
ABSTRACT
Beauverolides are hydrophobic cyclodepsipeptides that inhibit sterol O-acyltransferases and calmodulin, thereby reducing senile plaques in Alzheimer’s disease and preventing foam cell formation in atherosclerosis. Other sterol O-acyltransferase inhibitors suffer from low bioavailability, and evidence of the distribution of beauverolides in bodily fluids is lacking. We aimed to determine the optimal formulation of beauverolides for administration to experimental animals and to determine if the orally administered beauverolides could cross the gastrointestinal barrier and be secreted in urine. We found that beauverolides formed gels in aqueous solutions and we developed a formula for their peroral administration. Administration of the beauverolide gel pellets to experimental mice revealed that beauverolides cross the gastrointestinal barrier, circulate untouched in the blood, and are excreted in the urine. Using liquid chromatography-mass spectrometry analyses, we show that the administered beauverolides circulate in mouse blood and are excreted from the body 24 h after their administration.
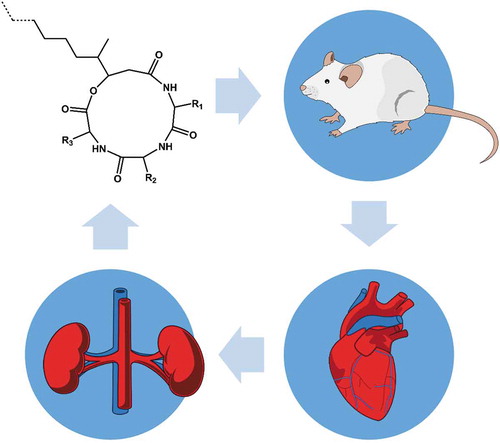
Graphical abstract
RESUMEN
Los beauverólidos son ciclodepsipéptidos hidrofóbicos que inhiben las O-actiltransferasas de esterol y la calmodulina, reduciendo las placas seniles en la enfermedad de Alzheimer y previniendo la formación de células espumosas en la aterosclerosis. Otros inhibidores de la O-aciltransferasa esterólica tienen baja biodisponibilidad y no existen pruebas sobre la distribución de los beauverólidos en los fluidos corporales. El presente estudio se propuso determinar la formulación óptima de los beauverólidos para su administración a animales de experimentación, de modo de establecer si administrados por vía oral podían atravesar la barrera gastrointestinal y ser secretados en la orina. En este sentido se constató que los beauverólidos forman geles en soluciones acuosas, por lo que se desarrolló una fórmula para su administración peroral. La administración de gránulos de gel de beauverólido a ratones experimentales confirmó que los mismos cruzan la barrera gastrointestinal, circulan sin ser modificados en la sangre y son excretados en la orina. Mediante análisis de cromatografía líquida y espectrometría de masas, demostramos que los beauverólidos administrados circulan en la sangre de los ratones y 24 horas después de su administración son excretados del cuerpo.
1. Introduction
Beauverolides (also known as beauveriolides or beauverolides) are peptidic secondary metabolites that are abundantly synthetized by entomopathogenic fungi, including Beauveria bassiana (Elsworth & Grove, Citation1977; Isogai et al., Citation1978), Beauveria brongniartii (syn. B. tenella; Jegorov et al., Citation2004), B. vermicoria (Kadlec et al., Citation1994), Isaria fumosorosea, Isaria japonica (Tuan et al., Citation2017), and Cordyceps militaris (Chen et al., Citation2018; Nakaya et al., Citation2012). Isaria spp. and Beauveria spp. are the anamorph stages of Cordyceps or Torrubiella. Ubiquitous fungi, such as Aspergillus versicolor and Penicillium chrysogenum (Arendowski et al., Citation2018), and the lichen Candelariella vitellina (Madariaga-Mazon et al., Citation2015) have been reported as additional sources of beauverolides. Recently, heterologous expression of beauveriolide-encoding genes was reported in Aspergillus nidulans (X. Wang et al., Citation2020). Some fungi of the genera Beauveria and Isaria, such as I. fumusorosea strain PFR 97-Apopka, are used as biological insecticides in agriculture, horticulture, and forestry. Beauverolides may be present in some fungal mycelia in mg/g amounts; however, beauverolides do not exhibit mycotoxic activities, and their roles in entomopathogenic processes are unknown. Concerning the effects of beauverolides on humans and experimental animals, they were previously reported to have a variety of biological activities, including the inhibition of sterol O-acyltransferase 1 (also known as ACAT-1) in the pathogenesis of Alzheimer’s disease (Doi et al., Citation2012; Ohshiro et al., Citation2017, Citation2007; Witter et al., Citation2009) and atherosclerosis (Namatame et al., Citation2004; Tomoda & Doi, Citation2008). Recently, beauverolides were reported to serve as potent calmodulin inhibitors (Madariaga-Mazon et al., Citation2015). Moreover, concurrent with the present study, a detailed in silico absorption, distribution, metabolism, and excretion (ADME) study of beauveriolides I, III and their derivatives for the selective inhibition of sterol O-acyltransferase 2 was reported by Ohshiro et al. (Citation2020). Nevertheless, the encapsulation of beauverolides, their capability of crossing the gastrointestinal barrier, and their distribution and metabolism in bodily fluids are still poorly understood. It is not clear if the structures of beauverolides remain untouched or are linearized and/or conjugated in the mammalian organism, and how beaverolides are excreted from the organism is also unknown.
The beauverolides have a cyclodepsipeptide structure consisting of L-phenylalanine, L-alanine, D-leucine, and either 3-(S)-hydroxy-4-(S)-methyldecanoic or 3-(S)-hydroxy-4-(S)-methyloctanoic acid, which are connected by three amide bonds and one ester bond. The ester bond is less stable than the peptide amide bond in vivo but promotes cell permeability before intracellular ester cleavage by esterases (Nielsen et al., Citation2017). Beauverolides are sparingly soluble in organic solvents, particularly in methanol, but their solubility in aqueous solutions is limited. The circulation of beauverolides has not been studied in human or animal fluids; therefore, data concerning the bioavailability of beauverolides are limited. However, numerous studies have administered beauverolides to cell lines and even animal models. Oral administration of beauveriolide III was shown to reduce atherosclerotic lesions in ApoE-KO and LDLR-KO mice (Namatame et al., Citation2004; Ohshiro et al., Citation2020; Tomoda & Doi, Citation2008). The atherosclerotic lesions of the aorta and heart were reduced by more than half when beauveriolide III was administered to the mice for a period of 2 months at a dose of ≥25 mg kg−1 day−1. Various experiments have reported the treatment of cell lines with beauverolides (). However, most of these studies used various methanolic or ethanolic solutions of beauverolides, which could have severely affected the observed outcomes unless proper controls were used. Some of the previous studies reporting the use of water-soluble compounds from methanolic extracts of beauverolides, as reported by Park et al. (Citation2018), are questionable as intact cyclic beauverolides are sparingly soluble in water. The limited solubility in water may explain the difficulties observed by Park et al. (Citation2018) when replicating their outcomes with the application of individual compounds, namely, beauverolide La, beauverolide L, beauveriolide V, and beauveriolide VI at 40 μg mL−1.
Table 1. Overview of previously published beauverolide formulations.
Tabla 1. Resumen de las formulaciones de beauverólido publicadas anteriormente.
In the present study, we determined the optimal formulation of beauverolides for administration to experimental animals, and we aimed to determine if the orally administered beauverolides could cross the gastrointestinal barrier and be secreted in the urine in their intact form. Beauverolides are hydrophobic peptides. However, the problem of water solubility affects ~90% of all compounds in drug delivery pipelines (Reintjes, Citation2011). Limited solubility and tissue permeability represent problems that are directly related to the classification of drugs according to the Biopharmaceutical Classification System (Karlsson et al., Citation2019). Solubility and tissue permeability are not mutually related and many hydrophobic compounds, such as β-carotene, can easily penetrate relevant physiological barriers despite their poor solubility in aqueous solvents. The use of surfactants, formation of complexes, or use of nanomaterials represent the most common solutions, which may improve the solubility. For solid compounds, such as beauverolides, the preparation of formulations that have a potential to form emulsions in the gastrointestinal tract is greatly facilitated by the self-emulsifying and self-microemulsifying drug delivery systems. However, the application of these delivery systems is limited by the ability of the compound to be dissolved in nontoxic oils, such as in olive oil; a typical example is the formulation of cyclosporine A that is marketed as Sandimmune or Neoral (Smith, Citation1996). Both, cyclosporine A and beauverolides are cyclic peptides that may have similarly limited bioavailability. Therefore, we employed previous experience from the preparation of bioavailable cyclosporine formulations in an attempt to solve the issue of the poor solubility of beauverolides. In the present study, we examined Kolliphor EL, which is synthetized by the reaction between castor oil and ethylene oxide, resulting in a mixture of triricinoleate esters of ethoxylated glycerol, polyethylene glycol ricinoleates, and their derivatives with polyethoxylated 12-hydroxyricinoleic acid residues. Kolliphor EL was successfully used to formulate cyclosporine A (Karlsson et al., Citation2019) and many other hydrophobic compounds.
2. Materials and methods
2.1. Solubilization of beauverolides
Beauverolides can be easily dissolved at 2 mg mL−1 in methanol (Elsworth & Grove, Citation1977). However, methanol is toxic to living organisms; therefore, an alternative formulation was needed. We tested the solubilization of a mixture of purified beauverolides, which contained predominantly (35%) beauverolide I. We isolated and purified this mixture of beauverolides from the I. fumosorosea strain PFR 97-Apopka (W. R. Grace & Co., Columbia, MD), the strain that is approved as the Bemisia tabaci biocontrol agent. We aimed to obtain solutions containing beauverolides at 2 mg mL−1, which can then be administered to cells and animals. We tested the solubility in DMSO, various concentrations of ethanol mixed with water, Tris pH 7.4, phosphate buffer pH 7.4, trehalose, or Kolliphor EL (BASF, Ludwigshafen, Germany). When beauverolides were insoluble at 2 mg mL−1 in the selected solvent, we tested lower concentrations. As the solubility was temperature-dependent and precipitation often occurred, we incubated the obtained solutions at various temperatures, ranging from 72°C to 4°C for the indicated time intervals (from one hour to one week) and checked for precipitation of beauverolides and formation of gels.
2.2. Gel formulations
To prepare a beauverolide pellet, we dissolved 2 mg of the mixture of beauverolides (Fig. S1) in 312.5 μL of 96% ethanol at 72°C and added 22 mg of Kolliphor EL formulated as a 50% solution (v/v) in 96% ethanol. Next, we added 32 mg of glycerol (Penta, Prague, Czech Republic) formulated as a 50% solution (v/v) to 50 mM Tris-HCl, pH 7.4. Finally, we added 87.5 μL of distilled water and facultative 1.6 mg of glucose (Penta, Prague, Czech Republic) or 17 mg of milk-based infant formula Nutrilon, vanilla flavor (Nutricia, Hoofddorp, Netherlands) with the aim of increasing the attractiveness of the gel capsules for the mice. We evaporated the ethanol from the solutions at 72°C until the content in the vials reached 80 to 90 mg; then, the content was allowed to air dry. The procedure resulted in the formation of gel capsules, which were immediately provided to the experimental mice.
2.3. Peroral administration of beauverolides
We offered the gel capsules containing beauverolides to the mice (n = 3). As a model, we used BALB/c females at 6 months of age. The BALB/c mice commonly serve as a model to study the development of atherosclerotic plaques, since these mice are resistant to high-fat diet-induced atherosclerosis but develop hyperglycemia-induced fatty streak lesions (Kunjathoor et al., Citation1996). The mice were bred in the specific pathogen‐free barrier area of the institutional animal facility and maintained in the clean, conventional part of the facility during the experiments. Prior to the administration of beauverolides, each mouse was starved for 24 h and then provided two gel capsules containing beauverolides (i.e. 4 mg of beauverolides in total per mouse). Each mouse was provided capsules containing the same amount of beauverolides but with different attractants (glucose, Nutrilon, or no attractants). All the mice ate the capsules readily. We collected the urine and peripheral blood from the mice 24 h after the oral administration of the beauverolides. We centrifuged the blood to obtain the serum. Both the urine and serum were frozen at −80°C until further processing. These experiments were approved by the Committee for Animal Care and Use of the First Faculty of Medicine, Charles University, Prague, and were designed in order to address the systemic bioavailability at a single time point following oral administration of the study compound sensu OECD TG 417 Toxicokinetics guidelines.
2.4. Stability assay
As we did not identify any linear forms of beauverolides in mouse urine or serum, we attempted to examine whether linear beauverolides were stable when stored in aqueous solutions. We opened the ester bonds in the isolated beauverolides as described (Šimčíková et al., Citation2020) and prepared solutions of linearized or cyclic beauverolides at concentration 2 mg mL−1 in methanol; then, we further dissolved them in methanol or human serum to a final concentration of 0.1 mg mL−1. We then incubated these solutions at 37°C and measured the content of beauverolides at the indicated time intervals for up to 24 h.
2.5. Detection of beauverolides
Beauverolides were detected in the mouse bodily fluids by LC-MS/MS analysis as described previously (Šimčíková et al., Citation2020). An Accela HPLC liquid chromatograph directly coupled to a linear ion trap mass spectrometer LTQ (all Thermo Fisher Scientific, Waltham, MA) equipped with an electrospray HESI2 ion source was operated in positive ion detection mode. Separation of the analytes was accomplished on a 150 × 3 mm, 2.6 µm reversed-phase Kinetex C18 (Phenomenex, USA). The data were processed by Xcalibur 4.3 software (Thermo Fisher Scientific, Waltham, MA).
3. Results
3.1. Solubilization of beauverolides
We aimed to develop bioavailable formulations that could allow the solubilization of beauverolides up to 2 mg mL−1. At this concentration, the beauverolides were insoluble in 80% DMSO and formed precipitates. However, the beauverolides were soluble in 96% ethanol at 72°C but precipitated out of the solution at 4°C or when the ethanol was dissolved with water to a lower content, such as 50% concentration (). The beauverolides were soluble at 2 mg mL−1 in 75% ethanol with Tris pH 7.4, and the solution of beauverolides remained stable at 37°C or 72°C. However, the solution started to form a gel at 20°C, which was also true for lower concentrations of beauverolides when those were incubated overnight at 20°C (). The use of 60% ethanol in Tris pH 7.4 resulted in the same effect even at lower concentrations of beauverolides. The dilution of 75% ethanol in phosphate buffer pH 7.4 yielded similar results. The supplementation of 40% or 75% ethanol with 0.25–2.0% trehalose allowed the solubilization of beauverolides only at 72°C, whereas at lower temperatures, we observed gel formation. Using various mixtures of Kolliphor EL with ethanolic solutions resulted in the solubilization of the beauverolides at 2 mg mL−1 at 5–25% Kolliphor EL in ethanol at 60°C but resulted in immediate or delayed gel formation at lower temperatures. The gels formed by the beauverolides in Kolliphor EL were solid and of a consistency that resembled 1% agarose. The addition of 0.45% NaCl to the ethanolic Kolliphor EL solutions, or the preparation of 1:1 mixtures of the ethanolic Kolliphor EL solutions with 50 mM Tris, pH 7.4, did not improve the solubility. The beauverolides formed solid gels at 1 mg mL−1 in all the tested ethanolic Kolliphor EL solutions that were supplemented with NaCl. The Tris-supplemented Kolliphor EL solutions remained fluid only at high temperatures, whereas gels were always formed when the formulations were incubated at 37°C or 20°C ().
Table 2. Overview of tested solubilization conditions for the beauverolides.
Tabla 2. Resumen de las condiciones de solubilización probadas para los beauverólidos.
3.2. Peroral administration of beauverolides
Reflecting the outcomes of the solubilization experiments, we prepared three formulations of beauverolides that consisted of gel pellets containing 2 mg of beauverolides in Kolliphor EL, glycerol, Tris pH 7.4, and water with facultatively added glucose or milk-based formula. We found that the beauverolides circulated in the mouse bodies 24 h following their peroral administration. Twenty-four hours after oral administration of beauverolides, the cyclic peptides were detected in both, the serum (Figs. S2-S4) and urine (Figs. S5-S7) of the mice that had been starved for 24 h and readily ate the gel pellets containing the beauverolides (). When analyzing the content of beauverolides in the mouse serum and urine, we did not identify any linear forms of beauverolides. Instead, we identified six to eight beauverolide species in each analyzed sample. Interestingly, the composition of the beauverolides in mouse serum and urine differed from the composition of the administered beauverolides. The most striking difference was in the content of beauverolide F, which was abundantly present in the administered mixture (17.6%) and was absent in all but one of the analyzed serum and urine samples. Similarly, another beauverolide containing two phenylalanine moieties, beauverolide C, was absent from the serum and urine samples. We did not detect the minor beauverolides because their putative concentrations in the serum and urine were below the limits of detection. The content of the other two beauverolides that dominated the orally administered mixture, beauverolide I and beauverolide M, was similar or higher in the serum and urine than in the orally administered mixture (). The total content of beauverolides in the serum and urine of each mouse was variable. In two mice, the urinary concentration of beauverolides was three-times higher than their serum concentration. In the mouse with the lowest serum levels of beauverolides, the concentration of beauverolides in urine was approximately equal to that in serum.
Table 3. Orally administered beauverolides from gel capsules are detectable in serum and excreted in intact form by urination.
Tabla 3. Los beauverólidos administrados oralmente mediante cápsulas de gel son detectables en el suero y son excretados intactos al orinar.
These data suggest that the beauverolides are bioavailable when administered orally; however, their availability is poor. The experiments that analyzed the content of beauverolides at a single time point alone did not allow us to calculate the total proportion of beauverolides that crossed the gastrointestinal barrier. However, based on the theoretically assumed volume of mouse urine per 24 h and the volume of mouse blood, we estimated that the analyzed mice internalized only 1.3 · 10−3% – 11.8 · 10−3% of the orally administered beauverolides (). The beauverolides were secreted from the bodies of the mice within 24 h, and only their cyclic forms were detected both in the serum and urine.
It was possible that we did not observe the linear beauverolides in the mouse serum or urine because they were unstable. Therefore, we further searched for the presence of the linear beauverolidesin which the ester bonds had opened; however, we did not identify these compounds in the analyzed biological materials. Therefore, we tested the stability of the linear beauverolides by storing them at 0.1 mg mL−1 at 37°C for up to 24 h in methanol or human serum. We found that the linear beauverolides were stable in methanol for the examined time period and that their content decreased by only 10–15% when the linear beauverolides were incubated in human serum during the analyzed 24 h period. Therefore, we assume that if the linear beauverolides were secreted into the urine, we would have been able to detect them using the techniques used in the present study. The LC-MS comparative analysis of the murine bodily fluids from the control and exposed animals did not reveal any other beauverolide metabolites in the examined biological samples.
4. Discussion
In the present study, we provide the first LC-MS data on the bioavailability of oral beauverolide formulations. Our findings complement those reported by Namatame et al. (Citation2004) and Ohshiro et al. (Citation2020), who conclusively showed that beauverolides serve as orally active inhibitors of ACAT-1 and ACAT-2. However, these studies did not determine whether the orally administered beauverolides penetrated the gastrointestinal barrier. Instead, these authors fed mice beauveriolide III at 50 mg kg−1 or 25 mg kg−1 supplemented in 0.05% sodium carboxymethyl cellulose daily for two months without examining the presence or the levels of beauverolides in the target organisms (Namatame et al., Citation2004); similar study was conducted with beauveriolide derivatives administered at the same dose (Ohshiro et al., Citation2020). In the present study, we confirmed that beauverolides are able to cross the gastrointestinal barrier and can be found in the organism for at least 24 h after ingestion. This finding is valuable, as currently available ACAT inhibitors, such as CP-113,818 and DuP 128, have characteristically low bioavailability, particularly because of their poor ability to cross the gastrointestinal barrier and their rapid metabolization in the blood and tissue (Hutter-Paier et al., Citation2004; Marzetta et al., Citation1994; Puglielli et al., Citation2001).
Beauverolides, similar to many other cyclic peptides, are hydrophobic compounds (Jegorov et al., Citation1990). Only trace amounts of beauverolides are soluble in water and aqueous buffers (). Therefore, the mechanism involved in the crossing of the gastrointestinal barrier remains unknown. We speculate that the crossing of the gastrointestinal barrier might be associated with the presence of the unique 3-hydroxy acid in the beauverolide structure; however, this suggestion deserves a more detailed follow-up study. Cyclic peptides with high lipophilicity may insert too readily into the cellular membrane, overly favoring the membrane-bound state and resulting in poor overall transport across biological membranes (C. K. Wang et al., Citation2015; Wang & Craik, Citation2016). Importantly, other hydrophobic cyclic peptides are known for their high protein binding capacity. For example, echinocandins have 84–99% protein binding capacity, which could also severely limit the amount of drug available for biological activity (Denning, Citation2003). Whether beauverolides are active when bound to their putative carrier proteins remains to be investigated.
Previous studies have suggested that some beauverolides are water-soluble (). When attempting to dissolve the mixture of beauverolides, Park et al. (Citation2018) reported the ability to dissolve 10.1% (w/w) of the analyzed beauverolide extract, and they identified the soluble compounds as beauverolides L and La, and beauveriolides V and VI. Beauverolide L was shown to be soluble at 1 mg mL−1 in saline with 10% ethanol (Vilcinskas et al., Citation1999). However, other studies provided conclusive evidence of the insolubility of cyclic beauverolides in aqueous solutions, which was also true for beauveriolides V and VI (Matsuda et al., Citation2004). In general, beauverolides are sparingly soluble in water and aquatic solutions; their solubility was demonstrated only for low (≤100 μM) concentrations (). In the present study, we have confirmed the poor solubility of beauverolides in water. However, we have identified a previously unreported characteristic of beauverolides, namely, that they can form gels. Peroral gels are scarcely used in pharmacological applications, although the first peroral gels were developed in the 1970s (Ueda et al., Citation1978). As gel formers, previous studies typically used cellulose ethers, carbohydrates, and other compounds, including betulin and phytosterols of plant origin (Bakhrushina et al., Citation2019). Here, we report a unique situation, where the active compound itself has gel-forming properties, which we demonstrated to be useful to exert therapeutic action in the gastrointestinal tract of the tested experimental animals. Under certain conditions, such as those that we used for the preparation of beauverolide formulations and subsequent peroral administration, beauverolides form solid gels that are easy to handle (). These formulations are nontoxic and allow the delivery of low hundreds of nanograms of beauverolides to experimental mice.
In conclusion, we have developed a novel gel formulation for the peroral administration of beauverolides and provided the first evidence of the uptake of orally administered beauverolides across the gastrointestinal barrier, their circulation in blood, and their secretion in urine.
Author contributions
PH and PŠ conceived and designed the experiments; AJ and PH acquired the data; PH and PŠ analyzed the data; PH wrote the paper and is responsible for the integrity of this work; all authors revised the article’s intellectual content and approved the final version.
Supplemental Material
Download MS Word (109.7 KB)Acknowledgments
We thank Petr Šimčík (www.petrsimcik.cz) for his help with graphical materials. The study was supported by the Czech Health Research Council project AZV 15-32432A. The funding body had no role in the design of the study, collection, analysis, and interpretation of data, or in writing the manuscript.
Disclosure statement
The authors declare no other conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
Related Research Data
References
- Arendowski, A., Szulc, J., Nizioł, J., Gutarowska, B., & Ruman, T. (2018). Metabolic profiling of moulds with laser desorption/ionization mass spectrometry on gold nanoparticle enhanced target. Analytical Biochemistry, 549(1), 45–52. https://doi.org/10.1016/j.ab.2018.03.016
- Bakhrushina, E. O., Anurova, M. N., Demina, N. B., & Ivannikov, G. Y. (2019). Main aspects of pharmaceutical gel development for peroral administration. Drug Synthesis Methods and Manufacturing Technology, 53(9), 838–844. https://doi.org/10.1007/s11094-019-02087-9
- Chen, L., Liu, Y., Guo, Q., Zheng, Q., & Zhang, W. (2018). Metabolomic comparison between wild Ophiocordyceps sinensis and artificial cultured Cordyceps militaris. Biomedical Chromatography, 11(9), e4279. https://doi.org/10.1002/bmc.4279
- Denning, D. W. (2003). Echinocandin antifungal drugs. The Lancet, 362(9390), 1142–1151. https://doi.org/10.1016/S0140-6736(03)14472-8
- Doi, T., Muraoka, T., Ohshiro, T., Matsuda, D., Yoshida, M., Takahashi, T., Omura, S., & Tomoda, H. (2012). Conformationally restricted analog and biotin-labeled probe based on beauveriolide III. Bioorganic & Medicinal Chemistry Letters, 22(1), 696–699. https://doi.org/10.1016/j.bmcl.2011.10.045
- Elsworth, J. F., & Grove, J. F. (1977). Cyclodepsipeptides from Beauveria bassiana Bals. Part 1. Beauverolides H and I. Journal of the Chemical Society, Perkin Transactions, 1(3), 270–273. https://doi.org/10.1039/p19770000270
- Hutter-Paier, B., Huttunen, H. J., Puglielli, L., Eckman, C. B., Kim, D. Y., Hofmeister, A., Moir, R. D., Domnitz, S. B., Frosch, M. P., Windisch, M., & Kovacs, D. M. (2004). The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron, 44(2), 227–238. https://doi.org/10.1016/j.neuron.2004.08.043
- Isogai, A., Kanaoka, M., Matsuda, H., Hori, Y., & Suzuki, A. (1978). Structure of a new cyclodepsipeptide beauverilide A from Beauveria bassiana. Agricultural and Biological Chemistry, 42(9), 1797–1798. https://doi.org/10.1080/00021369.1978.10863252
- Jegorov, A., Kadlec, Z., Novák, J., Maťha, V., Sedmera, P., Tříska, J., & Zahradníčkova, H. (1990). Are the depsipeptides of Beauveria brongniartii involved in the entomopathogenic process? In A. Jegorov & V. Maťha (Eds.), Biopesticides, theory and practice (pp. 71–82). House of Technics CSVTS.
- Jegorov, A., Paizs, B., Kuzma, M., Zabka, M., Landa, Z., Sulc, M., Barrow, M. P., & Havlicek, V. (2004). Extraribosomal cyclic tetradepsipeptides beauverolides: Profiling and modeling the fragmentation pathways. Journal of Mass Spectrometry, 39(8), 949–960. https://doi.org/10.1002/jms.674
- Kadlec, Z., Šimek, P., Heydová, A., Jegorov, A., Maťha, V., Landa, Z., & Eyal, J. (1994). Chemotaxonomic discrimination among the fungal genera Tolypocladium, Beauveria and Paecilomyces. Biochemical Systematics and Ecology, 22(8), 803–806. https://doi.org/10.1016/0305-1978(94)90083-3
- Kapojos, M. M., Abdjul, D. B., Yamazaki, H., Ohshiro, T., Rotinsulu, H., Wewengkang, D. S., Sumilat, D. A., Tomoda, H., Namikoshi, M., & Uchida, R. (2018). Callyspongiamides A and B, sterol O-acyltransferase inhibitors, from the Indonesian marine sponge Callyspongia sp. Bioorganic & Medicinal Chemistry Letters, 28(10), 1911–1914. https://doi.org/10.1016/j.bmcl.2018.03.077
- Karlsson, M., Pukenas, B., Chawla, S., Ehinger, J. K., Plyler, R., Stolow, M., Gabello, M., Hugerth, M., Elmér, E., Hansson, M. J., Margulies, S., & Kilbaugh, T. (2019). Neuroprotective effects of cyclosporine in a porcine pre-clinical trial of focal traumatic brain injury. Journal of Neurotrauma, 36(1), 14–24. https://doi.org/10.1089/neu.2018.5706
- Kunjathoor, V. V., Wilson, D. L., & LeBoeuf, R. C. (1996). Increased atherosclerosis in streptozotocin-induced diabetic mice. Journal of Clinical Investigation, 97(7), 1767–1773. https://doi.org/10.1172/JCI118604
- Madariaga-Mazon, A., Gonzalez-Andradeb, M., Toriello, C., Navarro-Barranco, H., & Mata, R. (2015). Potent anti-calmodulin activity of cyclotetradepsipeptides isolated from Isaria fumosorosea using a newly designed biosensor. Natural Product Communications, 10(1), 113–116. https://doi.org/10.1177/1934578X1501000128
- Marzetta, C. A., Savoy, Y. E., Freeman, A. M., Long, C. A., Pettini, J. L., Hagar, R. E., Inskeep, P. B., Davis, K., Stucchi, A. F., Nicolosi, R. J., & Hamanaka, E. S. (1994). Pharmacological properties of a novel ACAT inhibitor (CP-113,818) in cholesterol-fed rats, hamsters, rabbits, and monkeys. Journal of Lipid Research, 35(10), 1829–1838. https://www.jlr.org/content/35/10/1829.long
- Matsuda, D., Namatame, I., Tomoda, H., Kobayashi, S., Zocher, R., Kleinkauf, H., & Omura, S. (2004). Bew beauveriolides produced by amino acid-supplemented fermentation of Beauveria sp. FO-6979. The Journal of Antibiotics, 57(1), 1–9. https://doi.org/10.7164/antibiotics.57.1
- Nagai, K., Doi, T., Ohshiro, T., Sunazuka, T., Tomoda, H., Takahashi, T., & Omura, S. (2008). Synthesis and biological evaluation of a focused library of beauveriolides. Bioorganic & Medicinal Chemistry Letters, 18(15), 4397–4400. https://doi.org/10.1016/j.bmcl.2008.06.054
- Nagai, K., Doi, T., Sekiguchi, T., Namatame, I., Sunazuka, T., Tomoda, H., Omura, S., & Takahashi, T. (2006). Synthesis and biological evaluation of a beauveriolide analogue library. Journal of Combinatorial Chemistry, 8(1), 103–109. https://doi.org/10.1021/cc050084d
- Nakaya, S., Mizuno, S., Ishigami, H., Yamakawa, Y., Kawagishi, H., & Ushimaru, T. (2012). New rapid screening method for anti-aging compounds using budding yeast and identification of beauveriolide I as a potent active compound. Bioscience, Biotechnology, and Biochemistry, 76(6), 1226–1228. https://doi.org/10.1271/bbb.110872
- Namatame, I., Tomoda, H., Ishibashi, S., & Omura, S. (2004). Antiatherogenic activity of fungal beauveriolides, inhibitors of lipid droplet accumulation in macrophages. Proceedings of the National Academy of Sciences of the USA, 101(3), 737–742. https://doi.org/10.1073/pnas.0307757100
- Namatame, I., Tomoda, H., Si, S., Yamaguchi, Y., Masuma, R., & Omura, S. (1999b). Beauveriolides, specific inhibitors of lipid droplet formation in mouse macrophages, produced by Beauveria sp. FO-6979. The Journal of Antibiotics, 52(1), 1–6. https://doi.org/10.7164/antibiotics.52.1
- Namatame, I., Tomoda, H., Tabata, N., Si, S., & Omura, S. (1999a). Structure elucidation of fungal beauveriolide III, a novel inhibitor of lipid droplet formation in mouse macrophages. The Journal of Antibiotics, 52(1), 7–12. https://doi.org/10.7164/antibiotics.52.7
- Nielsen, D. S., Shepherd, N. E., Xu, W., Lucke, A. J., Stoermer, M. J., & Fairlie, D. P. (2017). Orally absorbed cyclic peptides. Chemical Reviews, 117(12), 8094–8128. https://doi.org/10.1021/acs.chemrev.6b00838
- Ohshiro, T., Imuta, S., Hijikuro, I., Yagyu, H., Takahashi, T., Doi, T., Ishibashi, S., & Tomoda, H. (2020). The Anti-atherOgenic activity of beauveriolide derivative BVD327, a SterOl O-acyltransferase 2-Selective InhibitOr, in ApOlipOprOtein E KnOckOut mice. Biological & Pharmaceutical Bulletin, 43(6), 951–958. https://doi.org/10.1248/bpb.b19-00913
- Ohshiro, T., Kobayashi, K., Ohba, M., Matsuda, D., Rudel, L. L., Takahashi, T., Doi, T., & Tomoda, H. (2017). Selective inhibition of sterol O-acyltransferase 1 isozyme by beauveriolide III in intact cells. Scientific Reports, 7(1), 4163. https://doi.org/10.1038/s41598-017-04177-8
- Ohshiro, T., Matsuda, D., Nagai, K., Doi, T., Sunazuka, T., Takahashi, T., Rudel, L. L., Omura, S., & Tomoda, H. (2009). The selectivity of beauveriolide derivatives in inhibit ion toward the two isozymes of acyl-CoA: Cholesterol acyltransferase. Chemical & Pharmaceutical Bulletin, 57(4), 377–381. https://doi.org/10.1248/cpb.57.377
- Ohshiro, T., Namatame, I., Nagai, K., Sekiguhi, T., Doi, T., Takahashi, T., Akasaka, K., Rudel, L. L., Tomoda, H., & Omura, S. (2006). Absolute stereochemistry of fungal beauveriolide III and ACAT inhibitory activity of four stereoisomers. The Journal of Organic Chemistry, 71(20), 7643–7649. https://doi.org/10.1021/jo0611667
- Ohshiro, T., Rudel, L. L., Omura, S., & Tomoda, H. (2007). Selectivity of microbial Acyl-CoA: Cholesterol acyltransferase inhibitors toward isozymes. The Journal of Antibiotics, 60(1), 43–51. https://doi.org/10.1038/ja.2007.6
- Park, Y. J., Lee, S. R., Kim, D. M., Yu, J. S., Beemelmanns, C., Chung, K. H., & Kim, K. H. (2018). The inhibitory effects of cyclodepsipeptides from the entomopathogenic fungus Beauveria bassiana on myofibroblast differentiation in A549 alveolar epithelial cells. Molecules, 23(10), E2568. https://doi.org/10.3390/molecules23102568
- Puglielli, L., Konopka, G., Pack-Chung, E., Ingano, L. A., Berezovska, O., Hyman, B. T., Chang, T. Y., Tanzi, R. E., & Kovacs, D. M. (2001). Acyl-coenzyme A: Cholesterol acyltransferase modulates the generation of the amyloid β-peptide. Nature Cell Biology, 3(10), 905–912. https://doi.org/10.1038/ncb1001-905
- Reintjes, T. (Ed). (2011). Solubility enhancement with BASF Pharma polymers. Solubilizer compendium. BASF SE.
- Šimčíková, D., Tůma, P., Jegorov, A., Jr., Šimek, P., & Heneberg, P. (2020). Rapid methods for the separation of natural mixtures of beauverolides, cholesterol acyltransferase inhibitors, isolated from the fungus Isaria fumosorosea. Journal of Separation Science, 43(5), 962–969. https://doi.org/10.1002/jssc.201901084
- Smith, S. G. (1996). Neoral®(cyclosporin) in dermatology: Technical aspects. British Journal of Dermatology, 135(48), 2–4. https://doi.org/10.1111/j.1365-2133.1996.tb00701.x
- Tomoda, H., & Doi, T. (2008). Discovery and combinatorial synthesis of fungal metabolites beauveriolides, novel antiatherosclerotic agents. Accounts of Chemical Research, 41(1), 32–39. https://doi.org/10.1021/ar700117b
- Tuan, N. N., Ngan, N. T., Kuo, P. C., Hung, H. Y., Lan, T. N., Thang, N. T., Xuan, D. T. T., Thang, T. D., & Wu, T. S. (2017). Characterization of cyclodepsipeptides from the mycelium of Isaria japonica from Vietnam. Natural Product Communications, 12(3), 377–378. https://doi.org/10.1177/1934578X1701200317
- Ueda, H., Ikeda, H., Sasaki, Y., & Shioji, R. (1978). Effect of oral administration of calcium carbonate, aluminium hydroxide gel and dihydrotachysterol on renal acidosis. Tohoku Journal of Experimental Medicine, 124(1), 1–11. https://doi.org/10.1620/tjem.124.1
- Vilcinskas, A., Jegorov, A., Landa, Z., Götz, P., & Maťha, V. (1999). Effects of beauverolide L and cyclosporine A on humoral and cellular immune response of the greater wax moth, Galleria mellonella. Comparative Biochemistry and Physiology. Part C, Pharmacology, Toxicology & Endocrinology, 122(1), 83–92. https://doi.org/10.1016/S0742-8413(98)10082-8
- Wang, C. K., & Craik, D. J. (2016). Cyclic peptide oral bioavailability: Lessons from the past. Biopolymers, 106(6), 901–909. https://doi.org/10.1002/bip.22878
- Wang, C. K., Northfield, S. E., Swedberg, J. E., Colless, B., Chaousis, S., Price, D. A., Liras, S., & Craik, D. J. (2015). Exploring experimental and computational markers of cyclic peptides: Charting islands of permeability. European Journal of Medicinal Chemistry, 97(1), 202–213. https://doi.org/10.1016/j.ejmech.2015.04.049
- Wang, X., Gao, Y.-L., Zhang, M.-L., Zhang, H.-D., Huang, J.-Z., & Li, L. (2020). Genome mining and biosynthesis of the Acyl-CoA: Cholesterolacyltransferase inhibitor beauveriolide I and III in Cordyceps militaris. Journal of Biotechnology, 309(1), 85–91. https://doi.org/10.1016/j.jbiotec.2020.01.002
- Witter, D. P., Chen, Y., Rogel, J. K., Boldt, G. E., & Wentworth, P., Jr. (2009). The natural products beauveriolide I and III: A new class of β-amyloid-lowering compounds. ChemBioChem, 10(8), 1344–1347. https://doi.org/10.1002/cbic.200900139