
ABSTRACT
This study addresses the crucial protein Internalin A in the development of a possible vaccination against Listeria monocytogenes. A meticulous in silico strategy was used which included physiochemical analysis, epitope prediction, and molecular interaction studies. Computational techniques shed light on potential development of a vaccine candidate against Listeria, with a focus on the Internalin A protein along with its immune simulations. Insights into molecular interactions and physiochemical features aid in understanding of prospective Listeria vaccine candidates. Ultimately, the computational analysis in this study sets the groundwork for the creation of a possible vaccination against Listeria. The discovered epitopes and insights into molecular interactions are useful for experimental validation. In vitro and in vivo investigations should be conducted in the future to confirm the effectiveness of these anticipated epitopes. These findings advanced our understanding of vaccine design by providing insights into battling Listeria infections and perhaps guiding tactics for other diseases.
Introduction
Listeria monocytogenes, sometimes known as Listeria, is a major foodborne pathogen. This Gram-positive bacterium causes listeriosis, a serious foodborne illness with a high fatality rate, particularly in pregnant women and others with impaired immune systems (Radoshevich & Cossart, Citation2018; Smith et al., Citation2019). The ability of Listeria to flourish at refrigeration temperatures poses a serious danger to the food business. To successfully limit the danger, strict cleanliness standards and frequent monitoring are required (Jemmi & Stephan, Citation2006). Numerous investigations have shown that Listeria can cause invasive gastroenteritis, encephalitis, meningitis, endocarditis, and sepsis in people with weakened immune systems, as well as in newborns. It has been linked to an increased risk of abortion or fetal loss in pregnant women (Elsayed et al. Citation2022; Mayer et al., Citation2022; Daniels et al., Citation2000). There are several types of listeriosis that may affect animals. The encephalitic type is recognized by neurological symptoms such as depression and incoordination, whereas the septicemic form is distinguished by symptoms such as depression, listlessness, emaciation, and diarrhea (Berrang et al., Citation2005; Elsayed et al., Citation2022).
According to the World Health Organization (WHO), the yearly incidence of listeriosis ranges from 0.1 to 10 cases per million persons. Despite the fact that it occurs seldom, it has a high death rate of 20%-30% (WHO, Citation2018). Foodborne pathogens have recently triggered a number of public health problems throughout the world, leading to the growth of foodborne illnesses with high death and morbidity rates. This tendency creates significant concerns, especially in underdeveloped nations (Rahman & Das, Citation2022). There have been several listeriosis outbreaks reported across the world, with one of the most lethal cases happening in the United States in 1985 as a result of tainted soft cheese. Another big epidemic connected to deli meat was reported in Canada in 2008. Similarly, in 2017–2018, South Africa saw a serious outbreak of deli meat (Jackson et al., Citation2018; Thomas et al., Citation2020). Despite the availability of modern medicines and safety precautions, multiple global outbreaks have shown that L. monocytogenes poses a substantial hazard to human health. This is due to its sophisticated pathogenic strategy, which includes an intracellular life cycle involving key proteins like internalin A (InlA), internalin B (InlB), listeriolysin O (LLO), phosphatidylinositol-specific phospholipase C (Pl-PLC), phosphatidylcholine-specific phospholipase C (PL-PLC), and surface actin assembly-inducing protein (Currie et al., Citation2015; Coelho et al., Citation2019).
This project aims to create and assess a new vaccine that targets Internalin A, a critical protein involved in the pathogenesis and cellular entry processes of Listeria monocytogenes, with the goal of enhancing vaccine development against infectious disorders. This intracellular bacterium causes listeriosis, which is quite dangerous for the general public’s health, especially for those with compromised immune systems (Shahabi et al., Citation2008). This work blends sequence retrieval, physiochemical analysis, antigenicity profiling, epitope prediction, and population coverage analysis to inform the development of a robust vaccine construct by utilizing computational tools and bioinformatics methodologies. The thorough evaluation of the suggested vaccine’s immunogenic potential and structural qualities aims to make a significant contribution to the field of vaccine design, possibly opening the door to improved methods of preventing Listeria infections and, consequently, advancing vaccine research in general (Akhtar et al., Citation2021; Larijani et al., Citation2018; Mayer et al., Citation2022)
Internalin A is a critical virulence factor that plays a role in cellular entrance processes and plays a major role in the pathogen’s capacity to infiltrate host cells (Schlech, Citation2019). Developing effective preventative strategies is imperative given the ongoing threat that Listeria infections represent to public health. Vaccines that specifically target virulence factors, such Internalin A, have demonstrated potential for enhancing host protection. By using computational tools to construct a vaccine that might lessen the effects of Listeria infections and advance the area of vaccine research, this work builds on the urgent need for novel approaches (Lecuit, Citation2020). The current research topic centers on the critical need for a vaccine against Listeria monocytogenes, a bacterium that can cause serious foodborne illnesses and high fatality rates, especially in susceptible groups. Internalin A, a crucial virulence component that aids in the bacterium’s cellular entrance, is the subject of particular attention. The study heavily relies on computational approaches in its quest to create a vaccine that conforms to the complex structural and immunological features of the target protein, filling a major vacuum in the present arsenal of defenses against Listeria infections.
This study examines the creation and assessment of a possible vaccination against Listeria monocytogenes, with a focus on Internalin A, a virulence component. The study uses a variety of computational techniques, including population coverage analysis, allergenicity profiling, epitope predictions, and sequence retrieval and physiochemical analysis. A thorough analysis is conducted of the vaccine’s development and characterization, including its physicochemical characteristics and predicted tertiary structure. Potential immunogenicity can be understood by immune simulations with the C-ImmSim server and molecular interaction investigations with the host receptor. The results are interpreted within the framework of vaccine research, highlighting the role of computational techniques in improving our knowledge of potential vaccines (Ding et al., Citation2023).
Finally, by focusing on Internalin A, this study establishes a basis for the potential creation of a vaccine against Listeria monocytogenes. The thorough computer assessments, which span everything from immunological simulations to sequence retrieval, offer insightful information about the vaccine’s possible immunogenicity and population coverage worldwide (Ji et al., Citation2022). Notwithstanding various constraints, such as presumptions in computer models, the research emphasizes how crucial it is to incorporate bioinformatics tools into vaccine development. Clinical studies and experimental validations should be a part of future research projects in order to confirm the suggested vaccine’s safety and effectiveness (Ding et al., Citation2023).
Materials and methods
Sequence retrieval
Internalin A, one of the key players in pathogenesis and entry processes, was selected as the target protein for vaccine development (Ireton et al., Citation2021). The UniProt ID for the selected protein is P0DJM0, and the protein sequence length is 800 amino acids (AA). Using the UniProt ID, the entry corresponding to Internalin A was located in the UniProt database (https://www.uniprot.org). The protein sequence information was obtained by reading the UniProt entry (P0DJM0). The 800 amino acid protein sequence was retrieved in FASTA format for additional in silico studies. The obtained sequence was cross-referenced with relevant literature and databases to check its alignment with the intended Interleukin A protein to verify correctness and dependability.
Physiochemical parameters of candidate protein
The ExPASy ProtParam tool (https://web.expasy.org/protparam) was used in the study to investigate the physiochemical characteristics of the Internalin A protein sequence. Several metrics were calculated, including molecular weight, theoretical isoelectric point (pI), amino acid content, extinction coefficient, and instability index. The theoretical isoelectric point denotes the pH at which a protein has no net electrical charge, whereas molecular weight offers information about the protein’s total mass, which aids in determining its size. Future investigations benefit from the computation of extinction coefficients and the breakdown of amino acid composition (Gulzar & Hussain, Citation2020). Additionally, evaluated was the instability index, which shows possible instability. This thorough physiochemical characterization improves knowledge of Internalin A’s structural and functional features and provides the groundwork for further in-depth in silico research.
Allergenicity and antigenicity profiling of proteins
To assess the potential of the Internalin A protein as a vaccine design candidate, antigenicity and allergenicity investigations were carried out using two different bioinformatics tools.
The antigenicity of the protein was predicted using VaxiJen 2.0, available at (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). This tool uses auto-cross covariance (ACC) alteration of protein sequences to classify proteins as antigenic or non-antigenic. The study sought to determine the possibility that the Internalin A protein would induce an immunological response, a critical factor in vaccine development (Doytchinova & Flower, Citation2007).
Simultaneously, the allergenic potential of the protein was determined using AllerTOP v2, which is available at (https://www.ddg-pharmfac.net/AllerTOP/method.html). AllerTOP v2 predicts allergenicity based on multiple physicochemical parameters using an alignment-free technique. The goal was to guarantee that the chosen protein would not offer allergic concerns, as allergenicity might jeopardize a vaccine’s safety and efficacy (Dimitrov et al., Citation2014).
Prediction of the linear B–cell epitopes
To anticipate B-cell epitopes, the study used the Bepipred linear epitope prediction tool, which is accessible on the Immune Epitope Database (IEDB) at (http://tools.iedb.org/main/bcell) (Ras-Carmona et al., Citation2022). To find probable epitopes inside a protein, the Bepipred linear epitope prediction tool uses the Hidden Markov Model approach, which relies on a hidden amino acid propensity scale. To estimate the sites of linear B-cell epitopes, this method uses a mix of the hidden Markov model and propensity scale algorithms.
Prediction of the MHC–specific epitopes
To anticipate conserved domain-based cytotoxic and helper T cell epitopes, the Immune Epitope Database (IEDB) was utilized. The MHC-I and MHC-II epitope prediction tools by IEDB are freely available at (http://tools.iedb.org/mhci) and (http://tools.iedb.org/mhcii). For projecting MHC-I restricted epitopes, the ANN 4.0 algorithm was used, which has the capacity to arrange findings based on IC50 values. For MHC-II restricted epitope prediction, NN-align 2.3 was used (Andreatta et al., Citation2018; Trevizani et al., Citation2022). To examine probable epitopes, allele sets (HLA-A, -B, -C for MHC-I and HLA-DRB, -DQB, and -DPB for MHC-II) from the IEDB database were used, with epitope lengths set at 9,10 for MHC-I and default for MHC-II.
Population coverage analysis
The Immune Epitope Database (IEDB) population coverage tool (http://tools.iedb.org/population) was used to determine the fraction of the worldwide population targeted by the epitopes. The analysis included distinct MHC-I and MHC-II modules, as well as a combined module. The population parameter was created to represent the whole planet. A total of 21 epitopes were evaluated, including all 9 from MHC-I and 12 from MHC-II, as well as their corresponding target alleles. This method attempted to assess the epitopes worldwide reach and possible impact.
Vaccine construction
Linkers like as AAY, EAAAK, and GPGPG are extensively used in vaccine development. In the present study, these linkers were used to connect epitopes, and an adjuvant was added to boost the host immune response. A poly-histidine tag (hexa-histidine tail) was also added to the construct for purification tests.The VaxiJen 2.0 web server (https://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) (Doytchinova & Flower, Citation2007) and AllerTop (https://www.ddg-pharmfac.net/AllerTOP) (Doytchinova & Flower, Citation2007) were used again in the study to determine the antigenicity and allergenicity of the construct. This comprehensive approach guarantees that many parameters are carefully considered in the creation and assessment of the vaccine construct.
Physiochemical properties assessment of vaccine construct
The physicochemical parameters of the vaccine construct were evaluated using the ExPASy ProtParam tool (https://web.expasy.org/protparam), a well-known online software for computing various protein properties. ExPASy ProtParam was critical in determining important physicochemical parameters such as molecular weight, theoretical isoelectric point (pI), amino acid content, extinction coefficient, and instability index. Using ExPASy ProtParam ensured a thorough investigation of the physicochemical parameters required to acquire insights into the intrinsic qualities of the vaccine design (Gulzar & Hussain, Citation2020).
Secondary structure prediction of the vaccine construct
PsiPred 2.0 (http://bioinf.cs.ucl.ac.uk/psipred) a widely known bioinformatics tool for evaluating protein structures, was used to predict the vaccine construct’s secondary structure. PsiPred predicts secondary structural features such as alpha helices, beta strands, and coils using a complex algorithm that incorporates information from position-specific scoring matrices and neural networks. The vaccine construct’s amino acid sequence was sent into the PsiPred 2.0 server, and the resulting output offered vital insight into the expected arrangement of secondary structural components inside the construct.
Tertiary structure prediction of vaccine construct
The TrRosetta online server (https://yanglab.nankai.edu.cn/trRosetta) was used to estimate the vaccine construct’s tertiary structure. TrRosetta uses a technique based on deep learning to infer a protein’s three-dimensional structure from its amino acid sequence. In order to forecast inter-residue distances and orientations, the system makes use of a neural network and a sizable library of protein structures. A three-dimensional model of the protein structure is then produced using the anticipated orientation and distance restrictions (Du et al., Citation2021). This sophisticated computational method improves the tertiary structure predictions accuracy and offers important new information about the vaccine construct’s atomic-level spatial organization.
Vaccine tertiary structure quality estimation
Two commonly used methods, ERRAT and PROCHECK (https://saves.mbi.ucla.edu) were used to evaluate the quality of the vaccine’s tertiary structure. ERRAT analyzes the statistics of non-bonded atom-atom interactions to assess the overall quality of protein structures. Better model quality is indicated by higher ERRAT scores. Furthermore, a Ramachandran Plot was utilized in conjunction with PROCHECK to appraise the stereochemical quality of the model by analyzing the distribution of phi (υ) and psi (ψ) dihedral angles inside the protein structure (Zhou et al., Citation2019). Reliability in the anticipated vaccine structure is ensured by a high-quality tertiary structure, which is shown by a favorable distribution within the plot’s permitted areas.
Molecular interaction studies with host receptor
The HDOCK SERVER (http://hdock.phys.hust.edu.cn) was used to predict vaccine and receptor docking. This server uses a hybrid approach that blends ab initio free docking techniques with template-based modeling. While ab initio free docking considers the intrinsic flexibility of molecules, template-based modeling makes use of pre-existing structural templates. The HDOCK SERVER offers a reliable method for predicting the spatial configurations of molecules that interact, making it possible to investigate molecular interactions that occur between proteins or between proteins and nucleic acids (Yan et al., Citation2020). The vaccine construct was docked against the E-Cadherin receptor for the interaction studies.
Molecular dynamic simulations
Immune simulations
The potential immunogenicity of the vaccine design was evaluated through the use of immune simulations on the C-ImmSim server (https://kraken.iac.rm.cnr.it/C-IMMSIM/index.php?page=1). A computational program called C-ImmSim models immune system interactions to forecast the immunological response that an antigen would elicit. C-ImmSim offers insights on the vaccine’s anticipated immunogenic qualities by simulating many immune system functions, such as antigen presentation, processing, and T-cell activation. In order to support the assessment of the vaccination’s potential effectiveness and capacity to elicit a strong immunological response, the simulations attempt to forecast how the immune system will react to the vaccine build (Ragone et al., Citation2021; Rapin et al., Citation2010; Stolfi et al., Citation2022).
Results
Sequence retrieval & physiochemical analysis
The sequence of Internalin A protein of Listeria monocytogenes was retrieved from UniProt KB under the UniProt ID of P0DJM0 was consisting of 800 AA. The protein sequence was downloaded in the FASTA format for further analysis. The protein sequence studied has a molecular weight of 86,492.91 Da. The determined theoretical isoelectric point (pI) is 4.93. A complete amino acid composition displays the percentage distribution of each amino acid, with Thr (13.4%), Leu (12.0%), and Ser (7.8%) standing out. There are 72 negatively charged residues (Asp + Glu), whereas there are 58 positively charged residues (Arg + Lys). The formula C3832H6084N1004O1251S8 consists of 3832 carbon (C), 6084 hydrogen (H), 1004 nitrogen (N), 1251 oxygen (O), and 8 sulfur (S) atoms. At 280 nm, the extinction coefficients in water are 96,260 M-1 cm-1, with an absorbance of 1.113 at 0.1%. The estimated half-life ranges from 30 hours (in vitro mammalian reticulocytes) to more than 10 hours (in vivo Escherichia coli). With an instability score (II) of 20.52, the protein is categorized as stable. The aliphatic index is 88.31, and the GRAVY (grand average of hydropathicity) is −0.298. This detailed study gives vital insights into the protein’s physicochemical properties, helping to a better understanding of its qualities.
Allergenicity and antigenicity profiling of internalin A
The allergenicity and antigenicity of Internalin A protein was profiled in order to assess its possible immunogenic characteristics. An in-depth investigation using the VaxiJen 2.0 web server and AllerTop found substantial antigenic features, indicating that Internalin A has the potential to provoke an immunological response. The VaxiJen 2.0 program predicted a high antigenic score of 0.6076 (Probable ANTIGEN), indicating that it has the potential to serve as an antigen. Furthermore, the AllerTop study revealed a low allergenicity probability, indicating a lower possibility of triggering allergic reactions. These findings highlight intriguing immunogenic character, indicating that it should be investigated further as a contender in vaccine development and immunotherapeutic techniques.
Prediction of the linear B-Cell epitopes
The B-Cell Epitopes were predicted using the IEDB Linear Epitope Prediction Tool v2.0 utilizing the Kolaskar & Tongaonkar Antigenicity technique. The study identified putative antigenic determinants with antigenicity scores ranging from 0.842 to 1.233. The average antigenicity was determined to be 1.009. Setting an antigenic determination threshold of 0.5, epitopes with scores greater than this threshold were deemed possible antigenic determinants. Finally, 5 antigenic epitopes were identified as matching the requirements for further evaluation in the study considering the antigenicity and allergenicity. The below represents the selected B-cell epitopes for vaccine construction and the below represents the graphical view with scoring of the sequence.
Figure 1. Graphical representation of scoring values in B-Cell epitopes prediction.
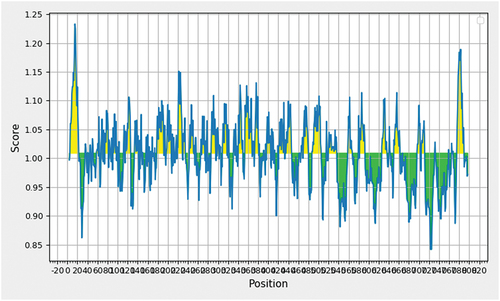
Table 1. Selected linear B-Cell epitopes for final vaccine construction.
Prediction of the MHC-Specific epitopes
Utilizing the ANN 4.0 algorithm and sorting by IC-50 values, the IEDB analysis found a total of 12 MHC Class-II and 9 MHC Class-I epitopes. lists these epitopes in detail. They are essential for triggering particular immune responses. In order for epitopes to effectively engage the immune system, it is crucial that they are chosen based on their affinity as shown by IC-50 values. This thorough prediction aids in the logical development of a strong and focused immune response against the designated antigens and offers insightful data for future investigation of the immunogenic potential of the vaccine formulation.
Table 2. Selected MHC class-I and MHC class-II epitopes for vaccine construction.
Population coverage analysis
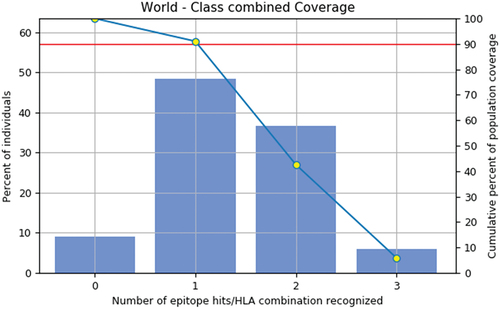
Using the IEDB population coverage analysis tool, the population coverage study showed that the projected epitopes for MHC Class I and Class II target around 91.08% of the global population. This thorough investigation offers insightful information about the epitopes’ possible global coverage, showing a high percentage of coverage across a variety of global populations. The population coverage map is shown in the below.
Figure 2. Population coverage map by IEDB population coverage analysis tool.
Vaccine construction
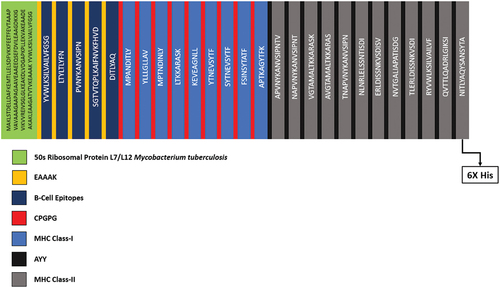
The vaccine construct was made up by joining the EAAK Linkers, CPGPG Linkers and AAY Linkers, and the predicted B-Cell Epitopes, MHC Class-I and MHC Class-II Epitopes. The whole vaccine construct was consisting of 570AA. The below depicts the map of vaccine construct indicating the adjuvant in the green color, B-Cell Epitopes in dark blue, MHC Class-I epitopes in light blue and MHC Class-II epitopes in grey color. While the yellow color in the figure indicates the EAAAK linkers, Red color indicates the CPGPG linkers and the black color indicates the AAY linkers.
Figure 3. Vaccine construct map showing the sequences of linkers, adjuvant and epitopes.
Physiochemical properties assessment of vaccine construct
The Expasy ProtParam tool was utilized to evaluate the vaccine construct’s physicochemical qualities. The sequence has a molecular weight of 60,887.58 Da and 570 amino acids. It is determined that the theoretical isoelectric point (pI) is 8.11. The distribution of amino acids is varied, with significant amounts of alanine (15.8%), leucine (8.1%), lysine (7.4%), and valine (7.5%). The atomic composition is C2770H4290N692O820S15, which is made up of carbon, hydrogen, nitrogen, oxygen, and sulfur. With various cysteine pairings, the extinction coefficients at 280 nm are 78,550 and 78,050. The calculated instability index of 22.30 indicates stability, while the projected half-life points to stability in a variety of cellular settings. The vaccine construct’s structural and biochemical characteristics are further characterized by the aliphatic index of 88.30 and the grand average of hydropathicity (GRAVY) of 0.019.
Secondary structure prediction of the vaccine construct
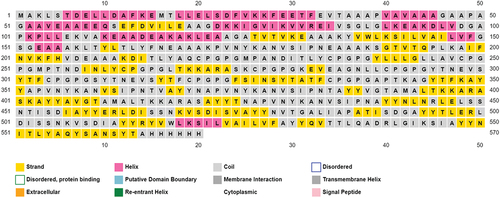
PsiPred 2.0 was used to anticipate the vaccine construct’s secondary structure. The distribution of coils, beta strands, and alpha helices within the protein sequence was revealed by the research. The anticipated secondary structural components advance our knowledge of the overall folding pattern and putative functional areas of the construct. The pink color in the results indicated the presence of Helices, the yellow color indicated the strands and grey color indicated the coils. The predicted secondary structure of the vaccine construct is given below in the .
Figure 4. Predicted secondary structure of vaccine construct by PsiPred 2.0.
Tertiary structure prediction of vaccine construct
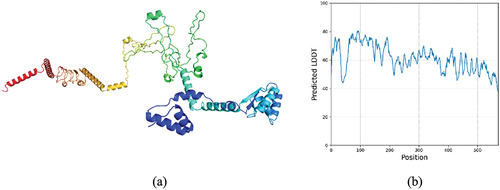
The TrRosetta predicted the tertiary structure of vaccine construct with 100% confidence and model coverage indicating it as the most authentic model of prediction. The predicted structure of the vaccine construct is given below in the and the predicted LDDT is shown below in the .
Figure 5. (a) predicted tertiary structure of the vaccine construct (B) predicted LDDT of the selected model 1.
Vaccine tertiary structure quality estimation
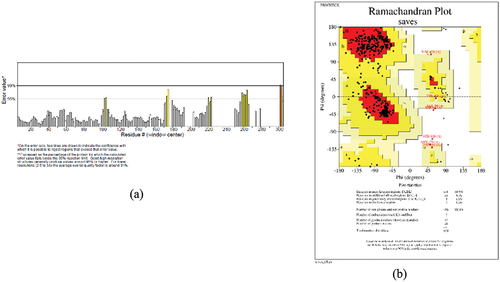
The vaccine’s tertiary structure quality was evaluated using two commonly used tools, ERRAT and PROCHECK. With a high score of 95.227%, ERRAT which focuses on non-bonded atom-atom interactions – showcased better overall structural quality. Furthermore, phi (υ) and psi (ψ) dihedral angles were used by PROCHECK to evaluate the stereochemical quality using a Ramachandran Plot. Plotting showed that 88.9% of dihedral angles were in the preferred locations, confirming the vaccine’s tertiary structure’s good distribution and dependability. All these findings support the high caliber and sound structure of the predicted vaccination model. The ERRAT quality chart is shown below in the and the Ramachandran Plot is shown in the .
Figure 6. (a) ERRAT quality chart (b) Ramachandran Plot by ProCheck.
Molecular interaction studies with Host receptor
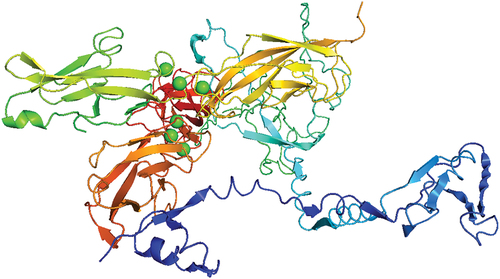
The results of molecular docking with the HDOCK server were encouraging; docking scores, confidence scores, ligand root-mean-square deviation (RMSD), and interface residues were used to select the top 10 docking models. The protein-protein or protein-DNA/RNA components exhibited good interactions, as shown by the docking scores, which varied from −213.15 (Rank 1) to −204.03 (Rank 10). The confidence ratings, which varied from 0.7795 to 0.7466, were likewise strong. The different ligand RMSD values in the docking models indicate different structural conformations. Each model’s interface residues are provided, revealing possible points of contact. These results imply strong connections and provide insightful knowledge for the protein complex’s future examination and improvement. The docking complex of the model 1 visualized in the PyMol visualization system is shown below in the .
Figure 7. The docking complex of the vaccine construct and the receptor E-Cadherin visualized by PyMol visualization system.
Molecular dynamic simulations
Immune simulations
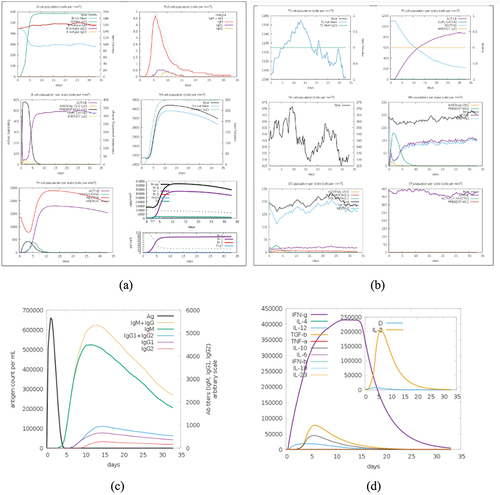
A thorough examination of the vaccine design’s potential immunogenicity was made possible by the immunological simulations run on the C-ImmSim server. The simulations were designed to predict the vaccine’s expected immunological response, and they covered a range of immune system processes, including antigen presentation, processing, and T-cell activation. Dendritic cells, macrophages, epithelial cells, B lymphocytes, CD4 T-helper lymphocytes, CD8 T-cytotoxic lymphocytes, natural killer cells, and cytokines were among the cell types whose movements were shown. The findings provide important information about the immune system’s anticipated reaction to the vaccination, laying the groundwork for future evaluations of the vaccine’s efficacy and ability to stimulate a strong immune response. The detailed results are shown below in the .
Figure 8. (a) cell counts shown. Legend: Act = active, Intern = internalized the ag, pres II = presenting on MHC II, Dup = in the mitotic cycle, anergic = anergic, resting = not active. (b) symbols as figure above. (c) the virus, the immunoglobulins and the immunocomplexes. (d) concentration of cytokines and interleukins. Inset plot shows danger signal together with leukocyte growth factor IL-2.
Discussion
The creation of a vaccine that targets Internalin A, a crucial component in the pathophysiology of Listeria monocytogenes, was the main goal of the study. The protein’s physiochemical study provided crucial details for further in silico research. Internalin A’s potential immunogenicity was revealed by antigenicity and allergenicity profiling, which showed low allergenicity and high antigenicity (Zhao et al., Citation2023). Widely distributed B-cell and MHC-specific epitopes were predicted. The adjuvants and linkers used in the vaccine design demonstrated good physicochemical characteristics as well as structural stability (Naveed et al., Citation2022). HDOCK-based molecular interaction experiments revealed strong binding to the E-Cadherin receptor. ERRAT and PROCHECK-validated tertiary structure predictions revealed a vaccine structure of superior quality (Chen et al., Citation2023; Zhu et al., Citation2021). Potential immunogenic responses were demonstrated by immune simulations on the C-ImmSim server, laying the groundwork for more analysis. From sequence extraction to immunological simulations, the all-encompassing strategy validates the vaccine’s viability as a candidate for additional development (Huang et al., Citation2022; Naveed et al., Citation2021; Shen et al., Citation2023; Zhang et al., Citation2023).
Internalin A was effectively identified by the study as a potential target for the creation of a vaccine against Listeria monocytogenes. Strong binding interactions with the E-Cadherin receptor, as well as excellent physicochemical qualities and structural stability, were demonstrated by the vaccine design. Widely distributed B-cell and MHC-specific epitopes were predicted by immunoinformatics analysis. Simulations of the immune system shed light on the possible immunogenic reaction. All things considered, the thorough methodology – which includes everything from sequence analysis to immunological simulations – supports the developed vaccine’s eligibility for additional research and development (He et al., Citation2023; Ikram et al., Citation2018; Zhang et al., Citation2023).
A strong contender for the immunization against Listeria monocytogenes is the discovered vaccine design that targets Internalin A. It may be effective given its advantageous physicochemical characteristics, proven antigenicity, and wide population coverage. Its candidacy is further strengthened by strong molecular interactions with the host receptor and insights from immunological simulations. These results highlight the vaccine’s encouraging potential in the fight against Listeria infections, indicating that more clinical and experimental research is necessary (Le et al., Citation2012).
This study has limitations even if the results are encouraging. Predictions made in silico might not accurately reflect the intricacies of the actual world. For anticipated immunogenicity to be confirmed, experimental validation is essential. Careful interpretation is also required due to the lack of in vivo evidence and possible individual differences in immune responses. Preclinical and clinical studies, among other types of research, are necessary to determine the safety and efficacy of the vaccine (Mayer et al., Citation2022).
Future investigations should concentrate on experimental validations, such as in vitro and in vivo tests, to verify the vaccine construct’s anticipated immunogenicity and safety. Furthermore, to evaluate the effectiveness of the vaccination and any possible adverse effects, clinical studies involving a variety of populations are essential. The efficacy and applicability of the vaccine in treating infectious illnesses will be improved by ongoing monitoring and design modifications based on new scientific findings.
Conclusion
To conclude, this extensive in silico investigation effectively discovered a possible vaccine candidate that targets Listeria monocytogenes’ Internalin A. Sequence retrieval, physiochemical analysis, antigenicity profiling, epitope prediction, population coverage analysis, and vaccine manufacture were all steps in the multi-step process. Molecular docking validated the produced vaccine construct’s good physicochemical features, structural stability, and interactions with host receptors. Important insights regarding the expected immunogenic properties were obtained from immune simulations. The absence of experimental validation is one of the study’s weaknesses, which highlights the need for caution when interpreting the results. Notwithstanding these drawbacks, the outcomes provide a strong platform for upcoming research and clinical development, indicating that the planned vaccine construct has the capacity to elicit a strong immune response and calling for additional study as a viable option in the fight against Listeria monocytogenes infections.
Acknowledgments
The authors greatly acknowledge and express their gratitude to the Researchers Supporting Project number (RSP2024R462), King Saud University, Riyadh, Saudi Arabia.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All the data generated in this research work has been included in the manuscript.
Additional information
Funding
References
- Akhtar, N., Joshi, A., Singh, J., & Kaushik, V. (2021). Design of a novel and potent multivalent epitope based human cytomegalovirus peptide vaccine: an immunoinformatics approach. Journal of Molecular Liquids, 335, 116586. https://doi.org/10.1016/j.molliq.2021.116586
- Andreatta, M., Trolle, T., Yan, Z., Greenbaum, J. A., Peters, B., & Nielsen, M. (2018). An automated benchmarking platform for MHC class II binding prediction methods. Bioinformatics, 34(9), 1522–1528. https://doi.org/10.1093/bioinformatics/btx820
- Berrang, M. E., Meinersmann, R. J., Frank, J. F., Smith, D. P., & Genzlinger, L. L. (2005). Distribution of Listeria monocytogenes subtypes within a poultry further processing plant. Journal of Food Protection, 68(5), 980–985. https://doi.org/10.4315/0362-028X-68.5.980
- Chen, L., He, Y., Zhu, J., Zhao, S., Qi, S., Chen, X., Zhang, H., Ni, Z., Zhou, Y., Chen, G. & Liu, S. (2023). The roles and mechanism of m6A RNA methylation regulators in cancer immunity. Biomedicine & Pharmacotherapy, 163, 114839. https://doi.org/10.1016/j.biopha.2023.114839
- Coelho, C., Brown, L., Maryam, M., Vij, R., Smith, D. F. Q., Burnet, M. C., Kyle, J. E., Heyman, H. M., Ramirez, J., Prados-Rosales, R., Lauvau, G., Nakayasu, E. S., Brady, N. R., Hamacher-Brady, A., Coppens, I., & Casadevall, A. (2019). Listeria monocytogenes virulence factors, including listeriolysin O, are secreted in biologically active extracellular vesicles. Journal of Biological Chemistry, 294(4), 1202–1217. https://doi.org/10.1074/jbc.RA118.006472
- Currie, A., Farber, J. M., Nadon, C., Sharma, D., Whitfield, Y., Gaulin, C., Galanis, E., Bekal, S., Flint, J., Tschetter, L., Pagotto, F., Lee, B., Jamieson, F., Badiani, T., MacDonald, D., Ellis, A., May-Hadford, J., McCormick, R., Savelli, C., & Sierpinska, U. (2015). Multi-province listeriosis outbreak linked to contaminated deli meat consumed primarily in institutional settings, Canada, 2008. Foodborne Pathogens & Disease, 12(8), 645–652. https://doi.org/10.1089/fpd.2015.1939
- Daniels, J. J., Autenrieth, I. B., & Goebel, W. (2000). Interaction of Listeria monocytogenes with the intestinal epithelium. FEMS Microbiology Letters, 190(2), 323–328. https://doi.org/10.1111/j.1574-6968.2000.tb09306.x
- Dimitrov, I., Bangov, I., Flower, D. R., & Doytchinova, I. (2014). AllerTOP v. 2—a server for in silico prediction of allergens. Journal of Molecular Modeling, 20, 1–6. https://doi.org/10.1007/s00894-014-2278-5
- Ding, Y., Shu, L., He, R., Chen, K., Deng, Y., Zhou, Z., Xiong, Y., & Deng, H. (2023). Listeria monocytogenes: A promising vector for tumor immunotherapy. Frontiers in Immunology, 14, 1278011. https://doi.org/10.3389/fimmu.2023.1278011
- Doytchinova, I. A., & Flower, D. R. (2007). VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics, 8(1), 1–7. https://doi.org/10.1186/1471-2105-8-4
- Du, Z., Su, H., Wang, W., Ye, L., Wei, H., Peng, Z., Anishchenko, I., Baker, D., & Yang, J. (2021). The trRosetta server for fast and accurate protein structure prediction. Nature Protocols, 16(12), 5634–5651. https://doi.org/10.1038/s41596-021-00628-9
- Elsayed, M. E., El-Hamid MI, A., El-Gedawy, A., Bendary, M. M., ELTarabili, R. M., Alhomrani, M., Alamri, A. S., Alghamdi, S. A., Arnout, M., Binjawhar, D. N., Al-Sanea, M. M., & Abousaty, A. I. (2022, October 21). New insights into Listeria monocytogenes antimicrobial resistance, virulence attributes and their prospective correlation. Antibiotics (Basel), 11(10), 1447. https://doi.org/10.3390/antibiotics11101447
- Gulzar, S., & Hussain, S. (2020). Bioinformatics study on structural protein of severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) for better understanding the vaccine development. bioRxiv. 2020.2004. 2021.053199.
- He, Z., Yue, C., Chen, X., Li, X., Zhang, L., Tan, S., Yi, X., Luo, G., & Zhou, Y. (2023). Integrative analysis identified CD38 as a key node that correlates highly with Immunophenotype, Chemoradiotherapy Resistance, and prognosis of head and neck Cancer. Journal of Cancer, 14(1), 72–87. https://doi.org/10.7150/jca.59730
- Huang, H., Wu, N., Liang, Y., Peng, X., & Shu, J. (2022). SLNL: A novel method for gene selection and phenotype classification. International Journal of Intelligent Systems, 37(9), 6283–6304. https://doi.org/10.1002/int.22844
- Ikram, A., Zaheer, T., Awan, F. M., Obaid, A., Naz, A., Hanif, R., Paracha, R. Z., Ali, A., Naveed, A. K., & Janjua, H. A. (2018). Exploring NS3/4A, NS5A and NS5B proteins to design conserved subunit multi-epitope vaccine against HCV utilizing immunoinformatics approaches. Scientific Reports, 8(1), 16107. https://doi.org/10.1038/s41598-018-34254-5
- Ireton, K., Mortuza, R., Gyanwali, G. C., Gianfelice, A., & Hussain, M. (2021). Role of internalin proteins in the pathogenesis of Listeria monocytogenes. Molecular Microbiology, 116(6), 1407–1419. https://doi.org/10.1111/mmi.14836
- Jackson, K. A., Gould, L. H., Hunter, J. C., Kucerova, Z., & Jackson, B. (2018). Listeriosis outbreaks associated with soft cheeses, United States, 1998–20141, emerg. Infectious Diseases, 24(6), 1116–1118. https://doi.org/10.3201/eid2406.171051
- Jemmi, T., & Stephan, R. (2006). La Listeria monocytogenes, un agente patógeno transmitido por los alimentos que también sirve de indicador de higiene. Revue scientifique et technique, 25(2), 571–580. https://doi.org/10.20506/rst.25.2.1681
- Ji, Q., Ma, J., Wang, S., & Liu, Q. (2022). Construction and immunological evaluation of live vector vaccine based on attenuated Listeria monocytogenes vector against Vibrio parahaemolyticus infection. Aquaculture, 560, 738560. https://doi.org/10.1016/j.aquaculture.2022.738560
- Larijani, M. S., Sadat, S. M., Bolhassani, A., Pouriayevali, M. H., Bahramali, G., & Ramezani, A. (2018). In silico design and immunologic evaluation of HIV-1 p24-nef fusion protein to approach a therapeutic vaccine candidate. Current HIV Research, 16(5), 322–337. https://doi.org/10.2174/1570162X17666190102151717
- Le, D. T., Brockstedt, D. G., Nir-Paz, R., Hampl, J., Mathur, S., Nemunaitis, J., Sterman, D. H., Hassan, R., Lutz, E., & Moyer, B. (2012). A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: Phase I studies of safety and immune induction. Clinical Cancer Research, 18(3), 858–868. https://doi.org/10.1158/1078-0432.CCR-11-2121
- Lecuit, M. (2020). Listeria monocytogenes, a model in infection biology. Cellular Microbiology, 22(4), e13186. https://doi.org/10.1111/cmi.13186
- Mayer, R. L., Verbeke, R., Asselman, C., Aernout, I., Gul, A., Eggermont, D., Boucher, K., Thery, F., Maia, T. M., & Demol, H. (2022). Immunopeptidomics-based design of mRNA vaccine formulations against Listeria monocytogenes. Nature Communications, 13(1), 6075. https://doi.org/10.1038/s41467-022-33721-y
- Naveed, M., Tehreem, S., Arshad, S., Bukhari, S. A., Shabbir, M. A., Essa, R., Ali, N., Zaib, S., Khan, A., & Al-Harrasi, A. (2021). Design of a novel multiple epitope-based vaccine: An immunoinformatics approach to combat SARS-CoV-2 strains. Journal of Infection and Public Health, 14(7), 938–946. https://doi.org/10.1016/j.jiph.2021.04.010
- Naveed, M., Yaseen, A. R., Khalid, H., Ali, U., Rabaan, A. A., Garout, M., Halwani, M. A., Al Mutair, A., Alhumaid, S., & Al Alawi, Z. (2022). Execution and design of an anti HPIV-1 vaccine with multiple epitopes triggering innate and adaptive immune responses: An immunoinformatic approach. Vaccines, 10(6), 869. https://doi.org/10.3390/vaccines10060869
- Radoshevich, L., & Cossart, P. (2018). Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nature Reviews, Microbiology, 16(1), 32–46. https://doi.org/10.1038/nrmicro.2017.126
- Ragone, C., Manolio, C., Cavalluzzo, B., Mauriello, A., Tornesello, M. L., Buonaguro, F. M., Castiglione, F., Vitagliano, L., Iaccarino, E., Ruvo, M., Tagliamonte, M., & Buonaguro, L. (2021). Identification and validation of viral antigens sharing sequence and structural homology with tumor-associated antigens (TAAs). The Journal for ImmunoTherapy of Cancer, 9(5), e002694. https://doi.org/10.1136/jitc-2021-002694
- Rahman, S., & Das, A. K. (2022). A subtractive proteomics and immunoinformatics approach towards designing a potential multi-epitope vaccine against pathogenic Listeriamonocytogenes. Microbial Pathogenesis, 172, 105782. https://doi.org/10.1016/j.micpath.2022.105782
- Rapin, N., Lund, O., Bernaschi, M., & Castiglione, F. (2010). Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PloS one, 5(4), e9862. https://doi.org/10.1371/journal.pone.0009862
- Ras-Carmona, A., Lehmann, A. A., Lehmann, P. V., & Reche, P. A. (2022). Prediction of B cell epitopes in proteins using a novel sequence similarity-based method. Scientific Reports, 12(1), 13739. https://doi.org/10.1038/s41598-022-18021-1
- Schlech, W. F., III. (2019). Epidemiology and clinical manifestations of Listeria monocytogenes infection. Microbiology Spectrum, 7(3). https://doi.org/10.1128/microbiolspec.GPP3-0014-2018
- Shahabi, V., Reyes-Reyes, M., Wallecha, A., Rivera, S., Paterson, Y., & Maciag, P. (2008). Development of a Listeria monocytogenes based vaccine against prostate cancer. Cancer Immunology Immunotherapy, 57, 1301–1313. https://doi.org/10.1007/s00262-008-0463-z
- Shen, W., Pei, P., Zhang, C., Li, J., Han, X., Liu, T., Shi, X., Su, Z., Han, G., Hu, L & Yang, K. (2023). A polymeric hydrogel to eliminate programmed death-ligand 1 for enhanced tumor radio-immunotherapy. Agricultural Science & Technology Nano, 17(23), 23998–24011. https://doi.org/10.1021/acsnano.3c08875
- Smith, A. M., Tau, N. P., Smouse, S. L., Allam, M., Ismail, A., Ramalwa, N. R., Disenyeng, B., Ngomane, M., & Thomas, J. (2019). Outbreak of Listeria monocytogenes in South Africa, 2017–2018: Laboratory activities and experiences associated with whole-Genome Sequencing analysis of Isolates. Foodborne Pathogens & Disease, 16(7), 524–530. https://doi.org/10.1089/fpd.2018.2586
- Stolfi, P., Castiglione, F., Mastrostefano, E., DiBiase, I., DiBiase, S., Palmieri, G., & Prisco, A. (2022). In-silico evaluation of adenoviral COVID-19 vaccination protocols: Assessment of immunological memory up to 6 months after the third dose. Frontiers in Immunology, 13, 998262. https://doi.org/10.3389/fimmu.2022.998262
- Thomas, J., Govender, N., McCarthy, K. M., Erasmus, L. K., Doyle, T. J., Allam, M., Ismail, A., Ramalwa, N., Sekwadi, P., Ntshoe, G., Shonhiwa, A., Essel, V., Tau, N., Smouse, S., Ngomane, H. M., Disenyeng, B., Page, N. A., Govender, N. P. … Smith, A. M. (2020). Outbreak of listeriosis in South Africa associated with processed meat. The New England Journal of Medicine, 382(7), 632–643. https://doi.org/10.1056/NEJMoa1907462
- Trevizani, R., Yan, Z., Greenbaum, J. A., Sette, A., Nielsen, M., & Peters, B. (2022). A comprehensive analysis of the IEDB MHC class-I automated benchmark. Briefings in Bioinformatics, 23(4), bbac259. https://doi.org/10.1093/bib/bbac259
- World Health Organization. Listeriosis. Retrieved February 20, 2018, from https://www.who.int/news-room/fact-sheets/detail/listeriosis.
- Yan, Y., Tao, H., He, J., & Huang, S.-Y. (2020). The HDOCK server for integrated protein–protein docking. Nature Protocols, 15(5), 1829–1852. https://doi.org/10.1038/s41596-020-0312-x
- Zhang, Y., Lian, B., Yang, S., Huang, X., Zhou, Y., & Cao, L. (2023). Metabotropic glutamate receptor 5-related autoimmune encephalitis with reversible splenial lesion syndrome following SARS-CoV-2 vaccination. Medicine, 102(7), e32971. https://doi.org/10.1097/MD.0000000000032971
- Zhang, Q., Wang, Y., Bai, R. T., Lian, B. R., Zhang, Y., & Cao, L. M. (2023). X-linked Charcot-Marie-tooth disease after SARS-CoV-2 vaccination mimicked stroke like episodes. A case report. World Journal of Clinical Cases, 11(2), 464–471. https://doi.org/10.12998/wjcc.v11.i2.464
- Zhao, Q., Wang, Y., Zhu, Z., Zhao, Q., Zhu, L. & Jiang, L. (2023). Efficient reduction of β-lactoglobulin allergenicity in milk using clostridium tyrobutyricum Z816. Food Science and Human Wellness, 12(3), 809–816. https://doi.org/10.1016/j.fshw.2022.09.017
- Zhou, J., Negi, A., Mirallai, S. I., Warta, R., Herold-Mende, C., Carty, M. P., Ye, X.-S., & Murphy, P. V. (2019). N-Alkyl-1, 5-dideoxy-1, 5-imino-l-fucitols as fucosidase inhibitors: Synthesis, molecular modelling and activity against cancer cell lines. Bioorganic Chemistry, 84, 418–433. https://doi.org/10.1016/j.bioorg.2018.12.003
- Zhu, Y., Huang, R., Wu, Z., Song, S., Cheng, L. & Zhu, R. (2021). Deep learning-based predictive identification of neural stem cell differentiation. Nature Communications, 12(1), 2614. https://doi.org/10.1038/s41467-021-22758-0