
ABSTRACT
The neuro-2a bioassay is considered as one of the most promising cell-based in vitro bioassays for the broad screening of seafood products for the presence of marine biotoxins. The neuro-2a assay has been shown to detect a wide array of toxins like paralytic shellfish poisons (PSPs), ciguatoxins, and also lipophilic marine biotoxins (LMBs). However, the neuro-2a assay is rarely used for routine testing of samples due to matrix effects that, for example, lead to false positives when testing for LMBs. As a result there are only limited data on validation and evaluation of its performance on real samples. In the present study, the standard extraction procedure for LMBs was adjusted by introducing an additional clean-up step with n-hexane. Recovery losses due to this extra step were less than 10%. This wash step was a crucial addition in order to eliminate false-positive outcomes due to matrix effects. Next, the applicability of this assay was assessed by testing a broad range of shellfish samples contaminated with various LMBs, including diarrhetic shellfish toxins/poisons (DSPs). For comparison, the samples were also analysed by LC-MS/MS. Standards of all regulated LMBs were tested, including analogues of some of these toxins. The neuro-2a cells showed good sensitivity towards all compounds. Extracts of 87 samples, both blank and contaminated with various toxins, were tested. The neuro-2a outcomes were in line with those of LC-MS/MS analysis and support the applicability of this assay for the screening of samples for LMBs. However, for use in a daily routine setting, the test might be further improved and we discuss several recommended modifications which should be considered before a full validation is carried out.
Introduction
Marine biotoxins are naturally occurring compounds mostly produced by certain algae. These toxins affect human health mainly through foodborne intoxications, i.e. consumption of contaminated seafood, and occasionally through direct exposure to seawater aerosols (James et al. Citation2010; Berdalet et al. Citation2016). Consumption of seafood contaminated with marine biotoxins may result in relatively mild symptoms, such as diarrhoea, dizziness, numbness and tingling of the mouth and digits, but also paralysis and in severe cases even death (Watkins et al. Citation2008; Visciano et al. Citation2016). Several major types of poisoning are described: amnesic shellfish poisoning (ASP), diarrhetic shellfish poisoning (DSP), neurologic shellfish poisoning (NSP) and paralytic shellfish poisoning (PSP) (Visciano et al. Citation2016). A fifth syndrome, azaspiracid poisoning (AZP), has been characterised during the last 20 years (Twiner et al. Citation2008). To avoid intoxications, monitoring is obligatory in many parts of the world. Within the EU, limits have been set by the European Commission (Regulation No 853/2004) for ASP and PSPs, as well as several lipophilic marine biotoxins (LMBs) (Regulation (EU) No 15/2011).
Worldwide, the main assay applied is the mouse bioassay (MBA), where mice are intraperitoneally injected with a sample extract, using lethality as the endpoint (Yasumoto et al. Citation1978, Citation1984; Kat Citation1983). Besides ethical issues, the MBA gives high rates of false-positive and false-negative results (EFSA Citation2009, Citation2010). In Europe, the use of the MBA has been banned for LMBs since 2015, but not for PSP analysis and not for the control of production areas, aiming at detection of possibly unknown LMBs (Regulation (EU) No 15/2011). The reference method for the detection of LMBs is now the LC-MS/MS method of the EURL on marine biotoxins (Regulation (EC) No 2074/2005; Gerssen, Pol-Hofstad et al. Citation2010; Regulation (EU) No 15/2011). LC-MS/MS based methods are fit for purpose, but for many toxins certified standards and reference materials are scarce or not available. This makes their use for detecting all marine biotoxins very difficult, if not impossible. For example, there are at least 24 saxitoxin analogues (Etheridge Citation2010), 13 okadaic acid-ester derivatives (Dominguez et al. Citation2010), 90 yessotoxin analogues (Paz et al. Citation2008), 15 brevetoxin analogues (Plakas and Dickey Citation2010) and around 30 azaspiracid analogues (EFSA Citation2008; Rehmann et al. Citation2008) and currently there are only 20–30 certified reference standards available. Furthermore, analytical methods are by definition unable to predict toxicity of complex mixtures or pick up new risks. As a result, many countries hesitate to rely solely on analytical methods and keep using the MBA.
Because of the drawbacks of the MBA and the analytical chemical methods, EFSA emphasised the need for developing animal friendly alternatives (EFSA Citation2009). There is thus an urgent need for in vitro tests that allow the detection of marine biotoxins that are currently known and those which might emerge (Peperzak Citation2005; Moore et al. Citation2008). Biochemical assays and especially cell-based bioassays have the potential to fulfil these requirements (Caillaud et al. Citation2009; Nicolas et al. Citation2014). The neuro-2a assay, using the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) or water-soluble tetrazolium salts (WST) as a viability end-point, is considered to be one of the most promising cell-based bioassays for the broad screening of marine neurotoxins (Cañete and Diogène Citation2008; Nicolas et al. Citation2014). It is already used to some extent for the detection of ciguatoxins (CTXs), tetrodotoxin (TTX) (Reverte et al. Citation2014) and the lipophilic brevetoxins (PbTxs) (Dechraoui et al. Citation2005; Empey Campora et al. Citation2008). However, although the neuro-2a assay has been tested intensively for PSPs, this is mainly with standards and very limited with routine samples (Jellett et al. Citation1995; Manger et al. Citation1995; Humpage et al. Citation2007, Citation2010). Thus far, the assay seems not widely applied routinely for PSP or DSP testing and certainly not within the EU. According to an extensive collaborative study that involved testing of OA, PTX-2 and AZA-1 in three cell lines, i.e. Caco-2, HepG2 and Neuro-2a, matrix effects seems to be an important issue that needs to be addressed before these assays are applied for real samples (Ledreux et al. Citation2012). Therefore, developing a suitable extraction method is critical to allow the routine use of the neuro-2a assay for shellfish samples (Aballay-Gonzalez et al. Citation2016).
The aim of the present study was therefore to evaluate and optimise the neuro-2a assay for routine testing of shellfish on the presence of LMBs. First, a comparison was made between the use of the murine neuro-2a neuroblastoma cells and another previously applied cell line, murine neuroblastoma × rat glioma hybrid cells (NG108-15) in order to establish the sensitivity of both cell-lines in our laboratory. We tested all EU regulated lipophilic marine biotoxins for which standards were available, i.e. okadaic acid (OA), dinophysistoxin-1 (DTX-1), dinophysistoxin-2 (DTX-2), pectenotoxin-2 (PTX-2), azaspiracid-1 (AZA-1), azaspiracid-2 (AZA-2), azaspiracid-3 (AZA-3), yessotoxin (YTX) and 1a-homo yessotoxin (hYTX). Subsequent testing of shellfish samples was carried out with the neuro-2a cells, as these turned out to be slightly more sensitive. Based on first test results with real samples, an improved clean-up procedure was developed by introducing an additional n-hexane wash step in order to reduce false positives due to matrix effects. Potential recovery losses due to this extra wash step were checked by LC-MS/MS analysis of blank sample extracts spiked just before and after the n-hexane wash step. Next, extracts of both blank and contaminated shellfish were tested and results were compared with LC-MS/MS outcomes in order to examine whether this bioassay is applicable to real samples in a daily routine setting and to identify possible issues for further improvement prior to a full validation. This is necessary, as, besides the extensive efforts needed, such a validation will be very expensive due to the high costs of the required marine toxin standards.
Materials and methods
Reagents and standards
Certified reference materials (CRMs) of OA (13.7 ± 0.6 µg ml−1), DTX-1 (15.1 ± 1.1 µg ml−1), DTX-2 (7.8 ± 0.4 µg ml−1), PTX-2 (4.40 ± 0.13 µg ml−1), AZA-1 (1.24 ± 0.07 µg ml−1), AZA-2 (1.28 ± 0.05 µg ml−1), AZA-3 (1.04 ± 0.04 µg ml−1), YTX (5.6 ± 0.2 µg ml−1), and hYTX (5.8 ± 0.3 µg ml−1) were purchased from the National Research Council, Institute for Marine Biosciences (NRC CNRC, Halifax, Canada). Stock solutions of these toxin standards were prepared in dimethyl sulfoxide (DMSO) after evaporation of the original solvent. DMSO, ammonium hydroxide (25%) and n-hexane were obtained from Merck (Darmstadt, Germany). Acetonitrile (Ultra LC/MS), methanol (Ultra LC/MS) and water (Ultra LC/MS) were purchased from Actu-All (Oss, the Netherlands).
Samples
In-house samples, both blank mussel samples from the Netherlands, and contaminated samples obtained from various locations and used for previous validation studies of the LC-MS/MS method, were tested (van den Top et al. Citation2011). In addition, 50 samples (crude methanol extracts) from various types of marine gastropods and bivalves, potentially naturally contaminated with LMBs, were kindly donated by Dr Carlos García from the Faculty of Medicine, Universidad de Chile, Santiago, Chile (Zamorano et al. Citation2013).
Table 1. Calculated EC50 values for the effect of lipophilic marine biotoxins on the viability of murine neuroblastoma neuro-2a cells, and murine neuroblastoma x rat glioma hybrid NG108-15 cells, the resulting TEF values in the neuro-2a assay, and recommended TEFs by FAO/WHO and EFSA.
Table 2. LC-MS/MS determined levels of lipophilic marine biotoxins in 16 naturally contaminated shellfish samples compared to the outcome of the neuro-2a assay. Samples (S1-S8) were mixed samples used for the validation of the LC-MS/MS method.
Table 3. Effect of Chilean seafood extracts on the viability of neuro-2a cells and levels of lipophilic marine neurotoxins measured by LC-MS/MS.
Preparation of extracts
Prior to extraction of the lipophilic marine biotoxins (i.e. DSPs and AZPs), shellfish material was homogenised with a T25 Ultra Turrax mixer at 24,000 rpm (IKA® Works Inc., Wilmington, NC, USA). One gram of shellfish homogenate was vortex mixed with 3 ml methanol for 1 min and centrifuged for 5 min at 2000 × g. The supernatant was transferred to a volumetric flask and the residue was extracted twice more with 3 ml methanol. After the third extraction the volume of the collected supernatant was adjusted to 10 ml with methanol. For the neuro-2a assay, additional clean-up steps using n-hexane and solid phase extraction (SPE) were applied (see below), which were not required for the LC-MS/MS analysis.
Clean-up by n-hexane wash step followed by SPE
A 4.8 ml aliquot of the crude methanol shellfish extract was diluted with 1.2 ml Milli-Q water and extracted twice with 6 ml n-hexane in order to remove matrix substances that led to false-positive test outcomes. The hexane layer was discarded and the aqueous methanolic extract was further diluted by adding 10 ml Milli-Q water and the total extract of 16 ml was transferred to an SPE StrataTM-X cartridge (200 mg/6 ml; Phenomenex, Utrecht, the Netherlands), previously conditioned with 4 ml methanol/water (30:70 v/v). Subsequently, the cartridge was washed with 8 ml methanol/water (20:80 v/v) and the toxins were eluted with 4.8 ml methanol. The eluate was evaporated to dryness under a stream of nitrogen gas and reconstituted in 20 µl DMSO.
Recovery of the n-hexane wash step
Blank mussel samples were pooled (10 g) and 1 g portions were extracted using the method described above. Fortification of extracts equivalent to 3, 1 and 1/3 of the maximum permitted level (MPL) of OA, DTX-1 and AZA-1 (i.e. 480 µg kg−1, 160 µg kg−1 and about 53 µg kg−1) and about 1/4 MPL for YTX (i.e. 1000 µg kg−1) before and after the n-hexane clean-up were carried out by adding a corresponding volume of a highly concentrated standard in methanol. The recovery was calculated by LC-MS/MS analysis of subsamples taken from the fortified extracts. The rest of the sample extracts fortified before and after the n-hexane extraction, were purified on SPE (see above) and analysed in the neuro-2a assay. The exposures were performed in three different experiments and all samples within an experiment were tested in triplicate. Due to the high costs and amounts needed, YTX experiments were performed twice and samples were tested in triplicate.
Cell culture and exposure
Neuroblastoma neuro-2a cells were purchased from the American Type Culture Collection (ATCC; CCL-131) and cultured in 75 cm2 culture flasks containing 15 ml RPMI-1640 medium (R0883, Sigma-Aldrich, Zwijndrecht, the Netherlands) supplemented with 10% (v/v) foetal bovine serum (FBS, Fisher Emergo, Landsmeer, the Netherlands), 1% (v/v) of a 100 mM sodium pyruvate solution (Sigma-Aldrich) and 1% (v/v) of a 200 mM L-glutamine solution (Sigma-Aldrich). NG108-15 cells were also obtained from ATCC (HB-12,317) and cultured in 75 cm2 culture flasks containing 30 ml Dulbecco’s modified Eagle’s medium (DMEM) obtained from Lonza (Verviers, Belgium) supplemented with 10% (v/v) FBS and 2% (v/v) of 50× HAT supplement (5 mM hypoxanthine, 20 µM aminopterin and 0.8 mM thymidine, Sigma-Aldrich). Both cell lines were routinely maintained in a humidified incubator at 37°C under 5% CO2 and sub-cultured three times per week (dilution 1/5) up to approximately 90% confluence.
The experimental conditions for the assay were adapted from Cañete and Diogène (Citation2008). Neuro-2a and NG108-15 cells were seeded separately into 96-well plates with an initial density of 25,000 and 14,000 cells/well, respectively. After growing the cells for 24 h, exposure to increasing concentrations of pure marine biotoxins or sample extracts was performed in quadruplicate in 200 μl medium for 24 h. The method of exposing the cells was changed in order to adjust it to our sample extracts and standards that were dissolved in DMSO, i.e. exposure media were prepared by using e.g. 3 µl standard or sample extract in DMSO and 1200 µl of culture medium. Dilutions, 5 or 10 times, were made in culture medium with 0.25% DMSO to keep the solvent concentration at 0.25%. At the end of the exposure time, cell viability was measured using the MTT assay.
Cell viability assay (MTT)
Briefly, MTT (Sigma-Aldrich) was prepared in PBS at 5 mg ml−1, and mixed with serum-free medium. Then, the media from the cells was removed and 60 µl of MTT mixed with serum-free medium was added to each well (final concentration of MTT in the well is 0.8 mg ml−1). After 30 min incubation at 37°C, the medium was removed and the formed formazan crystals were dissolved in 100 µl DMSO. Plates were placed in a plate shaker for 10 min at 600 rpm after which the absorbance was measured at 540 nm and corrected for background absorption at 650 nm. EC50 values were determined using a nonlinear regression model (GraphPad Prism software version 5.04, San Diego, CA, USA).
Chemical analysis
Chemical analysis was directly performed on crude methanol extracts. The method applied for the determination of lipophilic marine biotoxins (i.e. DSPs and AZPs) was previously described by Gerssen et al. (Citation2009) and Gerssen, van Olst et al. (Citation2010). Chromatographic separation was achieved using a Waters Acquity I-Class UPLC system (Waters, Milford, MA, USA). The system consisted of a binary solvent manager, sample manager and a column manager. The column temperature was kept at 60°C and the temperature of the sample manager was kept at 10°C. A 5 µl injection volume was used. Mobile phase A was water and mobile phase B was acetonitrile/water (9:1 v/v), both containing 6.7 mM ammonium hydroxide. A flow rate of 0.6 ml min−1 was used. A gradient started at 30% B and after 0.5 min was linearly increased to 90% B in 3 min. This composition was kept for 0.5 min and returned to 30% B in 0.1 min. An equilibration time of 0.9 min was allowed prior to the next injection. The effluent was directly interfaced in the electrospray ionisation (ESI) source of the AB Sciex QTrap 6500 mass spectrometer (Ontario, Canada). The mass spectrometer was operated in both negative and positive electrospray ionisation by rapid polarity switching. For each toxin two transitions were measured. For quantification of the toxins, so-called ‘matrix matched’ calibration curves were constructed. These calibration curves were constructed by fortifying blank shellfish extracts with different concentrations of toxin. The area of the toxin in the unknown sample is then calculated using the linear equation of the calibration curve. The concentration is expressed in µg kg−1 shellfish.
Results and discussion
Effects of individual lipophilic marine biotoxins
A number of different toxins were tested on both neuro-2a and NG108-15 cells using MTT reduction as measure for cell viability. Overall, all lipophilic marine biotoxins and their analogues induced a concentration-dependent decrease in viability of both neuro-2a and NG108-15 cells. shows the effect of the lipophilic marine biotoxins OA, DTX-1, DTX-2, AZA-1, AZA-2, AZA3, PTX-2, YTX and hYTX on the cell viability of neuro-2a cells. shows the calculated EC50 values for both cell lines, being the concentration reducing the MTT response by 50% of the maximal observed difference. Although AZAs and YTXs caused a decrease in cell viability at relatively low concentrations, MTT activity was only reduced to about 50% of the initial response, while OA, DTXs, and PTX-2 were able to further reduce the MTT activity. Using NG108-15 cells and a similar MTT protocol, Cañete and Diogène (Citation2010) also observed that AZA-1 induced a maximum reduction to about 40%. The EC50 values determined for OA, DTX-1, AZA-1 and YTX-1 in the NG108-15 assay are also in line with those published by Cañete and Diogène (Citation2010). Serandour et al. (Citation2012) also observed a levelling off for AZA-1, but around 10% and they also observed that AZA-1 is more potent than OA and PTX-2, but the EC50 value of 6.8 nM was sevenfold higher than the one we and Cañete and Diogène (Citation2010) obtained. The EC50 values determined for OA, DTX-1 and PTX-2 in the neuro-2a assay, being respectively 23.4, 5.5 and 76.4 nM, differ from those published by Cañete and Diogène (Citation2008), being respectively 21.9, 20.6 and 28.3 nM. Thus similar EC50 values were obtained for OA, but lower and higher values for DTX-1 and PTX-2 respectively. Serandour et al. (Citation2012) reported values of 41.2 and 35.5 nM for OA and PTX-2, respectively, being twofold higher for OA than obtained by us and Cañete and Diogène (Citation2008) and twofold lower for PTX-2 than observed in the present study. These differences are possibly caused by differences in quality of the standards, the applied solvents and solvent concentration, or the culturing and exposure conditions of the cells, e.g. 48 h compared to 24 h in our study.
Figure 1. Effect of several lipophilic marine biotoxins on the viability of neuro-2a cells as measured with the MTT assay compared to the average of the solvent control (0.25% DMSO): (a) OA: okadaic acid; DTX-1, DTX-2: dinophysistoxin −1, −2; PTX-2: pectenotoxin-2; (b) AZA-1, AZA-2, AZA-3: azaspiracid −1, −2, −3; YTX: yessotoxin; hYTX: 1a homo yessotoxin. Data are expressed as mean ± SD (n = 3).
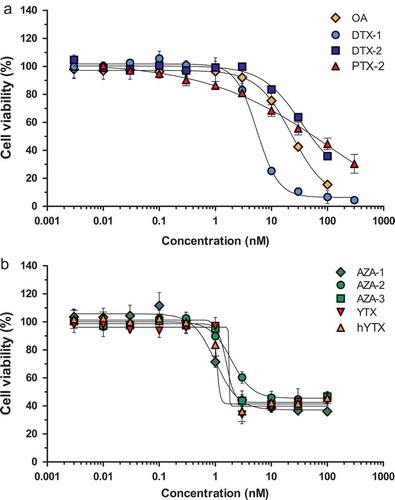
also shows the toxicity equivalency factors (TEFs) as derived from the EC50 values in the neuro-2a assay and the TEFs for the OA/DTX and the AZA class of toxins, as published in a joint FAO/WHO technical paper (FAO/WHO Citation2016), both the ones based on experimental data, as well as recommended TEFs. The latter are quite similar to those proposed by EFSA, which should be used in routine testing. No full dose–response curves were obtained for DTX-2 within the concentration range tested and the EC50 values determined for DTX-2 should therefore be considered as less accurate. However, demonstrates that DTX-2 is less toxic than DTX-1 (similar in NG108-15 cells, but curves not shown) and these observations are in line with those of the FAO/WHO and EFSA, and also with results obtained by Aune et al. (Citation2007) with intraperitoneal (i.p.) treated mice, showing a TEF of 0.6 for DTX-2 compared to OA. Still, the toxic potency of OA resembled more that of DTX-2 than that of DTX-1, which is in agreement with other in vitro data suggesting that DTX-1 is more potent than OA (Fernández et al. Citation2014; Ferron et al. Citation2014; Botana et al. Citation2017). However, eventually both EFSA and FAO/WHO assigned a similar TEF to OA as DTX-1 (EFSA Citation2008; FAO/WHO Citation2016).
In the case of AZA-1, AZA-2 and AZA-3 relative potencies of 1, 0.53 and 0.67 were observed, which seems to disagree with the TEFs assigned by EFSA (Citation2008) of 1, 1.8 and 1.4. However, based on more recent data from i.p. treated mice, FAO/WHO assigned TEFs of 1.0, 0.7 and 0.5, which are much more in line with our data. Overall, it is clear that there is still some uncertainty attached to the currently established and applied TEF values.
Overall, the present study shows that neuro-2a cells are slightly more sensitive than NG108-15 cells, respond to all tested LMBs and their analogues, and display toxic potencies which are reasonably in line with the TEFs for these marine biotoxins as established by EFSA and the FAO/WHO. The assay based on neuro-2a cells was therefore used for testing shellfish samples.
Sensitivity of the assay in relation to the maximal allowed levels
The current EU limits for the regulated LMBs and the above determined sensitivities of the neuro-2a cells for these toxins were used to calculate the required sample amount and the dilution of the prepared sample extract in the cell culture medium. All regulated LMBs should be detected at levels below their established limits. As such, the worst case scenario is the limit for the sum of OA, DTXs and PTXs, being 160 µg OA-eq kg−1, as this is the lowest maximum permitted level (MPL), whereas these toxins display the highest EC50 values. Following our extraction protocol (i.e. 0.48 g shellfish diluted in an equivalent of 8 ml medium), and assuming 100% recovery, a sample contaminated with 160 µg OA per kg would theoretically result in a medium concentration of 11.9 nM OA (Mw 805). This is just high enough to cause an effect in the neuro-2a cells (; ). This also applies for DTX-2 (11.9 nM, Mw 805), whereas a similar level of DTX-1 (11.7 nM, Mw 819) would cause an almost complete inhibition of the MTT-activity. On the other hand, PTX-2 (11.2 nM, Mw 859) would not be detected at this level. Whether the test would work in practice for pectenotoxins would depend on the actual combinations of toxins occurring in shellfish samples. As PTX-2 is produced by the same algae as DTX and OA, and any sample containing PTX-2 above the limit would contain even higher levels of DTX and OA (Vale Citation2004; Fernandez et al. Citation2006; Suzuki et al. Citation2009; Nagai et al. Citation2011), the relatively high EC50 value for PTX-2 is probably not limiting the suitability of the neuro-2a assay for selecting contaminated samples. Moreover, in shellfish PTX-2 is known to be converted into the non-toxic PTX-2 seco acid metabolite (Miles et al. Citation2004).
It is important to realise that as a consequence of the used sample amount and dilution factor required to detect OA, the test will be relatively sensitive for samples contaminated with yessotoxins. Since YTXs have a high EU limit of 3750 µg YTX-eq kg−1, this amount would theoretically correspond to a well concentration of 196.9 nM YTX (Mw 1143) with the applied protocol, and thus display clearly reduced MTT-activity in the neuro-2a assay (: EC50 1.6 nM). Therefore, samples containing YTX-eq levels well below 3750 µg kg−1 will also be screened as suspect. For azaspiracids, the limit is 160 µg per kg and this level would result in a medium concentration of 11.4 nM AZA-1 (Mw 842), 11.2 nM AZA-2 (Mw 856) and 11.6 nM AZA-3 (Mw 828). Given the low EC50 values, these toxins should easily be detected with the current test protocol.
Blank samples and the effect of the improved clean-up with n-hexane
An additional clean-up step using n-hexane (see experimental section) was introduced to eliminate matrix effects that would otherwise result in a high percentage of false-positive outcomes in the neuro-2a assay, as was observed before by Ledreux et al. (Citation2012). This additional cleaning step was introduced before the SPE clean-up was performed. The matrix effect is demonstrated in ), showing the results of 10 blank mussel samples without the additional n-hexane wash step, both without and with an additional 10-fold dilution. All undiluted blank sample extracts caused a marked reduction in the MTT activity and were thus falsely screened as suspect. A blank chemical control was included, and this extract caused no cytotoxicity. Tenfold-diluted sample extracts were not toxic anymore, but as explained above, this results in too low sensitivity of the assay for most toxins. ) shows the results of 20 blank mussel samples when the n-hexane wash step was included. Undiluted blank sample extracts no longer showed a strong decrease in MTT activity, implying that the n-hexane wash step worked very well in order to remove false positives due to matrix effects. The observed cytotoxic effects without the extra n-hexane wash step, i.e. matrix effects, are most probably caused by free fatty acids, also known to interfere with the outcome of the MBA (Suzuki et al. Citation1996).
Figure 2. Effect of the introduction of an additional n-hexane wash step in the sample extraction procedure: (a) undiluted and 10-fold diluted sample extracts of blank mussel samples prepared without the n-hexane wash step; (b) undiluted and fivefold diluted sample extracts of blank mussel samples prepared by introducing the n-hexane wash step. ADL is the ‘arbitrary’ decision limit, set at 75% cell viability. Data are expressed as mean ± SD (n = 3).
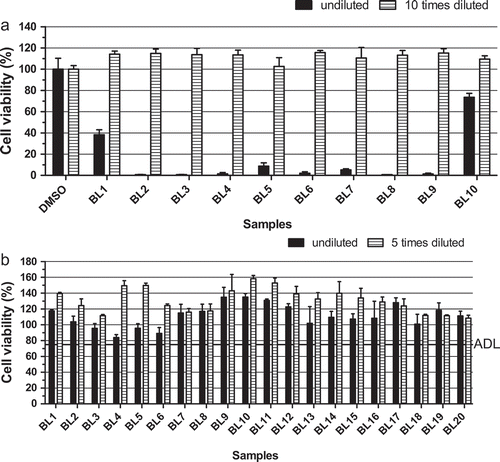
Based on the data obtained with the new procedure including the n-hexane step and blank mussel samples ()), an ‘arbitrary’ decision limit (ADL) was set at a reduction of the MTT activity of 25% or more. Samples with an MTT value above this decision limit are classified as negative (safe) and samples resulting in MTT values below this decision limit are classified as suspect (potentially unsafe).
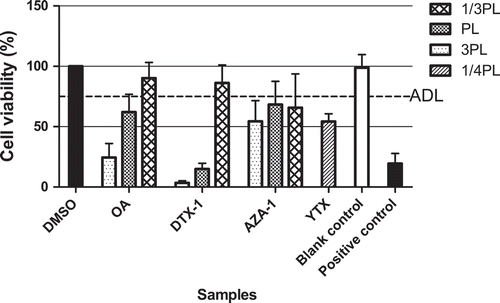
shows the results of blank mussel samples spiked at 1/3, 1 and 3 times the MPLs for OA, DTX-1 and AZA-1, i.e. 53, 160 and 480 µg kg−1, and at about 1/4 MPL for YTX, i.e. 1000 µg kg−1, using the n-hexane wash and the subsequent SPE clean-up. The spiking was performed to an aliquot of the methanol extract, i.e. in order to reduce the required amount of the expensive standards. Moreover, the methanol extraction is already known to result in high extraction efficiencies for the different LMBs (Gerssen, van Olst et al. Citation2010). The results show that all spiked samples reduced the MTT-activity to 75% or lower when spiked at or above their MPL in case of OA, DTX-1 and AZA-1 and in case of YTX even at 1/4 MPL, implying that all toxins could be detected at levels above their MPLs. This includes the above-described worst case of OA, the toxin with the lowest MPL (160 µg OA-eq kg−1) and relatively high EC50 value (23.4 nM, ). In addition, LC-MS/MS analyses of samples spiked at their MPL for OA, DTX-1, AZA-1 and at 1/4 MPL for YTX just before and after the n-hexane clean-up elicited recoveries of 90, 123, 96 and 104%, respectively. This shows that no toxins were lost in the n-hexane. Since the comparison is based on extracts spiked just before and after hexane extraction, recoveries above 100% point to the effectiveness of the n-hexane wash step for removing small matrix effects for LC-MS/MS analysis too, i.e. probably removing compounds that otherwise would cause a little bit of ion suppression. Although the recovery losses from the use of SPE were not determined in the present study, previous studies demonstrated that losses due to the SPE were lower than 15% (Gerssen et al. Citation2009).
Figure 3. Mussel samples spiked at 3, 1 and 1/3 times the maximal permitted levels (MPLs) of 160 µg kg–1 for OA, DTX-1 and AZA-1, and at about 1/4 MPL for YTX, i.e. 1000 µg kg–1, extracted with the procedure including the extra n-hexane wash step and analysed in the neuro-2a assay. An ‘arbitrary’ decision limit (ADL) of 75% was used. Positive control: DTX-1 12nM. Data are expressed as mean ± SD (n = 3).
Shellfish samples contaminated with lipophilic marine biotoxins
To allow a first evaluation of the performance of the newly developed clean-up method, extracts were prepared from eight samples (S1–S8) naturally contaminated with various LMBs that were previously used in an inter-laboratory validation study of the LC-MS/MS method (van den Top et al. Citation2011). These contaminated samples were prepared by blending naturally contaminated samples having various toxin profiles with blank samples in order to get a variety of materials with different profiles and levels. ) shows the results as obtained in the neuro-2a bioassay. A summary of the bioassay and LC-MS/MS results is given in , showing that these validation samples were contaminated with levels of OA/DTXs or AZAs above regulatory limits. As all samples elicited a decrease in cell viability below that of the ‘arbitrarily’ set decision limit, they were all correctly classified as suspect. Three additional blank control samples caused no decreased response. It should be pointed out that six of the eight samples contained YTXs, and S2 and S3 at relative high amounts of 1702 and 1110 µg kg−1 respectively. This would most likely result in a suspected response also when present alone. However, samples S4 and S8 do not contain YTXs and were also classified correctly as being suspect, due to the presence of OA/DTX alone (S8) or in the presence of AZAs (S4). Based on this, it seems likely that the response obtained with samples S1, S5, S6 and S7 are to a large extent caused by toxins other than YTXs.
Figure 4. Effect on the viability of neuro-2a cells (as measured with the MTT assay) of: (a) shellfish products (validation samples S1–S8) contaminated with okadaic acid/dinophysistoxins, yessotoxins and/or azaspiracids () and three blank shellfish samples (samples 9–11); (b) eight mussel samples (1–8) naturally contaminated with detectable levels of one or more LMBs (). An ‘arbitrary’ decision limit (ADL) of 75% was used and 0.25% DMSO was included as a control in each experiment. Data are expressed as mean ± SD (n = 3).
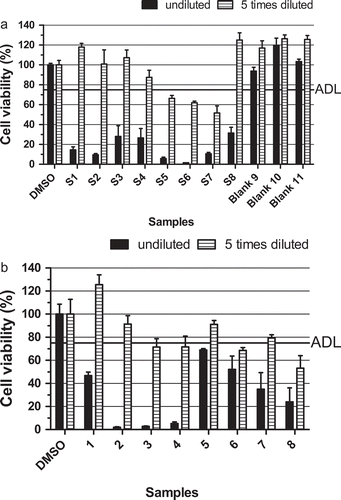
) shows the results of eight unblended samples naturally contaminated with LMBs that were previously analysed by LC-MS/MS (, lower part). All samples resulted in MTT values below the decision limit and were classified as suspect. As shown in , all samples contained detectable toxin levels, some well above (samples 3 and 4), or just below (samples 1 and 2) the EU-limits (160 µg OA-eq kg−1, 160 µg AZA-eq kg−1 and 3750 µg YTX-eq kg−1). Samples 6, 7 and 8 contained elevated levels of YTXs, but far below the limit, confirming the sensitivity of the assay for this class of toxins. Sample 5, which showed a response just below the ADL, only contained low amounts of AZAs, less than 1/5 of the regulatory limit. The bioassay classification of these samples was thus in line with the toxin levels measured by LC-MS/MS, taking into consideration the sensitivity of the assay for YTXs.
Next, 48 sample extracts obtained from Chile (derived from different species of bivalves and gastropods) were tested in the neuro-2a assay and also analysed by LC-MS/MS for lipophilic marine biotoxins (OA, DTXs, PTX-2, AZAs and YTXs). Although many samples concerned the hepatopancreas or viscera and as such levels should be divided by at least a factor of 3 (Garcia et al. Citation2012), this was not taken into account for the evaluation of the assay performance. shows the results of these Chilean samples as tested in the neuro-2a assay. Samples 4, 6, 17, 25, 28, 30, 34 and 45 decreased the cell viability as determined with the MTT assay and were screened as suspect in the neuro-2a assay. shows the LC-MS/MS results, revealing that all suspected samples, except sample 30, presented relatively high amounts of YTX-eq. The highest levels were observed in the viscera of a sea snail (17, 28, 45), followed by the hepatopancreas of two types of mussels (34, 38). Four of these samples (17, 28, 34 and 45) also contained low levels of OA-eq, unlikely to have contributed much to the response. None of the 48 samples contained PTXs or AZAs. From a qualitative point of view, there is a good correlation between the neuro-2a bioassay outcomes and the LC-MS/MS analysis. All 40 samples with no detectable toxins or only trace amounts (<10% of the limit) showed a negative result. Of the eight samples that tested suspected, seven contained relatively high levels of YTX (477–3472 µg YTX-eq kg−1).
Figure 5. Effect of extracts of 48 seafood products obtained from Chile and both positive and blank mussel sample controls, on the viability of neuro-2a cells as measured with the MTT assay. Positive sample control: mussels naturally contaminated with YTX. The decision limit was ‘arbitrarily’ set at 75% viability (ADL) and 0.25% DMSO was included as a control. Data are expressed as mean ± SD (n = 3).
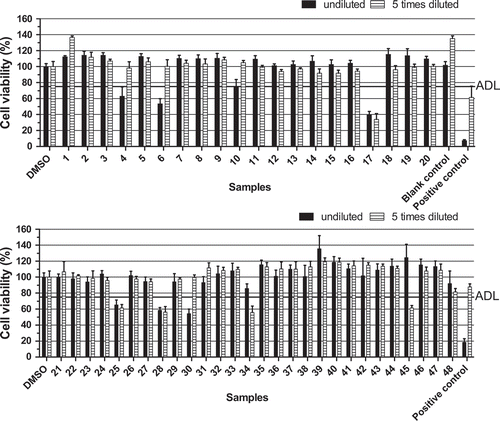
Nevertheless, there are some discrepancies when analysing the data on a semi-quantitative level, in particular when focussing on samples 23, 30, 38, and 45. Sample 45 was screened as suspect based on testing the diluted extract. A similar decrease in activity upon dilution was observed for sample 34 and this requires further investigation. Samples 23 and 38 were screened as negative, but contained relatively high amounts of YTX-eq, 701 and 1787 µg YTX-eq kg−1 respectively. Sample 30 was suspect in the bioassay, but according to the LC-MS/MS contained YTX only and at a low level (185 µg YTX-eq kg−1). This level would result in a medium concentration of 3 nM and thus could be enough to cause an effect in the neuro-2a assay (), but it cannot fully be ruled out that this sample may contain yet unknown lipophilic toxins, missed by LC-MS/MS. This might be unknown YTX analogues, or possibly another type of lipophilic toxin. Brevetoxins (BTxs) and palytoxins (PlTxs) are known to be extracted with methanol (Reverte et al. Citation2014; Turner et al. Citation2015). However, to detect BTxs, the addition of ouabain and veratridine (O/V) during exposure of the cells is needed. These compounds interact with the sodium voltage-gated channels in the cells, causing cell death (Watkins et al. Citation2008; Paredes et al. Citation2011). The addition of O/V in a concentration causing a 20% reduction of cell viability is needed to detect BTxs, which then cause a further decrease of the cell viability (Manger et al. Citation1993). PlTx is also able to decrease the MTT activity without the addition of O/V (EC50 for BTx-3, BTx-9 and PlTx in the neuro-2a assay of 8, 8.4 and 0.04 nM respectively, data not shown). Although dedicated LC-MS/MS methods for BTxs and PlTx are available, these methods are not routinely applied as there are no certified standards and legislation is lacking for these toxins. Overall the bioassay classification of these Chilean samples was in line with the lipophilic toxin levels measured by LC-MS/MS. However, it should be pointed out that samples contained primarily yessotoxins.
Conclusions and recommendations
Although the neuro-2a assay is regarded as suitable for PSPs and has occasionally been used to analyse samples for the presence of PSPs (Jellett et al. Citation1995; Manger et al. Citation1995; Humpage et al. Citation2007), its routine application for these toxins is still rather limited. And although the test has also been shown to detect various classes of LMBs, up till now it is not considered for routine testing of shellfish for LMBs. Lack of routine use is due to observed matrix effects, and as a result a lack of studies on performance with routine samples and (international) validation studies. The present paper is the first describing the performance of the neuro-2a assay for routine testing of seafood samples on the presence of LMBs using a newly developed clean-up procedure including an n-hexane wash step, thereby allowing identification of potential issues to be improved and whether the assay is worth the further intensive and expensive efforts needed for a full validation. Although the reason for the difference between the toxic effects of LMBs as observed in mouse and humans and the cell viability of neuro cells is missing, the study confirms previous reports showing that the neuro-2a assay allows the detection of the regulated LMBs at the required levels. Also the few commercially available analogues of these LMBs could be detected and there is a reasonable correlation between toxic potencies and TEFs established by EFSA and WHO/FAO. Compared to the current analytical and immuno-based alternatives, the neuro-2a assay will most likely be able to detect unknown analogues and as yet unknown marine biotoxins too.
The newly introduced n-hexane wash step is an important improvement in order to eliminate matrix effects causing too many false-positive screening outcomes, which would preclude its routine application. These effects were most probably caused by free fatty acids known to interfere also with the outcome of the MBA. The new clean-up procedure worked well for mussels, but was also successful with some cockles and oysters that we tested (latter data not shown) and the Chilean samples that included several species (this study).
An ‘arbitrary’ decision limit was set for the routine screening of real samples, which might be refined based on further experience and validation studies. In a qualitative way, the neuro-2a assay outcomes correlated well with the LC-MS/MS analysis. Among the 87 samples screened for the presence of lipophilic marine biotoxins by the neuro-2a assay, 24 were screened as suspect and 63 as negative, while LC-MS/MS identified 12 positives out of the 24 suspected samples (ignoring that some samples were viscera and not whole flesh). No false negative screening results were obtained. Many of the 12 false-positive samples turned out to contain elevated levels of toxins, some just below the regulatory limits.
However, in particular the relatively high sensitivity of the assay for yessotoxins resulted in a number of clearly false-positive results. Whether this is a problem in routine screening depends on the actual occurrence of these toxins in a certain production area. In the Netherlands for example, samples rarely contain any of the LMBs, and detection of samples with even low levels of marine biotoxins would probably be welcomed as an early warning. However, this may be different in areas where yessotoxins occur regularly. For such cases, a different clean-up procedure could be developed in order to separate the YTXs from the other marine toxin classes. Moreover, it should be noticed that the n-hexane step will remove esterified forms of okadaic acid and the dinophysis toxins, which are mentioned in the legislation under the generic term DTX-3. Thus for application to real samples in the future, a hydrolysis step should be incorporated prior to the n-hexane wash step and SPE clean-up (which is already performed prior to LC-MS/MS analysis for the determination of DTX-3). Finally, the suitability of the neuro-2a bioassay should be further assessed by testing samples that have been tested negative and positive for LMBs in the MBA.
In summary, the present data show that the neuro-2a assay is worth the intensive and expensive efforts needed for a full validation as this could result in a cheap and fast screening method for testing shellfish for the presence of DSPs and AZAs, i.e. separating negative samples from those potentially contaminated above MLs, which are then further analysed by LC-MS/MS. Since it can detect unknown analogues, it is the best alternative to the MBA, both for LMBs and other marine biotoxins.
Acknowledgments
Dr Carlos García from the Faculty of Medicine, Universidad de Chile (Santiago, Chile) is gratefully acknowledged for providing us with shellfish extracts from Chile.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Aballay-Gonzalez A, Ulloa V, Rivera A, Hernandez V, Silva M, Caprile T, Delgado-Rivera L, Astuya A. 2016. Matrix effects on a cell-based assay used for the detection of paralytic shellfish toxins in bivalve shellfish samples. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 33:869–875. Epub 2016/03/24.
- Aune T, Larsen S, Aasen John AB, Rehmann N, Satake M, Hess P. 2007. Relative toxicity of dinophysistoxin-2 (dtx-2) compared with okadaic acid, based on acute intraperitoneal toxicity in mice. Toxicon. 49:1–7.
- Berdalet E, Fleming LE, Gowen R, Davidson K, Hess P, Backer LC, Moore SK, Hoagland P, Enevoldsen H. 2016. Marine harmful algal blooms, human health and wellbeing: challenges and opportunities in the 21st century. J Mar Biol Assoc U.K. 96:61–91.
- Botana LM, Hess P, Munday R, Nathalie A, DeGrasse SL, Feeley M, Suzuki T, van den Berg M, Fattori V, Garrido Gamarro E, et al. 2017. Derivation of toxicity equivalency factors for marine biotoxins associated with Bivalve Molluscs. Trends Food Sci Technol. 59:15–24.
- Caillaud A, Canete E, de la Iglesia P, Gimenez G, Diogene J. 2009. Cell-based assay coupled with chromatographic fractioning: a strategy for marine toxins detection in natural samples. Toxicol In Vitro. 23:1591–1596. Epub 2009/09/02.
- Cañete E, Diogène J. 2008. Comparative study of the use of neuroblastoma cells (Neuro-2a) and neuroblastoma × glioma hybrid cells (NG108-15) for the toxic effect quantification of marine toxins. Toxicon. 52:541–550.
- Cañete E, Diogène J. 2010. Improvements in the use of neuroblastomaxglioma hybrid cells (NG108-15) for the toxic effect quantification of marine toxins. Toxicon. 55:381–389. Epub 2009/09/08.
- Dechraoui MY, Tiedeken JA, Persad R, Wang Z, Granade HR, Dickey RW, Ramsdell JS. 2005. Use of two detection methods to discriminate ciguatoxins from brevetoxins: application to great barracuda from Florida Keys. Toxicon. 46:261–270. Epub 2005/06/29.
- Dominguez HJ, Paz B, Daranas AH, Norte M, Franco JM, Fernandez JJ. 2010. Dinoflagellate polyether within the yessotoxin, pectenotoxin and okadaic acid toxin groups: characterization, analysis and human health implications. Toxicon. 56:191–217. Epub 2009/11/21.
- EFSA. 2008. Marine biotoxins in shellfish – azaspiracid group, scientific opinion of the panel on contaminants in the food chain. EFSA J. 723:1–52.
- EFSA. 2009. Scientific opinion of the panel on contaminants in the food chain. Marine biotoxins in shellfish - summary on regulated marine biotoxins. EFSA J. 1306:1–23.
- EFSA. 2010. Scientific opinion on marine biotoxins in shellfish – emerging toxins: ciguatoxin group. EFSA J. 8:1627.
- Empey Campora C, Dierking J, Tamaru CS, Hokama Y, Vincent D. 2008. Detection of ciguatoxin in fish tissue using sandwich ELISA and neuroblastoma cell bioassay. J Clin Lab Anal. 22:246–253. Epub 2008/07/16.
- Etheridge SM. 2010. Paralytic shellfish poisoning: seafood safety and human health perspectives. Toxicon. 56:108–122. Epub 2009/12/29.
- FAO/WHO. 2016. Technical paper on toxicity equivalency factors for marine biotoxins associated with Bivalve Molluscs. Rome: FAO/WHO. 108 pp.
- Fernández D, Louzao M, Fraga M, Vilariño N, Vieytes M, Botana L. 2014. Experimental basis for the high oral toxicity of dinophysistoxin 1: a comparative study of DSP. Toxins. 6:211–228.
- Fernandez L, Reguera B, Gonzalez-Gil S, Miguez A. 2006. Pectenotoxin-2 in single-cell isolates of Dinophysis caudata and Dinophysis acuta from the Galician Rias (NW Spain). Toxicon. 48:477–490. Epub 2006/08/22.
- Ferron PJ, Hogeveen K, Fessard V, Le Hegarat L. 2014. Comparative analysis of the cytotoxic effects of okadaic acid-group toxins on human intestinal cell lines. Mar Drugs. 12:4616–4634. Epub 2014/09/10.
- Garcia C, Rodriguez-Unda N, Contreras C, Barriga A, Lagos N. 2012. Lipophilic toxin profiles detected in farmed and benthic mussels populations from the most relevant production zones in Southern Chile. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 29:1011–1020. Epub 2012/03/20.
- Gerssen A, McElhinney MA, Mulder PP, Bire R, Hess P, de Boer J. 2009. Solid phase extraction for removal of matrix effects in lipophilic marine toxin analysis by liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem. 394:1213–1226. Epub 2009/04/24.
- Gerssen A, Pol-Hofstad IE, Poelman M, Mulder PP, van den Top HJ, de Boer J. 2010. Marine toxins: chemistry, toxicity, occurrence and detection, with special reference to the Dutch situation. Toxins (Basel). 2:878–904. Epub 2010/04/01.
- Gerssen A, van Olst EH, Mulder PP, de Boer J. 2010. In-house validation of a liquid chromatography tandem mass spectrometry method for the analysis of lipophilic marine toxins in shellfish using matrix-matched calibration. Anal Bioanal Chem. 397:3079–3088. Epub 2010/06/17.
- Humpage AR, Ledreux A, Fanok S, Bernard C, Briand JF, Eaglesham G, Papageorgiou J, Nicholson B, Steffensen D. 2007. Application of the neuroblastoma assay for paralytic shellfish poisons to neurotoxic freshwater cyanobacteria: interlaboratory calibration and comparison with other methods of analysis. Environ Toxicol Chem. 26:1512–1519. Epub 2007/08/02.
- Humpage AR, Magalhaes VF, Froscio SM. 2010. Comparison of analytical tools and biological assays for detection of paralytic shellfish poisoning toxins. Anal Bioanal Chem. 397:1655–1671. Epub 2010/01/27.
- James KJ, Carey B, O’Halloran J, van Pelt FN, Skrabakova Z. 2010. Shellfish toxicity: human health implications of marine algal toxins. Epidemiol Infect. 138:927–940. Epub 2010/04/24.
- Jellett JF, Stewart JE, Laycock MV. 1995. Toxicological evaluation of saxitoxin, neosaxitoxin, gonyautoxin II, gonyautoxin II plus III and decarbamoylsaxitoxin with the mouse neuroblastoma cell bioassay. Toxicol In Vitro. 9:57–65. Epub 1995/02/01.
- Kat M. 1983. Diarrhetic mussel poisoning in the Netherlands related to the dinoflagellate Dinophysis acuminata. Antonie Van Leeuwenhoek. 49:417–427.
- Ledreux A, Serandour AL, Morin B, Derick S, Lanceleur R, Hamlaoui S, Furger C, Bire R, Krys S, Fessard V, et al. 2012. Collaborative study for the detection of toxic compounds in shellfish extracts using cell-based assays. Part II: application to shellfish extracts spiked with lipophilic marine toxins. Anal Bioanal Chem. 403:1995–2007. Epub 2012/04/27.
- Manger RL, Leja LS, Lee SY, Hungerford JM, Hokama Y, Dickey RW, Granade HR, Lewis R, Yasumoto T, Wekell MM. 1995. Detection of sodium channel toxins: directed cytotoxicity assays of purified ciguatoxins, brevetoxins, saxitoxins, and seafood extracts. J AOAC Int. 78:521–527. Epub 1995/03/01.
- Manger RL, Leja LS, Lee SY, Hungerford JM, Wekell MM. 1993. Tetrazolium-based cell bioassay for neurotoxins active on voltage-sensitive sodium channels: semiautomated assay for saxitoxins, brevetoxins, and ciguatoxins. Anal Biochemistry. 214:190–194.
- Miles CO, Wilkins AL, Munday R, Dines MH, Hawkes AD, Briggs LR, Sandvik M, Jensen DJ, Cooney JM, Holland PT, et al. 2004. Isolation of pectenotoxin-2 from Dinophysis acuta and its conversion to pectenotoxin-2 seco acid, and preliminary assessment of their acute toxicities. Toxicon. 43:1–9. Epub 2004/03/24.
- Moore SK, Trainer VL, Mantua NJ, Parker MS, Laws EA, Backer LC, Fleming LE. 2008. Impacts of climate variability and future climate change on harmful algal blooms and human health. Environ Health. 7(Suppl 2):S4. Epub 2008/12/17.
- Nagai S, Suzuki T, Nishikawa T, Kamiyama T. 2011. Differences in the production and excretion kinetics of okadaic acid, dinophysistoxin-1, and pectenotoxin-2 between cultures of dinophysis acuminata and dinophysis fortii isolated from western Japan. J Phycol. 47:1326–1337. Epub 2011/12/01.
- Nicolas J, Hendriksen PJ, Gerssen A, Bovee TF, Rietjens IM. 2014. Marine neurotoxins: state of the art, bottlenecks, and perspectives for mode of action based methods of detection in seafood. Mol Nutr Food Res. 58:87–100. Epub 2013/12/07.
- Paredes I, Rietjens IM, Vieites JM, Cabado AG. 2011. Update of risk assessments of main marine biotoxins in the European Union. Toxicon. 58:336–354.
- Paz B, Daranas AH, Norte M, Riobo P, Franco JM, Fernandez JJ. 2008. Yessotoxins, a group of marine polyether toxins: an overview. Mar Drugs. 6:73–102. Epub 2008/08/30.
- Peperzak L. 2005. Future increase in harmful algal blooms in the North Sea due to climate change. Water Sci Technol. 51:31–36. Epub 2005/05/28.
- Plakas SM, Dickey RW. 2010. Advances in monitoring and toxicity assessment of brevetoxins in molluscan shellfish. Toxicon. 56:137–149. Epub 2009/11/21.
- Regulation (EC) No 2074/2005. Commission Regulation (EC) No 2074/2005 of 5 December 2005 laying down implementing measures for certain products under Regulation (EC) No 853/2004 of the European Parliament and of the Council and for the organisation of official controls under Regulation (EC) No 854/2004 of the European Parliament and of the Council and Regulation (EC) No 882/2004 of the European Parliament and of the Council, derogating from Regulation (EC) No 852/2004 of the European Parliament and of the Council and amending Regulations (EC) No 853/2004 and (EC) No 854/2004 (Text with EEA relevance). Off J Eur Union. L338:27–59.
- Regulation (EU) No 15/2011. Commission Regulation (EU) No 15/2011 of 10 January 2011 amending Regulation (EC) No 2074/2005 as regards recognised testing methods for detecting marine biotoxins in live bivalve molluscs. Off J Eur Union. L6:3–6.
- Rehmann N, Hess P, Quilliam MA. 2008. Discovery of new analogs of the marine biotoxin azaspiracid in blue mussels (Mytilus edulis) by ultra-performance liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 22:549–558. Epub 2008/01/30.
- Reverte L, Solino L, Carnicer O, Diogene J, Campas M. 2014. Alternative methods for the detection of emerging marine toxins: biosensors, biochemical assays and cell-based assays. Mar Drugs. 12:5719–5763. Epub 2014/11/29.
- Serandour AL, Ledreux A, Morin B, Derick S, Augier E, Lanceleur R, Hamlaoui S, Moukha S, Furger C, Bire R, et al. 2012. Collaborative study for the detection of toxic compounds in shellfish extracts using cell-based assays. Part I: screening strategy and pre-validation study with lipophilic marine toxins. Anal Bioanal Chem. 403:1983–1993. Epub 2012/04/28.
- Suzuki T, Miyazono A, Baba K, Sugawara R, Kamiyama T. 2009. LC–MS/MS analysis of okadaic acid analogues and other lipophilic toxins in single-cell isolates of several Dinophysis species collected in Hokkaido, Japan. Harmful Algae. 8:233–238.
- Suzuki T, Yoshizawa R, Kawamura T, Yamasaki M. 1996. Interference of free fatty acids from the hepatopancreas of mussels with the mouse bioassay for shellfish toxins. Lipids. 31:641–645. Epub 1996/06/01.
- Turner AD, Higgins C, Davidson K, Veszelovszki A, Payne D, Hungerford J, Higman W. 2015. Potential threats posed by new or emerging marine biotoxins in UK waters and examination of detection methodology used in their control: brevetoxins. Mar Drugs. 13:1224–1254. Epub 2015/03/17.
- Twiner MJ, Rehmann N, Hess P, Doucette GJ. 2008. Azaspiracid shellfish poisoning: a review on the chemistry, ecology, and toxicology with an emphasis on human health impacts. Mar Drugs. 6:39–72.
- Vale P. 2004. Differential dynamics of dinophysistoxins and pectenotoxins between blue mussel and common cockle: a phenomenon originating from the complex toxin profile of Dinophysis acuta. Toxicon. 44:123–134.
- van den Top HJ, Gerssen A, McCarron P, van Egmond HP. 2011. Quantitative determination of marine lipophilic toxins in mussels, oysters and cockles using liquid chromatography-mass spectrometry: inter-laboratory validation study. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 28:1745–1757. Epub 2011/10/15.
- Visciano P, Schirone M, Berti M, Milandri A, Tofalo R, Suzzi G. 2016. Marine biotoxins: occurrence, toxicity, regulatory limits and reference methods. Front Microbiol. 7:1051. Epub 2016/07/28.
- Watkins SM, Reich A, Fleming LE, Hammond R. 2008. Neurotoxic shellfish poisoning. Mar Drugs. 6:431–455. Epub 2008/11/14.
- Yasumoto T, Murata M, Oshima Y, Matsumoto K, Clardy J. 1984. Diarrhetic shellfish poisoning. Seafood Toxins. 44:207–214.
- Yasumoto T, Oshima Y, Yamaguchi M. 1978. Occurrence of a new type of toxic shellfish poisoning in the Tohuku District. Bull Jpn Soc Sci. 44:1249–1255.
- Zamorano R, Marin M, Cabrera F, Figueroa D, Contreras C, Barriga A, Lagos N, Garcia C. 2013. Determination of the variability of both hydrophilic and lipophilic toxins in endemic wild bivalves and carnivorous gastropods from the Southern part of Chile. Food Addit Contam Part A Chem. Sep;30:1660–1677.