
ABSTRACT
Lactiplantibacillus plantarum is a probiotic starter culture that improves the flavor and nutritional content of fermented foods. It also shows potential in enhancing mucosal and systemic immunity, which contributes to overall health. We aimed to examine the complex genetic composition of the L. plantarum 13–3 via a detailed analysis of comparative genomics and our findings discovered that this genome displays high efficacy and conciseness as a nutrition enhancer, encompassing a cumulative sum of 2921 genes encoding proteins and a GC content of 45%. This study clarifies the evolutionary lineage of L. plantarum strains by examining the level of gene order conservation and synteny across various strains. Our strain has been found to possess a range of genetic components, including bacteriocin gene clusters and prophage regions, which indicates its potential applicability in various fields, including biotechnology and medicine. The unique genetic components have the potential to reveal novel therapeutic and biotechnological applications in the future.
Introduction
Lactiplantibacillus plantarum is a gram positive bacteria that holds significant importance in both the food, biotechnological and pharmaceutical sectors. The genetic diversity and functional traits of this organism have garnered attention due to its probiotic capabilities and role in food fermentation. The utilization of comparative genomic analysis enables scholars to examine the genetic variability, CRISPR-Cas systems, and functional discoveries of L. plantarum (Aziz et al., Citation2020a; Yilmaz et al., Citation2022). Scholars conduct a comparative analysis of the genomes of various strains of L. plantarum to identify genetic variations, comprehend its evolutionary and adaptive characteristics, and ascertain its prospective applications in safety-related inquiries and typing (Mao et al., Citation2021). The present study undertakes an analysis of the genomic characteristics of L. plantarum, with a particular emphasis on its genetic diversity, CRISPR-Cas systems, and the potential functional implications of these features for future typing methodologies and safety-related applications. The present investigation enhances our comprehension of L. plantarum and unveils novel prospects for subsequent scholarly inquiry and pragmatic application (Nordström et al., Citation2021).
Despite the growing interest in L. plantarum and its potential usefulness, many questions remain. A comprehensive comparative genomic investigation of L. plantarum is needed to understand its genetic diversity and evolutionary dynamics. Despite extensive study on CRISPR-Cas systems in several species, their abundance, variety, and functional importance in L. plantarum are still unknown. L. plantarum strain’s metabolic pathways and genetic traits related to type and safety have not been fully studied (Surve et al., Citation2022). Thus, a comparative genomic study of L. plantarum is needed to overcome these gaps. This investigation should examine the organism’s genomic diversity, CRISPR-Cas systems, and functional implications for typing and safety. These shortcomings will improve our understanding of L. plantarum and lay the groundwork for typing and safety evaluation methods. Food, probiotics, and biotechnology will benefit (Villena et al., Citation2021).
This study examines L. plantarum genomic features, focusing on genetic diversity, CRISPR-Cas systems, and functional implications for typing and safety. Genome analysis will assess L. plantarum strains’ genetic diversity and identify genetic differences. This study examines CRISPR-Cas systems in L. plantarum strains to better understand their potential usefulness in genetic editing and immunological defense. The study aims to investigate L. plantarum strains’ functional implications. This involves identifying metabolic pathways and genetic features for typing and safety. The research examines genetic information’s possible uses in food production, probiotics, and biotechnology. This will aid in typing and safety evaluation. This comparative genomic study aims to fill information gaps, make discoveries, and guide L. plantarum research and implementation.
Comparative genomics of L. plantarum revealed its genetic diversity, CRISPR-Cas systems, and functional insights that may be used for typing and safety. Lactobacillus plantarum strains varied genetically. CRISPR-Cas is pervasive, and diversified, and may protect against diseases and manipulate genetic material. Functional investigation revealed metabolic pathways and genetic traits that might be used for categorization and safety across sectors (Mao et al., Citation2021). L. plantarum study has several possibilities. Investigating genetic differences and metabolic pathways may reveal strain-specific traits and uses in personalized diets and medications. Lactobacillus plantarum CRISPR-Cas systems may improve genome editing and probiotic effectiveness. Comparative genomic analysis, transcriptomics, and proteomics can help us understand L. plantarum gene expression patterns and functional dynamics in different contexts. This may help us understand its probiotic processes and build personalized therapies (Li et al., Citation2022; Min et al., Citation2020). The present study establishes a fundamental basis for forthcoming research undertakings in L. plantarum, accentuating the significance of comparative genomics in elucidating its genetic heterogeneity and functional discernments. The field of genomics and bioinformatics has witnessed a steady progression in technological advancements. This has opened new avenues for research, which could lead to the creation of innovative applications, more effective safety assessment techniques, and greater utilization of L. plantarum across diverse industries.
Materials & methods
Bacterial strain and culture condition
In 2015, L. plantarum 13–3 (L. plantarum 13–3) was obtained from Tibetan Kefir grains and subsequently preserved as frozen stocks at a temperature of −9°C in MRS broth that was supplemented with 20% (v/v) glycerol. The identification of these strains was primarily based on Gram staining, catalase testing, and cellular morphology. The identification of strains was carried out through the utilization of the API 50 CHL test (bio-Merieux, Marcy-l’Étoile, France) and analysis of 16S rDNA sequencing as previously described by (Zhang et al., Citation2017)
DNA extraction and whole genome sequencing
The Wizard® Genomic DNA Purification Kit from Promega, Madison, WI, U.S.A. was utilized to extract the genomic DNA, which was subsequently quantified using a TBS-380 fluorometer (Turner Bio Systems Inc., Sunnyvale, CA, U.S.A.). The insert size for the quantification was 15 kb. For further analysis, DNA of superior quality was utilized, with an OD260/280 ratio ranging from 1.8 to 2.0 and a quantity exceeding 20 micrograms. The sheared fragments were utilized to prepare Illumina sequencing libraries through the employment of the NEXTflex™ Rapid DNA-Seq Kit. The sheared fragments were utilized to prepare Illumina sequencing libraries through the employment of the NEXTflex™ Rapid DNA-Seq Kit. In brief, a total of 50 prime ends underwent initial end-repair and phosphorylation. Subsequently, the 30 termini were subjected to A-tailing and ligation with sequencing adapters. Subsequently, the third phase involved the amplification of the adapters-ligated products through PCR for enrichment purposes. Subsequently, the libraries that had been prepared were subjected to paired-end Illumina sequencing, with a read length of 150 bp for each end, utilizing an Illumina HiSeq X Ten platform. The genome of the chosen strain, L. plantarum 13–3, was sequenced using both single molecule real-time (SMRT) technology and Illumina sequencing platforms. The resulting sequence has been assigned the accession number GCA_004028315.1. The complexity of the genome was assessed utilizing the Illumina data (Churro et al., Citation2012). The genomic DNA was isolated following the manufacturer’s protocol, utilizing the Qiagen DNA extraction kit. The isolation process was carried out during the period spanning March to May of 2019 (Aziz et al., Citation2021).
Data collection
The genome was previously submitted to the National Centre for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov) under the accession number GCA_004028315.1. The FASTA file of the genome was imported for the analysis and further utilization.
Gene annotation and average nucleotide identity
The study employed MetaGeneMark, a tool accessible at (http://opal.biology.gatech.edu/GeneMark), to conduct gene coordinate prediction, as well as protein and nucleotide sequence prediction. Furthermore, the Prokka v1.12 software was utilized to establish pipelines for the prediction of tRNA, tmRNA, rRNA, and CRISPR sequences. This was accomplished through the utilization of Aragorn, which is accessible at (http://www.ansikte.se/ARAGORN). The genome sequence was subsequently submitted in the FASTA DNA file format to predict the presence of tRNA, tmRNA, and CRISPR sequences within the genome, as described in 2022 by Gemayel et al. (Citation2022). To examine the taxonomic delineations of the genomes, the Average Nucleotide Identity (ANI) was ascertained through the utilization of the Genome-to-Genome Distance Calculator (GGDC), which is available at (http://ggdc.dsmz.de) and employs BLAST+ as its similarity search algorithm. The 10 genomes that were obtained were subjected to analysis on the server, alongside the reference genome, as reported by Trimble et al. (Citation2012).
Genome-based taxonomy analysis
The Type (Strain) Genome Server (TYGS), which is publicly available at (https://tygs.dsmz.de), was employed for Genome Based Taxonomy analysis. The FASTA DNA file containing the complete genome sequence was submitted for analysis, and subsequent evaluation of the genome-based taxonomy was conducted on the obtained results (Meier-Kolthoff et al., Citation2022).
Pan-genomics analysis
The Pan Genome analysis was executed utilizing the PanX online server, which is accessible at no cost via the following website: (https://pangenome.org). The chosen species for analysis was Lactiplantibacillus plantarum, from which core genes, distribution ranking, and strain trees were derived (Ding et al., Citation2018).
Genomic plasticity analysis
Genome plasticity analysis is a powerful method for finding new therapeutic targets and coming up with different ways to combat bacterial infections. It is critical to have an appreciation for the role that bacterial genome evolution and adaptation play in this process. Insights on bacterial population diversity, virulence factor distribution, and antibiotic resistance gene dispersal can be gained by pinpointing the genomic areas implicated in gene plasticity. IslandViewer 4, available for free at (https://www.pathogenomics.sfu.ca), was used to conduct the genomic plasticity analysis. Analysis was performed on the genome of Lactobacillus plantarum strain 13–3, and the results were interpreted (Boluk et al., Citation2021).
CRISPR cas systems identification
Using the CRISPR Detect online tool, which is freely available at (http://crispr.otago.ac.nz/CRISPRDetect) CRISPR-Cas9 systems in genomes were found. A web-based computational tool called CRISPR Detect was developed especially for locating and characterizing CRISPR-Cas systems in genomic sequences. To precisely anticipate the presence of CRISPR-Cas9 systems, this method combines sequence alignment, pattern recognition, and machine learning techniques. The genome sequence was entered into the online CRISPR Detect program, which processed the information and carried out a thorough search for CRISPR-Cas9 systems (Wu et al., Citation2021). The CRISPR Detect tool uses a multistep process to recognize CRISPR arrays and the Cas proteins that go with them. To find probable CRISPR repeats within the genomic sequences, it first performs a sequence alignment. After that, it makes use of pattern recognition algorithms to determine whether any of the distinctive Cas protein motifs connected to CRISPR-Cas9 systems are present (Dronina et al., Citation2022; Zhao et al., Citation2022).
Annotation of the pathways
The Bioinformatics and Virtual Biology Resource Center (BVBRC) online server was utilized to perform pathway annotation for L. plantarum 13–3. The BVBRC is an online platform that provides an extensive range of bioinformatics tools for the analysis of genomes and the annotation of pathways (Olson et al., Citation2023). It utilizes a hybrid approach that integrates computational algorithms and comparative genomics methodologies to annotate pathways within the genome of interest. The process involves the utilization of diverse databases such as KEGG (Kyoto Encyclopedia of Genes and Genomes), MetaCyc, and BioCyc, to ascertain and allocate functional annotations to genes and their corresponding products (Olson et al., Citation2023). The sequence was prepared into the FASTA format and then submitted to BVBRC for the pathway annotations.
Prediction of the bacteriocin-producing genes
The BAGEL 4 (http://bagel4.molgenrug.nl) software was used to predict the genes that produce bacteriocin. It accurately predicts and annotates bacteriocin genes by combining sequence analysis, machine learning, and comparative genomics methods (Suryaletha et al., Citation2022). To find homologous sequences, the software first performed a sequence similarity search against a large database of known bacteriocin genes. To categorize and distinguish possible bacteriocin gene clusters from non-bacteriocin sequences, the tool utilized machine learning algorithms that have been trained on a varied range of bacteriocin genes (Abdulkarim et al., Citation2020).
Identification of prophage and other insertion sequences
The Phage Search Tool Enhanced Release (PHASTER) (http://phaster.ca) was employed to detect prophage regions. The prophages length, localization, GC content, and gene annotation were predicted as reported by reference (Arndt et al., Citation2016). The validation of the predicted intact prophages was carried out through a comparison with the Virus-Host DB database (https://www.genome.jp/virushostdb). The generation of a proteomics tree for the viral genome sequence was facilitated through the utilization of the VIPtree application. The construction of the tree was facilitated through the utilization of tBLASTx-computed genome-wide sequence similarities. The present analysis facilitated the elucidation of the evolutionary interrelationships among viral genomes.
Phylogenetic and comparative analysis
To authenticate the taxonomic classification of the L. plantarum 13–3 strain, an Average Nucleotide Identity (ANI) analysis was performed on various strains of L. plantarum. ANI analysis was conducted post plasmid removal, utilizing the JSpecies Web Server, which is accessible free at (Richter et al., Citation2016) (http://jspecies.ribohost.com/jspeciesws) [19]. The clustering and heatmap were generated using HemI version 2.0, which can be accessed at http://hemi.biocuckoo.cn (Ning et al., Citation2022).
Results
Data collection
The retrieved genomic data was analyzed and visualized for the assembly statistics. There were a total of 7 contigs and 7 scaffolds observed in the genome with a genome coverage of 145.0x. The total number of genes observed in the genome was 3218 of which 2921 were protein coding and 87 were non-coding. The Chromosome table depicting the size and GC content is given below in .
Table 1. The genome table for the Lactiplantibacillus plantarum 13–3 whole genome indicating the assembly accession, GC count, percentage, molecular type and sequence lengths of the genome under study.
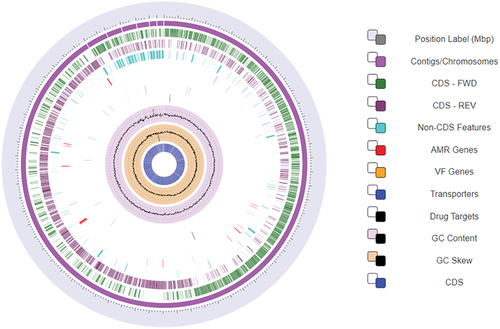
The quality analysis of the genome suggested a genome completeness of 99.02% with only 1.39% contamination. The Completeness of Lactiplantibacillus plantarum RefSeq assemblies is shown in Figure S1 and the circular view of Lactiplantibacillus plantarum 13–3 is shown in (Olson et al., Citation2022).
Figure 1. The circular view of the L. plantarum 13–3 strain along with the genomic features generated by BVBRC.
Gene annotation and average nucleotide identity
The genes of L. plantarum strain 13–3 microbial genome were predicted using MetaGeneMark. A set of 3081 genes, with an average length ranging from 100–2000 base pairs, were predicted. The genome exhibited gene dispersion, as evidenced by the presence of predicted genes with an average intergenic distance of 1000 bp. The examination conducted unveiled a diverse array of tRNA, tmRNA, and rRNA sequences within the genomes of the examined organisms. Several types of tRNA were identified, such as tRNA-Val, tRNA-Met, and tRNA-Ala, among others. The anticipated secondary structures of tRNA molecules were examined, revealing discrepancies in both the anticodon loops and stems.
In conjunction with transfer RNA, the presence of transfer-messenger RNA was also anticipated in the genomes that were scrutinized. The process of translation involves the degradation of anomalous proteins, in which tmRNA plays a crucial role. The anticipation of tmRNA can offer a valuable understanding of the mechanisms of protein quality control. The total count of tRNA genes was recorded as 69. A diverse array of tmRNA sequences was discovered, exhibiting variations in size, structure, and genomic location. The tRNA codon and tRNA anti-codon frequency are given in Table S1.
The examination conducted indicated a diverse spectrum of Average Nucleotide Identity (ANI) values within the genomes that were analyzed. The ANI values exhibited a range of 75.5 to 81.4%, which suggests notable variations in the genomic resemblance among the studied organisms. The ANI values were found to be higher among organisms belonging to the same genus, indicating a greater degree of genomic similarity. Conversely, organisms from distinct genera exhibited lower ANI values, indicating a lower degree of genomic similarity. Intra-species variability of ANI values was also observed in our study. In certain instances, ANI values approached 82%, signifying a substantial level of genomic resemblance, whereas, in other instances, ANI values were comparatively lower, indicating a reduced level of genomic similarity. The observed variability could potentially be attributed to dissimilarities in genomic composition or evolutionary processes occurring within a given species. presents a comprehensive breakdown of the outcomes obtained from the ANI analysis.
Table 2. Average nucleotide identity of the L. plantarum genomes under study.
Genome-based taxonomy analysis
The TYGS algorithm employed various metrics, including average nucleotide identity (ANI) and digital DNA-DNA hybridization (dDDH), to assess the genetic relatedness between the submitted genome and the reference genomes. The obtained results from TYGS were subsequently evaluated to determine the genome-based taxonomy of the analyzed sequence. The taxonomic classification was based on the closest matching reference genome(s) in the database, taking into consideration the ANI and dDDH values (Volpiano et al., Citation2021).
The genome-based taxonomy analysis conducted by TYGS provided valuable insights into the taxonomic classification of the submitted genome sequence. It enabled the identification and assignment of the strain to a specific species, genus, and potentially higher taxonomic ranks, depending on the available reference genomes. The genome-based taxonomy analysis results are given in Table S2.
Pan-genomics analysis
Based on this study, the pan-genome is comprised of a multitude of genes and exhibits significant size and diversity. It was observed that genomes can be classified into different types, namely, a set of genes that are universally shared among all living beings and another set of genes that are shared only among a specific subset of organisms. The variances in the sizes of auxiliary genomes among prokaryotes suggest dissimilarities in their genomic content and evolutionary paths. The functional annotation of the pan-genome has revealed a multitude of pathways that are implicated in various biological processes such as metabolism, transport, and regulation. The distribution of genes implicated in virulence and antibiotic resistance exhibited variability among the species under investigation (Chambers et al., Citation2020). The list of strains used in the pangenome analysis is given below in Table S1.
The construction of a phylogenetic strain tree utilizing the pan-genome yielded valuable insights into the evolutionary connections among the organisms. The data obtained from the strain tree analysis revealed distinct clades, which can be interpreted as distinct patterns of genomic diversity and evolutionary history. Except for a few noteworthy cases, our findings indicate that the taxonomic classification of the organisms is consistent with their evolutionary relationships. The distribution of gene count rank ranged up to 80 genes, with an approximate length of 6000 base pairs. Figure S1 displays the distribution of gene count rank and strain count rank and the strain tree of the pertinent genomes.
Genomic plasticity analysis
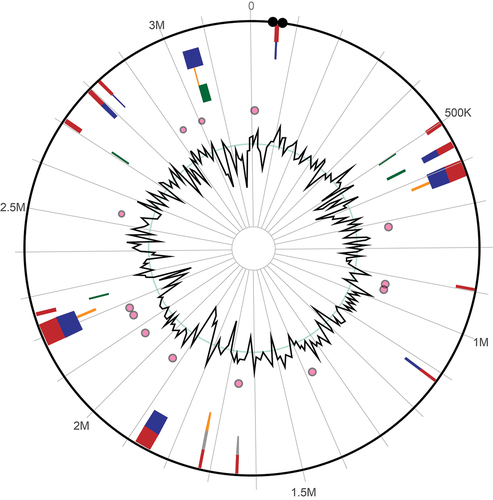
The present investigation demonstrated that the genomes of the specimens under scrutiny exhibited a diverse array of genomic islands and mobile genetic elements. The identification of genomic islands such as pathogenicity islands, prophages, and integrative conjugative elements has been observed. Various types of transposons and insertion sequences, which are genomic elements capable of mobility, were also identified in our study. Functional annotation of genomic islands and mobile genetic elements (MGEs) unveiled a diverse array of genes implicated in virulence, antibiotic resistance, and other cellular processes. The observed variations in the occurrence of genomic islands and mobile genetic elements among the taxa under investigation indicate divergent genomic compositions and evolutionary trajectories. depicts the map for predicting genome islands.
Figure 2. Genome islands of L. plantarum 13–3 predicted by IslandViewer4.
Annotation of the pathways
The utilization of the BVBRC online tool for the annotation of pathways about L. plantarum 13–3 has yielded significant insights into the metabolic capacity of this particular strain. The findings of the study indicate the existence of a comprehensive collection of annotated pathways that are linked to diverse metabolic processes.
The findings indicate that L. plantarum 13–3 exhibits a broad spectrum of metabolic functionalities. The study has identified several significant pathways that are involved in the metabolism of carbohydrates, which include glycolysis, the pentose phosphate pathway, and the citric acid cycle. The pathways in question are of paramount importance in facilitating energy generation and carbohydrate degradation in the bacterial organism.
Additionally, the analysis of annotations has indicated pathways linked to the metabolism of amino acids, underscoring the strain’s capacity to produce and employ diverse amino acids. The study identified the pathways implicated in the biosynthesis of crucial amino acids, namely lysine, methionine, and tryptophan. These findings suggest that the strain can thrive under conditions of limited nutrient availability.
Furthermore, pathways linked with lipid metabolism were annotated, indicating the capacity of L. plantarum 13–3 to catabolize and employ lipids as a source of carbon and energy. The strain’s lipid metabolism and membrane synthesis are influenced by various pathways, such as fatty acid biosynthesis, fatty acid degradation, and phospholipid metabolism.
Prediction of bacteriocins
Functional annotation of the bacteriocin gene clusters revealed several functional categories, including biosynthesis, regulation, and immunity genes. The study revealed the presence of specific clusters of bacteriocin genes, such as Plantaricin, Lacticin, and Plantacylin along with antibiotic resistance genes. This observation suggests that bacteriocins may serve a dual purpose of protecting the organism against external stressors and competing microorganisms. The bacteriocin gene cluster is illustrated in .
Figure 3. The gene cluster of bacteriocin-producing genes in Lactiplantibacillus plantarum 13–3.
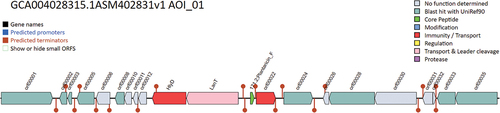
The present investigation has also detected multiple structural variations of bacteriocins within the genomic composition of the L. plantarum 13–3 strain. It could be deemed valuable to explore whether these variations exhibit distinct antibacterial properties and attributes. Table S2 depicts all the bacteriocins produced by the strain along with their function and motifs.
Identification of prophage and other insertion sequences
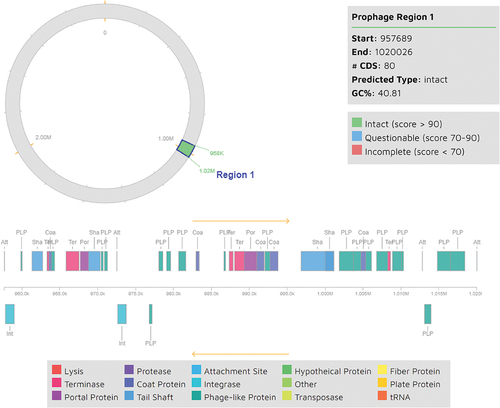
The process of identifying and characterizing prophage regions was carried out using the Phage Search Tool Enhanced Release (PHASTER) available at (http://phaster.ca) The aforementioned robust tool effectively forecasted various attributes of the prophages, including but not limited to their length, localization, GC content, and gene annotation. To authenticate the anticipated undamaged prophages, the ascertained regions were cross-referenced with the Virus-Host DB (https://www.genome.jp/virushostdb) database. This comparative analysis validated the presence of prophages and provided insights into their prospective host specificity. One prophage region was identified with a length of 62.3Kb including 76 proteins and intact completeness ranging from position 957,689–1020015, as indicated in .
Figure 4. Prophage region identified in the L. plantarum 13–3 genome by PHASTER.
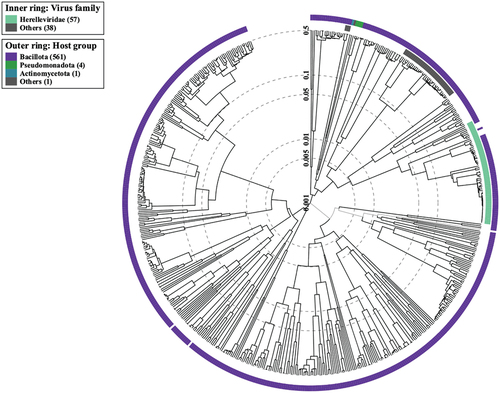
The proteomics tree of the viral genome sequence was generated utilizing the VIPtree application. The utilization of genome-wide sequence similarities in constructing this tree facilitated the visualization of the evolutionary connections among viral genomes. The study conducted a thorough analysis of prophage regions in the studied genomes and their potential impact on bacterial evolution and host-pathogen interactions. This was achieved through the use of the PHASTER tool, database comparisons, and the VIPtree application, resulting in a comprehensive understanding of the genetic elements present. depicts the Circular Proteomic Tree of the viral genome sequences.
Figure 5. Circular proteomic tree of the viral genomes. The outermost ring illustrates the host group of bacteriophages, while the innermost ring represents the virus family of phages.
Phylogenetic & comparative analysis
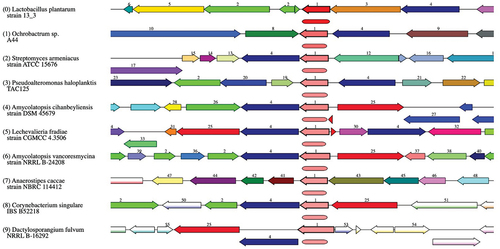
The comprehensive genome assembly of L. plantarum 13–3 was utilized to confirm its taxonomy and phylogenetic association through the application of ANI analysis. Recent studies have indicated that ANI analysis is a dependable metric for evaluating genetic and evolutionary divergence. The reason for this is that ANI analysis is dependent on a significant quantity of conserved genes. Consequently, the ANI analysis encompassed diverse strains of L. plantarum. According to the ANI analysis outcomes, LPG1 manifested a considerable level of resemblance to other L. plantarum strains, surpassing the recognized species identity threshold of > 95% for ANI value. Additionally, it was noted that Lactobacillus plantarum was present. illustrates the clustering and heat maps of different strains of L. plantarum. Compare region viewer of L. plantarum 13–3 with other strains is shown in .
Figure 6. Clustering and heatmaps of L. plantarum various strains indicating the ANIm percentage, aligned percentage, aligned bp, and total bp.
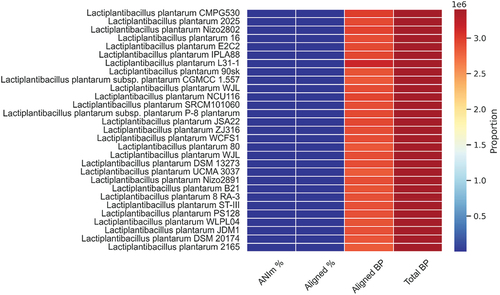
Figure 7. Compare region viewer of L. plantarum 13–3 with other strains.
Discussion
The present investigation centered on the examination of the genomic composition of L. plantarum 13–3 strain via comparative genomics. The results indicate that the genome is notably streamlined and proficient, encompassing 2921 genes that encode proteins, with a GC content of 45%. The investigation additionally examined the evolutionary ancestry of L. plantarum varieties, underscoring their closely intertwined evolutionary paths through the preservation of gene order and synteny (Tenea, Citation2022). Furthermore, the 13–3 strain was found to harbor several genetic elements, including bacteriocin gene clusters and prophage regions, which suggest promising prospects for its utilization in the fields of biotechnology and medicine. The distinctive genetic components exhibit potential for novel therapeutic and biotechnological remedies in the forthcoming times. In summary, this exhaustive examination illuminates the noteworthy genomic capacity of L. plantarum 13–3, presenting novel prospects for leveraging its unique genetic characteristics in diverse scientific domains (Aziz et al., Citation2023).
The study’s findings hold noteworthy implications across various domains. The genome of L. plantarum 13–3 exhibits a notable degree of efficiency and compactness, which suggests its successful adaptation to its ecological niche and capacity to flourish amidst diverse environmental circumstances. Comprehending the genetic characteristics that contribute to the efficiency can yield valuable insights into the metabolic capabilities of the strain and its potential applications in various domains, including but not limited to probiotics, food fermentation, and biotechnology. The detection of genetic elements, specifically bacteriocin gene clusters and prophage regions, within the L. plantarum 13–3 strain holds significant practical implications across multiple scientific disciplines. Bacteriocins are peptides with antimicrobial properties that exhibit potential as antibiotic substitutes (De Simone et al., Citation2022; Jiang et al., Citation2018). The existence of bacteriocin gene clusters implies that this particular strain may harbor antimicrobial characteristics. Furthermore, the detection of prophage regions suggests the possibility of horizontal gene transfer and the assimilation of new genetic components, thereby augmenting the strain’s versatility and prospective usefulness in biotechnology and medical contexts (Aziz et al., Citation2020c, Citation2022).
Although the present investigation has yielded significant findings regarding the genetic constitution and prospective utilities of L. plantarum 13–3, it is imperative to recognize certain constraints. Initially, the examination was centered on a solitary strain, which may not comprehensively embody the genetic variability and operational potentials of the complete species. Conducting additional research with a greater number of strains would yield a more exhaustive comprehension of the genetic diversity and possible applications of the species (Syaputri et al., Citation2023). The study predominantly utilized computational analysis and comparative genomics methodologies, which are susceptible to limitations and potential errors. These limitations may include incomplete or inaccurate genome annotations or the presence of mobile genetic elements that could impact genomic stability. The reliability of the findings could be enhanced through experimental validation and verification of the computational predictions. Finally, the research did not thoroughly examine the phenotypic traits or ecological relationships of L. plantarum 13–3. Additional research employing phenotypic assays, ecological investigations, or animal models would be advantageous in comprehensively elucidating the functional characteristics, ecological significance, and prospective practical utilities of the strain (Aziz et al., Citation2020b, Citation2023).
To summarize, the present study has provided insights into the complex genetic composition and possible practical uses of L. plantarum 13–3. The research findings indicate that the genome is characterized by a remarkable level of efficiency and compactness, which underscores its capacity to adapt and perform metabolic functions with great proficiency. The study of comparative genomics has yielded valuable insights into the evolutionary lineage and interrelatedness of various strains of L. plantarum. These findings have contributed significantly to our understanding of the taxonomy and classification of this species. The detection of genetic elements, such as bacteriocin gene clusters and prophage regions, holds potential for advancements in biotechnology and medicine. However, additional experimental verification is required. The present study offers significant insights; however, it is crucial to recognize its constraints and promote further investigation into the functional characterization, phenotypic traits, and ecological interactions of L. plantarum strains. In general, this study enhances the current pool of knowledge and establishes a foundation for future inquiries and pragmatic applications that exploit the distinctive genetic characteristics of L. plantarum in diverse scientific domains.
Conclusion
In summary, this study provides a comprehensive analysis of the genetic makeup and potential applications of L. plantarum 13–3. The findings highlight its efficient genome, evolutionary relationships, and genetic components with biotechnological and medical relevance. However, further experimental validation and exploration of phenotypic traits and ecological interactions are warranted. This research expands our understanding of L. plantarum and paves the way for future investigations and practical applications utilizing its unique genetic characteristics. Future recommendations include conducting functional characterization of identified genetic components in L. plantarum 13–3 and expanding the study to encompass a larger number of strains for a more comprehensive understanding of the species’ genetic diversity and functional capabilities. Additionally, investigating the phenotypic traits and ecological interactions of L. plantarum strains would provide valuable insights into their practical applications and ecological roles.
Author contributions
Conceptualization, T.A, M.N, M.A.S, A.S, A.W, and Y.Z; methodology, T.A, M.N, M.A.S, A.S, A.A.K, and Y.Z; software, M.A; validation, A.A.S; formal analysis, S.N, and N.N.; investigation, T.A, M.N, M.A., A.S, A.F.A, and Y.Z; resources, Y.Z; M.A and A.A.S.; data curation, A.Z, and I.S.; writing – original draft preparation, T.A, and M.N.; writing – review and editing, M.A., T.H.A, A.F.A; visualization, A.A.S and M.A.S; supervision, Y.Z.; project administration, M.J.R and AZ; funding acquisition, Y.Z.
Supplemental Material
Download MS Word (191.8 KB)Acknowledgments
The authors greatly acknowledge and express their gratitude to the Researchers Supporting Project number (RSPD2024R568), King Saud University, Riyadh, Saudi Arabia.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All the data generated during this research work has been included in this manuscript.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19476337.2024.2329753.
Additional information
Funding
References
- Abdulkarim, I., Mohammed, S., & Orukotan, A. (2020). Gene identification for bacteriocin production by lactic acid bacteria isolated from selected fermented foods. Journal of Biochemistry, Molecular Biology, and Biophysics, 3, 1–10. https://doi.org/10.9734/ajbgmb/2020/v3i430090
- Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., & Wishart, D. S. (2016). PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Research, 44(W1), W16–W21. https://doi.org/10.1093/nar/gkw387
- Aziz, T., Naveed, M., Jabeen, K., Shabbir, M. A., Sarwar, A., Zhennai, Y., Alharbi, M., Alshammari, A., & Alasmari, A. F. (2023). Integrated genome based evaluation of safety and probiotic characteristics of Lactiplantibacillus plantarum YW11 isolated from Tibetan kefir. Frontiers in Microbiology, 14, 1082. https://doi.org/10.3389/fmicb.2023.1157615
- Aziz, T., Sarwar, A., Fahim, M., Al-Dalali, S., Din, Z. U., Din, J. U., Fill, T. P., & Yang, Z. (2020b). Conversion of linoleic acid to different fatty acid metabolites by Lactobacillus plantarum 13-3 and in silico characterization of the prominent reactions. Journal of the Chilean Chemical Society, 65(3), 4879–4884. https://doi.org/10.4067/s0717-97072020000204879
- Aziz, T., Sarwar, A., Fahim, M., Al Dalali, S., Din, Z. U., Din, J. U., Xin, Z., Jian, Z., Fill, T. P., & Zhennai, Y. (2020a). In silico characterization of linoleic acid biotransformation to rumenic acid in food derived Lactobacillus plantarum YW11. Acta Biochimica Polonica, 67, 99–109. https://doi.org/10.18388/abp.2020_5095
- Aziz, T., Sarwar, A., Fahim, M., Din, J. U., Al Dalali, S., Din, Z. U., Khan, A. A., Jian, Z., & Zhennai, Y. (2020c). Dose-dependent production of linoleic acid analogues in food derived Lactobacillus plantarum K25 and in silico characterization of relevant reactions. Acta Biochimica Polonica, 67, 123–129. https://doi.org/10.18388/abp.2020_5167
- Aziz, T., Sarwar, A., Naveed, M., Shahzad, M., Shabbir, M. A., Dablool, A. S., Ud Din, J., Khan, A. A., Naz, S., & Cui, H. (2022). Bio-Molecular analysis of selected food derived Lactiplantibacillus strains for CLA production reveals possibly a complex mechanism. Food Research International, 154, 111031. https://doi.org/10.1016/j.foodres.2022.111031
- Aziz, T., Sarwar, A., Ud Din, J., Al Dalali, S., Khan, A. A., Din, Z. U., & Yang, Z. (2021). Biotransformation of linoleic acid into different metabolites by food derived Lactobacillus plantarum 12-3 and in silico characterization of relevant reactions. Food Research International, 147, 110470. https://doi.org/10.1016/j.foodres.2021.110470
- Boluk, G., Arizala, D., Dobhal, S., Zhang, J., Hu, J., Alvarez, A. M., & Arif, M. (2021). Genomic and phenotypic biology of novel strains of Dickeya zeae isolated from pineapple and taro in Hawaii: Insights into genome plasticity, pathogenicity, and virulence determinants. Frontiers in Plant Science, 12, 663851. https://doi.org/10.3389/fpls.2021.663851
- Chambers, J., Sparks, N., Sydney, N., Livingstone, P. G., Cookson, A. R., & Whitworth, D. E. (2020). Comparative genomics and pan-genomics of the myxococcaceae, including a description of five novel species: Myxococcus eversor sp. nov., Myxococcus llanfairpwllgwyngyllgogerychwyrndrobwllllantysiliogogogochensis sp. nov., Myxococcus vastator sp. nov., Pyxidicoccus caerfyrddinensis sp. nov., and Pyxidicoccus trucidator sp. nov. Genome Biology and Evolution, 12(12), 2289–2302. https://doi.org/10.1093/gbe/evaa212
- Churro, C., Pereira, P., Vasconcelos, V., & Valério, E. (2012). Species-specific real-time PCR cell number quantification of the bloom-forming cyanobacterium planktothrix agardhii. Archives of Microbiology, 194(9), 749–757. https://doi.org/10.1007/s00203-012-0809-y
- De Simone, N., Rocchetti, M. T., la Gatta, B., Spano, G., Drider, D., Capozzi, V., Russo, P., & Fiocco, D. (2022). Antimicrobial properties, functional characterisation and application of Fructobacillus fructosus and Lactiplantibacillus plantarum isolated from artisanal honey. Probiotics and Antimicrobial Proteins, 15(5), 1–18. https://doi.org/10.1007/s12602-022-09988-4
- Ding, W., Baumdicker, F., & Neher, R. A. (2018). PanX: Pan-genome analysis and exploration. Nucleic Acids Research, 46(1), e5–e5. https://doi.org/10.1093/nar/gkx977
- Dronina, J., Samukaite-Bubniene, U., & Ramanavicius, A. (2022). Towards application of CRISPR-Cas12a in the design of modern viral DNA detection tools (review). Journal of Nanobiotechnology, 20(1), 1–15. https://doi.org/10.1186/s12951-022-01246-7
- Gemayel, K., Lomsadze, A., & Borodovsky, M. (2022). MetaGeneMark-2: Improved gene prediction in metagenomes. bioRxiv, 2022.07.25.500264.
- Jiang, Y., Zhang, J., Zhao, X., Zhao, W., Yu, Z., Chen, C., & Yang, Z. (2018). Complete genome sequencing of exopolysaccharide-producing Lactobacillus plantarum K25 provides genetic evidence for the probiotic functionality and cold endurance capacity of the strain. Bioscience, Biotechnology, and Biochemistry, 82(7), 1225–1233. https://doi.org/10.1080/09168451.2018.1453293
- Li, K., Wang, S., Liu, W., Kwok, L.-Y., Bilige, M., & Zhang, W. (2022). Comparative genomic analysis of 455 Lactiplantibacillus plantarum isolates: Habitat-specific genomes shaped by frequent recombination. Food Microbiology, 104, 103989. https://doi.org/10.1016/j.fm.2022.103989
- Mao, B., Yin, R., Li, X., Cui, S., Zhang, H., Zhao, J., & Chen, W. (2021). Comparative genomic analysis of Lactiplantibacillus plantarum isolated from different niches. Genes, 12(2), 241. https://doi.org/10.3390/genes12020241
- Meier-Kolthoff, J. P., Carbasse, J. S., Peinado-Olarte, R. L., & Göker, M. (2022). TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Research, 50(D1), D801–D807. https://doi.org/10.1093/nar/gkab902
- Min, Z., Xiaona, H., Aziz, T., Jian, Z., & Zhennai, Y. (2020). Exopolysaccharides from Lactobacillus plantarum YW11 improve immune response and ameliorate inflammatory bowel disease symptoms. Acta Biochimica Polonica, 67, 485–493. https://doi.org/10.18388/abp.2020_5171
- Ning, W., Wei, Y., Gao, L., Han, C., Gou, Y., Fu, S., Liu, D., Zhang, C., Huang, X., Wu, S., Peng, D., Wang, C., & Xue, Y. (2022). HemI 2.0: An online service for heatmap illustration. Nucleic Acids Research, 50(W1), W405–W411. https://doi.org/10.1093/nar/gkac480
- Nordström, E. A., Teixeira, C., Montelius, C., Jeppsson, B., & Larsson, N. (2021). Lactiplantibacillus plantarum 299v (LP299V®): Three decades of research. Beneficial Microbes, 12(5), 441–465. https://doi.org/10.3920/BM2020.0191
- Olson, R. D., Assaf, R., Brettin, T., Conrad, N., Cucinell, C., Davis, J. J., Dempsey, D. M., Dickerman, A., Dietrich, E. M., Kenyon, R. W., Kuscuoglu, M., Lefkowitz, E. J., Lu, J., Machi, D., Macken, C., Mao, C., Niewiadomska, A., Nguyen, M., Olsen, G. J., & Stevens, R. L. (2022). Introducing the bacterial and viral bioinformatics resource center (BV-BRC): A resource combining Patric, IRD and ViPR. Nucleic Acids Research, 51(D1, D678–D689. https://doi.org/10.1093/nar/gkac1003
- Olson, R. D., Assaf, R., Brettin, T., Conrad, N., Cucinell, C., Davis, J. J., Dempsey, D. M., Dickerman, A., Dietrich, E. M., Kenyon, R. W., Kuscuoglu, M., Lefkowitz, E., Lu, J., Machi, D., Macken, C., Mao, C., Niewiadomska, A., Nguyen, M., & Scheuermann, R. (2023). Introducing the bacterial and viral bioinformatics resource center (BV-BRC): A resource combining PATRIC, IRD and ViPR. Nucleic Acids Research, 51(D1), D678–D689. https://doi.org/10.1093/nar/gkac1003
- Richter, M., Rosselló-Móra, R., Oliver Glöckner, F., & Peplies, J. (2016). JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics, 32(6), 929–931. https://doi.org/10.1093/bioinformatics/btv681
- Surve, S., Shinde, D. B., & Kulkarni, R. (2022). Isolation, characterization and comparative genomics of potentially probiotic Lactiplantibacillus plantarum strains from Indian foods. Scientific Reports, 12(1), 1940. https://doi.org/10.1038/s41598-022-05850-3
- Suryaletha, K., Savithri, A. V., Nayar, S. A., Asokan, S., Rajeswary, D., & Thomas, S. (2022). Demystifying bacteriocins of human microbiota by genome guided prospects: An impetus to rekindle the antimicrobial research. Current Protein and Peptide Science, 23(12), 811–822. https://doi.org/10.2174/1389203724666221019111515
- Syaputri, Y., Lei, J., Hasegawa, T., Fauzia, S., Ratningsih, N., Erawan, T. S., & Iwahashi, H. (2023). Characterization of plantaricin genes and lactic acid production by Lactiplantibacillus plantarum strains isolated from Ishizuchi-Kurocha. Applied Food Biotechnology, 10(1), 21–31. https://doi.org/10.22037/afb.v10i1.39166
- Tenea, G. N. (2022). Decoding the gene variants of two native probiotic Lactiplantibacillus plantarum strains through whole-genome resequencing: Insights into bacterial adaptability to stressors and antimicrobial strength. Genes, 13(3), 443. https://doi.org/10.3390/genes13030443
- Trimble, W. L., Keegan, K. P., D’Souza, M., Wilke, A., Wilkening, J., Gilbert, J., & Meyer, F. (2012). Short-read reading-frame predictors are not created equal: Sequence error causes loss of signal. BMC Bioinformatics, 13(1), 1–10. https://doi.org/10.1186/1471-2105-13-183
- Villena, J., Li, C., Vizoso-Pinto, M. G., Sacur, J., Ren, L., & Kitazawa, H. (2021). Lactiplantibacillus plantarum as a potential adjuvant and delivery system for the development of SARS-CoV-2 oral vaccines. Microorganisms, 9(4), 683. https://doi.org/10.3390/microorganisms9040683
- Volpiano, C. G., Sant’anna, F. H., da Mota, F. F., Sangal, V., Sutcliffe, I., Munusamy, M., Saravanan, V. S., See-Too, W.-S., Passaglia, L. M. P., & Rosado, A. S. (2021). Proposal of carbonactinosporaceae fam. nov. within the class actinomycetia. Reclassification of Streptomyces thermoautotrophicus as carbonactinospora thermoautotrophica gen. nov., comb. nov. Systematic and Applied Microbiology, 44(4), 126223. https://doi.org/10.1016/j.syapm.2021.126223
- Wu, H., Chen, X., Zhang, M., Wang, X., Chen, Y., Qian, C., Wu, J., & Xu, J. (2021). Versatile detection with CRISPR/Cas system from applications to challenges. TrAc Trends in Analytical Chemistry, 135, 116150. https://doi.org/10.1016/j.trac.2020.116150
- Yilmaz, B., Bangar, S. P., Echegaray, N., Suri, S., Tomasevic, I., Manuel Lorenzo, J., Melekoglu, E., Rocha, J. M., & Ozogul, F. (2022). The impacts of Lactiplantibacillus plantarum on the functional properties of fermented foods: A review of current knowledge. Microorganisms, 10(4), 826. https://doi.org/10.3390/microorganisms10040826
- Zhang, J., Zhao, W., Guo, X., Guo, T., Zheng, Y., Wang, Y., Hao, Y., & Yang, Z. (2017). Survival and effect of exopolysaccharide-producing Lactobacillus plantarum YW11 on the physicochemical properties of ice cream. Polish Journal of Food and Nutrition Sciences, 67(3), 191–200. https://doi.org/10.1515/pjfns-2017-0002
- Zhao, Y., Zhang, M., & Yang, D. (2022). Bioinformatics approaches to analyzing CRISPR screen data: From dropout screens to single-cell CRISPR screens. Quantitative Biology, 10(4), 307. https://doi.org/10.15302/J-QB-022-0299