
ABSTRACT
High-throughput sequencing was used to elucidate the microbial community of strong-flavor Daqu (SFD) (surface and central parts) produced in different Chinese regions, moisture and other six kinds of quality indicators of SFD were detected. In addition, Spearman correlation coefficient was used to identify the relationships between microbial community and quality indicators. In different sample groups of surface and central parts, the differential fungal genus was Alternaria, whereas differential bacteria genera were Weissella, Pediococcus and Leuconostoc. Spearman analysis showed that fungal genera Rhizopus, Stemphylium, and Alternaria were significantly positively correlated with saccharifying ability (p < .01), whereas Rasamsonia was significantly positively correlated with esterification ability (p < .01). Weissella, Pediococcus and Burkholderia were the bacterial genera significantly positively correlated with saccharification ability of SFD samples (p < .01). This study provides a reference for investigating the relationship between microbial community and quality indicators of SFD.
Introduction
Baijiu is a traditional Chinese distilled liquor with approximately 1,000 years of manufacturing history. Historical evidence indicates that the earliest distillation tools for Baijiu production (distillation pan) emerged during the Song dynasty (Zheng & Han, Citation2016). Strong-flavor Baijiu (SFB) is one of the 12 major aromatic classes of Baijiu which employs strong-flavor Daqu (SFD) as the fermentation starter. SFD contains raw ingredients, microorganisms, enzymes, aroma compounds and flavor precursors which are also found in SFB (Zheng et al., Citation2011). SFB accounts for over 70% of Chinese Baijiu production (Zhao et al., Citation2012), and the main production areas of SFB are concentrated in certain Chinese provinces, among which Sichuan, Anhui, Shandong, Jiangsu, and Henan. Wuliangye (Yi-Bin) and Luzhou Laojiao (Lu-Zhou) produced in Sichuan province are widely known SFB. Similarly, Gujinggongjiu (Bo-Zhou) manufactured in Anhui province is another remarkable SFB, whereas the Ji-Ning city in Shandong province is famous for its SFD production.
The microbial community of SFD has an important influence on its quality indicators and consequently on SFB (Yang et al., Citation2017; Zou et al., Citation2018). High-throughput sequencing technology has been widely used to study the microbial community structure of Daqu, which is conducive to obtaining more representative results (Wang, Ban, Hu, et al., Citation2017; Xie et al., Citation2020). Chen et al. (Citation2021) found significant differences in microbial community structure and distribution between the surface and central parts of special-flavor Daqu produced in Zhangshu City (N28°07, E115°53’). Currently, there has been a growing interest in investigating the relationship between microbial community structure and quality indicators of Daqu with the aim of establishing a classification for different Daqu types (Ma et al., Citation2022; Mao et al., Citation2022). In this context, microbial species correlated with Daqu quality indicators could be isolated and further applied to produce fortified Daqu (Fan et al., Citation2019; Li et al., Citation2017). Among these microbial species, Bacillus subtilis, Bacillus licheniformis, and Bacillus amyloliquefaciens were isolated from sesame-flavor Baijiu and used to leverage its fermentation (Li et al., Citation2017). However, only few studies have investigated the association between microbial community structure of SFD and the quality indicators of its surface and central parts.
This study aimed to investigate the structure of the microbial community of SFD (surface and central parts) obtained from different producing regions in China as well as investigate its association with quality indicators of SFB. The findings discussed herein provided reliable information to ensure and improve the quality of Daqu by bioaugmentation. In addition, the results of the present study will serve as the basis for future studies on the isolation of functional microorganisms from Daqu in terms of improving the design and/or the selection of culture methods. Finally, the information provided herein contribute to enlarge the current understanding of microbial community diversity of Daqu produced in different Chinese regions.
Materials and methods
Sample collection
SFD samples were collected in major Chinese producing regions of SFB, which included the following cities: Yi-Bin (N28°77’, E104°62’), Lu-Zhou (N28°52’, E105°26’), Bo-Zhou (N33°54’, E115°47’), Ji-Ning (N34°25’, E115°54’), Fu-Yang (N32°89’, E115°81’) and Lin-Quan (N32°87’, E115°19’). The macroscopical aspect of SFD samples included in the study is shown in . SFD from Yi-Bin (YB), Bo-Zhou (BZ), Fu-Yang (FY), and Lin-Quan (LQ) were rectangular-shaped (termed Pingban-qu), whereas SFD from Lu-Zhou (LZ) and Ji-Ning (JN) were rectangular-shaped with a central protuberance (termed Baobao-qu). All SFD samples were produced with freshly cultivated ingredients, including barley, wheat, and peas. In addition, the Daqu manufacturing process applied to Pingban-qu and Baobao-qu SFD samples was virtually identical, which included the following steps: material infiltration with water; crushing; water infiltration; incorporation of an aliquot of Daqu from a previous batch; mechanical or manual molding; temperature raise by brick accumulation in the culture room for 1 week, which was maintained within the range of 55–60°C by gyrating the bricks and covering with straw or sprinkler for 1 week, followed by drying for 2 weeks and storage for 2 or 3 months until further use. Then, SFD samples were transported to laboratory within 24 h after collection for genomic DNA extraction (refrigerated transport on dry ice) and detection of enzymatic activity (non-refrigerated transport in common packaging).
Figure 1. Appearance of Daqu samples from different geographical origins. a) Strong-flavor Daqu (SFD) from Yi-Bin city (YB), b) SFD from Lu-Zhou city (LZ), c) SFD from Ji-Ning city (JN), d) SFD from Bo-Zhou city (BZ), e) SFD from Fu-Yang city (FY), f) SFD from Lin-Quan city (LQ).

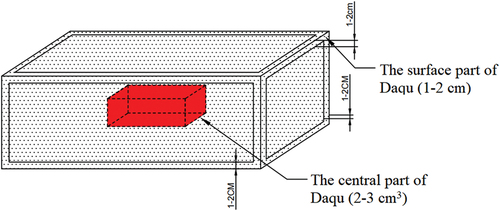
SFD samples were sampled following the proposed method: i) a 1–2 cm thick portion of the surface layer of SFD samples was obtained (hereafter referred to as group S); and ii) a 2–3 cm3 portion of the core of SFD samples was obtained (hereafter referred to as group I). A schematic representation of the relative positions of SFD sample collection is shown in . Collectively, 12 samples were included in the below-described experiments: LZS (surface layer of SFD collected in Lu-Zhou city), YBS (surface layer of SFD collected in Yi-Bin city), JNS (surface layer of SFD collected in Ji-Ning city), BZS (surface layer of SFD collected in Bo-Zhou city), FYS (surface layer of SFD collected in Fu-Yang city), LQS (surface layer of SFD collected in Lin-Quan city), LZI (central part of SFD collected in Lu-Zhou city), YBI (central part of SFD collected in Yi-Bin city), JNI (central part of SFD collected in Ji-Ning city), BZI (central part of SFD collected in Bo-Zhou city), FYI (central part of SFD collected in Fu-Yang city), LQI (central part of SFD collected in Lin-Quan city). Each sample was obtained in duplicate. All samples were divided into two parts: one part was stored at −80°C for amplicon sequencing analysis; the remainder part was stored at 4°C for the determination of SFD quality indicators.
Figure 2. Schematic representation of sampling positions in Daqu.
Amplicon sequencing analysis
One-third of each of the three parallel SFD samples was obtained and combined to obtain a pooled sample, which was employed in amplicon sequencing analysis (Chen et al., Citation2021). Genomic DNA was extracted from SFD samples using the E.Z.N.A. Soil DNA Kit (Omega Bio-tek, Norcross, GA, USA) in accordance with manufacturer’s instructions and the method described previously (Zhang et al., Citation2014). Integrity and purity of obtained DNA were verified by agarose gel electrophoresis. PCR amplifications were conducted using specific primers; for bacteria, the V3–V4 hypervariable region of the 16S rRNA gene was amplified using the forward primer 515F (GTGYCAGCMGCCGCGGTAA) and the reverse primer 806 R (GGACTACNVGGGTWTCTAAT); for fungi, the Internal Transcribed Spacer (ITS) 1(a) region was amplified using the forward primer ITS5-1737F (GGAAGTAAAAGTCGTAACAAGG) and the reverse primer ITS2-2043 R (GCTGCGTTCTTCATCGATGC).
PCR products were submitted to horizontal gel electrophoresis, and PCR amplicons of an appropriate size were recovered from agarose gels and pooled in equal amounts for DNA sequencing library preparation. The samples were diluted to 20 ng/μL, and the original DNA sample was used directly if sample quality and concentration were insufficient. Simultaneously, certain adjustments were performed to dilutions based on sample amplification quality. Amplifications were performed as follows (25 μL final volume): 5 × reaction buffer, 5 μL; 5 × GC buffer, 5 μL; dNTP (2.5 mm), 2 μL; forward primer (10 uM), 1 μL; reverse primer (10 uM), 1 μL; DNA template, 2 μL; ddH2O, 8.75 μL; Q5 DNA polymerase, 0.25 μL. Amplification parameters were as follows: initial denaturation at 98°C for 2 min; followed by 25–30 cycles of denaturation at 98°C for 15 s (Wang, Ban, Hu, et al., Citation2017), annealing at 55°C for 30 s, and extension at 72°C for 30 s; and a final extension at 72°C for 5 min. PCR-free library construction was conducted in a Miseq PE250 platform (Illumina, San Diego, CA, USA) by Suzhou Bio Novo Gene Co., Ltd., China.
Analysis of amplicon sequencing data
Amplicon sequencing data was processed using QIIME v.1.9.1 (http://qiime.org/scripts/split_libraries_fastq. html). Specific adapter sequences were trimmed from raw sequencing data using the cut adapt plugin (Bokulich et al., Citation2013; Caporaso et al., Citation2010). Subsequently, reads were filtered, denoised, and merged, and chimeras were removed using DADA2. Firstly, trimmed-paired primers were called, and unmatched primers were discarded. DADA2 was used for quality control, denoising, concatenation, and chimerization. The above steps were conducted separately for each library. After library denoising, the ASVs feature sequences and ASV tables were combined, and singletons ASVs were removed.
Operational taxonomic units (OTUs) were defined based on sequences with 97% similarity. All sequences were aligned against the Silva library (version SILVA_138.1) to obtain the taxonomic information. Alpha-diversity (including observed species, Chao1, Shannon, and Good’s coverage indices) and weighted UniFrac distance were calculated based on read counts normalized to the smallest read number per sample for fungi and bacteria (de Lipthay et al., Citation2004). Taxonomic assignment of sequences was conducted using Greengenes and UNITE databases for bacterial and fungal identification, respectively (McDonald et al., Citation2012; Nilsson et al., Citation2019). Bioxin cloud platform (http://www.biodeep.cn/), R software v3.4.1 (Mathsoft, Inc. USA) and Adobe Illustrator CS6 (Adobe Systems Incorporated, USA) were used to draw dilution curves and accumulation histogram, etc.
Determination of quality indicators of SFD and correlation analysis
Quality indicators of SFD samples were determined based on the Chinese standard methods (QB/T, Citation2011). The esterification ability of SFD samples at 72 h of production was determined, and results were expressed as mg/g·72 h. The determination of quality indicators of SFD samples was performed, which included physicochemical parameters and the enzymatic activity of each sample measured in triplicate, and the results were expressed as the mean of obtained measurements. Spearman’s rank correlation coefficients between dominant microbial genera (relative abundance >1%) and quality indicators of SFD samples were calculated using SPSS v25 (SPSS Inc., Chicago, IL, USA).
Results and discussion
Validity of sample preparation and sequencing data
Raw sequences were obtained with the Illumina platform, and after splicing and quality control on each sample, an average of 83,437 fungal sequences were obtained with a total of 1,375 OTUs after clustering, whereas an average of 82,932 bacterial sequences were obtained with a total of 699 OTUs. Chao 1 index calculation at the OTU level of 97% similarity was conducted to estimate the total number of species (Fu et al., Citation2020). From the dilution curve and Shannon curve (Figure S1), as well as based on alpha-diversity indices (), it can be stated that the metagenomic sequencing data obtained herein is representative of the SFD samples.
Table 1. Alpha diversity indices of fungal metagenomes of Strong-flavor Daqu samples from different geographical locations in China.
Table 2. Alpha diversity indices of bacterial metagenomes of Strong-flavor Daqu samples from different geographical locations in China.
Alpha diversity
The analysis of microbial community of SFD samples was used to investigate the abundance and diversity of microorganisms in each sample (He et al., Citation2019). Alpha-diversity indices reflect the diversity of a microbial community. In contrast, the Chao1 and ACE indices reflect the richness of the microbial community, while Shannon and Simpson indices reflect the richness and evenness of microbial species in a community (Wang et al., Citation2020). In the present study, Shannon’s and Simpson’s indices of fungal sequences in sample JNS were higher than those in other samples (), indicating that richness, evenness and diversity of fungal species in JNS were the highest.
The Ace index is used to determine microbial community abundance, and the higher the Ace index, the higher the microbial abundance in a sample (Zuo et al., Citation2020). Ace indices showed that microbial abundance of fungal species in sample BZS was higher than that in other samples (). The Chao1 index is often used to estimate the total number of species (OTUs) in a sample (Thomas et al., Citation2015). Considering alpha diversity of fungal sequences, Chao1 index of sample BZS was the highest (), indicating that the sample BZS had a higher total number of fungal species compared to other samples. Considering alpha diversity of bacterial sequences, Chao1 index of sample FYS was the highest (), indicating that it contained a higher total number of bacterial species compared to other samples.
OTU clustering analysis
OTUs obtained from SFD samples were classified, and shared and unique OTUs in different samples were identified to determine OTU distribution (Wang et al., Citation2012). The total number of fungal OTUs in samples of group S was 651, of which shared OTUs were 43 (Figure S2(a)), accounting for 6.6% of the total number of OTUs; shared OTUs accounted for 45.57% of total OTUs in sample FYS, of which proportion was the highest among group S samples. The total number of fungal OTUs in group I samples was 719, of which shared OTUs were 47 (Figure S2(b)), accounting for 6.5% of the total number of OTUs; shared OTUs accounted for 54.02% of total OTUs in sample BZI, of which proportion was the highest among group I samples.
The total number of bacterial OTUs in group S samples was 622, of which shared OTUs were 75 (Figure S2(c)), accounting for 12.06% of total OTUs. The number of OTUs in sample FYS was 117, accounting for 16.74% of the total number of OTUs, which was the highest among group S samples. The shared 75 OTUs accounted for 78.79% of the total OTUs in sample BZS, which was the highest among group S samples. Additionally, the total number of bacterial OTUs in group I samples was 478, of which shared OTUs were 46 (Figure S2(d)), thus accounting for 9.62% of total OTUs. The number of OTUs in sample FYI was 112, which accounted for 23.43% of total OTUs, the highest among group I samples. The shared 46 OTUs accounted for 79.31% of the total OTUs in sample LZI, which was the highest among group I samples.
Taxonomic classification of the microbial community of SFD samples at the phylum level
Obtained representative sequences of various OTUs were compared against the fungal UNITE database to assign taxonomic information to each OTU at different taxonomic levels. Among fungal OTUs, three phyla were identified, namely Ascomycota, Mucoromycota and Basidiomycota, whose relative abundances were 52.52%, 41.6% and 0.6% in group I, and 87.41%, 9.2% and 0.9% in group S, respectively (). The dominant phylum in groups S and I was Ascomycota, whose relative abundance was greater than 50%. Zhang (Citation2017) also found that the most abundant fungal phyla in SFD were Ascomycota, Mucoromycota and Basidiomycota, which agreed with the results of the present study.
Figure 3. Histogram of relative abundance at phylum level of microorganisms found in Daqu. a) Fungal phyla across the surface (S) and central (I) parts of Daqu samples, b) Fungal phyla across different Daqu samples, c) Bacterial phyla across S and I parts of Daqu, d) Bacterial phyla across different Daqu samples.
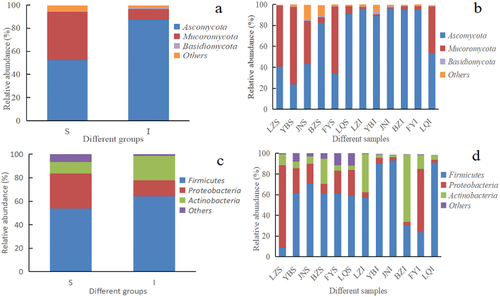
As shown in , the relative abundance of Mucoromycota in samples BZS and LQS was 5.46% and 7.21%, respectively, which was lower than that in other group S samples. Moreover, the relative abundance of Ascomycota in samples BZS and LQS accounted for 90.94% and 82.45%, respectively, which was higher than that in another group I samples. In contrast, the relative abundance of Ascomycota in sample LQI was 63.3%, which was lower than that in another group I samples. In addition, the relative abundance of Mucoromycota in sample LQI was 44.48%, which was higher than that in another group I samples. Collectively, differences in the relative abundance of fungal phyla were found in samples of groups S and I.
As shown in , bacterial phyla with higher relative abundances in SFD samples mainly included Firmicutes (53.71% in group S and 64.15% in group I), Proteobacteria (29.89% in group S and 13.66% in group I), and Actinobacteria (9.69% in group S and 20.83% in group I). Xie et al. (Citation2020) identified the three phyla Firmicutes, Actinobacteria and Proteobacteria in high-temperature Daqu used to produce sesame-flavor Baijiu, among which Firmicutes was the most dominant phylum during Daqu production, and the research results of (Xie et al., Citation2020) about the bacterial phyla with higher relative abundances in high-temperature Daqu were highly agreed with the present findings. In addition, the relative abundance of Firmicutes in samples JNS, YBS and LQS was 70.89%, 61.63% and 58.87%, respectively (). It is worth noting that OTUs assigned to Proteobacteria in sample LZS accounted for 79.39% of total OTUs, which was higher than in other group S samples. In contrast, the relative abundance of Firmicutes in samples JNI, YBI and LQI was 93.13%, 90.07% and 90.91%, respectively. Additionally, the relative abundance of Proteobacteria in sample FYI was 23.69%, which was higher than in other group I samples. Thus, the microbiota of Daqu was mainly composed of bacteria belonging to the phyla Firmicutes and Proteobacteria. Within the phylum Firmicutes, the main representatives were included within the class Bacilli, primarily in the order Bacillales (Fan et al., Citation2018), which agreed with the results obtained in the present study.
Taxonomic classification of the microbial community of SFD samples at the genus level
Daqu is one of the main sources of microorganisms for Baijiu fermentation. Molds and yeasts are mainly responsible for saccharification and alcoholic fermentation of Baijiu, and such microorganisms are also found in Daqu.
As shown in , fungal genera with higher relative abundances in group S samples were Rhizopus (19.91%), Rhizomucor (19.4%), Blumeria (17.57%) and Eurotium (12.8%). In contrast, fungal genera with higher relative abundances in group I samples were Thermomyces (26.04%), Eurotium (21.69%), and Thermoascomycetes (19.37%). The relative abundance of Thermomyces in group S samples was higher than that in group I samples, which could be mainly due to the highest temperature commonly used (50°C) during SFD manufacturing, considering also that the temperature of the SFD core is generally higher than 50°C. Therefore, a high number of heat-resistant and thermophilic fungal species survive in the core of SFD samples.
Figure 4. Histogram of relative abundance at genus level of microorganisms found in Daqu. a) Fungal genera across the surface (S) and central (I) parts of Daqu samples, b) Fungal genera across different Daqu samples, c) Bacterial genera across S and I parts of Daqu samples, d) Bacterial genera across different Daqu samples.
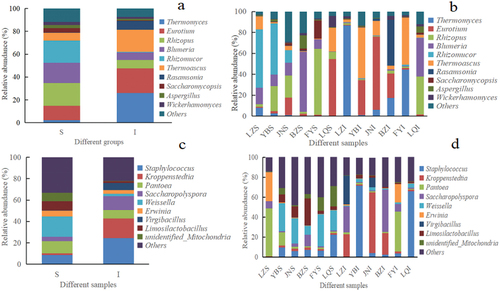
In addition, the relative abundance of Saccharomycopsis in samples of groups S and I was 0.36% and 0.05%, respectively. This difference could be mainly related to the difference in the temperature between the central and surface parts of SFD during manufacturing (Chen et al., Citation2021). Moreover, humidity, ventilation, and oxygen content in different parts of SFD samples differed. It has been previously reported that microorganisms of the genera Acetobacter and Pichia predominated on the surface of Daqu, whereas microorganisms belonging to the genera Aspergillus, Kroppenstedtia, Oceanobacillus and Bacillus were enriched in the central parts of Daqu (Chen et al., Citation2021).
As shown in , the relative abundance of Rhizopus (62.77%) was found to be highest in sample FYS of group S, followed by samples YBS (24.2%) and JNS (21.02%), which could be attributed to the temperature employed during SFD manufacturing in Fuyang City, which was lower than the temperature employed in the other five locations. Members of the genus Rhizopus secrete large amounts of various hydrolyzing enzymes which degrade macromolecules into low-molecular-weight metabolites and volatile compounds. The relative abundance of Thermomyces (86.66%) was found to be highest in sample LZI, followed by samples FYI (44.61%) and BZI (17.38%). Similarly, the relative abundance of Thermomyces (7.86%) in sample LZS was the highest in group S samples. Therefore, the differences in the relative abundance of Thermomyces may be related to the higher temperatures employed during SFD manufacturing in Luzhou City compared to SFD samples produced in the other five locations. Similarly, in addition to temperature, several other factors, such as raw material composition, Daqu moisture level, and Daqu acidity, can affect the structure of the autochthonous microbial community.
In the present study, the relative abundance of Saccharomycopsis in sample FYS was 17.1%, which was higher than that in other samples. However, it has been reported low levels of yeast on both the surface and central parts of Daqu, and that yeast was primarily present on the surface of Daqu (Chen et al., Citation2021; He et al., Citation2019). Thus, it can be inferred that an improved process may be employed in SFD manufacturing in Fuyang City, Anhui province, especially related to the application of yeasts to SFD.
As shown in , bacterial genera with higher relative abundances in samples of group S included Weissella (18.94%), Pantoea (11.30%), Staphylococcus (8.56%) and Lactobacillus (8.54%). Bacterial genera with higher relative abundances in group I samples included Staphylococcus (24.49%), Kroppenstedtia (18.38%), Saccharopolyspora (13.18%) and Pantoea (7.72%). The proportion of Staphylococcus in samples was correlated with the biosynthesis of the main organic acids produced during Baijiu fermentation. It has been described that Staphylococcus has an important role in the synthesis of four main esters, namely ethyl acetate, ethyl lactate, ethyl butyrate and ethyl caproate, which greatly affect the quality of Baijiu (Fan et al., Citation2020).
As shown in , the relative abundance of Weissella in samples JNS, YBS, FYS and LQS was 26.26%, 27.71%, 31.64% and 20.05%, respectively. The relative abundance of Staphylococcus in samples YBI and LQI was 71.44% and 65.84%, respectively. Moreover, the relative abundance of Kroppenstedtia (61.17%) in sample JNI was higher than in other samples. It has been reported that Kroppenstedtia dominates in the microbiota of high-temperature-treated sesame-flavor Daqu and medium-temperature-treated Jiannanchun Daqu (Fan et al., Citation2020). Moreover, it has been shown that the local environment, traditional manufacturing processes, and cultural environment contribute greatly to the differences in the microbial community of Daqu (Ma et al., Citation2022; Zuo et al., Citation2020).
Differences in Daqu microbiota at the genus level
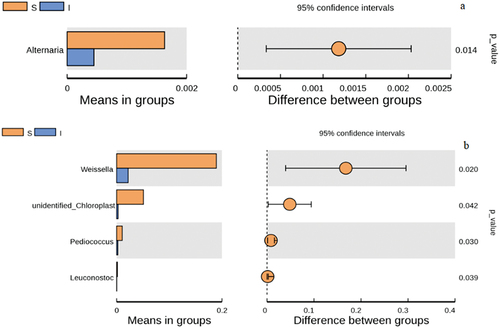
Differences between alpha-diversity indices of samples can enable the determination of significant discrepancies in the microbial community structure of samples. In this context, the analysis of these differences can indicate differential species which contributed to the observed differences. In addition, biomarkers can be associated with the abundance of differential species using conventional statistical tests. In the present work, Alternaria was the fungal species whose abundance significantly differed among sample groups (). In contrast, bacterial species whose abundances significantly differed among sample groups were Weissella, Pediococcus and Leuconostoc ().
Figure 5. Inter group T-test at genus level of microorganisms found in Daqu samples. a) Fungi, b) Bacteria. Each bar in represents the mean of species with significant differences in abundance between sample groups (surface part of Daqu, S, central part of Daqu, I). The leftmost endpoint of each circle represents the mean difference of the lower limit of the 95% confidence interval, the rightmost end of the circle represents the upper limit of the 95% confidence interval of the mean difference, the center of the circle represents the difference between means, the group represented by the color of the circle is the group with the highest mean, and the far right end of the displayed result is the corresponding difference between p values for species.
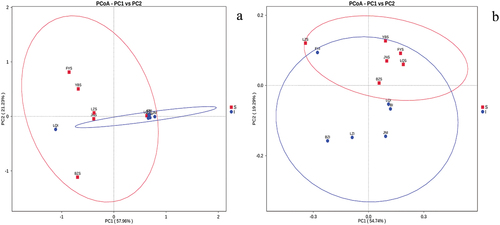
Principal coordinate analysis (PCoA)
In PCoA analysis, the distance between sample points belonging to the same sample group indicates great repeatability, while the distance between sample points belonging to different sample groups reflects the differences among samples. PCoA of fungal and bacterial communities of SFD samples was carried out based on weighted UniFrac distances. Overall, the two axes explained 74.03% of the total variance in bacterial community, and 79.19% of the total variance in fungal community of SFD samples (). The microbial community structure of group I samples was clearly distinct from that of group S samples. Moreover, the fungal community structure of samples FYI, BZI and JNI was like that of samples LZI and YBI, which differed from that of sample LQI (). In addition, the bacterial community structure of samples FYS, BZS, LQS and JNS was like that of sample YBS, which differed from that of sample LZI (). Collectively, the observed differences in the microbial community structure among sample groups may be attributed to distinct producing regions.
Figure 6. Principal coordinate analysis (PCoA) of microbial communities based on weighted UniFrac distances. a) Fungi, b) Bacteria. Data points are colored by sample type.
Analysis of Daqu quality indicators
Physical, chemical, and biochemical indicators are important parameters for evaluating the quality of Daqu. (Chen et al., Citation2014). Moisture, acidity, starch, and reducing sugar are parameters of microbial fermentation, which are important physicochemical indices used to classify Daqu (Liu et al., Citation2019). Simultaneously, Daqu fermentation ability is related to the capacity of sugar to ferment into alcohol, and esterase activity is a primary indicator of volatile ester content. In contrast, Daqu saccharification power reflects the ability of amylase to hydrolyze starch into glucose, which is related to the ability to degrade starch found in Daqu raw materials into reducing sugars by Daqu saccharifying enzymes (Chen et al., Citation2014; Liu et al., Citation2019). In addition, Daqu esterification power is the ability of esterification enzymes found in Daqu to catalyze alcohols and acids to form esters (He et al., Citation2022). Therefore, fermentation ability, saccharifying power and esterase activity are important biochemical indices used to classify Daqu.
As shown in , moisture, acidity, reducing sugar content, and esterification ability of group I samples were generally higher than those of group S samples. However, starch content, fermentation and saccharification abilities of group S samples were generally higher than those of group I samples. In addition, the moisture of sample BZI was up to 16.20 ± 0.16 g/100 g, the acidity of samples JNI and LQI were 1.00 ± 0.01 mmol/10 g and 1.00 ± 0.03 mmol/10 g, which were the highest in samples. The reducing sugar of sample YBS was 1.1 ± 0.19 g/100 g, which was the lowest in samples. However, the starch of sample YBS was 60.86 ± 3.33 g/100 g, which was the lowest in samples. The difference of those quality indicators was related to making-process, kinds of materials, and microbial activity of SFD during cultivation (Liu et al., Citation2019; Zeng et al., Citation2021).
Table 3. Quality indicator parameters of Strong-flavor Daqu samples from different geographical locations in China.
Previous studies have shown that filamentous fungi can improve Daqu saccharification ability (Chen et al., Citation2014; Fan et al., Citation2020). In the present study, the saccharification ability of sample JNS was up to 972 ± 10.33 mg/g·h, indicating that the abundance of filamentous fungi in this sample was likely higher than that in other samples. The relative abundances of Rhizopus and Rhizomucor in sample JNS were 21.02% and 11.78%, respectively (); it has been reported that Rhizopus sp. is able to saccharify and synthesize lipase, thus contributing positively to Baijiu flavor (Long et al., Citation2013). Moulds can produce a variety of enzymes, including cellulases, saccharifying and liquefying enzymes, proteases, lipases, esterifying enzymes, pectinases and laccases, which play important roles in Baijiu fermentation (Wang, Ban, Qiu, et al., Citation2017).
The esterifying ability of SFD is related to the formation of ester compounds during SFB fermentation, which include ethyl hexanoate, ethyl butyrate, ethyl acetate and ethyl lactate, which are important flavor compounds in Baijiu (Xu et al., Citation2022). Herein, the sample BZI had the highest fermentation and esterification abilities, 0.22 ± 0.02 g CO2/g·72 h and 13 ± 1.76 mg/g·72 h, respectively (). Fermentation and esterification abilities of sample FYS were 0.21 ± 0.01 g CO2/g ·72 h and 12 ± 2.47 mg/g·72 h, respectively, which were both at high levels and may be attributed to the relative abundance of Saccharomycopsis (17.1%).
Correlation analysis of genus-level microbial community structure and Daqu quality indicators
In Baijiu fermentation, saccharification is achieved by molds, whereas yeasts are necessary for fermentation, whereas raw aroma is produced by bacteria, which are the roles of Daqu microorganisms (Chen et al., Citation2021). Zeng et al. (Citation2021) found that water content and fermentation ability were positively correlated with the abundances of Aspergillus and Wickerhamomyces during Daqu cultivation, but water content was negatively correlated with the abundance of Rhizopus. As shown in , the abundance of Thermomyces was significantly positively correlated with water content (p < .05), whereas Mortierella was significantly positively correlated with reducing sugar content (p < .01); Rhizopus, Stemphylium and Alternaria were significantly positively correlated with saccharification ability, whereas Rasamsonia was highly significantly positively correlated with esterification ability (p < .01). Kluyveromyces was significantly negative correlated with reducing sugar (p < .01). In addition, Rhizopus was negative correlated with moisture and esterification ability (p < .05). Aspergillus was negative correlated with fermentation power (p < .05).
Figure 7. Spearman correlation analysis of genus-level microbial community structure and physicochemical parameters. a) Fungi, b) Bacteria. Environmental factor information is given vertically, species information is given horizontally. Values in the middle of the heat map are Spearman correlation coefficients (r), which are between −1 and 1, r < 0 indicates a negative correlation, r > 0 indicates a positive correlation, * indicates p values < .05, ** indicates p values < .01. M: moisture content, A: acidity, S1: starch, Rs: reducing sugar content, Fa: fermentation power, Sa: saccharification ability, Ea: esterification ability.
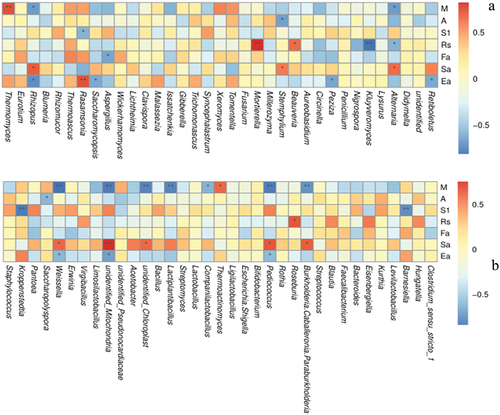
It has been reported that Rhizopus produces amylase, esterase, glucoamylase, lipase, and protease (Li et al., Citation2018). Moreover, the abundance of some fungal communities was negatively correlated with Daqu quality indicators, for instance, Saccharomycopsis was significantly negatively correlated with esterification ability (p < .01). However, it has been reported that Saccharomyces is the main producer of ethanol and important flavor compounds, such as esters and higher alcohols, during Baijiu fermentation (Liu et al., Citation2017). Environmental conditions and microbial composition of Daqu and Baijiu fermentation differed significantly. In addition, non-Saccharomyces species dominate in SFD, whereas Saccharomyces only represented a minority of microbes in Daqu due to their relatively poor tolerance to temperatures above 50°C (Pu et al., Citation2021). Thus, based on the above-described reasons may be related to the significant negative correlation between Saccharomycopsis and esterification ability of SFD, which differed from Baijiu fermentation.
As shown in , Thermoactinomyces in the bacterial community of Daqu was significantly positively correlated with water content (p < .01); Roseburia was significantly positively correlated with reducing sugar content (p < .01); and Weissella was significantly positively correlated with water content (p < .01). Moreover, Pediococcus and Burkholderia were significantly positively correlated with saccharification ability (p < .01). Pediococcus and Weissella are lactic acid bacteria whose major product of carbohydrate fermentation is lactic acid, while also provide substrates for esterification by yeasts (Wang et al., Citation2018). In addition, Weissella, Lactiplanti bacillus, and Pediococcus were significantly negative correlated with moisture (p < .01). Kroppenstedtia and Barnesiella were significantly negative correlated with starch (p < .01).
Conclusions
Herein, amplicon sequencing was employed to investigate the microbial community structure of SFD. Considering the surface and central parts of SFD, the differential fungal genus was Alternaria, whereas differential bacterial species were Weissella, Pediococcus and Leuconostoc. Spearman’s correlation coefficients of Daqu quality indicators and microbial community structure indicated that Thermomyces was very significantly positively correlated with moisture content; Mortierella was very significantly positively correlated with reducing sugar content; Rhizopus, Stemphylium and Alternaria were very significantly positively correlated with saccharification ability; Rasamsonia was significantly positively correlated with esterification ability. In addition, Thermoactinomyces was significantly positively correlated with moisture content, Roseburia was significantly positively correlated with reducing sugar content, Weissella, Pediococcus and Burkholderia were significantly positively correlated with saccharification ability.
Collectively, further research is necessary to identify. In addition, we are playing to study on spatial heterogeneity of active microbial community in SFD based on meta transcriptome. Future studies will encompass the isolation, purification, and identification of microorganisms in Daqu at species level which could be significantly related to quality indicators of SFD. In addition, further research will focus on identifying the correlations between microorganisms and enzyme activity in SFD, as well as exploring the spatial heterogeneity of active microbial communities in SFD based on transcriptome analysis. Ultimately, identified differential strains could be used in SFD production to obtain fortified Daqu.
Author contributions
Wei Cheng: Conceptualization, Writing – Original Draft. Xuefeng Chen and Huawei Zeng: Supervision. Xijia Xue: Data Curation. Xuefeng Chen: Writing – Review & Editing. All authors read and approved the final manuscript.
Supplemental Material
Download MS Word (485 KB)Acknowledgments
The authors thank Shaanxi University of Science and Technology, Jinzhongzi Distillery Co., Ltd., for their support in this work. We would also like to thank TopEdit (www.topeditsci.com) for its linguistic assistance during the preparation of this manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19476337.2022.2162974.
Additional information
Funding
References
- Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., Mills, D. A., & Caporaso, J. G. (2013). Quality-filtering vastly improves diversity estimates from illumina amplicon sequencing. Nature Methods, 10(1), 57–59. https://doi.org/10.1038/nmeth.2276
- Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., Fierer, N., Pea, A. G., Goodrich, J. K., Gordon, J. I., Huttley, G. A., Kelley, S. T., Knights, D., Koenig, J. E., Ley, R. E., Lozupone, C. A., McDonald, D., Muegge, B. D., Pirrung, M., & Knight, R. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7(5), 335–336. https://doi.org/10.1038/nmeth.f.303
- Chen, Y. R., Li, K. M., Liu, T., Li, R. Y., Fu, G. M., Wan, Y., & Zheng, F. P. (2021). Analysis of difference in microbial community and physicochemical indices between surface and central parts of Chinese special-flavor Baijiu Daqu. Frontiers in Microbiology, 11, 3340–3349. https://doi.org/10.3389/fmicb.2020.592421
- Chen, B., Wu, Q., & Xu, Y. (2014). Filamentous fungal diversity and community structure associated with the solid state fermentation of Chinese Maotai-flavor liquor. International Journal of Food Microbiology, 179, 80–84. https://doi.org/10.1016/j.ijfoodmicro.2014.03.011
- de Lipthay, J. R., Enzinger, C., Johnsen, K., Aamand, J., & Sørensen, S. J. (2004). Impact of DNA extraction method on bacterial community composition measured by denaturing gradient gel electrophoresis. Soil Biology & Biochemistry, 36(10), 1607–1614. https://doi.org/10.1016/j.soilbio.2004.03.011
- Fan, G. S., Fu, Z. L., Teng, C., Liu, P. X., Wu, Q. H., Rahman, M. K. R., & Li, X. T. (2020). Effects of aging on the quality of roasted sesame-like flavor Daqu. BMC microbiology, 20(1), 1–16. https://doi.org/10.1186/s12866-020-01745-3
- Fan, G. S., Fu, Z. L., Teng, C., Wu, Q. H., Liu, P. X., Yang, R., Minhazul, K. A. H., & Li, X. T. (2019). Comprehensive analysis of different grades of roasted-sesame-like flavored Daqu. International Journal of Food Properties, 22(1), 1205–1222. https://doi.org/10.1080/10942912.2019.1635154
- Fan, G. S., Sun, B. G., Fu, Z. L., Xia, Y. Q., Huang, M. Q., Xu, C. Y., & Li, X. T. (2018). Analysis of physicochemical indices, volatile flavor components, and microbial community of a light-flavor Daqu. Journal of the American Society of Brewing Chemists, 76(3), 209–218. https://doi.org/10.1080/03610470.2018.1424402
- Fu, W. H., Rao, H., Tian, Y., & Xue, W. T. (2020). Bacterial composition in sourdoughs from different regions in China and the microbial potential to reduce wheat allergens. LWT-Food Science and Technology, 117, 108669–108677. https://doi.org/10.1016/j.lwt.2019.108669
- He, G. Q., Huang, J., Zhou, R. Q., Wu, C. D., & Jin, Y. (2019). Effect of fortified Daqu on the microbial community and flavor in Chinese strong-flavor liquor brewing process. Frontiers in Microbiology, 10, 56–62. https://doi.org/10.3389/fmicb.2019.00056
- He, M. W., Jin, Y., Zhou, R. Q., Zhao, D., Zheng, J., & Wu, C. D. (2022). Dynamic succession of microbial community in Nongxiangxing daqu and microbial roles involved in flavor formation. Food Research International, 10, 111559–111567. https://doi.org/10.1016/j.foodres.2022.111559
- Li, P., Lin, W. F., Liu, X., Wang, X. W., Gan, X., Luo, L. X., & Lin, W. T. (2017). Effect of bioaugmented inoculation on microbiota dynamics during solid-state fermentation of Daqu starter using autochthonous of Bacillus, Pediococcus, Wickerhamomyces and Saccharomycopsis. Food Microbiology, 61, 83–92. https://doi.org/10.1016/j.fm.2016.09.004
- Liu, P. L., Xiong, X. M., Wang, S., & Miao, L. H. (2017). Population dynamics and metabolite analysis of yeasts involved in a Chinese miscellaneous-flavor liquor fermentation. Annals of Microbiology, 67(8), 553–565. https://doi.org/10.1007/s13213-017-1286-y
- Liu, P. H., Zhang, L. H., Du, X. W., Zhao, J. L., Gao, G., & Zhang, X. H. (2019). Dynamic analysis of physicochemical and biochemical indices and microbial communities of light-flavor Daqu during storage. Journal of the American Society of Brewing Chemists, 77(4), 287–294. https://doi.org/10.1080/03610470.2019.1629238
- Li, S., Yang, Q., Tang, B., & Chen, A. (2018). Improvement of enzymatic properties of Rhizopus oryzae α-amylase by site-saturation mutagenesis of histidine 286. Enzyme and Microbial Technology, 117, 96–102. https://doi.org/10.1016/j.enzmictec.2018.06.012
- Long, K., Zhao, Z. K., Ma, Y. Y., & Yang, J. G. (2013). Progresses of researches on Rhizopus for liquor-making. Modern Food Science and Technology, 29(2), 443–447. https://doi.org/10.13982/j.mfst.1673-9078.2013.02.043
- Ma, S. Y., Luo, H. B., Zhao, D., Qiao, Z. W., Zheng, J., An, M. Z., & Huang, D. (2022). Environmental factors and interactions among microorganisms drive microbial community succession during fermentation of Nongxiangxing Daqu. Bioresource Technology, 345, 126549–126558. https://doi.org/10.1016/j.biortech.2021.126549
- Mao, J. J., Liu, X. L., Gao, T., Gu, S. B., Wu, Y., Zhao, L. N., Ma, J. L., Li, X., & Zhang, J. (2022). Unraveling the correlations between bacterial diversity, physicochemical properties and bacterial community succession during the fermentation of traditional Chinese strong-flavor Daqu. LWT - Food Science and Technology, 154, 112764–112773. https://doi.org/10.1016/j.lwt.2021.112764
- McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., Andersen, G. L., Knight, R., & Hugenholtz, P. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. The ISME Journal, 6(3), 610–618. https://doi.org/10.1038/ismej.2011.139
- Nilsson, R. H., Larsson, K. H., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S., Schigel, D., Kennedy, P., Picard, K., Glöckner, F. O., Tedersoo, L., Saar, I., Kõljalg, U., & Abarenkov, K. (2019). The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Aesearch, 47(D1), D259–264. https://doi.org/10.1093/nar/gky1022
- Pu, S. C., Zhang, Y., Lu, N., Shi, C., & Yan, S. B. (2021). Yeasts from Chinese strong flavour Daqu samples: Isolation and evaluation of their potential for fortified Daqu production. AMB Express, 11(1), 1–12. https://doi.org/10.1186/s13568-021-01337-y
- QB, People’s Republic of China Professional Standard. (2011). General methods of analysis for Daqu (QB/T 4257-2011). In China (Ed.), Ministry of Industry and Information Technology of the People’s Republic of China QB/T 4257-2011 (p. 1–9). China Light Industry Press Ltd.
- Thomas, S. B., Schmidt, F., & Matias, R. (2015). Limits to robustness and reproducibility in the demarcation of operational taxonomic units. Environmental microbiology, 17(5), 1689–1706. https://doi.org/10.1111/1462-2920.12610
- Wang, X. D., Ban, S. D., Hu, B. D., Qiu, S. Y., & Zhou, H. X. (2017). Bacterial diversity of Moutai‐flavour Daqu based on high‐throughput sequencing method. Journal of the Institute of Brewing, 123(1), 138–143. https://doi.org/10.1002/jib.391
- Wang, X. D., Ban, S. D., & Qiu, S. Y. (2017). Analysis of the mould microbiome and exogenous enzyme production in Moutai‐flavor Daqu. Journal of the Institute of Brewing, 124(1), 91–99. https://doi.org/10.1002/jib.467
- Wang, Y., Sheng, H. F., He, Y., Wu, J. Y., Jiang, Y. X., Tam, N. F. Y., & Zhou, H. W. (2012). Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Applied and Environmental Microbiology, 78(23), 8264–8271. https://doi.org/10.1128/AEM.01821-12
- Wang, Z., Su, Z. X., Yang, Q., Liu, Y. C., Lin, B., Chen, S. X., & Yang, H. (2020). Characterizing relationship of microbial community in Xiaoqu and volatiles of light-aroma-type Xiaoqu Baijiu. Food Science and Technology Research, 26(6), 749–758. https://doi.org/10.3136/fstr.26.749
- Wang, J., Zhong, Q. P., Yang, Y. Y., Li, H. R., Wang, L., Tong, Y. G., Fang, X., & Liao, Z. L. (2018). Comparison of bacterial diversity between two traditional starters and the round-koji-maker starter for traditional cantonese chi-flavor liquor brewing. Frontiers in Microbiology, 9, 1053–1063. https://doi.org/10.3389/fmicb.2018.01053
- Xie, M. W., Lv, F. X., Ma, G. X., Farooq, A., Li, H. H., Du, Y., & Liu, Y. (2020). High throughput sequencing of the bacterial composition and dynamic succession in Daqu for Chinese sesame flavour liquor. Journal of the Institute of Brewing, 126(1), 98–104. https://doi.org/10.1002/jib.592
- Xu, Y. Q., Zhao, J. R., Liu, X., Zhang, C. S., Zhao, Z. G., Li, X. T., & Sun, B. G. (2022). Flavour mystery of Chinese traditional fermented Baijiu: The great contribution of ester compounds. Food Chemistry, 369, 1–9. https://doi.org/10.1016/j.foodchem.2021.130920
- Yang, J. G., Dou, X., Han, P. J., Bai, F. Y., Zhou, J., Zhang, S. Y., Qin, H., & Ma, Y. Y. (2017). Microbial diversity in Daqu during production of luzhou flavored liquor. Journal of the American Society of Brewing Chemists, 75(2), 136–144. https://doi.org/10.1094/ASBCJ-2017-2879-01
- Zeng, H. W., Jiang, X. J., Wang, Z. Q., Zeng, X., Xin, B. Y., Wang, Y. J., Zhang, X. X., Yang, H. L., Qiao, J., Dong, R. Y., Huang, M. Q., & Zhang, J. L. (2021). Environmental and physicochemical characterization and fungal community of two batches of Chinese Luzhou-Flavored Daqu. Journal of the American Society of Brewing Chemists, 1–8. https://doi.org/10.1080/03610470.2021.1968230
- Zhang, H. M. (2017). Study on the microbial community structure of Gujing tribute wine and its correlation with main flavor compounds. Harbin Institute of Technology.
- Zhang, L. Q., Wu, C. D., Ding, X. F., Zheng, J., & Zhou, R. Q. (2014). Characterisation of microbial communities in Chinese liquor fermentation starters Daqu using nested PCR-DGGE. World Journal of Microbiology & Biotechnology, 30(12), 3055–3063. https://doi.org/10.1007/s11274-014-1732-y
- Zhao, J. S., Zheng, J., Zhou, R. Q., & Shi, B. (2012). Microbial community structure of pit mud in a Chinese strong aromatic liquor fermentation pit. Journal of the Institute of Brewing, 118(4), 356–360. https://doi.org/10.1002/jib.52
- Zheng, X. W., & Han, B. Z. (2016). Baijiu (白酒), Chinese liquor: History, classification and manufactures. Journal of Ethnic Foods, 3(1), 19–25. https://doi.org/10.1016/j.jef.2016.03.001
- Zheng, X. W., Tabrizi, M. R., Nout, M. R., & Han, B. Z. (2011). Daqu-a traditional Chinese liquor fermentation starter. Journal of the Institute of Brewing, 117(1), 82–90. https://doi.org/10.1002/j.2050-0416.2011.tb00447.x
- Zou, W., Zhao, C. Q., & Luo, H. B. (2018). Diversity and function of microbial community in Chinese strong-flavor Baijiu ecosystem: A review. Frontiers in Microbiology, 9, 671–680. https://doi.org/10.3389/fmicb.2018.00671
- Zuo, Q. C., Huang, Y. G., & Guo, M. (2020). Evaluation of bacterial diversity during fermentation process: A comparison between handmade and machine-made high-temperature Daqu of Maotai-flavor liquor. Annals of Microbiology, 70(1), 1–10. https://doi.org/10.1186/s13213-020-01598-1