
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The present work reports on the design, execution and evaluation of results of an interlaboratory validation study aimed at verifying the fitness-for-purpose of a LC-MS/MS method for the detection of polar pesticides in food of animal origin in official control and monitoring programmes. To this scope, five participant laboratories, with relevant expertise, were recruited. After passing a pre-trial test, the participants were asked to analyse test samples of bovine fat, chicken eggs and cow’s milk, contaminated with 11 polar pesticides (group A: Aminomethyl phosphonic acid (AMPA), cyanuric acid, ethephon, glyphosate, fosetyl aluminium, 2-hydroxyethyphosphonic acid (HEPA), maleic hydrazide, N-acetyl-glyphosate, group B: N-acetyl glufosinate (NAG), 3-methylphosphinicopropionic acid (MPP) and glufosinate ammonium) at two different levels (0.05 and 0.25 mg/kg−1 and 0.01 and 0.05 mg/kg−1 for group A and B respectively. The method was based on acidified methanol/water extraction followed by dSPE clean up with C18 sorbent. For LC-MS/MS analysis isotopically labelled standards were used for all targeted analytes. With a couple of exceptions, average recoveries ranged from 85% to 110%, with repeatability (RSDr) ranging from 3% to 25%, and reproducibility (RSDR) from 4% to 26%. The assessment by different laboratories provided also insights on key factors impacting method performance characteristics and its implementation by new users.
Introduction
Glyphosate is a chemical used worldwide in plant protection, as well as the most known pesticide from the so-called “highly polar pesticides”, due to the controversial debate about its toxicity in the last years.
Within the renewal process (EC 2023) glyphosate has been thoroughly assessed by Member States, the European Chemicals Agency (ECHA) and the European Food Safety Authority (EFSA) (EFSA and ECHA Citation2023). The assessment led to the adoption of Implementing Regulation (EC) 2022/2364 by the Commission in December 2022 (EC 2022), which extended the five-year approval of this active substance by one year until 15 December 2023.
Similarly, there are many other highly polar pesticides, such as glufosinate, fosetyl, ethephon and their metabolites which form an extremely challenging group of residues to be analysed due to their physical-chemical properties (Verdini and Pecorelli Citation2022). For the majority of these polar pesticides, Maximum Residue Levels (MRLs) have already been established including metabolites in the residue definition (Regulation (EC) No 396/2005), while for glyphosate no metabolites are yet included. For this reason, a revision of MRLs has been suggested, considering inclusion of metabolites (EFSA Citation2019; EFSA Citation2022). In the case of monitoring of animal commodities, the sum of glyphosate and N-acetyl glyphosate, expressed as glyphosate, was required in order to consider also future MRL-setting procedure. (EFSA Citation2018; EFSA Citation2022)
To cope with the gap of information about the presence of glyphosate and its metabolites in products of animal origin, EFSA recommended the development of confirmatory methods for glyphosate, aminomethyl phosphonic acid and N-acetyl-glyphosate in fat, liver and kidney. The results from the monitoring programmes are a valuable source of information for estimating the dietary exposure of EU consumers. Regulation (EC) Citation2021/601 for the multi-year control plan (2022–2024) requires the analysis of glyphosate and glufosinate ammonium, specifically the analysis of cow’s milk and swine fat for the year 2022, poultry fat and bovine liver for 2023, and bovine fat and chicken eggs for 2024 (EC 2021). Therefore, robust analytical methods are needed for the above target matrices to support official control requirements and to evaluate the consumer exposure to polar pesticide residues. A method based on QuPPE (Quick Polar Pesticides) for the determination of polar pesticides in food of animal origin (QuPPE-AO) (Anastassiades et al. Citation2019) was developed by the EU Reference Laboratory for Single Residue of Pesticides (EURL SRM) and assessed by a collaborative study involving 17 laboratories (EURL-SRM EU Reference Laboratories for Residues of Pesticides Citation2021). To the best of our knowledge, no other collaborative studies concerning polar pesticides in food of animal origin have been reported so far. One trial only, covering glufosinate and its main metabolites (NAG and MPP) in 11 foods of plant origin, was recently published by Wu et al. (Wu et al. Citation2023).
Having regard to the lack of inter-laboratory studies in the field of polar pesticides, the only attempt to harmonise performances among the official EU laboratories is represented by the proficiency tests (PTs) organised by the EURLs.
Two European Union Proficiency tests (EUPTs), dealing with polar pesticides in commodities of animal origin, were scheduled over time: the EUPT-SRM-9, organized in the year 2014 for the determination of glyphosate and other pesticides such as glyphosate, cyanuric acid and maleic hydrazide in cow’s milk (EURL-SRM Citation2014) and the EUPT-SRM-14, organized in the year 2019, for the determining of highly polar pesticides (including AMPA, glyphosate and N-acetyl glyphosate) in bovine liver homogenate (EURL-SRM Citation2019). In the EUPT-SRM-9, the analysis of glyphosate and maleic hydrazide was mandatory whereas cyanuric acid was voluntary. Since test samples provided in the EUPT-SRM-9 were not contaminated with glyphosate and cyanuric acid, the z- scores could not be calculated. Only one participant laboratory (out of 69) reported a false positive result for glyphosate. In the case of maleic hydrazide, 30 out of 69 laboratories reported results, of which 87% were acceptable (z-score ≤ 2) and 13% were not acceptable (2 laboratories obtained z-score > 3 and 2 laboratories reported false negative results). Overall, a need for performance harmonization among the EU National Reference Laboratories participating in the EUPT-SRM-9 emerged from this collaborative exercise.
In the case of EUPT-SRM-14, many polar pesticides were included in the list of mandatory (glyphosate) or voluntary (AMPA, MPP, N-acetyl glyphosate, glufosinate, N-acetyl lugfosinate) ones. Acceptable performance results (z-score ≤ 2) were provided by 88% of laboratories for glyphosate, 90% for AMPA, 81% for N-acetyl glyphosate and 85% for MPP.
The aim of this work was to assess the fitness-for-purpose of monitoring and compliance testing by official control laboratories of a LC-MS/MS method for the determination of 11 polar pesticides - namely AMPA, cyanuric acid, ethephon, glyphosate, fosetyl aluminium, 2-hydroxyethyphosphonic acid (HEPA), maleic hydrazide, N-acetyl-glyphosate, N-acetyl glufosinate (NAG), 3-methylphosphinicopropionic acid (MPP) and glufosinate ammonium in bovine fat, chicken eggs and cow milk. The method, previously in-house validated and accredited (Verdini et al. Citation2023), was challenged in an interlaboratory-validation study, involving 5 laboratories, to verify its robustness, estimating method performance characteristics and to get insights into critical issues in method transferability.
Materials and methods
Chemicals and reagents
The following chemicals and reagents were provided by the study organizers: EDTA (Ethylenediaminetetraacetic acid disodium salt dihydrate) ≥ 99% (Merck, Darmstadt, Germany); C18 sorbent Polygoprep™ 300-30 C18 (Macherey-Nagel GmbH & Co. K G,Düren, Germany); ready to use pesticide solutions in acetonitrile (for blind spiking experiments and preparation of matrix matched calibrants) purchased from Lab Instruments Srl (Castellana Grotte, Italy):
spike mix 1, mixed stock solution to be used for spiking procedure: 5 µg mL−1 AMPA, cyanuric acid, ethephon, fosetyl Al, glyphosate, HEPA, maleic hydrazide, N-acetyl- glyphosate)
spike mix 2, mixed stock solution to be used for spiking procedure: 5 µg mL−1 glufosinate ammonium, MPP, NAG
spike mix 3 and spike mix 4, pure acetonitrile to be used for blind spiking of blank samples
IS spike 1, mixed stock solution of labelled internal standard (ISTD) to be used for spiking procedure: 5 µg/mL AMPA-13C,15N cyanuric acid 3C3, ethephon D4, fosetyl Al D15, glyphosate 2-13C,15N, HEPA D4, N-Acetyl-glyphosate 13C2,15N and maleic hydrazide D2, 1 µg mL−1 glufosinate ammonium D3, MPP D3 and NAG D3
For sample extraction and analysis, participants in the trial study were requested to use syringe filters (13 mm, 0.2 µm, PTFE) and reagents of recognized analytical grade and specifically the following: water, acetonitrile, methanol and formic acid.
Test materials
Three commodities - bovine fat, cow milk and chicken eggs (about 500 g each) were selected including samples purchased from an Italian retail market. Prior to their use in this study, the absence of contamination was verified by analysis of the pesticides content according to the in-house validated method (Verdini et al. Citation2023). In the case of bovine fat and chicken, samples were ground by a knife mill (GRINDOMIX GM 300, Retsch GmbH, Haan, Germany) with dry ice prior to the analysis. For each commodity, aliquots of blank samples to be spiked for recovery evaluation and to prepare matrix matched calibration standards were dispensed in plastic bottles (2.0 ± 0.1 g each) that were labelled, sealed, and stored at −20 °C until dispatch for trial study.
Study layout
Pre-trial
Prior to the full validation study, laboratories had to participate in a pre-trial study to become familiar with the correct execution of the method protocol and to optimize the LC-MS/MS conditions for the detection of the target pesticides in terms of required instrumental sensitivity. For the pretrial step, each participant was asked to analyse – as blind samples - a sample fortified at the limit of quantification (LOQ) and two matrix-matched calibrants, corresponding to LOQ and 2 x LOQ for each of the three matrices included in the study. The LOQ was equal to or lower than the MRL for ethephon, fosetyl Al, glyphosate, maleic hydrazide and equal to the LOQ of the method for the other pesticides as defined in the Regulation EC 396/2005 and subsequent amendments (European Union Citation2005). The LOQ was previously estimated via in house validation study (Verdini et al. Citation2023) using the same protocol. In the report form, each participant was asked to report the area and the signal-to-noise ratio (S/N) related to each pesticide/matrix combination including the two-matrix matched calibrants.
For pre-trial each participant received the following samples, consumables and documents:
2 aliquots of blank sample (2.0 g each) for each commodity comprised of 1 aliquot to be used for recovery evaluation by spiking and 1 aliquot to be used to prepare one blank (unspiked) sample and two matrix matched calibration solutions;
Mixed pesticides standard solutions in acetonitrile (spike mix 1 and spike mix 2, see Section 1d) and a mixed ISTD solution in acetonitrile (IS spike 1), both to be used for spiking purposes and calibrants preparation. Pesticides concentrations in the provided solutions were blind.
5 Plastic tubes with C-18 sorbent (100 mg)
1 plastic tube with EDTA (500 mg), to be used for the preparation 5 mM EDTA solution in water (100 ml)
Method protocol in SOP (Standard Operating Procedure) format and reporting sheets
After completion of the pre-trial phase, the laboratories participated in the validation study to estimate the precision of the candidate method under repeatability and reproducibility conditions (as defined in SANTE 11312/2021). The accuracy of the method was evaluated by spiking experiments at two different levels: a low level (LOQ – labelled as Level C) and a high level (5xLOQ – labelled as Level A). Five public laboratories involved in the official controls participated in the trial. Participants received the following materials:
16 aliquots of blank sample (2.0 g each) for each commodity comprised of 5 aliquots labelled as level A, 5 aliquots labelled as level B, 5 aliquots labelled as level C. After spiking (according to the procedure described in the following) samples labelled as level A and C resulted to be spiked at LOQ and 5 x LOQ and were used for recovery evaluation, whereas samples labelled as level B resulted to be blank, one aliquot labelled as BLANK was also provided to be used to prepare one blank (unspiked) sample and the matrix matched calibrant solutions used for the calibration curves (6 points for chicken egg and cow milk and 5 points for bovine fat)
1 additional blank sample (20 g), for each commodity, to be used to repeat the experiments in case of necessity;
Mixed pesticides standard solution in acetonitrile (spike mix 1, spike mix 2) and pure acetonitrile (spike mix 3 and spike mix 4) and ISTD solution in acetonitrile (mix IS cal 1) to be used for spiking purposes and calibrants preparation (see Section Pesticide Solutions). Pesticides’ concentrations in the solutions provided were blind to participants.
20 plastic tubes with C-18 sorbent (100 mg).
1 plastic tube with EDTA (500 mg) to be used for the preparation of 100 mL of a 5 mM EDTA solution in water.
Analytical method protocol in SOP (Standard Operating Procedure) format and reporting sheets
Pesticide solutions
Participants were asked to prepare blind working solutions by diluting the spike mix solutions provided by the study organizers (section 1b) according to the following protocols:
spike mix 5, mixed pesticide solution to be used for spiking purposes, to be prepared by 5 times dilution of spike mix 2
mix cal 1 and mix cal 2, working solutions to be used for preparation matrix-matched calibration standards. Mix cal 1 was prepared by 25-time dilution of spike mix 1 and spike mix 5 in 10% ACN in water (v/v), mix cal 2 was prepared by 4-time dilution of mix cal 1.
mix IS cal, working solutions to be used for preparation of matrix-matched calibration solutions, was prepared by adding 30 µL of IS spike 1 to 970 uL of 10% ACN in water (v/v)
Each participant laboratory was asked to prepare matrix-matched calibrant solutions by adding appropriate volumes of mix cal 1 and mix cal 2 and internal standard mix (mix IS) to the blank sample extract, more details are given in the supplementary material (Table S1). Blank sample extracts were prepared as described in section Sample preparation.
Sample preparation
Bovine fat
Bovine fat was analysed according to the following steps.
To the test sample (2.0 g), 10 mL of water and 10 mL of MeOH containing 1% formic acid (v/v) were added. After vortexing for 30 s, the sample was placed in a water bath at 80 °C for 3 min until the fat was completely melted to permit quantitative pesticide’s extraction. Afterwards the sample was mechanically shaken for 2 min and placed in the freezer at −80 °C for 15 min. After 10 min of centrifugation (15000 × g, 10 °C) the extract was passed through a 0.22 µm PTFE filter. Finally, 0.25 mL of the filtered extract was combined with 0.25 mL of water directly into plastic vials for instrumental analysis. The final matrix equivalent concentration was 0.05 g mL−1.
Chicken eggs
Chicken eggs were analysed as follows.
Test sample (2.0 g) was first mixed with 8 mL of water. After vortexing for 30 s, 10 mL of MeOH containing 1% of formic acid (v/v) were added. The sample was mechanically shaken for 5 min. Subsequently, the sample was placed in a freezer at -80° C for 15 min and centrifuged for 10 min (15000 × g, 10° C). For purification purpose 2 mL of supernatant and 2 mL of acetonitrile were added in the centrifuge tube containing 100 mg of C18 sorbent, vortexed for 1 min and centrifuged for 10 min (15000 × g, 0 °C). The supernatant (2 mL) was collected with a plastic syringe and filtered through a 0.22 µm PTFE filter. 0.5 mL of the final extract (equals to 0.05 g mL−1 of matrix concentration) was injected into the LC-MS/MS instrument.
Cow’s milk
Cow’s milk was analysed as follows.
To the test sample (2.0 g), 6 mL of water, 2 mL of EDTA solution (5 mM) and 10 mL of MeOH containing 1% formic acid (v/v) were added. The samples were mechanically shaken for 5 min. Successively, the samples were placed in a freezer at -80 °C for 15 min and centrifuged, still cold, for 10 min. (15000 × g, 10° C). The supernatant (2 mL) was withdrawn with a plastic syringe and filtered through a 0.22 µm PTFE filter. Finally, 0.25 mL of the filtered extract was combined with 0.25 mL of water and dispensed directly into plastic vial. The final matrix equivalent concentration was 0.05 g mL−1.
Spiking procedure
For the determination of the recoveries and precision, the laboratories spiked the blank samples with the mixed stock solutions spike mix1, spike mix2, spike mix3, spike mix 4, spike mix5 and IS spike 1, as described in Table S2. Samples were spiked just prior to the extraction.
LC- MS/MS analysis
The choice and optimization of the LC–MS/MS set up, including the chromatographic column was left to the participants. Examples of suitable LC-MS/MS settings were provided with the SOP, more details are given in the supplementary material (Table S3–S7). In an example of chromatographic separation for the different columns are shown.
Figure 1. Reports an LC–MS/MS chromatograms of a chicken eggs sample spiked at 0.005 mg/kg for AMPA, cyanuric acid, ethephon, fosetyl Al, glyphosate, HEPA, maleic hydrazide, N-Acetyl- glyphosate; at 0.001 mg/kg for glufosinate ammonium, MPP, and NAG for three different chromatographic columns. Raptor polar X (30x2.1 mm, 2.7 µm); Hypercarb (100x2.1 mm, 5 µm); Anionic Polar Pesticides (APP, 100x2.1 mm, 5 µm).
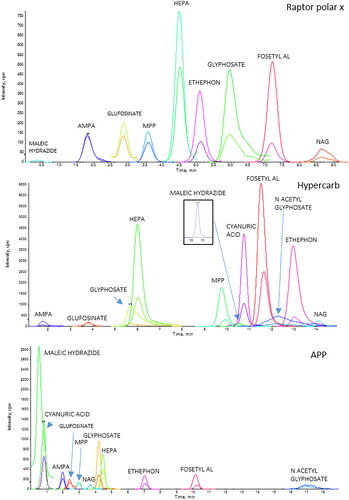
A mandatory requirement was to not use ion exchange chromatography to separate target pesticides, but only columns based on the partition principle. The use of the same type of chromatographic separation column for all laboratories, based on the partition principle, was required to ensure greater homogeneity of results and, considering that partition columns are the most widely used, this type of separation was selected. For pesticide identification, it was requested to fulfil the criteria defined in the SANTE/11312/2021 document (European Commission Citation2021).
Calculations
To provide more reliable results an internal standard calibration was selected. For this purpose, the ratio of the peak area of each analyte to the peak area of the related labelled analogue was calculated for each analyte. These ratios (RF – response factor) were then used in all subsequent calculations. A calibration curve, for each analyte, was prepared by plotting the RF calculated in the calibration solutions reported in Table S1 (Y-axis), against the corresponding amount (ng mL−1) of analyte injected on column (X axis).
Then mass fraction was calculated according to the following equation:
(1)
(1)
RF is the response factor (adimensional)
a is the slope of the calibration curve from calibration data (mL ng−1);
b is the intercept of the calibration curve from calibration data;
1000 is a conversion factor
DF is the dilution factor of the method, here: 20 (mL g−1)
The DF was calculated according to formula (2):
(2)
(2)
V1 is the final volume of the test sample, here: 0.5 mL;
is the sample equivalent weight in the final test sample, here: 0.025 g, calculated following the formula (3):
m is the mass of the extracted test portion, here: 2.0 g
V2 is the volume of the extraction mixture, here: 20 mL
V3 is the volume of extract collected, in mL, here: 0.25 mL
Result and discussion
Pretrial results
Prior to the full validation study, the laboratories were involved in the pre-trial study. The aims of the pre-trial were to check the sensitivity of the instrumentation and verify whether it was able to detect each pesticide at the LOQ level, corresponding to the lowest level proposed for validation. Overall, all participants report that they were able to reproduce the method’s protocol without significant deviations from the stated steps. The trickiest step was reported to be the filtration of the milk extract on PTFE filter, which was sometimes was hampered by filter blockage, thus requiring careful handling or the use of centrifuge filters. Matrix effects for early eluting compounds (cyanuric acid and maleic hydrazide) were reported by the participant using the Anion Polar Pesticide column (Waters), which is a column operating in hydrophilic interaction liquid chromatography (HILIC) and weak anion exchange modes. The results forms were returned by five laboratories (1,2,3,4,5) out of six participating in the pre-trial. One laboratory (lab 6) was not able to achieve the necessary instrumental sensitivity and was therefore withdrawn from the study. One participant reported acceptable sensitivity only for few molecules (4/11) in all matrices. The issue was solved by changing the LC conditions. Specifically, the participant finally chose a Hypercarb™, column instead of the formerly used Raptor Polar X as suggested by the interlaboratory study coordinator. The Raptor Polar X column was also tested by the study coordinator to propose different alternatives to separate the target polar pesticides. This column was identified as a suitable candidate to retain polar compounds due to its new stationary phase combining both hydrophilic interaction chromatography (HILIC) and ion exchange approach on a single ligand, but showed poor signal stability and a double peak for glyphosate in the egg matrix. The final optimised instrumental set-up for each participant is shown in , more details are given in the supplementary material (Table S8–S9).
Table 1. Overview of LC-MS/MS systems used by the trial participants (*column dedicated to analyse maleic hydrazyde).
Trial study
All five laboratories passing the pre-trial test, participated in the full trial providing complete report forms.
Based on the data provided, recoveries, repeatability and reproducibility standard deviations (SDr and SDR) and relative standard deviations (RSDr and RSDR) of the method were determined for each pesticide and for each spiked level. Robust statistical methods were used for the estimation of repeatability and reproducibility as described in ISO 5725 Part 5 (ISO 5725-5, 1994). Since the EXCEL macro used for statistics calculation required an equal number of results per laboratory, only laboratories with a complete set of 5 analytical values were included in the calculation of the robust statistics. If a value provided by a laboratory was considered as an extreme one (f.i. extremely low detected mass fractions – below 20% of the spiking level) the whole set of data, provided by the specific participant, was eliminated prior to the calculation of the method performances. The overview of the processed results is in and . The method was developed and validated with the perspective to be applied for monitoring and compliance testing purposes by official control laboratories, therefore method performances were assessed according to SANTE/11312/2021 criteria.
Figure 2. Graphic overview of the study results: relative recovery rate (%) ± RSDR for polar pesticides in bovine fat (A), chicken egg (B) and cow milk (C). The dotted lines indicate the range of acceptability according to SANTE/11312/2021 criteria.
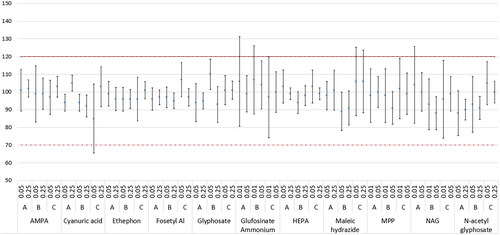
Table 2. Method performance characteristics for all analyte/matrix. The samples were spiked at two concentration levels: equal to or lower than the MRLs (spiking level A) and at levels corresponding to 5 times the LOQs (spiking level C). For the other pesticides were no MRL in food of animal origins are already sets, the lowest level (LOQ) was set at the same concentration set for LOQ during in-house validation study (Verdini et al. Citation2023). n.i.: not included in the MRL definition. * = LOQ could be equal to MRL. Average RSDr (%) obtained in the house validation study (Verdini et al. Citation2023) were also reported **validation level used in-house validation.
Overall recoveries and repeatability were compliant with the acceptability criteria.
Specifically, average recoveries ranged from 85% to 110%. Analysis of samples contaminated at high level resulted in RSDr ranging from 3% to 18%, and RSDR from 4% to 26%. The RSDr values obtained for samples contaminated at the lowest evaluated level, ranged from 5% to 25% whereas RSDR from 8% to 41%. Higher values for the RSDR were obtained for samples contaminated at the lowest studied level, specifically high values were obtained at 0.01 mg/kg for NAG in bovine fat matrix (41%) and glufosinate ammonium in bovine fat and milk, 26 and 30% respectively.
Only for NAG, MPP and glufosinate-ammonium, the investigated levels in the in-house study differed slightly from those used in the test study. In particular, for NAG in bovine fat, a better repeatability value was obtained at the lowest level studied, which was double the lowest level in the test study (9% RSDr at 0.025 mg/kg vs. 41% RDSR at 0.01 mg/kg) (Verdini et al. Citation2023).
Overall, average recovery and method precision obtained in the trial study confirmed data obtained in house validation study (Verdini et al. Citation2023)
The most challenging analyte was cyanuric acid in all matrices. Two laboratories (lab 2 and lab 5) using a LC column other than the Hypercarb™ reported no results, probably because the cyanuric acid was poorly retained and eluted too close to the solvent front. On the other hand, lab 3 was able to achieve satisfactory recoveries using APP column but only after to a careful optimization of the chromatographic conditions (see the final optimized conditions in Table S8 – supplementary material). An example of suitable LC-MS/MS setting obtained by APP column for the three matrices were provided in .
Figure 3. Selected ion chromatogram of (A) bovine fat, (B) chicken egg and (C) cow milk sampled spiked with 0.25 mg kg -1 AMPA, cyanuric acid, ethephon, glyphosate, fosetyl aluminum, HEPA, maleic hydrazide and N-acetyl-glyphosate, 0.01 mg kg -1 of glufosinate ammonium, MPP, N-acetyl glufosinate (NAG) and relevant isotopically labelled internal standard (sum of quantifier and qualifier transition for each analyte - upper line, ISTD quantifier transition—lower lines) analyzed with Anionic Polar Pesticides (APP, 100x2.1 mm, 5 µm) column.

N-acetyl glyphosate resulted to be challenging in the egg matrix, since only 3 out of 5 participants reported a full data set for this residue. This was tentatively attributed to challenges in the LC separation. N acetyl glyphosate shows a broad and late-eluting peak on the HILIC stationary phase (used by Lab 2, 3, 5) affecting its reliable detection and quantification. Lab 5, could not report any data for all matrices, probably because the selected chromatographic conditions did not allow the elution of N-acetyl glyphosate, which is strongly retained in this column. This behaviour was also in agreement with the study by Dias et al. (Dias et al. Citation2021) on the influence of three different HILIC columns on the determination of N acetyl glyphosate and other anionic pesticide in feed matrix. To improve the retention factor of N acetyl glyphosate on the Anionic Polar Pesticide column, Lab 3 changed the mobile phase composition by using a mixture of methanol and acetonitrile for eluent B to increase the eluting strength on the HILIC column. Finally, lab 2 reported unsatisfactory results only for egg matrix –for N acetyl glyphosate – probably due to some matrix-related interference.
Conclusions
A previously in-house validated and accredited LC-MS/MS method for the determination of polar pesticides in food of animal origin was challenged in an interlaboratory validation study to confirm its fitness-for-purpose of monitoring and compliance testing by official control laboratories. Moreover, assessing the sample preparation and analysis protocol across five participant laboratories provided some insights on key factors impacting method performance characteristics.
The interlaboratory comparison confirmed that instrumental performances are definitely impactful, and the use of highly sensitive LC-MS/MS and their set up is crucial for the determination of polar pesticides at regulatory levels. Achieving a suitable LC separation has proven to be the most challenging step in the detection of polar pesticides, therefore a careful optimisation of chromatographic conditions is essential for a reliable detection of either early eluting compounds (cyanuric acid and maleic hydrazide) or the broad and late-eluting peak of N - acetyl glyphosate. Overall, the method complies with official criteria (SANTE 11312/2021) and with the appropriate precautions has proven to be suitable for the purpose of monitoring and compliance testing.
Supplemental Material
Download MS Word (81.8 KB)Acknowledgments
The authors thank all participants of the study including: Istituto Zooprofilattico Sperimentale delle Venezie; Istituto Zooprofilattico Sperimentale dell’Abruzzo e del Molise "Giuseppe Caporale"; Istituto Zooprofilattico Sperimentale della Sardegna " Giuseppe Pegreffi"; University of Chemistry and Technology, Prague; Wageningen Food Safety Research.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Anastassiades M, Wachtler K, Kolberg DI, Eichhorn E, Benkenstein A, Zechmann S, Mack D, Barth A, Wildgrube C, Sigalov I, et al. 2019. Quick method for the analysis of numerous highly polar pesticides in foods of plant origin via LC-MS/MS involving simultaneous extraction with methanol II. Food of Animal Origin (QuPPe-AO-Method). EU Reference Laboratory for pesticides requiring Single Residue Methods (EURL-SRM). Fellbach: CVUA Stuttgart. www.eurl-pesticides.eu
- Dias J, López SH, Mol H, de Kok A. 2021. Influence of different hydrophilic interaction liquid chromatography stationary phases on method performance for the determination of highly polar anionic pesticides in complex feed matrices. J Sep Sci. 44 (11):2165–2176. doi:10.1002/jssc.202001134.
- [EC] European Commission. 2022. Commission IMPLEMENTING REGULATION (EU) 2022/2364 of 2 December 2022 amending Implementing Regulation (EU) No 540/2011 as regards the extension of the approval period of the active substance glyphosate. Off J Eur Union. L312:99–100.
- [EC] European Commission. 2021. COMMISSION IMPLEMENTING REGULATION (EU) 2021/601 of 13 April 2021 concerning a coordinated multiannual control programme of the Union for 2022, 2023 and 2024 to ensure compliance with maximum residue levels of pesticides and to assess the consumer exposure to pesticide residues in and on food of plant and animal origin. Off J Eur Union. L127:29–41.
- [EC] European Commission. 2023. European commission extension of the approval period of glyphosate. https://food.ec.europa.eu/plants/pesticides/approval-active-substances/renewal-approval/glyphosate_en
- EFSA, ECHA. 2023. European food safety authority. https://echa.europa.eu/it/-/glyphosate-efsa-and-echa-update-timelines-for-assessments).
- [EFSA] European Food Safety Authority. 2018. Reasoned Opinion on the review of the existing maximum residue levels for glyphosate according to Article 12 of Regulation (EC) No 396/2005. EFSA J. 16(5):230. doi:10.2903/j.efsa.2018.5263.
- [EFSA] European Food Safety Authority. 2019. Review of the existing maximum residue levels for glyphosate according to Article 12 of 2005/2005 – revised version to take into account omitted data. EFSA J. 17(10):5862. doi:10.2903/j.efsa.2019.5862.
- [EFSA] European Food Safety Authority. 2022. Report of pesticide peer review TC 80. https://www.efsa.europa.eu/sites/default/files/2023-01/glyphosate-peer-review-minutes-nov-dec-2022.pdf
- European Union. 2005. Regulation (EC) No 396/2005 of the European Parliament and of the council of 23 February 2005 on maximum residue levels of pesticides in or on food and feed of plant and animal origin amending Council Directive 91/414/EEC. Off J Eur Union. L70:1–16.
- EURL-SRM EU Reference Laboratories for Residues of Pesticides. 2021. Interlaboratory validation study polar pesticidesQuPPe Method “Gly&Co”. https://www.eurl-pesticides.eu/userfiles/file/EurlFV/Joint2021/Wachtler-Zipper.pdf.
- EURL-SRM. 2014. EU proficiency test on the analysis of spiked pesticides requiring single residue methods in cow’s milk. 1–67. https://www.eurl-pesticides.eu/library/docs/srm/EUPT_SRM9_FinalReport.pdf
- EURL-SRM. 2019. EU proficiency test on the analysis of bovine liver homogenate for residues of pesticides requiring single residue methods. 1–43. https://www.eurl-pesticides.eu/library/docs/srm/EUPT_SRM14_FinalReport.pdf
- European Commission. 2021. Guidance document on analytical quality control and method validation for pesticide residues analysis in food and feed SANTE 11312/2021. Sante/11312/2021. 1–7. https://ec.europa.eu/food/system/files/2022-02/pesticides_mrl_guidelines_wrkdoc_2021-11312.pdf.
- ISO 5725-5. 1994. Accuracy (trueness and precision) of measurement method and results. Part 5: alternative methods for the determination of the precision of a standard measurement method. Geneva: international Organization for Standardization.
- Verdini E, Pecorelli I. 2022. The current status of analytical methods applied to the determination of polar pesticides in food of animal origin: a brief review. Foods. 11(10):1527. doi:10.3390/foods11101527.
- Verdini E, Lattanzio VMT, Ciasca B, Fioroni L, Pecorelli I. 2023. Improved method for the detection of highly polar pesticides and their main metabolites in foods of animal origin: method validation and application to monitoring programme. Separations. 10(1):44. doi:10.3390/separations10010044.
- Wu Y, Zhou Y, Jiao X, She Y, Zeng W, Cui H, Pan C. 2023. Development and inter-laboratory validation of analytical methods for glufosinate and its two metabolites in foods of plant origin. Anal Bioanal Chem. 1–12. doi:10.1007/s00216-023-04542-9.