Abstract
Automatic and manual sampling for ochratoxin A (OTA) in barley grain was compared under industrial conditions considering sampling uncertainty as well as practical and technical aspects. Ten tonnes of barley inoculated with Penicillium verrucosum were incubated until the OTA concentration reached approximately 15 µg kg−1 and sampled with manual and automatic sampling. A nested experimental design and ANOVA was used to estimate variance components from sampling, sample reduction, sample preparation and analysis. Manual sampling resulted in a high sampling uncertainty and OTA concentrations in aggregate samples ranged from 2 to 80 µg kg−1. When aggregate samples were formed by automatic sampling the uncertainty arising from nugget effects and spatial distribution was practically eliminated. Results from this study show that an automatic sampler mounted after a mixer or conveyer can provide representative samples of OTA from a moving stream of barley. Automatic sampling might present a practical and economical alternative to manual sampling for feed mill operators when monitoring low levels of mycotoxins in grain or other commodities. Despite careful precautions, sample preparation and analysis resulted in a relative uncertainty of ±40% (p = 0.95), which was attributed to the sub-sampling following the two grinding steps. Size fractionation of the coarsely ground barley showed that 40% of the total amount of OTA was present in a small fraction of fine particles with a strong tendency to aggregate or stick to equipment and containers. Thus, in order to take advantage of the automatic sampling, it is crucial to apply an appropriate sub-sampling to prevent segregation of particles which may affect the OTA measurements.
Introduction
Ochratoxin A (OTA) has been shown to be a mycotoxin of considerable concern for animal as well as human health. In a large number of animal studies nephrotoxic, carcinogenic, teratogenic and immunotoxic effects were demonstrated (Kuiper-Goodman Citation1996). Exposure of OTA to pigs which are considered to be among the most sensitive animal species has been extensively studied and resulted in clinical symptoms such as porcine nephropathy or immune suppression by reduction of phagocyte activity (Pfohl-Leszkowicz and Manderville Citation2007). Cereals are usually the most important source of animal or human OTA intake in Europe and for that reason limits of OTA in raw cereals were regulated in the European Union. A highest permitted level of 5 µg kg−1 in cereals for human consumption was implemented in European legislation (European Commission Citation2006b) and for feedstuffs for pigs the guidance value was set to 50 µg kg−1 (European Commission Citation2006a).
Penicillium verrucosum is the mould species responsible for OTA production in cereals in Europe (Lund and Frisvad Citation2003). OTA is produced when moist grain is stored (Denli and Perez Citation2010), and the source of conidial infection is agricultural machinery as well as residues in drying and storage facilities. The minimum water content allowing growth of P. verrucosum is 16–17% (Northolt et al. Citation1979), while a slightly higher water content in the grain is required for OTA production (Richter et al. Citation2001). Thus, low water content is the most important factor for controlling OTA formation in cereals. In large-scale storage of grain, variations in moisture content in the commodity or seasonal thermal changes will induce moisture migration (Holmberg Citation1993; Scudamore Citation2005) and may lead to formation of ‘hot spots’ where P. verrucosum will grow and subsequently produce OTA.
Sampling uncertainty for OTA in cereals may arise from spatial distribution as well as the nugget effect, i.e. localisation of the toxin to a low proportion of highly contaminated kernel (Nowicki and Roscoe Citation2010). Thus, in order to form a representative sample, it is necessary to take a large number of incremental samples and also grind an aggregate sample of substantial mass to ensure that the number of infected kernels in the sample is representative for the lot. Considering those conditions, estimations of OTA can be challenging due to the heterogeneous distribution observed for the mycotoxin (Biselli et al. Citation2008; Nowicki and Roscoe Citation2010).
In the framework of Biotracer (http://www.biotracer.org), with the aim to create tools and models for the improvement of tracing contaminations of feed and food, a questionnaire was sent out to several European feed companies about sampling for feed contaminants. The results showed that very few companies were using statistically based sampling plans or very few increments for the monitoring of mycotoxins in feed ingredients (unpublished data). Alternative sampling methods for contaminants that are easy to use, are cost effective and require limited manpower and give accurate results that are important especially for small and medium-sized companies in order to maintain a high quality of their products in a competitive environment (Andersson and Häggblom Citation2009).
Sampling for contaminants in grain is commonly performed with manual sampling using a chute, but there is currently little information on the performance of these sampling methods for OTA in grain. Forming a representative sample by manual sampling is challenging and time consuming and if the sampling results in non-representative samples it may result in incorrect decisions about the rejection or acceptance of the grain lot. Thus, it can be expected that the application of simple and reliable automatic sampling equipment could contribute to more dependable and cost-effective sampling of feed commodities.
The aim of the present study was to compare automatic sampling with manual sampling of OTA in barley grain under industrial conditions more specifically to study the sampling uncertainty for OTA and also practical and technical aspects. A second aim was to evaluate if the recommendations regarding the minimum mass of grain to analyse are fit for purpose, and to investigate the importance of sample preparation procedures including grinding and sub-sampling as sources of uncertainty.
Materials and methods
Production of OTA contaminated barley grain
A 4 kg inoculum was prepared by seeding gamma-irradiated barley grain with conidia from an OTA producing strain of Penicillium verrucosum (IBT 22626) obtained from Dr J. Frisvad (DTU, Denmark). Conidia were harvested by washing two malt extract agar Petri dishes of P. verrucosum, grown at 25°C for 10 days, with 5 ml each of salt water buffer with 0.1% Tween. The conidia were added to the grain at a moisture content of 22% and the axenic culture was cultivated for 8 days at 25°C. The grain culture was kept at +4°C until inoculation of the barley in the container.
During the harvest season incoming barley grain from farms was screened by the feed company for batches with a high water content using NIR and one batch of 10 tonnes was identified. The water content was elevated to approximately 20.5% by the addition of water followed by mechanical mixing. The grain was inoculated with 4 kg of the P. verrucosum grain culture and mechanically mixed and placed in a container (length = 6 m, width = 2.5 m, to a height of 1.30 m) equipped with four ventilation pipes (diameter = 125 mm) in order to prevent low oxygen tension in the bottom layers and placed at ambient temperature during the experiment. The experiment was started on 6–7 October 2008 and was terminated after 9 weeks. The temperature and water content in the centre of the container and close to the walls were measured weekly. Water content was also measured on a weekly basis in two aggregate samples from the centre of the container as well as in surface samples and near the walls.
To verify that the formation of analytical samples () would not be associated with a very large uncertainty and dominate other sources of uncertainty in the ANOVA, sub-sampling by spooning and fractional shovelling (International Organization for Standardization (ISO) Citation2001) was evaluated. Sub-sampling using a spoon was performed by mixing the test sample and taking different parts of the sample with a spoon. For fractional shovelling, a cone of the mixed test sample was formed. Sub-samples of approximately 1 g were taken around the base of the cone and added to one out of n analytical samples until the entire cone was distributed. Depending on the mass of the test sample, n was selected so that the mass of each analytical sample was approximately 25 g.
Sampling and physical sample preparation
Manual sampling was performed using a 30 mm sampling spear with eight chutes. One hit with the spear collected approximately 350 g of grain.
In the monitoring of ochratoxin A formation, duplicate aggregate samples of approximately 4 kg were formed every week during the 9-week incubation period. Each aggregate sample was formed from 11 incremental samples (). A sub-sample from each aggregate sample was analysed for moisture content using NIR. The entire aggregate sample was finely ground using a SLAGO-200-A beater wheel mill (Kamas Kvarnmaskiner AB, Malmö, Sweden) with a 1 mm sieve and one analytical sample was analysed for OTA content.
Figure 1. Sampling schemes for a comparison of manual and automatic sampling of OTA in barley. Figures illustrate the positions of incremental samples in the container. (a) Increments during the monitoring of OTA formation where open and filled circles indicate aggregate samples 1 and 2, respectively. (b) End-point manual sampling where aggregate samples are formed from incremental samples collected at positions 1–8. (c) Formation of eight aggregate samples by interpenetrating automatic sampling.
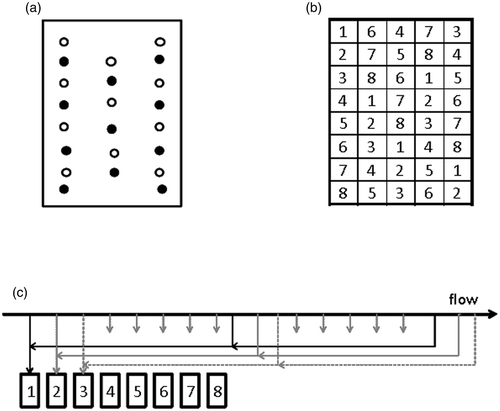
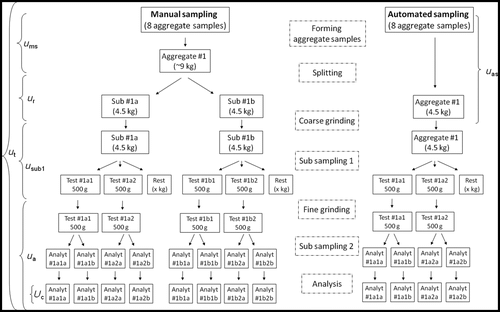
Figure 2. OTA estimations in barley, experimental design and procedures for sampling and sample preparation. Aggregate samples were formed by manual and automatic sampling. After sample splitting, milling and reduction, four test samples were obtained in the manual sampling procedure and two test samples in the automatic sampling, respectively. Each test sample was analysed twice for OTA concentration by means of immunoaffinity clean up and HPLC-FLD. Brackets indicate total uncertainty (u t) and uncertainty from sampling with automatic (u as) or manual (u ms) sampling, sample reduction (u r), subsampling (u sub1) and analysis (u a) (analysis = subsampling2 + extraction + HPLC).
The end-point sampling was carried out in week 10. Eight aggregate samples were formed from incremental samples taken according to different sampling patterns as illustrated in . To form an aggregate sample 6 × 350 g incremental samples were taken with spear at each of five locations (). The total weight of the aggregate sample was approximately 9 kg. The aggregate samples formed by manual sampling were split into two sub-samples (each approximately 4.5 kg) to simulate sample reduction (). Eight aggregate samples were formed by interpenetrating sampling (ISO Citation2003; (FAO/WHO) Citation2006a, Citation2006b) () using an automatic sampler type PP (Tagumatic A/S, Vedbæk, Denmark) from the same lot that was previously manually sampled. The grain was removed from the container into a hopper and the automatic sampler was fitted to a conditioner (length = 2400 mm, diameter = 600 mm, total volume = 675 L) where the grain was partly mixed at a speed of 160 rpm. During the sampling study approximately 5 tons h−1 were processed. Approximately 8 × 130 incremental samples were collected and the weight of each aggregate sample was approximately 4.5 kg. Following sampling, the barley samples were stored at –20°C until dried for 16 h at 55°C to a water content of 12–14%.
Both sub-samples from manual sampling and the entire sample from the automatic sampling were coarsely ground using a RAS® (Romer Analytical Sampling, Romerlabs, Tulln, Austria) mill where 96% of the particles passed a 2 mm sieve. Two test samples of approximately 500 g were formed by serial splits using a riffle splitter (, Sub-sampling1). Fine milling (particle size <0.5 mm) of laboratory samples was performed using a universal cutting mill (Pulverisette 19, Fritsch, Idar-Oberstein, Germany). From each test sample two analytical samples (approximately 25 g) were formed by fractional shovelling (, Sub-sampling2).
Sample extraction, clean-up and analysis
Analytical samples of approximately 25 g were extracted with ACN/water (60 : 40, v/v) with a sample to solvent ratio of 1 : 4. The extract was filtered through a pre-folded filter (595½; Ø185 mm; Schleicher & Schuell, Dassel, Germany), an aliquot (4 ml) was diluted with 150 ml PBS with 0.05% Tween 20 and applied on the immunoaffinity columns (OtaCLEAN™, LC-Tech, Dorffen, Germany). The columns were rinsed with 10 ml distilled water and the OTA eluted with 2 ml MeOH. The eluates were evaporated to dryness and redissolved in mobile phase and an aliquot was injected in the HPLC-FLD system.
HPLC
The HPLC system consisted of a pump (LC6A, Shimadzu, Tokyo, Japan) connected to an autosampler with a 100 µl loop (SIL-6B, Shimadzu) and controller (SCL-6B, Shimadzu) A guard column (LiChrospher 100-5 RP-18, Macherey-Nagel, Düren, Germany) was placed between the sample injection and the analytical column (LiChrospher 100 RP 18, 250 × 4 mm, 5 µm, Macherey-Nagel Düren). The determinations were carried out using a fluorescence detector (Merck-Hitachi, 1050 F, Tokyo, Japan) with an excitation wavelength of 333 nm and emission wavelength of 467 nm. The signals were visualized with Stratos® (Polymer Laboratories, Version 4.5, UK). The mobile phase which consisted of an MeOH : ACN : 3.3% aqueous solution of glacial acetic acid (35 : 35 : 30, v/v/v) permitted good separation with a flow rate of 0.8 ml min−1.
Standards
OTA stock solution containing 49.2 µg ml−1 OTA was obtained from Biopure (Tulln, Austria). Working solutions were prepared. OtaCLEAN™ immunoaffinity columns were purchased from LC-Tech (Dorfen, Germany). The expected recovery according to the manufacturers’ specifications is 92% ± 2%.
Distribution of OTA in coarsely ground barley
In order to study the distribution of OTA in coarsely ground barley, a sub-sample from the sampling experiment collected after grinding with a RAS® mill () was sieved using a test screening machine (J. Engelsmann AG, Ludwigshafen, Germany) with serial sieves and different mesh openings. Ground barley with a particle size >2, >1, >0.5, >0.25 and <0.25 mm were collected and weighed. After fractional shovelling, two analytical samples from each fraction were analysed for OTA.
Estimation of uncertainty using nested ANOVA
Variance and variance components were estimated by robust ANOVA using the software Roban (downloaded from http://www.samplersguide.com/sampling%20quality%20control.htm) according to recommendations in the Eurachem/EUROLAB/CITAC/Nordtest/AMC guide (Ramsey and Ellison Citation2007), where the variance components were estimated using a multilevel approach (see Appendix D of Ramsey and Ellison Citation2007). The variance from the sample preparation and analysis was estimated using data from all sub-samples formed by automatic or manual sampling (). Variance from automatic sampling was estimated using data from the eight aggregate samples and variance from the manual sampling, including sample reduction, was estimated using data from the ‘a’ sub-samples from each aggregate sample ().
In order to separate the variance resulting from the formation of aggregate samples from the sample reduction data from the manual sampling were reanalysed, using the ‘a’ analytical sample from each test sample (). The calculations were also repeated using an ANOVA spreadsheet (downloaded at http://www.samplersguide.com/sampling%20quality%20control.htm).
In order to study the impact of different sub-sampling techniques, three 25 g analytical samples were taken by spoon from each of eight randomly selected test samples. The variance was analysed with a modification of the ANOVA spreadsheet allowing for the analysis of triplicate samples.
The standard uncertainty u is estimated from the standard deviation from the measurement process, where u t is the total uncertainty from sampling, sample preparation and analysis. The coverage factor (k) was set to 2 representing a confidence level of approximately 95%. The uncertainty is expressed as the relative expanded uncertainty defined as:
Results
Monitoring of OTA formation
After 3–4 weeks an increase in the temperature in the centre of the grain was recorded with a maximum at week 7 with 23°C while the temperature near the surface remained between 10°C and 13°C. The grain temperature dropped significantly in both the centre and surface of the grain during the last weeks because of decreased outdoor temperature (). The moisture content in the grain was in the range 20.4% ± 0.6% throughout the experimental period.
Figure 3. Monitoring of temperature, moisture content and concentration of OTA (µg kg−1) in barley inoculated with Penicillium verrucosum at ambient temperature. Bars represent OTA concentration of aggregate samples (A, B), respectively. Graphs indicate the temperature (°C) near the surface and in the centre of the lot and the average moisture content (%).
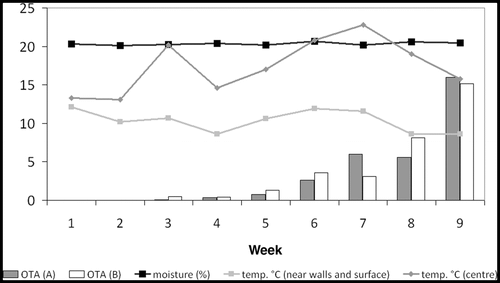
Traces of OTA were detected 2 weeks after the start of the experiment and increased rapidly from week 6 to approximately 15 µg kg−1 at week 9 (). The OTA concentrations from duplicate samples were not corrected for water content as the dry matter in all samples was in the range 86–88% throughout the experiment.
In the monitoring of OTA formation duplicate aggregate samples were analysed in order to get an idea of the total measurement uncertainty including the uncertainty from sampling. When fine milling was performed according to common practice by weighing 25 g of the ground material using a spoon, it was observed that the test result could differ with up to an order of magnitude between two analytical samples (results not shown). For that reason a pre-study was conducted to establish a reproducible sub-sampling procedure. The results indicated that sub-sampling by fractional shovelling resulted in acceptable errors (results not shown).
Comparison of manual and automatic sampling
Total uncertainty from automatic and manual sampling as well as the components of uncertainty corresponding to the different sources of variability were estimated by robust and standard ANOVA, as described by Ramsey and Ellison (Citation2007).
The following relations apply:
| u t | = |
total uncertainty; |
| u ms | = |
uncertainty from manual sampling; |
| u as | = |
uncertainty from automatic sampling; |
| u r | = |
uncertainty from sample reduction with splitter; |
| u sub1 | = |
uncertainty from sub-sampling after coarse grinding; |
| u sub1+a | = |
uncertainty from sample preparation and analysis; |
| u a | = |
analytical uncertainty including sub-sampling 2, extraction, clean-up and HPLC; and |
| U c | = |
uncertainty from the HPLC. |
The results from ANOVA are summarized in . Manual sampling resulted in high sampling uncertainty () and OTA concentrations in the different aggregate samples ranged from 2.65 to 76.96 µg kg−1 (), whereas sample preparation and analysis contributed marginally to total uncertainty (). When automatic sampling was applied the uncertainty from sample preparation and analysis prevailed, while the uncertainty from sampling was drastically reduced, making (u sub1+a) > 75% of the total uncertainty (). The uncertainty from the automatic sampling (u as) (±13%) was in the same range as the uncertainty arising from reduction of the manual samples (u r) (±17%).
Table 1. Concentrations of OTA (µg kg−1) at the end-point sampling (week 10) in individual analytical samples from manual and automatic sampling.
Table 2. ANOVA results for manual and automatic sampling of OTA in barley.
The uncertainty from sample preparation and analysis was approximately ±40% with a slightly higher contribution from the sub-sampling after coarse grinding (u sub1) (). The results in were obtained using robust ANOVA and when the same data set was analysed using standard ANOVA the estimated total uncertainty from sample preparation and analysis was similar, but the relative importance of sub-sampling and analysis was shifted (relative uncertainty = 23% and 32% for sub-sampling and analysis respectively). As expected, the estimates from the standard ANOVA were very sensitive to the inclusion or exclusion of the extreme samples from the manual sampling (M2 and M6) (results not shown).
In order to compare alternative sub-sampling techniques, analytical samples were taken in triplicate by spooning finely ground barley from a mixed test sample. The results indicated that this procedure resulted in a higher uncertainty from analysis (u a-spoon) (). By combining results from the two experiments using the formula:
Distribution of OTA in aggregate samples
In order to test whether segregation of particles of different size after milling could contribute to the sub-sampling uncertainty, particles from an aggregate sample were separated through serial sieves and the size fractions were weighed and analysed for OTA (). The result showed that the largest size fraction (>2 mm) contained very low concentration of OTA, whereas more than 40% of the toxin was present in 10% of the material with a particle size <0.25 mm ().
Table 3. Distribution of OTA in fractions of coarsely ground barley after sieving.
Discussion
In, for example, European Commission Regulation 401/2006 for official control of mycotoxins in foodstuffs, it is recommended that a sampling plan of 100 incremental samples and an aggregate sample of 10 kg is used for lots >50 tonnes, whereas for smaller lots the recommended number of increments is approximately proportional to the square-root of the lot size. That means for a lot size of 10 tonnes a recommendation of 40 increments and an aggregate sample of 4 kg. Such extensive manual sampling of contaminants is time consuming, costly and also dependent on human factors compared with on-line monitoring of, for example, other feed parameters using, for example, near infrared (NIR) technology. In practice these sampling plans may be used only in official control whereas sampling for other purposes generally relies on simplified protocols. However, the number of incremental samples needed to form a representative sample will not decrease with lot size (CAC Citation2004). Thus, if less stringent sampling is applied to small and medium-sized lots, the risk of misclassifying a lot due to sampling uncertainty may be large. Experience from the present study shows that in order to reduce costs and labour while maintaining the quality of mycotoxin sampling, automatic sampling may serve as a valuable alternative to manual sampling at, for example, feed mill operators for the monitoring of OTA.
In the present experiment 10 tonnes of OTA-contaminated barley were compared by manual and automatic sampling. In a commercial scenario, sampling of grain from an individual farmer could be in a similar range, while large-scale storage may be in the range of >1000 tonnes. In the present study the automatic sampler was attached to the moving stream of grain in the end of a short mixer. The sampler was operated by compressed air and an incremental sample was collected every 7–8 s for 2 h, and by adjusting the frequency of sampling a representative sample with the specified mass can be formed from an arbitrarily sized lot (http://www.tagumatic.dk). According to specifications the maximum sampling capacity is 80 tons h−1. For smaller lots where the time delay between samples may be a limiting factor, it is possible to use a sampler with a larger probe to form the aggregate sample from, for example, 50 incremental samples each of 100 g. For larger lots the small probe used here makes it possible to form an aggregate sample from >100 incremental samples without the need for subsequent sample reduction.
The complete cost for the present installation was approximately US$6000. Automatic samplers are available from several suppliers for an estimated cost of approximately US$4000. The costs for manual sampling are difficult to estimate depending on the costs for labour and equipment, the number of grain lots to be sampled, etc.
The results from the manual sampling clearly showed that not only the nugget effect, but also the spatial distribution may influence sampling uncertainty for OTA. In the automatic sampling each 4.5 kg aggregate sample was formed from approximately 130 incremental samples of approximately 27 g each and the results from the analysis of variance showed that the uncertainty arising from sampling is almost eliminated. These results are in agreement with the model of Casado et al. (Citation2009) where it was predicted that the collection of more than 50–60 incremental samples from a lot will not significantly reduce sampling uncertainty. However, the automatic sampling equipment was fitted to a paddle mixer and the grain was filled into a hopper situated above the mixer and thus it cannot be excluded that also the mixing contributed to the lower sampling uncertainty. For the manual sampling the lot analysed in this study had not been previously mixed, which may have contributed to the large effects of spatial distribution.
The results from the automatic sampling and the splitting of the manual samples indicated that the nugget effect was relatively modest (u r = 13–17%) using a sample mass of 4–5 kg. The impact of sample mass on the relative uncertainty was, in the present study, smaller than what was previously reported by Nowicki and Roscoe (Citation2010), where the OTA concentration was 2.3–3.5 µg kg−1 with the higher uncertainty at the lower concentrations.
When the pairs of aggregate samples were compared during the OTA formation, we observed that U appeared to be relatively constant at concentrations below 1 µg kg−1, whereas at higher concentrations U was approximately proportional to the OTA concentration (results not shown). According to sampling theory, the uncertainty from sample reduction (U r) is inversely proportional to the square-root of the average number of critical particles in the sample (Minkkinen Citation2004). Accordingly, the results from the present study and the results of Nowicki and Roscoe (Citation2010) indicate that the ‘nugget effect’ is a major source of uncertainty at lower concentrations, where at higher concentrations other sources including spatial distribution and sample preparation are more important.
Other important factors for accurate estimations of OTA are sample reduction and sample preparation. (For practical reasons the unprocessed sample is usually reduced to a manageable size before it is sent to the laboratory.) The nugget effect may result in the variation in toxin content in the test sample and for that reason the recommended procedure for OTA analysis includes two grinding steps. According to Minkkinen (Citation2004) the grinding of the grain in a preparatory mill (e.g. RAS mill) would reduce the uncertainty from the first sub-sampling as the number of critical particles increases. However, in agreement with Nowicki and Roscoe (Citation2010), we found that the total relative uncertainty (U t) was ±40% even when the nugget effect and the effect of spatial distribution was eliminated by automatic sampling. It is well known that OTA is present in certain parts of the grain, e.g. the chaff and bran, and that after milling OTA will accumulate in certain fractions of the material (Scudamore et al. Citation2003). It is also well known that particles of different size and shape may segregate and introduce sub-sampling errors (Brittain Citation2002). The effect of particle segregation was demonstrated in the present study where after milling the grain in the RAS® mill a large fraction of the material consisted of fine particles, which tended to form aggregates and to stick to the equipment. In a pilot experiment we observed that formation of analytical samples from the fine-milled sub-sample introduced extremely large variability in the test results.
In this study a nested experimental design (ISO Citation2003; FAO/WHO Citation2006b) based on aggregate samples was used to estimate the uncertainty from sampling, sample preparation and analysis. Due to the uneven distribution of mycotoxins in many commodities the normal assumption is not valid for incremental samples from a lot and for this reason studies of sampling uncertainty for mycotoxins are often based on statistical modelling (Berry and Day Citation1973; Johansson et al. Citation2000; Casado et al. Citation2009). However, an aggregate sample is the average of the concentration of the increments if sample size and mass are large enough. In this case the central limit theorem of statistics states that the concentration will approximately follow a normal distribution (Blom Citation1989) and in this work results from Biselli et al. (Citation2008) were used to design the sampling plan.
Acknowledgements
This work was supported by the European Union Integrated Project BIOTRACER (Contract Number 036272) under the 6th RTD Framework. Thanks to Pelle Bäckström, Lars-Göran Andersson and Eva Jonsson for technical support; and to Erik Nordkvist for valuable comments on the manuscript.
Related Research Data
References
- Andersson , G and Häggblom , P . 2009 . Sampling for contaminants in feed – estimating the concentration of mycotoxins found in bulk samples can be a complicated process . Feed Int , 3 : 16 – 19 .
- Berry , G and Day , NE . 1973 . The statistical analysis of the results of sampling an environment for a contaminant when most samples contain an undetectable level . Am J Epidemiol , 97 ( 3 ) : 160 – 166 .
- Biselli , S , Persin , C and Syben , M . 2008 . Investigation of the distribution of deoxynivalenol and ochratoxin A contamination within a 26 t truckload of wheat kernels . Mycotox Res , 24 ( 2 ) : 98 – 104 .
- Blom , G . 1989 . Sannolikhetsteori med tillämpningar , 4. Lund (Sweden) : Studentlitteratur .
- Brittain , HG . 2002 . Particle-size distribution II: the problem of sampling powdered solids . Pharm Technol , 7 : 67 – 73 .
- CAC. 2004. General guidelines on sampling, No. 69. Rome, Italy: FAO/WHO Joint Publications
- Casado , MR , Parsons , DJ , Weightman , RM , Magan , N and Origgi , S . 2009 . Modelling a two dimensional spatial distribution of mycotoxin concentration in bulk commodities to design effective and efficient sample selection strategies . Food Addit Contam A , 26 ( 9 ) : 1298 – 1305 .
- Denli , M and Perez , JF . 2010 . Ochratoxins in feed, a risk for animal and human health: control strategies . Contr Strateg , 2 : 1065 – 1077 .
- European Commission . 2006a . Commission Recommendation (2006/576/EC) of 17 August 2006 on the presence of deoxynivalenol, zearalenone, ochratoxin A, T-2 and HT-2 and fumonisins in products intended for animal feeding . Off J Eur Union , L229 : 7 – 9 .
- European Commission . 2006b . Commission Regulation (EC) No. 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuff . Off J Eur Union , L 364 : 5 – 24 .
- Food and Agricultural Organization/World Health Organization (FAO/WHO). 2006a. Uncertainty of sampling. Joint FAO/WHO Food Standards Programme. Codex Committee on methods of analysis and sampling. Twenty-Seventh Session, Budapest, Hungary, 15–19 May 2006
- Food and Agricultural Organization/World Health Organization (FAO/WHO). 2006b. Uncertainty of sampling. CX/MAS 06/27/10. Joint FAO/WHO Food Standards Programme. Codex Committee on methods of analysis and sampling. Twenty-Seventh Session, Budapest, Hungary, 15–19 May 2006
- Holmberg , T . 1993 . Ochratoxin in cereal grain and its potential effects on animal health , Uppsala (Sweden) : Swedish University of Agricultural Sciences .
- International Organization for Standardization (ISO) . 2001 . ISO 11648-2. Statistical aspects of sampling from bulk materials – Part 2: Sampling of particulate materials , Geneva (Switzerland) : ISO .
- International Organization for Standardization (ISO) . 2003 . ISO11648-1. Statistical aspects of sampling from bulk materials – Part 1: General principles , Geneva (Switzerland) : ISO .
- Johansson , AS , Whitaker , TB , Hagler Jr , WM , Giesbrecht , FG , Young , JH and Bowman , DT . 2000 . Testing shelled corn for aflatoxin, Part I: Estimation of variance components . J AOAC Int , 83 ( 5 ) : 1264 – 1269 .
- Kuiper-Goodman , T . 1996 . Risk assessment of ochratoxin A: an update . Food Addit Contam , 13 ( Suppl ) : 53 – 57 .
- Lund , F and Frisvad , JC . 2003 . Penicillium verrucosum in wheat and barley indicates presence of ochratoxin A . J Appl Microbiol , 95 ( 5 ) : 1117 – 1123 .
- Minkkinen , P . 2004 . Practical applications of sampling theory . Chemometr Intell Lab , 74 ( 1, Special Issue: 50 years of Pierre Gy's Theory of Sampling Proceedings: First World Conference on Sampling and Blending (WCSB1) Tutorials on sampling: Theory and Practice ) : 10
- Northolt , M , van Egmond , HP and Paulsch , WE . 1979 . Ochratoxin A production by some fungal species in relation to water activity and temperature . J Food Prot , 42 ( 6 ) : 485 – 490 .
- Nowicki , T and Roscoe , M . 2010 . An alternative to slurry mixing to minimise sample preparation variance for determination of ochratoxin A in wheat . World Mycotoxin J , 3 ( 2 ) : 147 – 156 .
- Pfohl-Leszkowicz , A and Manderville , RA . 2007 . Ochratoxin A: an overview on toxicity and carcinogenicity in animals and humans . Mol Nutr Food Res , 51 ( 1 ) : 61 – 99 .
- Ramsey MH, Ellison SLR. 2007. Eurachem/EUROLAB/CITAC/Nordtest/AMC guide: Measurement uncertainty arising from sampling: A guide to methods and approaches. ISBN: 978 0 948926 26 6
- Richter , WIF , Baranowski , A , Schuster , M and Scholz , W . 2001 . Bildung von Ochratoxin A bei der Lagerung von Triticale nach künstlicher Infektion mit Penicillium verrucosum in Abhängigkeit unterschiedlicher Feuchtegehalte . Mycotox Res , 17 : 193 – 197 .
- Scudamore , KA . 2005 . Prevention of ochratoxin A in commodities and likely effects of processing fractionation and animal feeds . Food Addit Contam , 22 ( Suppl. 1 ) : 17 – 25 .
- Scudamore , KA , Banks , J and MacDonald , SJ . 2003 . Fate of ochratoxin A in the processing of whole wheat grains during milling and bread production . Food Addit Contam , 20 ( 12 ) : 1153 – 1163 .