MAbs. 2021;13(1):1893888. doi:10.1080/19420862.2021.1893888.
- Ghetie V, Hubbard JG, Kim JK, Tsen MF, Lee Y, Ward ES. Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. Eur J Immunol. 1996;26(3):690–96. doi:10.1002/eji.1830260327.
- Tesar DB, Tiangco NE, Bjorkman PJ. Ligand valency affects transcytosis, recycling and intracellular trafficking mediated by the neonatal Fc receptor. Traffic. 2006;7(9):1127–42. doi:10.1111/j.1600-0854.2006.00457.x.
- Brandsma AM, Bondza S, Evers M, Koutstaal R, Nederend M, Jansen JHM, Rösner T, Valerius T, Leusen JH, Ten Broeke T. Potent Fc receptor signaling by IgA Leads to superior killing of cancer cells by neutrophils compared to IgG. Front Immunol. 2019;10:704. doi:10.3389/fimmu.2019.00704.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9(2):182–212. doi:10.1080/19420862.2016.1268307.
- Kumar Bharathkar S, Parker BW, Malyutin AG, Haloi N, Huey-Tubman KE, Tajkhorshid E, Stadtmueller BM. The structures of secretory and dimeric immunoglobulin A. Elife. 2020;9. doi:10.7554/eLife.56098.
- Wei H, Wang JY. Role of polymeric immunoglobulin receptor in IgA and IgM transcytosis. Int J Mol Sci. 2021;22(5):2284. doi:10.3390/ijms22052284.
- Renegar KB, Small PA, Boykins LG, Wright PF. Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J Immunol. 2004;173(3):1978–86. doi:10.4049/jimmunol.173.3.1978.
- Herr AB, Ballister ER, Bjorkman PJ. Insights into IgA-mediated immune responses from the crystal structures of human FcalphaRI and its complex with IgA1-Fc. Nature. 2003;423(6940):614–20. doi:10.1038/nature01685.
- Ramsland PA, Willoughby N, Trist HM, Farrugia W, Hogarth PM, Fraser JD, Wines BD. Structural basis for evasion of IgA immunity by Staphylococcus aureus revealed in the complex of SSL7 with Fc of human IgA1. Proc Natl Acad Sci U S A. 2007;104(38):15051–56. doi:10.1073/pnas.0706028104.
- Yu X, Chan HTC, Fisher H, Penfold CA, Kim J, Inzhelevskaya T, Mockridge CI, French RR, Duriez PJ, Douglas LR, et al. Isotype switching converts anti-CD40 antagonism to agonism to elicit potent antitumor activity. Cancer Cell. 2020;37(6):850–66 e7. doi:10.1016/j.ccell.2020.04.013.
- Spreter Von Kreudenstein T, Lario PI, Dixit SB. Protein engineering and the use of molecular modeling and simulation: the case of heterodimeric Fc engineering. Methods. 2014;65(1):77–94. doi:10.1016/j.ymeth.2013.10.016.
- Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Kotts CM, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A. 1992;89(10):4285–89. doi:10.1073/pnas.89.10.4285.
- Barash S, Wang W, Shi Y. Human secretory signal peptide description by hidden Markov model and generation of a strong artificial signal peptide for secreted protein expression. Biochem Biophys Res Commun. 2002;294(4):835–42. doi:10.1016/S0006-291X(02)00566-1.
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66(2):125–32. doi:10.1107/S0907444909047337.
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(4):658–74. doi:10.1107/S0021889807021206.
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(12):2126–32. doi:10.1107/S0907444904019158.
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68(4):352–67. doi:10.1107/S0907444912001308.
- Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, Jain S. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315.