
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Clinical anti-drug-antibody (ADA) responses represent a substantial obstacle to the development of efficacious therapeutic antibodies. The enhanced ADA production against the idiotype (Id) often displayed by cancer immunotherapy antibodies (CitAbs) can lead to exposure loss and subsequently affect anti-tumor efficacy and cause undesired effects on safety. Thus, ADA responses contribute to prolonged clinical development and high attrition rates. Most conventional therapeutic antibodies are now of human origin or humanized proteins, and are hence immunologically tolerized in most patients. In contrast, the contribution of additional factors, other than the protein sequence, to the higher rates of clinical ADA to certain CitAbs, remains poorly understood. Here, we used human immunoglobulin gamma 1 (IgG1) transgenic mice (named “hIgG1 transgenic mice” or “TG”), which are immunologically tolerant to human IgG1, to study the immunogenicity of 13 conventional antibodies and 2 CitAbs. We found that tolerance to non-germline encoded Ids is maintained in part by the function of neonatal Fc-receptor (FcRn). Additionally, the incorporation of T cell-engaging moieties like an interleukin 2 (IL-2)-based immunocytokine or a CD3ε-specific antigen-binding fragment (Fab) was sufficient to revert tolerance and trigger ADA production directed to the Id of these compounds. We postulate that T cell receptor or IL-2 receptor activation may result in activation of unresponsive T cells specific for the crystallizable fragment (Fc) that typically inactivate Id-specific B cells and mediate “linked-antigen tolerance”. Reversal of this unresponsiveness by the action of CitAbs on T cells may be the cause of undesired ADA responses.
Abbreviations
ADA Anti-Drug Antibodies; BCR B Cell Receptor; BId Idiotype-specific B Cell; BiTE Bispecific T cell Engager; BMC Bone Marrow Chimeric Mice; BSA Bovine Serum Albumin; CDR Complementary Determining Region; CEA Carcinoembryonic Antigen; CIT Cancer Immunotherapy; CitAbs Cancer Immunotherapy Antibodies; DC Dendritic Cell; ELISA Enzyme-Linked Immunosorbent Assay; FcRn Neonatal Fc Receptor; FcyR Fc gamma Receptor; GM-CSF Granulocyte-Macrophage Colony Stimulating Factor; gMFI Geometric Mean Fluorescence Intensity; H Heavy Chain; IC Immune Complex; Id Idiotype; IgA Immunoglobulin alpha; IgG1 Immunoglobulin gamma 1; IL-2 Interleukin 2; IL-2R Interleukin 2 Receptor; IL2v Interleukin 2 Variant; IVIG1 Intravenous Immunoglobulin 1; KLH Keyhole Limpet Hemocyanin; L Light Chain; MAPPs MHC-associated Peptide Proteomics; MHC Major Histocompatibility Complex; PBMC Peripheral Blood Mononuclear Cells; PBS Phosphate Buffered Saline; SHM Somatic Hypermutation; scFv Single-chain Variable Fragment; TCR T cell Receptor; TFc Fc-specific T cell; TId Id-specific T cell; UV Ultraviolet; V Variable.
Introduction
The broad clinical use of therapeutic antibodies has revealed that in some cases this treatment results in unwanted immune responses and the production of anti-drug antibodies (ADA). In some cases, ADA can cause substantial exposure loss that affects efficacy, with the potential to provoke undesired adverse events.Citation1 Presently, it is unknown which factors, in addition to foreign immunogenic protein sequences, contribute to the onset of ADA production by some therapeutic antibodies and not by others. In a recent survey, the collected ADA data from 60 clinical antibodies revealed that the majority (76%) of these antibodies display an immunogenicity rate of less than 10% and the minority (10%) of the antibodies tested cause an ADA rate higher than 20%.Citation2 Thus, most human or humanized conventional therapeutic antibodies are weakly immunogenic in humans. In contrast, bispecific, immunomodulatory antibodies frequently used in cancer immunotherapy (CIT) – herein referred to as CitAbs – appear to cause ADA with a higher incidence than expected, given their generally human/humanized origin.Citation1,Citation3 Therefore, it is crucial to better understand the mechanisms involved in breaking tolerance toward CitAbs, leading to ADA responses.
CitAbs are engineered bispecific antibodies bearing binding arms directed at multiple targets.Citation4 They can cross-link agonistic receptors like the T cell receptor (TCR) or interleukin 2 receptor (IL-2R) on immune cells and a tumor antigen or checkpoint inhibitor on the respective target cancer cells in order to induce tumor cell killing by the immune cell.Citation5 Here, we aimed to study the immunogenic properties of CitAbs by making use of a transgenic system that provides immune tolerance to a broad range of human antibodies of the immunoglobulin gamma 1 (IgG1) isotype, the hIgG1 transgenic mouse.Citation6 As this transgenic system has previously been shown to sense immunogenic modifications of an otherwise tolerated human therapeutic antibody,Citation7 we reasoned that it would also help with the study of the immunogenicity of CitAbs pre-clinically. To this end, we first conducted a study to validate this transgenic mouse system using 13 commercially available conventional therapeutic antibodies with known clinical immunogenicity rates. The study revealed that the immunogenicity in hIgG1 transgenic mice is substantially reduced as compared to control mice. Then, we tested the immunogenic potential of CEA-IL2v (cergutuzumab amunaleukin) and CEA-TCB (cibitasamab), two prototypical CitAbs,Citation8,Citation9 and investigated possible mechanisms responsible for the enhanced ADA responses they elicit in the hIgG1 transgenic mice.
In current models of immune tolerance, early expression of germline-encoded self-antigens in the bone marrow and the thymus predisposes B and T lymphocytes, respectively, for a lifelong state of unresponsiveness toward most self-proteins. The case of antibodies is peculiar because, although germline-encoded, the amino acid composition of the antigen-binding site can be further altered during the process of gene rearrangement (to assemble the variable (V) region) and of somatic hypermutation (SHM), which increases the affinity to antigen during maturation of antibody responses. Thus, the resulting array of complementary-determining regions (CDR) of mature, high-affinity antibodies that bind defined antigens can diverge substantially from corresponding germline-encoded sequences. Nonetheless, the variant amino acid segments of secreted antibodies during immune responses normally do not provoke antibody responses to the newly formed CDR combinations or idiotypes (Id), even if they deviate from the tolerated V regions. Similarly, the iterative clinical infusion of large quantities of antibody-based therapeutics does not generally result in immunogenic ADA responses. In a simplified experimental system, we made use of the mouse immune system, transgenically rendered tolerant to human antibodies, and asked the two basic questions: 1) How is tolerance to unforeseen V region epitopes (idiotypes) maintained? and 2) What causes the enhanced immunogenic properties of CitAbs, as compared to conventional antibodies?
The results of our study revealed two regulatory pathways associated with the Fc region of antibodies that down-modulate anti-Id ADA responses. We propose a model to explain the predominant tolerance to non-germline Id determinants and its breakdown by CitAbs and discuss mitigation strategies derived thereof.
Results
Immunogenicity of conventional antibodies
As described previously, hIgG1 transgenic mice are immunologically tolerant to a broad range of human IgG1 antibodies.Citation6 Here, we interrogated if this transgenic system could reproduce experimentally the clinical ADA incidence of 13 therapeutic antibodies of the IgG1 isotype surveyed by Dingman and Balu-Iyer.Citation2 We selected 5 antibodies with an immunogenicity rate higher than 10% and 8 antibodies with an immunogenicity rate lower than 10% (). According to regulatory documents and patents, cross-reactivity to the target is absent or only low in mice. Hence, ADA data in transgenic mice refer mostly to the intrinsic antigenicity, and not to the function-related immunogenic properties, of the antibodies analyzed.
Table 1 Comparison of clinical ADA rates and experimental in vivo immunogenicity of 13 selected human IgG1 clinical mAbs. Table 1 lists the 13 clinical mAbs with reported clinical ADA rates and experimental in vivo immunogenicity (Incidence; IGF) observed in hIgG1 transgenic (TG) and wild-type (WT) mice. In addition, the target, format, and highest V gene identity to the closest VH and VL gene of the transgene or germline is indicated in percent.
Clinical-grade preparations of the selected antibodies were tested in groups of 10 hIgG1 transgenic and 10 littermate wild-type mice except for avelumab, where only 9 wild-type mice were used. The mice were immunized following the established immunization protocol with seven consecutive subcutaneous applications of 10 μg IgG antibody without adjuvant over a three-week period.Citation6 Supplementary Figure 1 gives an overview on the general study outline of all in vivo immunogenicity experiments in this work. The results of the individual immunization experiments are shown in Supplementary Figure 2 and are summarized in . As indicated in , the ADA incidence in wild-type mice varies from 20% to 90% of treated mice. These differences probably reflect intrinsic immunogenic differences of the V region sequences used, since all compounds tested bear the same human constant region. Two of the three chimeric antibodies bear murine V regions with a high degree (over 90%) of sequence identity with endogenous mouse germline V gene sequences (brentuximab, cetuximab) for both the heavy (H) and the light (L) chain. The finding that two (infliximab, brentuximab) of the three antibodies displaying higher clinical ADA scores are chimeric human/mouse antibodies might suggest that the murine V sequences increase their immunogenic potential in humans.
In contrast, hIgG1 transgenic mice display a low ADA incidence that is generally reminiscent of that observed in humans with these compounds. The clinical ADA incidence reported for eight antibodies analyzed range between 0 and 10%, but is very variable for two of the remaining five antibodies studied (brentuximab and infliximab), ranging between 7% and 51% (). Compared with the large case numbers contributing to the clinical ADA rates of the compounds listed in , the ADA frequency in hIgG1 transgenic mice stems from experimental groups of 10 mice. This explains the better granularity of the clinical ADA rates, especially for the small numbers, as ADA incidence values between 0% and 10% effectively cannot be reproduced in hIgG1 transgenic mice (less than one mouse). In fact, all compounds with a clinical ADA incidence up to 13% cause an ADA rate between 0% and 10% in hIgG1 transgenic mice (). Thus, given the group size limitation, the hIgG1 transgenic mice consistently reproduce the range of clinical immunogenicity reported for the compounds tested here.
Tolerance to human antibodies does not depend on V sequence similarity to resident V gene repertoire
Given the observed fluctuation of the measured ADA titer values within immunization groups (Supplementary Figure 2), and in order to provide a tool reflecting both the incidence and the intensity of individual immune responses, we introduced the concept of the “Immunogenicity Factor” (IGF in ). This value integrates the frequency of responders and the respective titer values as described in the materials and methods section.
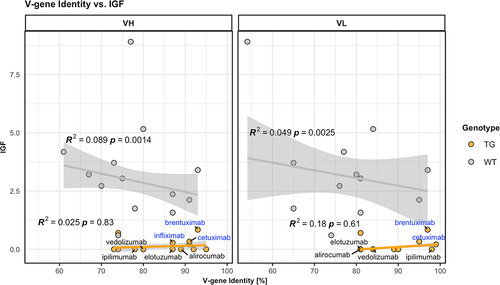
We determined the level of identity between the V sequence of the 13 tested antibodies and the endogenous murine or the five transgenic human V gene sequences for the H and the L chain in each case. These values are given in for the H and the L chain V gene of each antibody as sequence identity (in %) to the closest resident transgenic or endogenous murine V genes. For example, for alirocumab, a maximum of amino acid identity of 89% with one of the five transgenic VH genes and of 66% with one of the two transgenic V kappa (Vκ) genes is indicated. displays the IGF of the responses to each compound as a function of the sequence identity of its V gene. Interestingly, this comparison in wild-type mice denotes a trend toward increased tolerance when V sequence identity increases (, gray symbols). However, this systematic effect only accounts for a few percent of the overall variability in the data, based on R2 values, therefore not constituting a pivotal factor determining the IGF. In hIgG1 transgenic mice, V sequence identity does not correlate at all with observed immunogenicity (, orange symbols). The data suggest an “equalizing principle” in hIgG1 transgenic mice causing a general flattening of IGF irrespective of their V sequence identity to resident transgenic or endogenous V sequences. In summary, we conclude that tolerance to human antibodies has a broader coverage than expected by the five transgenic V genes expressed in hIgG1 transgenic mice. Therefore, we next asked if the common Fc domain has an influence in maintaining unresponsiveness to V sequences.
Figure 1. Low identity of antibody V genes to endogenous V genes does not correlate with increased immunogenicity. V gene identity is given as percent amino acid sequence identity of the variable (V) domain of each antibody listed with the best matching hIgG1 transgenic (TG, orange circles) or endogenous murine wild-type (WT, gray circles) V heavy (VH, left panel) and V light (VL, right panel) gene elements. The Immunogenicity Factor (IGF) as a degree of immunogenicity is correlated with the V gene identity for hIgG1 transgenic (orange lines) or wild-type mice (gray lines) and R2 values, as well as p-values are displayed in the plot. A robust linear model was used, based on Huber’s M-estimator, which downweighs the influence of single outlying data points, as well as deviations from non-normality. R2 coefficients of determination were calculated using modified residual and total sums of squares, i.e., weighing the squared deviations with the respective weights assigned by the robust estimator. Compounds showing higher identity to endogenous V genes compared to transgenic V genes are labelled and in addition, chimeric compounds are labelled in blue.
Fab fragments display enhanced ADA responses
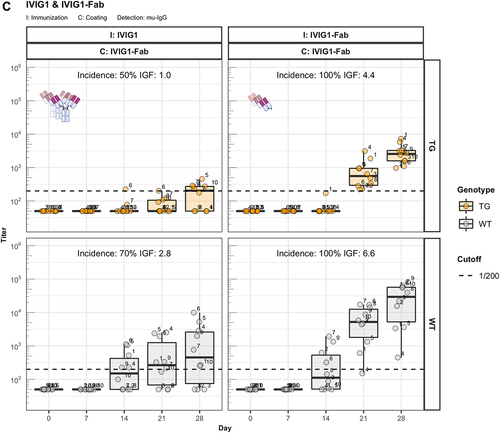
We analyzed the immunogenic properties of purified antigen-binding fragments (Fab) lacking the crystallizable fragment (Fc) of three representative IgG1 antibodies: the therapeutic antibody bevacizumab, the scaffold antibody CEA-IgG containing the same sequence as the CitAbs CEA-IL2v and CEA-TCB, and the experimental antibody DP47-IgG bearing VH and Vκ regions encoded by the transgenic human VH3-23 and Vκ3-20 genes, respectively. The data in illustrate that the immunogenic potential of these antibodies observed in wild-type mice is strongly reduced or abolished in hIgG1 transgenic mice. In contrast, the Fab preparations of bevacizumab (beva.-Fab) and CEA-IgG (CEA-Fab), but not of DP47-IgG (DP47-Fab), display enhanced immunogenicity both in hIgG1 transgenic and wild-type mice (). The results imply that Fab-specific B cells exist in hIgG1 transgenic mice that are silenced upon immunization with full antibody, but are unleashed when triggered with Fab preparations. Interestingly, the Fab of the antibody DP47-IgG fails to elicit this “reactivation” response (). Given that the same human VH3-23, joining (J)H4, Vκ3-20, and Jκ1 genes of DP47 are also expressed in hIgG1 transgenic mice,Citation6 we interpret the absence of a corresponding antibody response to DP47-Fab as resulting from central tolerance-induced deletion of DP47-specific lymphocytes. Immunization with IgG1 fraction of intravenous immunoglobulin (IVIG1) and Fab preparations thereof (IVIG1-Fab) extend the validity of this assumption over the two cases with beva.-Fab and CEA-Fab. As shown in , the incidence and the intensity of the ADA response to IVIG1-Fab is enhanced when compared with IVIG1, both in hIgG1 transgenic and in wild-type mice.
Figure 2. Fc contributes to the unresponsiveness towards conventional mAbs. Murine IgG anti-drug antibody (ADA) titers in hIgG1 transgenic (orange, upper panels) and wild-type (gray, lower panels) are measured over time before (day 0) and after the first immunization. Groups of 10 hIgG1 transgenic and 10 wildtype mice were immunized with the full antibody and the respective Fab preparation in the same experiment. The immunization compound (I:) and the coating compound for the ELISA (C:) are displayed above each plot. ADA titers above the arbitrary threshold of 200 are considered as positive and hence used to calculate the ADA incidence displayed in percent and the Immunogenicity Factor (IGF). Panel A) displays ADA titers against the full IgG1 antibodies bevacizumab, CEA-IgG, DP47-IgG and panel B) shows ADA titers against the respective Fab fragments beva.-Fab, CEA-Fab, and DP47-Fab. Panel C) shows ADA titers against the Intravenous Immunoglobulin subclass 1 Fab fragment (IVIG1-Fab) induced by immunization with the full IVIG1 or IVIG1-Fab preparation.
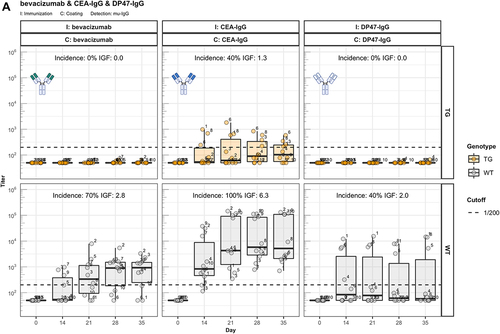
Figure 2. (Continued).
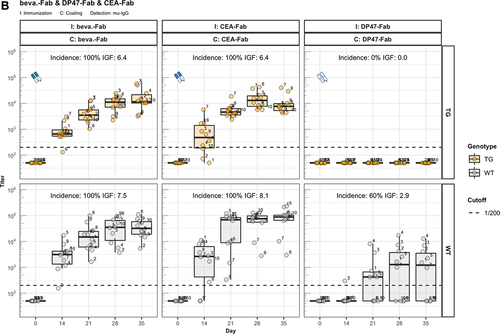
Figure 2. (Continued).
Reitan and HannestadCitation10 discovered that the immunogenicity of Id depends on the isotype of the antibody. To assess the influence of the constant region on immunogenicity of the CEA-binder, we generated antibody constructs composed of this particular human V region associated with the murine IgA constant region (CEA-mIgA). Immunization of hIgG1 transgenic and wild-type littermate mice demonstrates that the V region associated with the mIgA isotype elicits a similar ADA response as the corresponding CEA-Fab preparation (). Further, the results in demonstrate that the observed ADA response to Fab is not caused by cryptic epitopes exposed in the Fab, but results from the absent tolerogenic effect of IgG-Fc.
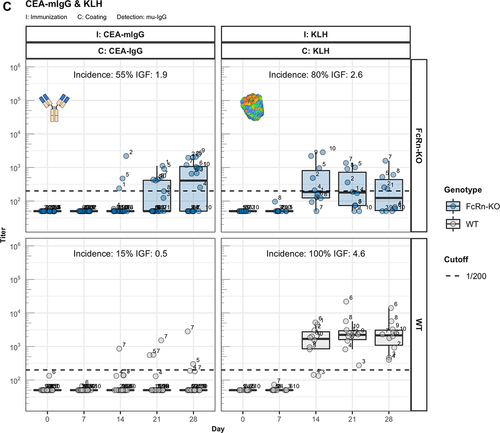
Figure 3. The role of FcRn in immunogenicity. Murine IgG anti-drug antibody (ADA) titers in hIgG1 transgenic (orange), wild-type (WT, gray) and FcRn-KO (blue) mice are plotted over time before (day 0) and after the first immunization. For each immunization, groups of 10 mice were used. The immunization compound (I:) and the coating compound for the ELISA (C:) are displayed above each plot. ADA titers above the arbitrary threshold of 200 are considered as positive and hence used to calculate the ADA incidence displayed in percent and the Immunogenicity Factor (IGF). Panel A) shows the ADA response of 10 hIgG1 transgenic and 10 wild-type mice immunized with a CEA-mIgA against the full molecule (C: CEA-mIgA), the variable domain (C: CEA-Fab) or the constant region (C: DP47-Fab). Panel B) shows ADA responses against the CEA-binder in 10 hIgG1 transgenic and 10 wild-type mice immunized with CEA-mIgG and CEA-mIgG-AAA. In panel C), FcRn-KO and wild-type mice were immunized with CEA-mIgG and KLH. The immunization with CEA-mIgG was repeated to confirm the result leading to the duplicated sample size (n=20 each).
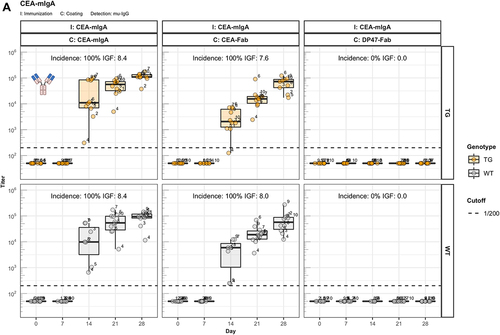
Figure 3. (Continued).
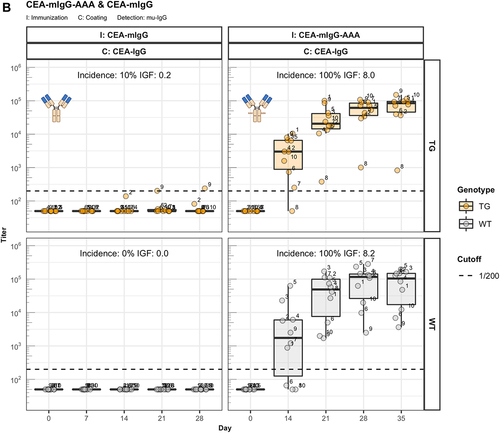
Figure 3. (Continued).
IgG binding to FcRn is required for immune tolerance
One major difference between IgG and IgA is the binding capacity to the neonatal Fc receptor (FcRn).Citation11 To investigate the contribution of this receptor, we generated a variant of CEA-IgG bearing a murine IgG1 constant region (CEA-mIgG) and an AAA variant thereof (CEA-mIgG-AAA) containing a triple mutation (I253A, H310A, H435A) that abolishes binding to the FcRn.Citation12 The elevated ADA response of CEA-mIgG-AAA clearly links FcRn to the Fc-mediated unresponsiveness with CEA-mIgG ().
Elevated ADA response in mice deficient for FcRn
Immunization experiments in mice deficient for the FcRn receptor (FcRn-KO) further corroborated this assumption. Because this mouse strain does not express human IgG1 as self-protein, it requires immunizations with the mouse surrogate CEA-mIgG antibody. In analogy to the enhanced ADA response found with CEA-mIgG-AAA, the CEA-mIgG antibody elicits a stronger ADA response in FcRn-KO mice as compared to wild-type mice (), despite the expected strongly reduced exposure of the injected antibody (Supplementary Figure 3A) and the overall reduced endogenous IgG levels (Supplementary Figure 3B) in mutant mice due to the lack of FcRn-mediated recycling. The control immunization with the T cell-dependent antigen keyhole limpet hemocyanin (KLH) in this experiment demonstrates that FcRn-KO mice are capable to mount ADA responses, but the titers are generally reduced in comparison to wild-type mice (). Taken together, we conclude that tolerance to idiotype components of antibodies is mediated by the Fc domain of IgG (but not by IgA isotype) through interaction with FcRn.
IL-2 immunocytokine and T-cell bispecific antibody display enhanced immunogenicity
The identification of Fc-associated mechanisms preserving unresponsiveness to conventional antibodies prompted us to address the widely observed strong immunogenic potential of bispecific therapeutic antibodies.Citation1 Thus, we extended our studies to two forms of CitAbs, based on the same CEA-specific scaffold antibody. The structure of CEA-based CitAbs used is shown in . The T cell bispecific (TCB) cibisatamab (CEA-hTCB) is composed of two Fab arms specific for CEA and one arm specific for the CD3ε chain of the TCR protein complex.Citation8, In our experiments, we also used a variant with a V domain of the hamster-anti mouse CD3ε clone 145-2C11 (CEA-mTCB), in the similar, though not identical, 2:1 format of clinical CEA-hTCB. The Fc-distal position of the CD3ε binder in CEA-mTCB, as compared to its Fc-proximal position in CEA-hTCB, results from protein expressibility and activity limitations of the murine version. The immunocytokine cergutuzumab amunaleukin (CEA-IL2v) is composed of the same CEA-binding scaffold antibody fused to a modified human IL-2 molecule at the carboxy terminal of one of the Fc moieties by a linker.Citation9 This IL-2 variant (IL2v) used in this immunocytokine contains mutations that abolish binding to the high-affinity IL-2Rα chain (CD25), but preserve binding to the intermediate affinity IL-2Rβγ heterodimer receptor.Citation9 Therefore, it preferentially stimulates CD25dull effector T cells and NK cells, while avoiding activation of CD25high regulatory T cells.Citation9 In addition, the CEA-IgG backbone bears the PGLALA modification within the Fc region that eliminates the Fc-gamma receptor (FcyR) and complement C1q binding, while retaining FcRn binding.Citation13 In the experiments presented here, the CEA-IgG scaffold antibody served as control of the baseline immunogenic potential. As shown in , CEA-IgG and CEA-hTCB are poorly immunogenic, whereas CEA-mTCB and CEA-IL2v elicit immune responses with high incidence and titer in hIgG1 transgenic mice. It is particularly significant that the fusion with the human CD3ε-specific Fab in CEA-hTCB does not raise the immunogenic potential of CEA-IgG, whereas addition of the mouse CD3ε-reactive Fab in CEA-mTCB and of the human IL2v protein in CEA-IL2v results in increased immunogenicity. As both CEA-mTCB and CEA-IL2v can engage the corresponding receptors in murine T cells (CD3ε and IL-2R), the data support a role for this interaction in reverting the pre-existing unresponsiveness to CEA-IgG. Furthermore, shows that immunization with CEA-IL2v induces ADA responses directed against CEA-Fab, indicating the involvement of Fab-specific B cell clones. In contrast to wild-type, hIgG1 transgenic mice usually do not produce ADA against the IgG1 Fc portion, suggesting an elimination of Fc-specific B cells mediated by the expression of the transgene.
Figure 4. The enhanced immunogenicity of CitAbs. Panel A) ADA response of hIgG1 transgenic mice (TG, orange, n=10) immunized with either CEA-IgG, CEA-hTCB, CEA-mTCB or CEA-IL2v against the scaffold antibody (CEA-IgG) or the injected compound (CEA-hTCB) is shown. Panel B) shows the specific ADA response of hIgG1 transgenic mice (orange, n=10) immunized with either CEA-IL2v directed against the scaffold antibody (CEA-IgG), the F(ab’)2 fraction (CEA-Fab2) and the Fc domain (Fc-PGLALA). Panel C) depicts the specific ADA response of hIgG1 transgenic mice (orange, n=10) immunized with either CEA-IL2v (as in A)) or DP47-IL2v directed against the scaffold antibody, CEA-IgG or DP47-IgG, respectively. Panel D) shows the specific ADA response of hIgG1 transgenic mice immunized with CEA-mTCB or DP47-mTCB specific for the parental molecule or the murine CD3 binding moiety (mCD3-IgG).
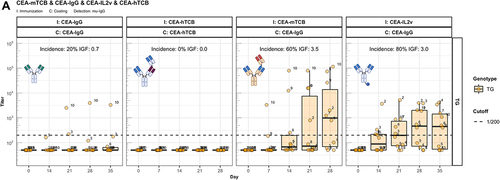
Figure 4. (Continued).
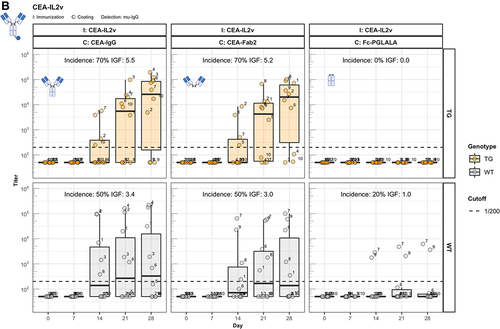
Figure 4. (Continued).
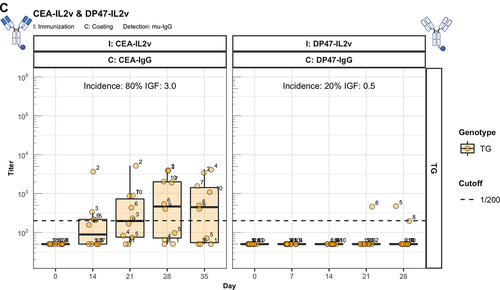
Figure 4. (Continued).
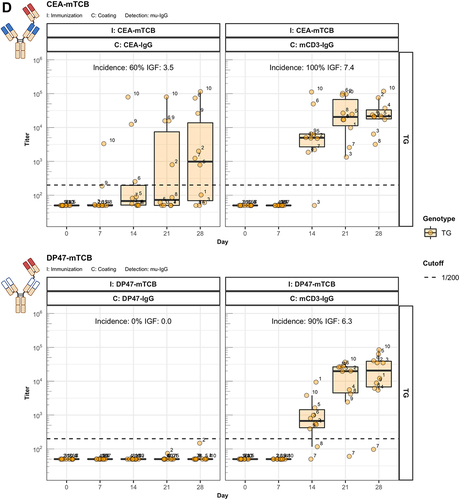
When similarly modified variants of the germline antibody DP47-IgG are tested for their immunogenicity, a different picture emerges. It is evident that no ADA against DP47 are elicited with DP47-IL2v () and DP47-mTCB () despite the addition of IL2v or mouse CD3ε binder, respectively. This demonstrates that the absence of anti-DP47 ADA reflects the lack of germline-specific B cell clones. Interestingly, the detection of mTCB-specific ADA in mice immunized with DP47-mTCB (, lower panels) suggests the presence of T and B cell epitopes in the hamster-anti mouse CD3ε binder.
Enhanced immunogenicity of CitAbs does not correlate with dendritic cell uptake
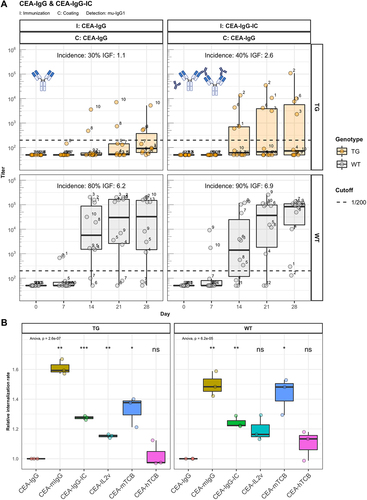
It is known that pre-existing ADA, even below the detection limit, can contribute to the formation of immune complexes (ICs) with infused antibodies, thus increasing their immunogenicity.Citation14 The underlying mechanism could be enhanced uptake, processing, and presentation by dendritic cells (DC).Citation15,Citation16 Concerns were raised that structural modifications of CEA-IgG, such as the fusion of IL2v or TCB and the knob-into-hole amino acid changes for engineering of bispecific antibodies,Citation17 could promote binding of pre-existing ADA, and hence increase immunogenicity or cause stimulation of DCs via other mechanisms. Thus, we generated large ICs formed by CEA-IgG and mouse IgG2a antibodies specific to the idiotype of CEA-IgG (CEA-IgG-IC) to investigate this hypothesis (Supplementary Figure 4A). Immunizing hIgG1 transgenic and wild-type mice with CEA-IgG-IC leads to a distribution of intact ICs in the serum (Supplementary Figure 4B) but causes only a slight increase of the ADA response against CEA-IgG (). This slight increase caused us to question the contribution of DC internalization to the immunogenicity of CitAbs. To that end, we differentiated DCs from the bone marrow of three hIgG1 transgenic and three wild-type mice using recombinant murine granulocyte-macrophage colony stimulating factor (GM-CSF). Subsequently, we measured by flow cytometry the internalization rate of the compounds labeled with a pH sensitive fluorescent dye, which is brighter in the acidic environment of the lysosome. As expected, DCs internalized CEA-IgG-IC more efficiently than monomeric CEA-IgG (), which could contribute to the slightly enhanced immunogenicity. The internalization rate of monomeric CEA-IL2v and CEA-mTCB is also in the range of CEA-IgG-IC, even though these compounds induce a stronger ADA response in hIgG1 transgenic mice (). Interestingly, CEA-mIgG displays the highest DC internalization despite its low immunogenicity in hIgG1 transgenic and wild-type mice (). Similarly, CEA-hTCB is not immunogenic in hIgG1 transgenic mice (), but shows an intermediate DC internalization rate. From the combination of these data, we conclude that there is no simple correlation between the rate of uptake and internalization and the immunogenic attributes of the analyzed compounds. Thus, other explanations are needed to explain the enhanced immunogenicity of CitAbs.
Figure 5. IC formation leads to slightly enhanced immunogenicity. Panel A) shows murine IgG ADA titers against the scaffold antibody (CEA-IgG) of hIgG1 transgenic (TG, orange, n=10) and wild-type (WT, gray, n=10) mice immunized with either CEA-IgG or CEA-IgG in immune complex with a mouse IgG2a anti-idiotype (CEA-IgG-IC). Panel B) displays the internalization rate of compounds with a pH sensitive labelling into the acidic lysosome of CD11c+/CD11b+ DCs cultivated from bone marrow of three hIgG1 transgenic and three wild-type mice.
The assumption that ADA responses to CEA-mTCB and CEA-IL2v are stimulated by activation of T cells targeted by their mTCB or the IL2v moieties is further supported by immunization experiments with mixtures of scaffold CEA-IgG with purified IL2v or a one-arm variant of the mCD3ε-specific antibody (mCD3-OA). These immunization studies show an incremental difference in immunogenicity comparable to that found with the corresponding T cell engager antibodies (Supplementary Figure 5). From these results, we conclude that idiotype-specific B cell clones exist that are indirectly reactivated by the action of IL2v or CD3ε binder molecules. Given the T cell-activating nature of the IL2v and anti-CD3ε Fab, the question arises whether this reactivation is an antigen-cognate process, or the sole result of bystander responses mediated by nonspecific T cells.
ADA responses elicited by CitAbs require cognate T-B cell interaction
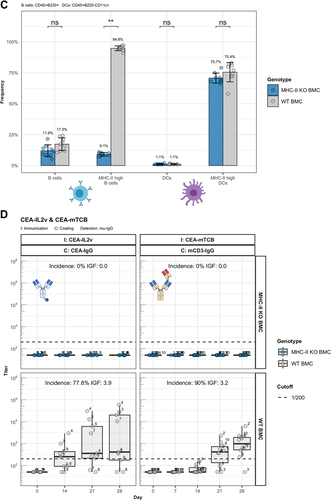
To address the question of cognate T-B cell activation, we studied the role of antigen-presentation by B cells in the formation of ADA. To do so, we used an experimental system of lethally irradiated mice reconstituted with a mix of bone marrow cells. The mix is composed of 80% bone marrow cells from mice homozygous for the Igh-JtmCitation1Cgn targeted mutation (JhT−/−), and thus lacking functional B cells, and of 20% bone marrow cells derived from MHC-II deficient mice (MHC-II−/−) in order to create a system where only B cells are deficient for MHC-II expression. A mix containing 20% bone marrow cells from wild-type mice serves as a control. The generation of the bone marrow chimeric mice (BMC) is described in and in the materials and methods section (Mice and Immunization). The B cells in such reconstituted mice are MHC-II−/−, while all other immune cells bear the JhT−/− mutation but express MHC-II (, B and C). The system is apt to test if the CitAbs CEA-IL2v and CEA-mTCB were able to induce antigen presentation-independent, non-cognate T cell activation, and hence MHC-II-independent ADA production. As shown in , the experiment evinced that only mice reconstituted with the mix containing wild-type bone marrow cells (WT BMC) produced ADA to CEA-IL2v and CEA-mTCB upon immunization. In contrast, chimeric mice containing MHC-II-deficient B cells (MHC-II KO BMC) were unable to respond to the immunization with CEA-IL2v and CEA-mTCB. Since the bone marrow reconstitution generated similar numbers of lymphocytes in the two groups of chimeras (), we totally exclude a non-cognate type of T cell activation and ADA production as explanation for the immunogenic potential of CEA-IL2v and CEA-mTCB. Although MHC-II KO BMC displays reduced endogenous IgG titer (Supplementary Figure 6A), both groups can be considered equally immunocompetent. This was also evident from the comparable antibody response elicited by the T cell-independent antigen NP-Ficoll in both groups of chimeric mice (Supplementary Figure 6B). Therefore, reversal of unresponsiveness to CEA-IgG by the bispecific versions CEA-IL2v and CEA-mTCB requires both extracellular binding of IL-2 or TCB to their receptors on the T cell surface and intracellular processing and presentation by MHC-II.
Figure 6. ADA responses to CitAbs require MHC-II dependent T/B collaboration. Panel A) Graphical abstract of the generation of bone marrow chimeric mice using JhT−/−, MHC-II−/− and WT mice. Panel B) Representative FACS plots showing the MHC-II expression on B cells and DCs in spleens of WT BMC and MHC-II KO BMC on day 28. Panel C) Quantification of key populations as shown in Panel B). Mean frequencies for each population are given above the bar. Panel D) ADA response induced by CEA-IL2v and CEA-mTCB in WT BMC (gray) and MHC-II KO BMC (blue). The ADA response against the parental CEA-IgG molecule and the ADA response against the T cell binding moiety is shown for CEA-IL2 and CEA-mTCB, respectively. I: Immunization; C: Coating; D: Detection
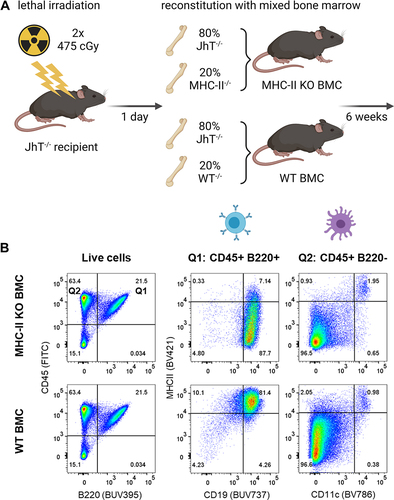
Figure 6. (Continued).
Discussion
Despite intensive efforts in the development of bispecific antibody therapeutics specifically designed for cancer immunotherapy, few have reached the market.Citation1 Doubtless, antibody therapy based on CitAbs and related biotherapeutics has the potential to be highly efficacious in cancer treatment.Citation5,Citation18 Given that immunogenicity represents one of the major liabilities of this class of anti-tumor drugs, the preclinical assessment of their immunogenic properties is the objective of intensive research efforts.Citation19–22 Methods are in place aiming to identify potential antigenic epitopes of defined biotherapeutic proteins in silico (e.g., EpiVax, netMHC-pan), in vitro (MAPPs), ex vivo (e.g., PBMC and DC-TC assays) or in vivo with transgenic mouse systems.Citation23,Citation24 Yet, the basic question of the mechanism(s) causing break of tolerance to antibody proteins in general, and more specifically to CitAbs in particular, was often raised but insufficiently addressed experimentally.Citation1 Immunogenicity of antibodies is centered around anti-Id responses.Citation25–27 Anti-Id ADA responses have been studied in detail in experimental murine systems based on the transgenic expression of Id-specific B and T cell receptors.Citation28–30 These studies demonstrated that, upon administration of Id-bearing antibodies, Id-specific naive B and T cells collaborate in vivo to the formation of germinal centers and plasma cells, resulting in isotype-switched anti-Id ADA. This process occurs in the absence of adjuvants and does not require antigen presentation by DCs.Citation31 These mechanisms differ significantly from conventional antibody responses to foreign antigens and could also apply to Id-specific ADA responses to human antibodies in clinical trials. Using these transgenic tools, it became evident that self-reactive B and T cell clones specific for germline-encoded Id are deleted centrally.Citation28,Citation30 However, lymphocytes with specificity for non-germline Ids can mature and persist in the periphery and could respond to challenge by idiotypic determinants arising during SHM of antibodies or appearing upon infusion of exogenous therapeutic antibodies.
Nonetheless, the accumulated clinical data of numerous marketed therapeutic antibodies suggest a low to moderate immunogenicity potential for most human therapeutic antibodies clinically tested so far (, ref. 2). In contrast, CitAbs tend to elicit ADA more frequently, often causing exposure and efficacy loss and even adverse events, which contribute to high attrition rates.Citation1
Here, we addressed the apparent broad tolerance to Ids encoded by non-germline V genes of exogenous conventional therapeutic antibodies and the comparably pronounced immunogenic attributes of CitAbs. We used the hIgG1 transgenic mouse previously shown to display immunological tolerance to a broad range of human IgG1 antibodies and to be sensitive to immunogenic modifications thereof.Citation6,Citation7
Of course, this model cannot evaluate patient-related factors contributing to ADA occurrence in clinical trials, including cross-reacting pre-existing antibodies, dosing regimens, pre-medication, administration route, biodistribution, and density of the tumor target antigen(s). While all these factors, along with genetic factors, can influence the ADA outcome to a defined treatment with CitAbs, our study addresses fundamental immunological mechanisms governing immune tolerance and its reversal, that are shared by the human and mouse immune systems. In this study, we have not addressed other questions potentially adding to the immunogenic attributes, such as the affinity of CitAbs to the T cell target antigens or the different formats of CitAbs. However, given the tightly coordinated interaction between T and B cells, anti-CD3ε with higher affinities could in turn lead to stronger B cell help and thus be more immunogenic.
Our results confirm the previously reported broad range of tolerance to human IgG1 antibodies in the hIgG1 transgenic mice.Citation6 Nonetheless, this is somehow surprising considering that only five human V sequences are included in the germline of hIgG1 transgenic mice, in comparison to the marked diversity of V gene sequences of the human antibodies tested ( and ). The lack of correlation between immunogenicity and sequence identity to resident V sequences in hIgG1 transgenic mice suggests a mechanism ensuring tolerance to V sequences not encoded in the germline. We hypothesized that the IgG1 constant region plays this tolerizing role. Indeed, we found that Fc-devoid preparations of two prototypical antibodies (bevacizumab and CEA-IgG) and of IVIG1 elicit strong ADA responses in hIgG1 transgenic mice (), indicating that Id-specific B and T cells exist in these mice, but are kept silent toward Fc-containing antibodies. Taken together, these results support the general tolerogenic role of the Fc moiety of IgG1 antibodies, but not of antibodies of other isotypes like IgA () in hIgG1 transgenic mice. Even considering that Fab are unnatural, artificial substances used here for experimental purposes, the results clearly suggest that the Fc has a role in modulating immune responses toward Id determinants of antibodies. Interestingly, the few published immunogenicity studies on bispecific T cell engagers (BiTEs) have revealed the immunogenic character of this class of small therapeutic antibodies composed of two different single-chain variable fragment (scFv) binding one tumor antigen and an activating receptor on T cells.Citation32,Citation33 According to our findings, it should be expected that appendage of an Fc moiety would abrogate or mitigate the immunogenic properties of BiTEs in humans.
Using FcRn-deficient mice we demonstrated that the tolerogenic role of Fc is related to the function of FcRn (). Based on these data, we propose that FcRn-directed recycling diverts internalized antibodies away from the proteasomal degradation pathway to peptide presentation and immune activation.Citation12
Similar observations have been made for murine anti-Id ADA responses. Indeed, certain murine Id-bearing antibodies proved to be very immunogenic and efficient in inducing anti-Id ADA responses even when administered adjuvant-free in syngeneic mice.Citation34 In that study, Reitan et al. showed that the immunogenicity of these mouse idiotypes depends on the associated isotype. Indeed, the same idiotype is tolerized or can elicit vigorous anti-Id responses when borne by IgG isotypes or by IgM, IgE and IgA isotypes, respectively. Furthermore, this IgG-associated tolerogenic Id becomes immunogenic when administered Fc-free, as Fab fragments.Citation10
Here, using hIgG1 transgenic mice, we demonstrate that germline encoded V regions of human antibodies promote central elimination of corresponding Id-specific lymphocytes, as exemplified by the antibody DP47-IgG, while unresponsiveness to non-germline human antibodies (bevacizumab, CEA-IgG) is aided by a recycling pathway mediated by FcRn. Assuming efficient binding of bevacizumab and CEA-IgG to the murine FcRnCitation35 and the lack of binding to murine FcyRs of the CEA-Fc with the PGLALA mutations,Citation13 we conclude that FcRn, but not FcyR, substantially contributes to the observed unresponsiveness to infused antibodies. Upon B cell receptor (BCR) ligation, antigen-specific B cells are also able to present BCR-derived Id determinants and receive help from Id-specific T cells.Citation29 However, naive B cells initially bind antigen with low affinity via non-mutated germline BCR. These B cells are not likely to encounter T cells specific for such germline-encoded Id, as they are clonally deleted in the thymus.Citation28 In contrast, whenever Id-bearing antibodies are generated through SHM (or are exogenously added) and capable of ligating a corresponding anti-Id BCR, ADA can resultCitation29 from direct Id-specific B-T cell interaction.Citation31 Here, we describe that this process is down-modulated by the action of FcRn () and by an additional suppressive and unresponsive state conveyed by putative Fc-specific, suppressor, or anergic T cells.
In contrast to the poorly immunogenic scaffold antibody CEA-IgG, the derivatives CEA-IL2v and CEA-mTCB elicit vigorous anti-Id ADA responses in hIgG1 transgenic mice (). This fundamental finding identifies the IL2v and TCB moieties as responsible for the immunogenicity increment. This assumption is further supported by the finding that the fusion of a human CD3ε-binding Fab, which does not cross-react with mouse CD3ε, does not cause an increase in immunogenicity (). The fact that germline V-based DP47-IL2v was immunologically inert () and DP47-mTCB elicited exclusively an anti-mTCB ADA response () further substantiates the deletion of DP47-Id B cell clones during central tolerance. The absence of ADA against DP47 excludes the formation of new epitopes in CEA-IL2v and CEA-mTCB as the primary reason for the onset of immunogenicity.
The possible formation of IC composed of CEA-IL2v or CEA-mTCB and pre-existing low levels of ADA was simulated with antibodies directed against the Id of CEA-IgG, yet the immunogenicity rate of this preparation failed to explain the typical enhancement of immunogenicity of the CitAbs. The measurement of antibody internalization in antigen-presenting DCs has been proposed as a tool for immunogenicity risk assessment because a higher degree of internalization generally correlates with higher clinical immunogenicity.Citation36 However, in a similar assay, the internalization rate of these compounds in murine DC revealed no simple correlation with their corresponding immunogenic potential in hIgG1 transgenic mice. Finally, the artificial mixture of CEA-IgG with soluble IL2v or a one-armed antibody specific for mCD3ε confirmed T cell activation as the mechanism causing the ADA responses of CEA-IL2v and CEA-mTCB, respectively ( and Supplementary Figure 5).
Taken together, these results add another level of control of anti-Id responses in addition to FcRn that is sensitive to T cell engagement. Binding of IL-2R or TCR-CD3 by CEA-IL2v and CEA-TCB, respectively, seemingly causes the reversal of a state of unresponsiveness not compensable by the recycling pathway driven through FcRn. This unresponsiveness has features reminiscent of T cell anergy, as it is sensitive to T cell stimulation by activating molecules like IL-2 or TCR-binding moieties of certain CitAbs (). Indeed,Citation37–39 FcRn relates to a salvage pathway of internalized antibody while reversal of unresponsiveness by CitAbs like CEA-IL2v and CEA-TCB occurs through the extracellular triggering of T cell activation surface receptors like IL-2R and TCR-CD3. The immunomodulatory property of the Fc region discussed before seems to be overruled by direct engagement of surface T cell activators. Thus, the questions arise, if the reversal of the unresponsive state requires extracellular ligation of the affected T cell surface receptors alone, or if cognate antigen presentation by the Id-specific B cell is also needed. We answered these questions with bone marrow chimeric mice designed to lack expression of MHC-II in B cells, but not in other antigen-presenting cells likeCitation40 DCs ). These animals are apt to discern the need for antigen presentation by B cells in the process of breaking tolerance to CEA-IgG by immunization with CEA-IL2v and CEA-mTCB. The experiment clearly demonstrated the need for MHC-II-dependent antigen presentation by B cells ().
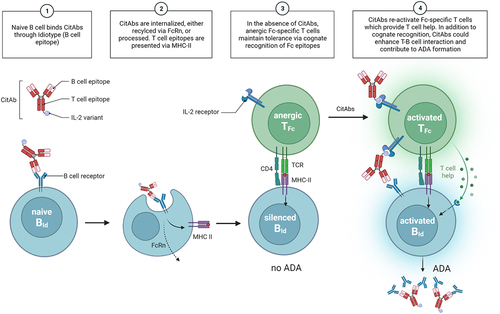
In summary, we conclude that tolerance to antibodies, in addition to clonal deletion, is also the result of a peripheral state of T cell unresponsiveness that can be broken if the CitAb includes additional T cell-activating entities like the IL2v or TCR-CD3-binding moieties. This could lead to the activation of hitherto inactive Id-specific B cells, and to ADA production. We postulate that immunomodulatory T cells specific for common “public” epitope(s) of the Fc region (TFc cells), do maintain this unresponsive state on B cells of different “private” Id specificities. Hence, reversal of the unresponsive status of TFc cells with CitAbs will result in activation of all possible Id-specific B cell clones (BId) ligated by the corresponding CitAb. This type of linked-antigen presentation would then lead to unresponsiveness rather than to activation, and hence it is termed “linked-antigen tolerance”. Interestingly, the immunogenicity study of commercial antibodies shown in and additionally supports the idea that expression of the human Fc in hIgG1 transgenic mice creates an immunological resort ensuring tolerance to associated V sequences independent of their degree of identity to (immune tolerized) resident V sequences.
summarizes schematically the features of a T-B cell cooperation set in motion by CitAbs that would break the postulated status of linked-antigen tolerance toward IgG antibodies and provoke the observed ADA production to the involved CitAbs. This model invokes a status of broad immune tolerance to non-germline encoded V regions as achieved by unresponsive TFc cells specific for the common Fc region. The linked-antigen tolerance model postulates a frequency of the unresponsive (anergic, suppressive) TFc cells emerging from the thymus being theoretically higher than that of functional, rare Id-specific T cells (TId). Upon binding of the CitAb (, Panel 1), BId cells would process and present Id epitopes on MHC-II, possibly under the influence of FcRn-mediated recycling (, Panel 2). Of course, rare TId cells can eventually interact with, and activate, BId cells to produce anti-Id ADA. Yet, more abundant Fc-specific, unresponsive TFc cells are more probable to interact with BId cells and maintain their unresponsive state. Therefore, TFc cells mediating unresponsiveness and silencing of BId cells is the predominant result (, Panel 3). Such a mechanism of linked-antigen tolerance would explain the extended low immunogenicity of conventional therapeutic antibodies in humans and hIgG1 transgenic mice (). In contrast, the ligation of the BCR of BId cells by CitAbs is accompanied by the triggering of activating surface receptors (IL-2R, TCR-CD3) on TFc cells (, Panel 4). The ensuing reversal of unresponsiveness and re-activation of TFc cells would set in motion the program of helping the cognate BId cells for anti-Id ADA production. This model could explain the high rates of ADA observed with many of the CitAbs.Citation1 If confirmed, the hypothesis proposed here of breakage of linked-antigen tolerance to CitAbs represents an intrinsic hurdle asking for specific mitigation strategies. Confirmation of the linked-antigen tolerance hypothesis requires identification of the putative TFc cells and of the predicted Fc epitopes involved in unresponsiveness. If verified, the model would open a door for mitigation strategies through elimination of the putative Fc epitopes recognized by the unresponsive TFc cells, such as to cancel their cognate reactivation by CitAbs. This Fc amino acid modification(s) would result in mitigation of all immunogenic CitAbs. In essence, this Fc mutation would restrict anti-Id ADA production to that resulting from interaction of rare BId with rare TId cells, to an extent possibly comparable to other, low immunogenic antibodies (), thus reducing their high attrition rate.
Figure 7. Mechanistic model. A graphical summary of how CitAbs can lead to the break of antigen-linked tolerance and formation of ADA response. Adapted from the figure template “B Cells Internalize and Process Antigens Only if Their BCR is Specific to the Antigen”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates
Materials and methods
Antibodies and compounds
All marketed therapeutic antibodies () are clinical-grade products purchased from pharmacies. All other therapeutic antibodies (overview in Supplementary Table 1) were transiently expressed in HEK293 cells or produced in stable CHO clones and subsequently purified using Protein A affinity chromatography and size exclusion chromatography. All Fabs were prepared by enzymatic digestion from IgG fractions and subsequently purified using Protein A affinity chromatography or Kappa select capture resin, followed by size exclusion chromatography. The IVIG1 and enzymatically digested IVIG1-Fab preparations were obtained from Athens Research & Technology. Fab preparations of bevacizumab were generated at Genscript by papain digestion, Protein A/G Fc removal, and subsequent endotoxin removal. All compounds were assessed for purity and endotoxin after manufacturing.
NP-Ficoll (Biosearch Technologies; Cat: F-1420) and keyhole limpet hemocyanin (KLH) subunits (Thermo Scientific; Cat: 77649) served as T cell independent and T cell dependent immunogens, respectively.
Immune complexes were prepared and characterized as described previously.Citation41 In short, a mixture containing 2 mg/ml CEA-IgG and 3 mg/ml monoclonal mIgG2a anti-idiotype antibody was prepared in histidine buffer (20 mM histidine, 140 mM NaCl, pH 6.0) and incubated for 1 h at room temperature on a shaker at 500 rpm. The ICs were characterized as previously described.Citation41 In brief, a Waters XBridge Protein BEH SEC Guard Column, 450 Å, 3.5 μm, 7.8 mm × 30 mm and a XBridge Protein BEH SEC Column, 450 Å, 3.5 μm, 7.8 mm × 300 mm were used in a Dionax UltiMate 3000 system from Thermo Fisher Scientific GmbH (ultraviolet (UV) detector MWD-3000, auto sampler, automated fraction collector). 20 μl of centrifuged dosing solutions were injected for analysis. Phosphate-buffered saline (PBS) with 5% ethanol (v/v) was used as running buffer with a flow rate of 0.5 ml/min. Online UV detection was performed at 280 nm.
Antibodies for the DC internalization assay were labeled using the SiteClick Antibody Azido Modification Kit (Invitrogen; Cat: S20026) according to manufacturer’s instructions. Briefly, antibodies were labeled with a molar dye excess of 3.5, which was subsequently removed using the Amicon Ultra-2 Centrifugal Filter with a MWCO of 50 kD and re-buffered in 20 mM histidine 140 mM NaCl (pH 5.5). The dye-to-antibody ratio is calculated from the absorbance at 280 nm and 532 nm, the extraction coefficient of the dye and a correction factor of 0.36. ICs for DC internalization were generated as described above using labeled CEA-IgG and unlabeled anti-idiotype antibody.
Mice and immunization
Animal experiments were conducted in AAALAC certified facilities of F. Hoffmann-La Roche in Basel or at the Department for Biomedicine of the University of Basel (Switzerland) in accordance with local rules and regulations of the veterinary authorities under cantonal licence number 2634 and 3125. Throughout the studies, mice were kept under specific-pathogen-free conditions with continuous health monitoring.
The hIgG1 transgenic mouse model (C57BL/6-Tg(hIgg1,k,l)ait) breeds heterozygous at Taconic in Denmark and the resulting wild-type C57BL/6J littermates serve as controls. The FcRn-KO mice (B6.129X1-Fcgrttm1Dcr/DcrJ; Strain #003982), and wild-type C57BL/6J control mice (Strain #000664) originate from the Jackson Laboratory (USA).
To generate B cell-specific MHC-II deficient bone marrow chimeras, lethally irradiated JhT−/− mice (B6.129P2-Igh-Jtm1Cgn/J) were reconstituted with 80% bone marrow of JhT−/− mice and 20% bone marrow of MHC-II−/−mice (B6.129S2-H2dlAb1-Ea/J). Control mice were reconstituted with 20% bone marrow of wild-type C57BL/6 mice instead of MHC-II KO mice. One day prior to reconstitution, recipient mice were irradiated twice in an interval of 4 h with 475 cGy using a Gammacell 40 137Cs irradiator. The bone marrow of donor mice was extracted by crushing femur and tibia, depleted from red blood cells using Lysing Buffer (BD Biosciences; Cat: 555899), depleted from T cells using CD90.2 beads on LD columns (Miltenyi; Cat: 130-121-278) and mixed in the previously mentioned ratio prior to intravenous injection of 10 × 106 cells per recipient. All mice used for studies employing bone marrow chimeric mice were bred and maintained in an animal facility at the Department of Biomedicine, University of Basel.
Unless otherwise mentioned, five female and five male mice between 6 and 15 weeks of age were carefully assigned to treatment groups in order to balance relevant factors such as litter, sex, body weight and exact age at the time of the experiment, reducing the potential danger of confounding the treatment effect with these factors. Mice were immunized two times per week with a total of seven subcutaneous injections into the abdominal region, alternating between the left and right sides. A dose of 10 µg full IgG molecule or the equimolar equivalent in the case of antibody fragments or conjugates was used per injection. The dose of ICs was adjusted to 10 µg CEA-IgG per mouse and the KLH subunits (Thermo Scientific; Cat: 77649) were injected twice on day 0 and 7 at a dose of 125 μg/mouse. The test items were freshly diluted in a total volume of 100 µl DPBS (Gibco; Cat: 12559069) or histidine buffer (20 mM histidine, 140 mM NaCl, pH 6.0) at dosing dates prior to administration.
Blood was collected prior to dosing on day 0 (naive), and weekly on day 7, 14, 21, and 28 by tail vein sampling into serum gel micro-sample tubes (Sarstedt; Cat: 41.1500.005). After clotting, the samples were centrifuged as indicated and stored at -20°C until analysis by enzyme-linked immunosorbent assay (ELISA). In case drug exposure was assessed, additional blood was sampled 7 and 24 h after the first injection.
Sample analysis
ADA was detected by ELISA as previously described.Citation6 In short, 96-well Nunc MaxiSorp™ plates (Thermo Scientific; Cat: 44-2404-21) were coated overnight with 5 µg/ml of the indicated antigens in 0.1 M sodium bicarbonate buffer, pH 8.5 (Alfa Aesar; Cat: J60408.AP). Subsequently, the plates were washed three times with DPBS + 0.05% Tween (Sigma; Cat: P9416-100ML), blocked for 2 h with DPBS + 2% bovine serum albumin (BSA; Thermo Scientific; Cat: 37520), and washed again. Serially diluted serum samples (1:50, then 1:3 for a total of eight dilutions) were incubated for 2 h at room temperature followed by further washing. Binding ADA was detected using alkaline phosphatase-conjugated goat anti-mIgG-Fcy (Jackson ImmunoResearch; Cat: 115-055-071) for 1 h and OD 450 nm values were measured after washing and 10 min incubation with p-Nitrophenyl Phosphate (Merck Millipore; Cat: P7998).
The drug exposure and endogenous IgG concentrations were measured similarly by coating with anti-Id antibody (in house production) or goat anti-mIgG light chain specific (Jackson ImmunoResearch; Cat: 115-005-174) antibody, and detection with alkaline phosphatase-conjugated goat anti-mIgG Fcy fragment-specific antibody.
The baseline for titer determination was calculated as the mean OD value at 1:50 dilution of all naive samples plus six times the standard deviation of those values (6 sigma cutoff). OD values greater than the upper quartile plus 1.5×IQR (interquartile range) were classified as outliers and excluded from baseline calculation. For a few cases, due to high background signals, cut-off values were adjusted manually.
The titration curve was fitted using the 5th degree polynomial and intersected with the baseline to obtain titer values. If no intersection occurred within the range of applied dilutions and the titration curve stayed consistently below baseline, we fixed the titer at the value corresponding to an intersection at the lowest dilution (1:50). If the intersection was not reached until the highest dilution, we allowed extrapolation of the titration curve up to a dilution of 1:328050, i.e., three times as high as the actual highest one used, applying a more stable 2nd degree polynomial fit. In any case, the titer was bounded by a maximum value corresponding to a 1:328050 dilution, and a flag was retained to indicate whether the result was obtained by proper interpolation or whether extrapolation/bounding was applied instead. Titers above the arbitrary threshold of 200 are considered ADA positive results.
The Immunogenicity Factor (IGF) integrates the response frequency and the intensity using the following equation:
Responder: Mice with titer > 200 at any time point after first immunization
Group Size: Number of mice per group
MeanPosMaxTiterInc: Increase of the average of highest titer of all responses
MaxTiter: Maximum possible titer (50×38= 328050 (extrapolated))
MinTiter: Lowest possible titer (50×30= 50)
Sequence analysis
The V gene identity displays the amino acid sequence identity of the variable domains of the therapeutic antibodies with the best matching transgenic or endogenous murine variable heavy- or light-chain genes. Sequence identity was obtained by pairwise alignment (EMBOSS water) of antibody V sequences with the five transgenic VH and two VL sequences or by blasting against mouse genomic sequences in the PRO ensmusp-Ensembl database, release 99.Citation42
DC internalization assay
DCs for the use in the internalization assay were cultured from isolated bone marrow of hIgG1 transgenic mice and wild-type littermates as previously described.Citation43 In short, the cells were seeded into 100 mm Ultra-Low Attachment Culture Dishes (Corning; Cat: 4615) at a concentration of 2×105 cells/ml in a total volume of 10 ml complete RPMI-1640 medium (Gibco; Cat: 61870-010) with 100 mM sodium pyruvate (Gibco; Cat: 11360-070), 50 mM 2-mercaptorethanol (Gibco; Cat: 31350-010), 10ʹ000 U/ml Penicillin/Streptomycin (Gibco; Cat: 15140-122), 10% FBS (Seradigm; Cat: 97068-085), and containing 20 ng/ml recombinant mouse GM-CSF (Gibco; Cat: PMC2016). The medium was replenished on day 3 and 6 and the cultures were harvested on day 9 by pipetting. This mouse DC internalization assay is designed according to a protocol developed in-house for the validated human DC internalization assay. The GM-CSF differentiated mouse DCs were seeded in ultra-low attachment 96-well plates at a concentration of 1×105 cells/well and incubated with a final concentration of 200 nM of the labeled antibodies for 120 min and 240 min in complete medium. Subsequently, FcyRs were blocked using TruStain FcX Plus (Biolegend; Cat: 156604) and the cells were stained using antibodies against CD11c (N418, BUV737) from BD Biosciences, and B220 (RA3-6B2, BV421), CD11b (M1-70, PE-Cy7), CD8a (53-6.7, APC), CD86 (GL-1, PE), and I-A/I-E (M5/114.15.2, BV786) from Biolegend. After dead cell staining with Zombie NIR (Biolegend; Cat: 423106), the events were acquired on BD LSR Fortessa. Cells were gated based on their morphology, aggregation, viability, and expression of CD11c+, CD11b+. The geometric mean fluorescence intensity (gMFI) in the FITC channel (excitation at 532 nm, filter at 572/35 nm) is recorded as a measure of the internalization of the dye into the acidic lysosome. gMFI values of a medium-stimulated control were subtracted, followed by normalization to the dye-to-antibody ratio. The normalized gMFI from each antibody is plotted as a linear regression curve to extract the slope (gMFI/min for 120 min and 240 min).
Characterization of bone marrow chimeras
The expression of MHC-II on B cells and DCs in bone marrow chimeric mice was assessed by flow cytometry. For this, the spleens of the bone marrow chimeras were sampled and splenocytes were extracted by mechanical dissociation in RPMI-1640 medium (Gibco; Cat: 61870-010) using the gentleMACS Octo Dissociator (Miltenyi). Red blood cells were depleted using Lysing Buffer (BD Biosciences; Cat: 349202), Fc block (Biolegend; Cat: 101320) was applied and extracellular markers were stained using antibodies against CD45 (30-F11, FITC), B220 (RA3-6B2, BV421), IgD (11-26c.2a, PerCP-Cy5.5), TCR-β (H57-597, BV605), CD4 (GK1.4, PE-Cy7), CD8 (53-6.7, APC), MHC-II (M5/114.15.2, BV786), CD25 (PK136, PE) from Biolegend and CD11c (N418, BUV737) and IgM (II/41, BUV395) from BD Biosciences. Subsequently, dead cells were stained using Zombie Aqua (Biolegend; Cat: 423102) and events were acquired on BD LSR Fortessa and analyzed using FlowJo software.
Statistical analysis
Statistical significance of differences in titers between groups in exposure and endogenous IgG assessments were calculated by Wilcoxon test (non-parametric) followed by p-value adjustment (Holm method). For DC internalization assay, statistically significant differences were calculated with ANOVA and paired t-test. Statistical analysis was performed using R.Citation44 Significance level: p < 0.0001= ****; p < 0.001= ***; p < 0.01= **; p< 0.05= *; not significant= ns.
Supplemental Material
Download Zip (4.8 MB)Acknowledgements
The authors would like to thank Laura Schröder, Michael Otteneder, Jasmin Messmer, David Waiz, Mailin Ashoff-Ciminski, Monique Farny, Lucien Rey, and Jule Koch for their support with in vivo studies at Roche. Furthermore, we would like to thank Anne Freimoser-Grundschober and Hubert Kettenberger from Roche for providing tool compounds. We are grateful to Michael Reth from the Department of Biology at the University of Freiburg for valuable input and discussion. Moreover, we thank Edith Horvath as well as Angelika Offinger and her team from the animal facility at the Department of Biomedicine at the University of Basel for support with the generation of bone marrow chimeras. All drawings and icons were created with BioRender.com.
Disclosure statement
J. Egli, S. Heiler, J. Bessa, F. Weber, G. Steiner, T. Schwandt, K. Bray-French, C. Klein, S. Fenn, G.P. Lotz, E. Opolka-Hoffmann, M. Siegel, T.E. Kraft and J. DeGeer are employees of the Roche Group and L. Petersen, R. Moser, and A. Iglesias were employees of Roche at the time the study was conducted. Additionally, F. Weber, J. Bessa, S. Fenn, G.P. Lotz, K. Bray-French, T. Schwandt, T.E. Kraft and C. Klein hold stock/options and/or patents with Roche. J. Egli and S. Heiler received funding from the Roche Postdoctoral Fellowship program.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2143009
Additional information
Funding
References
- Zhou Y, Penny HL, Kroenke MA, Bautista B, Hainline K, Chea LS, et al. Immunogenicity assessment of bispecific antibody-based immunotherapy in oncology. J Immunother Cancer. 2022;10(4):e004225. doi:10.1136/jitc-2021-004225.
- Dingman R, Balu-Iyer SV. Immunogenicity of Protein Pharmaceuticals. J Pharm Sci. 2019;108(5):1637–21. doi:10.1016/j.xphs.2018.12.014.
- Davda J, Declerck P, Hu-Lieskovan S, Hickling TP, Jacobs IA, Chou J, et al. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. Journal for ImmunoTherapy of Cancer. 2019;7:105. doi:10.1186/s40425-019-0586-0.
- Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs. 2017;9(2):182–212. doi:10.1080/19420862.2016.1268307.
- Ma J, Mo Y, Tang M, Shen J, Qi Y, Zhao W, et al. Bispecific Antibodies: From Research to Clinical Application. Front Immunol. 2021;12:626616. doi:10.3389/fimmu.2021.626616.
- Bessa J, Boeckle S, Beck H, Buckel T, Schlicht S, Ebeling M, et al. The immunogenicity of antibody aggregates in a novel transgenic mouse model. Pharm Res. 2015;32(7):2344–59. doi:10.1007/s11095-015-1627-0.
- Boll B, Bessa J, Folzer E, Ríos Quiroz A, Schmidt R, Bulau P, et al. Extensive Chemical Modifications in the Primary Protein Structure of IgG1 Subvisible Particles Are Necessary for Breaking Immune Tolerance. Mol Pharm. 2017;14(4):1292–99. doi:10.1021/acs.molpharmaceut.6b00816.
- Bacac M, Klein C. Umana P. CEA TCB: A novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology. 2016;5(8):e1203498. doi:10.1080/2162402X.2016.1203498.
- Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology. 2017;6(3):e1277306–e. doi:10.1080/2162402X.2016.1277306.
- Reitan SK, Hannestad K. Immunoglobulin heavy chain constant regions regulate immunity and tolerance to idiotypes of antibody variable regions. Proc Natl Acad Sci U S A. 2002;99(11):7588–93. doi:10.1073/pnas.052150899.
- Ghetie V, Hubbard JG, Kim J-K, Tsen M-F, Lee Y, Ward ES. Abnormally short serum half-lives of IgG in β2-microglobulin-deficient mice. Eur J Immunol. 1996;26(3):690–96. doi:10.1002/eji.1830260327.
- Qiao SW, Kobayashi K, Johansen FE, Sollid LM, Andersen JT, Milford E, et al. Dependence of antibody-mediated presentation of antigen on FcRn. Proc Natl Acad Sci U S A. 2008;105(27):9337–42. doi:10.1073/pnas.0801717105.
- Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29(10):457–66. doi:10.1093/protein/gzw040.
- Arnoult C, Brachet G, Cadena Castaneda D, Azzopardi N, Passot C, Desvignes C, et al. Crucial Role for Immune Complexes but Not FcRn in Immunization against Anti–TNF-α Antibodies after a Single Injection in Mice. J Immunol. 2017;199(2):418–424. doi:10.4049/jimmunol.1601246.
- Opolka-Hoffmann E, Jordan G, Otteneder M, Kieferle R, Lechmann M, Winter G, et al. The impact of immunogenicity on therapeutic antibody pharmacokinetics: A preclinical evaluation of the effect of immune complex formation and antibody effector function on clearance. mAbs. 2021;13(1):1995929. doi:10.1080/19420862.2021.1995929.
- Hoffmann E, Kotsias F, Visentin G, Bruhns P, Savina A, Amigorena S. Autonomous phagosomal degradation and antigen presentation in dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(36):14556–61. doi:10.1073/pnas.1203912109.
- Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, et al. An efficient route to human bispecific IgG. Nat Biotechnol. 1998;16(7):677–81. doi:10.1038/nbt0798-677.
- Suurs FV, Lub-de Hooge MN, de Vries Ege, de Groot Dja. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol Ther. 2019;201:103–19. doi:10.1016/j.pharmthera.2019.04.006.
- Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. mAbs. 2010;2(3):256–65. doi:10.4161/mabs.2.3.11641.
- Filipe V, Jiskoot W, Basmeleh AH, Halim A, Schellekens H, Brinks V. Immunogenicity of different stressed IgG monoclonal antibody formulations in immune tolerant transgenic mice. mAbs. 2012;4(6):740–52. doi:10.4161/mabs.22066.
- Bi V, Jawa V, Joubert MK, Kaliyaperumal A, Eakin C, Richmond K, et al. Development of a Human Antibody Tolerant Mouse Model to Assess the Immunogenicity Risk Due to Aggregated Biotherapeutics. J Pharm Sci. 2013;102(10):3545–55. doi:10.1002/jps.23663.
- Jawa V, Terry F, Gokemeijer J, Mitra-Kaushik S, Roberts BJ, Tourdot S, et al. T-Cell Dependent Immunogenicity of Protein Therapeutics Pre-clinical Assessment and Mitigation–Updated Consensus and Review 2020. Front Immunol. 2020;11:11. doi:10.3389/fimmu.2020.00011.
- Bray-French K, Hartman K, Steiner G, Marban-Doran C, Bessa J, Campbell N, Martin-Facklam M, Stubenrauch K-G, Solier C, Singer T, et al. Managing the Impact of Immunogenicity in an Era of Immunotherapy: From Bench to Bedside. J Pharm Sci. 2021;110(7):2575–84. doi:10.1016/j.xphs.2021.03.027.
- Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, De Groot AS. T-cell dependent immunogenicity of protein therapeutics: Preclinical assessment and mitigation. Clinical Immunology. 2013;149(3):534–55. doi:10.1016/j.clim.2013.09.006.
- van Schouwenburg Pa, van de Stadt La, de Jong Rn, van Buren Eel, Kruithof S, de Groot E, et al. Adalimumab elicits a restricted anti-idiotypic antibody response in autoimmune patients resulting in functional neutralisation. Ann Rheum Dis. 2013;72(1):104. doi:10.1136/annrheumdis-2012-201445.
- Stubenrauch K, Künzel C, Vogel R, Tuerck D, Schick E, Heinrich J. Epitope characterization of the ADA response directed against a targeted immunocytokine. J Pharm Biomed Anal. 2015;114:296–304. doi:10.1016/j.jpba.2015.05.029.
- Cassotta A, Mikol V, Bertrand T, Pouzieux S, Le Parc J, Ferrari P, et al. A single T cell epitope drives the neutralizing anti-drug antibody response to natalizumab in multiple sclerosis patients. Nat Med. 2019;25(9):1402–07. doi:10.1038/s41591-019-0568-2.
- Bogen B, Dembic Z, Weiss S. Clonal deletion of specific thymocytes by an immunoglobulin idiotype. EMBO J. 1993;12(1):357–63. doi:10.1002/j.1460-2075.1993.tb05664.x.
- Jacobsen JT, Lunde E, Sundvold-Gjerstad V, Munthe LA, Bogen B. The cellular mechanism by which complementary Id+ Id + and anti-Id antibodies communicate: T cells integrated into idiotypic regulation. Immunol Cell Biol. 2010;88(5):515–22. doi:10.1038/icb.2009.118.
- Jacobsen JT, Sundvold-Gjerstad V, Skjeldal FM, Andersen J-T, Abrahamsen G, Bakke O, et al. B-cell tolerance to the B-cell receptor variable regions. Eur J Immunol. 2013;43:2577–87. doi:10.1002/eji.201243203.
- Jacobsen J, Haabeth OA, Tveita AA, Schjetne KW, Munthe LA, Bogen B. Naive idiotope-specific B and T cells collaborate efficiently in the absence of dendritic cells. J Immunol. 2014;192(9):4174–83. doi:10.4049/jimmunol.1302359.
- Hummel H-D, Kufer P, Grüllich C, Seggewiss-Bernhardt R, Deschler-Baier B, Chatterjee M, et al. Pasotuxizumab, a BiTE® BiTE ® immune therapy for castration-resistant prostate cancer: Phase I, dose-escalation study findings. Immunotherapy. 2021;13(2):125–41. doi:10.2217/imt-2020-0256.
- You G, Won J, Lee Y, Moon D, Park Y, Lee SH, et al. Bispecific Antibodies: A Smart Arsenal for Cancer Immunotherapies. Vaccines. 2021;9(7):724. doi:10.3390/vaccines9070724.
- Reitan SK, Hannestad K. The primary IgM antibody repertoire: a source of potent idiotype immunogens. Eur J Immunol. 2001;31(7):2143–53. doi:10.1002/1521-4141(200107)31:7<2143::AID-IMMU2143>3.0.CO;2-1.
- Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody–FcRn interactions across species: implications for therapeutic antibodies. Int Immunol. 2001;13(12):1551–59. doi:10.1093/intimm/13.12.1551.
- Melendez R, Ordonia B, Guerrero J, Hassanzadeh A, Tran P, Low J, Wong M, Brumm J, Chung S, Kamen L, et al. Introducing dendritic cell antibody internalization as an immunogenicity risk assessment tool. Bioanalysis. 2022;14(10):703–13. doi:10.4155/bio-2022-0024.
- Colombetti S, Benigni F, Basso V, Mondino A. Clonal Anergy Is Maintained Independently of T Cell Proliferation. The Journal of Immunology. 2002;169(11):6178. doi:10.4049/jimmunol.169.11.6178.
- Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21(1):305–34. doi:10.1146/annurev.immunol.21.120601.141110.
- Yamamoto T, Hattori M, Yoshida T. Induction of T-cell activation or anergy determined by the combination of intensity and duration of T-cell receptor stimulation, and sequential induction in an individual cell. Immunology. 2007;121(3):383–91. doi:10.1111/j.1365-2567.2007.02586.x.
- Bessa J, Jegerlehner A, Hinton HJ, Pumpens P, Saudan P, Schneider P, et al. Alveolar Macrophages and Lung Dendritic Cells Sense RNA and Drive Mucosal IgA Responses. The Journal of Immunology. 2009;183(6):3788. doi:10.4049/jimmunol.0804004.
- Hoffmann E, Jordan G, Lauer M, Ringler P, Kusznir EA, Rufer AC, et al. Generation, Characterization, and Quantitative Bioanalysis of Drug/Anti-drug Antibody Immune Complexes to Facilitate Dedicated In Vivo Studies. Pharm Res. 2019;36(9):129. doi:10.1007/s11095-019-2661-0.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi:10.1016/S0022-2836(05)80360-2.
- Madaan A, Verma R, Singh AT, Jain SK, Jaggi M. A stepwise procedure for isolation of murine bone marrow and generation of dendritic cells. Journal of Biological Methods. 2014;1(1):e1. doi:10.14440/jbm.2014.12.
- R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2021.