
ABSTRACT
Critically ill patients are physiologically unstable and recent studies indicate that the intestinal microbiota could be involved in the health decline of such patients during ICU stays. This study aims to assess the intestinal microbiota in critically ill patients with and without sepsis and to determine its impact on outcome variables, such as medical complications, ICU stay time, and mortality. A multi-center study was conducted with a total of 250 peri-rectal swabs obtained from 155 patients upon admission and during ICU stays. Intestinal microbiota was assessed by sequencing the V3-V4 hypervariable regions of the 16S rRNA gene. Linear mixed models were used to integrate microbiota data with more than 40 clinical and demographic variables to detect covariates and minimize the effect of confounding factors. We found that the microbiota of ICU patients with sepsis has an increased abundance of microbes tightly associated with inflammation, such as Parabacteroides, Fusobacterium and Bilophila species. Female sex and aging would represent an increased risk for sepsis possibly because of some of their microbiota features. We also evidenced a remarkable loss of microbial diversity, during the ICU stay. Concomitantly, we detected that the abundance of pathogenic species, such as Enterococcus spp., was differentially increased in sepsis patients who died, indicating these species as potential biomarkers for monitoring during ICU stay. We concluded that particular intestinal microbiota signatures could predict sepsis development in ICU patients. We propose potential biomarkers for evaluation in the clinical management of ICU patients.
Introduction
The human gut hosts a massive and diverse number of microorganisms, known as the intestinal microbiota (IM), which coexists in a dynamic equilibrium with the host, relying on molecular cross-talk supported by the exchange of nutrients, metabolites, and protein–protein interactions.Citation1–Citation3 The identification of such microbes and their abundance changes associated with diseases have been broadly described in the latest years. Population-based assessments have revealed a vast inter-individual variability in the human IM. Besides, the human IM is involved in the protection against pathogen arrival, regulation of the intestinal endocrine function, and acting as biofactories for the synthesis of vitamin and cofactors.Citation4,Citation5
Critically ill patients are physiologically unstable given their multiple organ dysfunctions, leading to a risk of death or severe systemic sequelae and thus requiring advanced and specialized life support with continuous monitoring. In critical illness, the gut is considered the driver of associated infectious complications because of the underlying deterioration of the intestinal epithelium, alteration of immune function, and infiltration of the endogenous IM.Citation6 Moreover, these patients exhibit a complex stress response mediated by neuronal, endocrine, and immune mechanisms that is fine balanced through cellular and physiological adaptations.Citation7 Notwithstanding, a dysregulated inflammatory response can produce organ damage and systemic infection, i.e., sepsis, hence increasing the risk of medical complications, longer hospital stays, mortality and additional cost for public health systems.Citation8,Citation9
When compared with healthy subjects, critically ill patients harbor pathogenic microorganisms that can out-compete indigenous species of the human IM, thus resulting in the loss of beneficial microbial species.Citation10 That unbalance between harmful and healthy microbes in the IM of critically ill patients is thought to be promoted by the host response to physiological stress and invasive and pharmacological medical care treatments prescribed (e.g. antibiotics, insertion of feeding tubes, surgery, etc.).Citation11,Citation12 The changes in the IM diversity of critically ill patients after ICU admission seem to combine a reduction in strict anaerobes and increase in pathogenic species, thus associated with an increased mortality.Citation3,Citation13 Furthermore, a previous report indicated that prolonged critical illness in ICU patients produces profound alterations in the structure of the IM leading to the microbial diversity vanishing to almost-null levels, with concomitant boosting of the virulence of those ultra-low diversity communities by opioid and antibiotic administration.Citation14
The management of the IM on critically ill patients is a current trend of clinical research. All in all, it is considered as a pivotal factor that under control and surveillance could improve survival rates in ICU patients. Consequently, different IM-associated strategies to tackle adverse events in ICU patients have been proposed, mostly to prevent the growth of pathogens and improve patient prognosis;Citation15 e.g. the decontamination of the digestive tract (SDD), administration of probiotics and prebiotics alone or in combination (synbiotics), and fecal microbiota transplantation (FMT).Citation3,Citation16,Citation17
The present study aims to describe the IM profiles in critically ill ICU patients, with and without sepsis, and try to shed light on the role of IM as contributing factor in the evolution of these patients and its influence on ICU stay time, development of different medical complications, and mortality. Additionally, we explored several variables recorded upon ICU admission and stay to determine IM covariates and potential confounding variables in this cohort. Longitudinal evaluation of patients with sepsis was aimed at determining predictive variables from different descriptors of the IM assessment.
Results
summarizes the initial assessment of patients grouped according to sepsis status. Important differences were found in age and ICU stay time. As expected, differences in the CRP concentration and SOFA score were indicative of sepsis and supported the ICU patient grouping. Thirty-two out of the nonseptic patients (38.6%) developed a hospital-acquired infection with multiple origins. On the other hand, the pulmonary (56%) and abdominal (17%) infections were determined as the predominant origin of sepsis in such patients.
Table 1. Demographic, biochemical, and clinical evaluation of ICU patients.
The IM of ICU patients upon admission
Initial assessment of the microbial diversity of M1 samples and the microbial community structure between groups was performed with alpha- and beta-diversity approaches, respectively, using the abundance and prevalence information of OTUs recovered. Taking into account all four alpha diversity parameters evaluated, we observed that sepsis samples had no meaningful differential distributions of diversity descriptors compared to their no-sepsis counterparts (Figure S1).
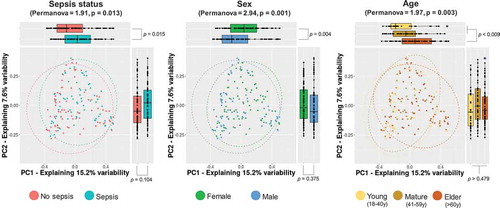
Beta-diversity assessment indicated that sepsis conditions could drive changes in the microbial community structure (PERMANOVA = 1.91, p = .013). However, we found that other variables could modulate the IM as well. As a result, sex and age seemed to influence the microbiota profiles to a larger extent than other parameters (PERMANOVA = 2.94, p < .001 and PERMANOVA = 1.97, p = .003 for sex and age, respectively) (). Moreover, we found that variables such as housing (PERMANOVA = 1.29, p = .083) and ICU venue (PERMANOVA = 1.22, p = .081) could also shape the IM to a lesser extent. An extra matched case–control analysis was done to try replicating the above patterns when comparing homogenous groups (N = 140) in terms of age (p = .243) and sex distributions (p = .061). Consequently, we retained significant microbiota structures only when discriminated by sex (PERMANOVA = 2.18, p = .002) and age (PERMANOVA = 1.81, p = .006). Conversely, no differential IM structure was distinguished in M1 samples by sepsis status (p = .098).
Figure 1. Microbial community structure of the study groups. Principal coordinate analysis (PCoA) of multidimensional data is drawn to display changes in microbial communities according to major variables retrieved to shape the IM, sepsis status, sex, and age. The x- and y-axes represent the two most informative principal coordinates (PCs) of the PCoA, and marginal boxplots describe the distribution of those values for the different groups. Color legends represent the respective variables under analysis. Blue-shaded points show outliers. A pairwise Wilcoxon rank-sum test was used to compare PC1 or PC2 values between groups, and p-values are shown beside marginal boxplots. The results of the permutation-based test (PERMANOVA) to compare dissimilarity indexes among samples are shown on top of plots accordingly.
The application of an LMM for the integration of demographic, biochemical, and clinical records with microbiota data produced results similar to those from beta-diversity analysis, thus indicating that age and sex were essential covariates of the IM in the study subjects. Furthermore, we identified several OTUs tightly associated with other variables in the metadata (p < .01) (). For instance, housing, SOFA score, APACHE score, and ICU venue exhibited a strong association with a large number of the most abundant OTUs explored (). Minor associations with the IM were found for variables such as ICU stay, ICU discharge, BMI, CRP level, breastfeeding status during infancy, and pet ownership. No associations with medical complications or hospital-acquired infections were detected. A Venn analysis was used to discern overlapping OTUs influenced (according to the LMM results) by related clinical variables such as sepsis status, CRP level, SOFA score, and APACHE score. We identified a high specificity, indicating that OTUs are almost strictly associated with each variable except for sepsis status and SOFA comparison, for which 19 OTUs were found to be simultaneously influenced by both variables. Such a relationship was expected because of the dependence of sepsis diagnosis on SOFA scoring.
Table 2. Interaction among the most prevalent OTUs and recorded variables.
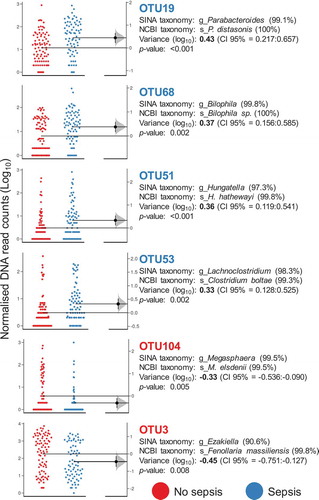
Finally, we attempted to discern a specific IM signature associated with sepsis status by removing the potential effect of all latent confounding variables (see methods). Consequently, after performing a new LMM analysis including significant covariates as random effects, we could retrieve a set of 24 OTUs specifically associated with the sepsis condition, and 7 out of them (OTU3 = Ezakiella spp., OTU104 = Megasphaera spp., OTU276 = Allisonella spp., OTU394 = unclassified, OTU453 = Peptoniphilus spp., OTU458 = Prevotella spp., OTU523 = Prevotella copri) were found to be in a lower proportion in samples from patients with sepsis than in those from patients without sepsis. shows the OTUs with extreme variance between groups (4 associated with sepsis and 2 with no sepsis). Moreover, we also identified that Fusobacterium species (OTU48, variance = 0.29, p = .003) were associated with ICU patients with sepsis, whereas Prevotella species (OTU458 and OTU523, variance = −0.27, p < .001) seemed to be more abundant in ICU patients without sepsis.
Figure 2. OTUs associated with the sepsis condition. Gardner–Altman estimation plots showing the distribution of the number of rarefied DNA reads obtained for OTUs with extreme variation between groups. In all cases, the variance is reported on a log scale and is referred to as observed in the “No sepsis” group (p ≤ 0.01) and accompanied by confidence intervals (CI 95%). The color legend represents the primary variable of the study, sepsis status. SINA aligner (https://www.arb-silva.de/aligner/) with the SILVA database and a Blast-based search against the non-redundant NCBI 16S database (https://blast.ncbi.nlm.nih.gov/Blast.cgi?) were used as methods to disclose the taxonomy of selected OTUs. The sequence identity percentage is shown within parentheses. The distribution of unpaired mean differences between groups (based on 5000 replicates) is shown on the right of the respective Gardner–Altman plots.
Short-term microbiota evolution in sepsis patients during ICU stays
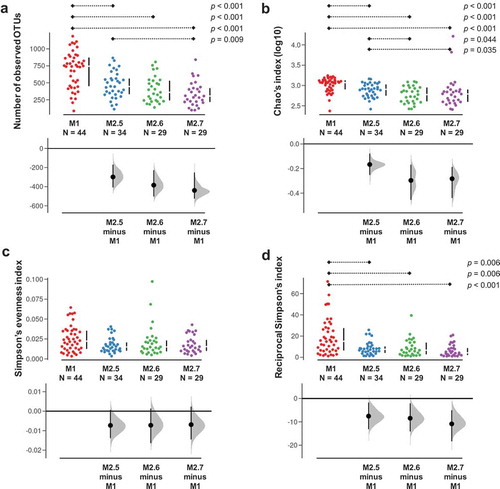
A complementary LMM-based analysis was conducted to survey the evolution of the IM during an ICU stay. A total of 44 sepsis patients with 135 longitudinal samples (M1 = 44 samples and M2 = 91 samples) were evaluated to discern potential changes in the diversity of samples across the ICU stay and to detect covariates in the metadata recorded during the stay. The alpha diversity assessment strongly suggested that diversity profoundly declined in M2 samples from that observed in M1 specimens (). A progressive pattern of microbial species loss was observed when observed OTUs and Chao’s index were evaluated, reaching a decline of 850 phylotypes in M2.7 samples ()). We calculated an average loss of 278 phylotypes from the 692 estimated average for M1 samples. A predictive death model based on logistic regression using the delta-observed richness (M2 minus M1 richness) indicated no exacerbated loss of microbial richness in patients that were dead upon ICU discharge.
Figure 3. Alpha diversity analysis of longitudinal samples from the sepsis groups. The observed OTUs (a), Chao’s index (b), Simpson’s evenness (c), and Simpson’s reciprocal index (d) were assessed across the M1, M2.5, M2.6, and M2.7 samples. Statistical assessment was carried out with the pairwise Wilcoxon rank-sum test for unpaired samples with the post hoc Benjamini–Hochberg method for multiple testing correction, and p-values derived from respective tests are depicted on top of Gardner–Altman estimation plots accordingly (p ≤ 0.05). Distributions at the bottom of the plots show the unpaired median difference based on 5000 replicates.
In contrast, the main variables likely to shape the IM during follow-up were age, with 49 OTUs associated; the antibiotic treatment (grouped into cephalosporins, glycopeptides, macrolides, oxazolidinones, penicillins, and carbapenems), with 24 different OTUs associated; and the caloric intake reached by nutritional support, with 17 different OTUs associated (). The impact of sex seemed to influence the IM to a lesser extent in the longitudinal cohort.
Table 3. Covariates influencing the IM during follow-up of sepsis patients.
We found only three OTUs associated with the ICU discharge variable (p ≤ 0.01), whose association was retained after expanding the set of random variables in the LMM model to those that exhibited a large influence on microbiota data in either the initial assessment or the longitudinal assessment (see methods). Consequently, Enterococcus (OTU45, variance = 0.63, p = .006 and OTU46, variance = 059, p = .008) and Salmonella (OTU110, variance = 0.41, p = .010) species were strongly associated with death as result for ICU discharge. ) shows the abundance of OTU45 and OTU46 in samples across the ICU stay and shows a higher abundance and prevalence of such OTUs in samples of patients with sepsis who died than in samples of patients with sepsis who lived. Logistic regression was used to build a potential predictive model of death in the ICU based on changes associated with the abundance of Enterococcus species (OTU45 and OTU46). We observed that changes in the abundance of OTU46 better predicted the outcome than changes in OTU45 abundance, suggesting that an increase of one logarithmic unit in the abundance of this phylotype leads to an increase of 3.14-fold in the probability of death in the ICU by sepsis (p = .029) ()).
Figure 4. OTUs associated with death. A – The normalized DNA read counts (log10) for OTU45 and OTU46 are depicted in a boxplot manner for sepsis samples across the time (M1 to M2.7) of stay in the ICU. The color legend discriminates the samples from patients with ICU discharge as “alive” and “dead”. Blue-shaded points indicate outliers. B – A logistic regression with data regarding OTUs potentially related to death of ICU patients with sepsis, based on their abundance changes across the ICU stay. Abundance changes were calculated as log[average(M2 samples)] – log[M1 samples]. The OTU information and the associated taxonomy as well as main parameters retrieved after logistic regression, such as the odds ratio (OR), Akaike information criterion (AIC), and p-values, are shown inside the plot.
![Figure 4. OTUs associated with death. A – The normalized DNA read counts (log10) for OTU45 and OTU46 are depicted in a boxplot manner for sepsis samples across the time (M1 to M2.7) of stay in the ICU. The color legend discriminates the samples from patients with ICU discharge as “alive” and “dead”. Blue-shaded points indicate outliers. B – A logistic regression with data regarding OTUs potentially related to death of ICU patients with sepsis, based on their abundance changes across the ICU stay. Abundance changes were calculated as log[average(M2 samples)] – log[M1 samples]. The OTU information and the associated taxonomy as well as main parameters retrieved after logistic regression, such as the odds ratio (OR), Akaike information criterion (AIC), and p-values, are shown inside the plot.](/cms/asset/da21e65f-45f9-41b7-850a-8fc74970b9de/kgmi_a_1707610_f0004_oc.jpg)
Alternatively, we used the abundance changes of OTU110 as well as of OTU19 and OTU68, the last two exhibited extreme associations with sepsis status (), to test alternative models, but their longitudinal variation fitted poorly to a predictive model (OR = 1.12, 1.47 and 0.89; AIC = 40.4, 44.9 and 45.7; and p = .053, 0.362 and 0.835, respectively). We observed no differences between the ICU time, medical complications, and ICU discharge criteria in this longitudinal cohort.
Discussion
Critically illness promotes severe physiology alterations in patients, and it has been reported to drive drastic changes in the IM composition.Citation8 Currently, the IM is indicated as a key player in maintaining metabolic and immune homeostasis in humans, which indicates that any alteration during critical illness regarding the proper balance of species inhabiting the human gut could have devasting effects on ICU patient progression.Citation18 In the present study, we found no apparent differences in terms of alpha diversity descriptors of the IM between patients with and without sepsis upon ICU admission.
Unlike alpha diversity, beta diversity evaluation suggested that critically ill patients with sepsis have an intestinal microbial community structure that is distinct from that of those without sepsis (p = .013). Nevertheless, we found that variables such as age and sex definitively influence the IM to a larger extent than the sepsis condition itself ( and ), this last discarded to modulate, in a specific manner, the IM structure as a whole according to the matched control-case analysis. The overlapping distribution in IM profiles among samples of sepsis patients, women, and elderly people in the PCoA () suggests that, for this Colombian population, the women older than 60 years condition would represent an elevated risk of developing sepsis after ICU admission.
Our results are consistent with previous findings of different IM surveys in elderly individuals that outlined a drastic change in their IM as a consequence of the intestinal physiology deterioration, poor diet, and permanent medication.Citation19,Citation20 Therefore, a loss of microbial diversity and change in the microbiome functionality is frequently associated with age,Citation21,Citation22 making this population more disease prone. However, a disparate IM between men and women has been previously reported, as well as its covariation with BMI.Citation23,Citation24 Interestingly, this IM dissimilarity between men and women is proposed to underlie the gender-specific response to dietary interventionsCitation25 and to potentially modulate disease onset.Citation26 Consequently, our data indicate that the IM of women could also be involved in sepsis predisposition in ICU patients. In any case, the co-variability of sex and age with the IM detected in our study reinforces the idea to take into account such information in the context of association-based research involving human microbiota data, with the aim to minimize the confounding factors generating spurious associations.Citation27
After proper control of several covariates found to influence the IM of ICU patients, we were able to isolate a set of 24 OTUs with differential abundance between sepsis and non-sepsis subjects upon ICU admission. Nineteen of them were associated with sepsis samples, including OTU19 (Parabacteroides distasonis) and OTU68 (Bilophila spp.), which were the phylotypes exhibiting the most extreme association with sepsis samples (). Whereas P. distasonis has been associated with endotoxin production, increased risk of mortality, antibiotic resistance,Citation28 and surface antigen-mediated attenuation of the immune response,Citation29 the genotoxic sulfide-producing Bilophila species are consistently associated with inflammation across studies.Citation30–Citation32 Furthermore, the higher abundance of Fusobacterium species (OTU48) in sepsis samples was already reported for ICU patients with septic shock.Citation33 Accordingly, taking into account the association of Bilophila spp. and Fusobacterium spp. with the onset and progression of colorectal cancer,Citation34–Citation36 we found that the IM of ICU patients with sepsis was enriched in harmful microbial species that would magnify the disruption of metabolic and immune homeostasis in these critically ill patients. Conversely, we found that 3 out of the 7 OTUs associated with non-sepsis samples were microbial species that are abundant in non-Westernized populations with traditional lifestyles. Therefore, the increased abundance of Ezakiella, the butyrate producer MegasphaeraCitation37 () and Prevotella (and particularly P. copri) species could confer a protective role against sepsis. Consequently, strategies of plant-based enteral nutrition could raise the proportion of beneficial microbial species associated with complex carbohydrate metabolism in the IM of critically ill patients, thus ameliorating the intestinal barrier and function as a whole, reducing the risk of septic shock and improving prognosis during the ICU stay.Citation38
We followed up a total of 44 out of the 72 sepsis patients (135 samples in sum) to determine the short-term evolution of their IM and its main shaping factors during the ICU stay. Strikingly, our follow-up survey in sepsis patients replicated previous results indicating a substantial loss of microbial diversity during ICU stay ().Citation14 As a result, while being aware of the controversy on the definition,Citation39 we observed a clear dysbiosis understood as a drastic reduction in the proportion of microbial species along the time between samples from the same donor. Although we observed no acute diversity depletion as Zaborin and coworkers described, we did detect loss of up to 850 phylotypes (~71% of the initial observed OTUs for a specific patient) after a maximum 7-day ICU stay and an average loss of 278 (~40%) phylotypes of the 692 observed for paired M1 samples on average. This loss of microbial diversity is expected given that the IM of sepsis patients is exposed to adverse effects, such as antibiotic administration, and drastic changes in dietary patterns during lengthened ICU stays. Globally, such a reduction in the effective number of microbial species in the ICU patient intestine is thought to break the equilibrium among species, reducing the number of beneficial commensals and promoting the arrival/proliferation of harmful and pathogenic bacteria.Citation40–Citation42 In this regard, some reports have previously suggested the use of variations in the intestinal microbial diversity and its longitudinal variation as a biomarker for the prognosis of ICU patients.Citation43
The data presented here strengthen the idea that a dramatic decrease in microbial diversity precedes the arrival of harmful bacteria in ICU patients. In our case, we detected the loss of microbial species, such as Megasphaera spp., Prevotella spp. and Ezakiella spp., in sepsis patients, gut microbes associated with a traditional diet, metabolism of complex carbohydrates, and being SCFAs producers. Notwithstanding, we disclosed unprecedently that such conditions would constitute a hallmark for the proliferation of pathogenic bacteria, such as Parabacteroides distasonis, Bilophila spp., Fusobacterium spp., and Enterococcus spp., and that the increased abundance over time would constitute a serious risk of mortality for ICU patients with sepsis ().
Among the different variables explored to influence the IM of sepsis patients, we again found that age is the main variable to determine the evolution of the IM during an ICU stay (). The above finding was evidenced by retrieving a total of 49 different OTUs influenced by this variable (p < .01). We found a unique OTU associated positively with age, which indicates that Klebsiella (OTU2) species become more abundant as patients increase in age. In contrast, the abundance of species associated with complex carbohydrate metabolism and traditional diets, such as Prevotella spp.Citation44,Citation45 (OTU10), were found to be less abundant in older patients than in younger patients. As expected, antibiotic treatment also seemed to exert an important influence in the IM of sepsis patients, and we found a total of 24 OTUs differentially associated with the seven classes of antibiotics recorded. Surprisingly, a higher abundance of Pseudomonas spp. (OTU26) and Klebsiella (OTU2) was correlated with the administration of oxazolidinones and glycopeptides, respectively. This result suggests the possibility of acquisition of respective resistant and pathogenic strains during treatment in the ICU. Contrary to that observed in the assessment of M1 samples, sex had a lesser extent impact on the IM in the follow-up data (). Nonetheless, Fusobacterium spp. (OTU1070) abundance was positively associated with women, thus emphasizing the risk of such condition to develop sepsis in the ICU in our study cohort.
Similarly, we could associate microbiota evolution with the main variables that were explored in the present study. Consequently, we retrieved particular associations between the microbiota and ICU stay time as well as between the microbiota and mortality, but no associations were observed for the microbiota with medical complications during an ICU stay. Remarkably, after controlling all possible covariates detected to influence the microbiota in the follow-up data, we retained the 3 OTUs associated with mortality, and two of them matched with Enterococcus species (OTU45 and OTU46) (). An increased abundance of vancomycin-resistant Enterococcus (VRE) has been previously reported in critically ill patients when prior depletion of SCFA-producing bacteria occurs.Citation46 Indeed, the reduction in the concentration of butyrate, an effector molecule mediating anti-inflammatory signals and glucose and lipid metabolism, in the intestine has been specifically associated with the arrival of Enterococcus species.Citation10
Enterococcus species have been reported to be recurrent members of the human IM, but they are also well-known nosocomial pathogens associated with urinary tract infections, bacteremia, and infectious endocarditis with elevated rates of mortality.Citation47 Although our study did not have the resolution to determine variability at the species or strain level, therefore failing to unveil if the enterococci boost was of endogenous or acquired origin, we did detect a progressive and significant increase in Enterococcus species abundance in the intestine of ICU patients with sepsis who died, making it feasible to use it as a predictive biomarker, as previously stated.Citation42 Moreover, we could also infer some resistance patterns for such microbes because all deceased patients with sepsis (N = 8) were treated with beta-lactam antibiotics; 3 of them were additionally treated with glycopeptides (vancomycin), and 4 of them were additionally treated with macrolides or lincosamides. Accordingly, future studies should be conducted to profile the precise identity and resistance traits of the Enterococcus species exhibiting progressive growth in the intestine of ICU patients with sepsis.
Conclusions
Exhaustive metadata recording is essential to outline reliable associations and minimize the impact of confounding factors in human-associated microbiota assessments. Our multiple covariate-controlled multi-center study indicated that the IM in ICU patients could be informative to determine their progression during an ICU stay, as well as for calculating the risk for sepsis and mortality. We concluded that upon ICU admission, a microbiota evaluation could help to predict the risk of sepsis and mortality by measuring the abundance of certain harmful gut microbes. We observed that such a pathobiont-prone profile in sepsis ICU patients would worsen immune and metabolic homeostasis and exacerbate health deterioration. Overall, we concluded that declining IM diversity during an ICU stay deeply alters the delicate microbial equilibrium in critically ill patients and allows the emergence and proliferation of specific pathogens, aggravating their already weakened condition, even resulting in death. Finally, we encourage health-care institutions to adopt molecular protocols to follow up the evolution of the IM in critically patients during their ICU stay for monitoring the efficacy of treatments and making prognosis more reliable.
Methods
Study design and subjects
This is a descriptive observational, multi-center study carried out with patients from five hospitals located in Medellín and Rionegro (Colombia, South America). We used samples and data from a cohort of 155 adult patients admitted to the respective ICUs from fourth-level hospitals (the top-level at the Colombian health-care system) namely, Hospital Pablo Tobón Uribe, Hospital San Vicente Fundación venues in Medellín and Rionegro, Hospital General de Medellín and Clinica Las Américas. Patients or their relatives were personally informed of the study with the presence of the responsible clinician and signed informed consent forms. Eligible men and women were at least 18 years old and admitted to the ICU of their respective hospitals. ICU patients with terminal illnesses as well as those undergoing colostomy or ileostomy were excluded. Pregnant women or those with breastfed infants and homeless people were also excluded from this study. The sepsis criterion to discern the groups of study was adopted according to The Third International Consensus Definitions for Sepsis and Septic Shock.Citation48 During a 6-month window, a total of 83 ICU patients without sepsis and 72 diagnosed with sepsis were included in the study according to inclusion and exclusion criteria. Then, biological samples were obtained, and metadata were recorded to complete this assessment.
Sampling and data recording
Peri-rectal swabs were obtained at ICU admission upon inclusion criteria verification and before nutritional support, using brand-sealed sterile, polystyrene, and RNAse and DNAse free swabs (Deltalab, Barcelona, Spain. Cat# 300252). These rectal swabs were DNA-free certified (sterilized by ethylene oxide) and provided with a polystyrene-made protection tube containing no storage buffer that could alter the microbiota profiles. The peri-rectal swabs were immediately stored at −20ºC in the respective hospitals and then transported in dry ice to a DNA extraction laboratory. Samples were stored at −80ºC until DNA extraction. The initial sampling moment (M1) was set for all subjects included in the “no-sepsis” and “sepsis” groups. Follow-up sampling was performed for 44 out of the 72 sepsis patients who remained in the ICU until M2 sampling. The M2 samples consisted of peri-rectal swabs obtained on the fifth (M2.5), sixth (M2.6), and/or seventh (M2.7) day in the ICU, whereas the M3 samples consisted of peri-rectal swabs collected at ICU discharge whenever possible (N = 3). Demographic, biochemical and clinical variables of interest were collected with respective M1 samples as patient-associated metadata, such as age, weight, height, BMI, sex, marital status, housing, pets, glucose, CRP level, sibling number, whether they were breastfed in infancy, ICU venue, sepsis status and the scores obtained from SOFA and APACHE-II assessments. Additional variables associated with the follow-up, such as the type of nutritional support, formula of nutritional support, antibiotic administration, and outcome variables (ICU stay [days], complications in the ICU, discharge condition), were also recorded. The staff from different ICUs collected the samples and information following consensus protocols agreed upon and approved by the clinicians and researchers involved in this study.
Fecal microbiota
The fecal DNA was obtained from peri-rectal swabs by using a QIAamp® DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Total DNA was quantified by UV absorbance methods (NanoDrop, Thermo Scientific, Wilmington, USA) and stored at −20ºC until processing. The V3-V4 hypervariable regions of the 16S ribosomal ribonucleic acid (rRNA) gene were amplified using 1 μL of fecal DNA (25 ng on average) and 27 PCR cycles consisting of the following steps: 95ºC for 20 sec., 55ºC for 20 sec. and 72ºC for 20 sec. Phusion High-Fidelity Taq Polymerase (Thermo Scientific, Wilmington, USA) and the barcoded primers S-D-Bact-0341-b-S-17 (TAGCCTACGGGNGGCWGCAG) and S-D-Bact-0785-a-A-21 (ACTGACTACHVGGGTATCTAATCC) that target a wide variety of bacterial 16S rRNA genesCitation49 were used for PCR. Dual-barcoded PCR products were purified from triplicate reactions with an Illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare, UK) and quantified by fluorometric methods (Qubit 3.0 – Thermo Fisher Scientific, Waltham, MA, USA). A total of 250 samples were multiplexed in one sequencing run by combining equimolar quantities of amplicon DNA (~50 ng per sample) and sequenced in one lane of an Illumina MiSeq platform with a 2 × 300 PE configuration (Eurofins Genomics GmbH, Germany). Raw data were delivered in fastq files, and paired ends with quality filtering were assembled using FLASH software.Citation50 Sample de-multiplexing was carried out using sequence information from the respective DNA barcodes and MOTHUR v1.39.5 suite of analysis.Citation51 After assembly and barcode and primer removal, the sequences were processed for chimera detection and removal using the UCHIME algorithmCitation52 and the SILVA reference set of 16S sequences (release 128).Citation53 A rarefied subset of 12,000 sequences per sample was randomly selected after multiple shuffling (10,000X) from the original dataset for downstream analyses. Operational taxonomic unit (OTU) counts were retrieved by using this rarefied set of sequences and the UCLUST algorithm (clustered at 97% sequence identity) implemented in USEARCH v8.0.1623.Citation54 Common alpha diversity descriptors, including the observed OTUs, Chao’s richness, Simpson’s evenness, and Simpson’s reciprocal index, were computed using QIIME v1.9.1Citation55 and the OTU abundance information. The beta diversity was also assessed with the respective algorithms implemented in QIIME v1.9.1, and evaluation of the community structure across the sample groups was assisted by principal coordinate analysis (PCoA) and Bray–Curtis dissimilarity indexes retrieved from sample pairwise comparisons.
Statistical analysis
Descriptive statistics were applied to assess compositional differences between groups regarding demographic, biochemical and clinical variables. The Shapiro–Wilk test was used to measure normality of the explored continuous variables before comparison using parametric (t-test) or non-parametric (Wilcoxon rank-sum test) methods. Similar approaches were adjusted for pairwise comparison of alpha diversity descriptors. Group differences in the distribution of categorical variables were analyzed using Pearson’s chi-squared test. Permutation-based methods (PERMANOVA) were applied to measure changes in microbial communities associated with variables recorded as metadata. A matched case–control approach was additionally executed to detect the impact of gut microbiota on sepsis condition by homogenizing sepsis and no-sepsis groups in terms of sex and age variables. Proper selection of controls (no sepsis) was carried out using the matchControls function of the e01071 R package. A total of 140 samples (70 sepsis and 70 no-sepsis samples) were included in this re-analysis. A linear mixed model (LMM – nlme R package) was used to measure the association of log-transformed OTU data, with individual variables recorded in the metadata. This analysis was performed on the most abundant OTUs (N = 377 with an average abundance >0.02%), accounting for 84% of the full diversity observed in the sample cohort. Idiosyncratic variation due to individual differences was set as a random effect for each variable analyzed (fixed effect). Covariates of the fecal microbiota were identified upon differential distribution of variables among groups supported by a p-value ≤0.01. OTUs specifically associated with the sepsis condition were retrieved by including age, sex, BMI, ICU venue (hospital center), APACHE, and housing as random effects in the LMM, in addition to individual variation. OTUs categorically associated with the ICU discharge condition were retrieved by including age, sex, BMI, caloric intake depending on nutritional support, ICU venue (hospital center), antibiotic treatment, and nutritional support as random effects in the LMM, in addition to individual variation. To establish possible relationships between changes in the abundance of OTUs and ICU discharge condition (alive or dead), a logistic regression model was applied, using the glm [family = “binomial”(link = “logit”))] function of R v3.5; ICU discharge condition was used as a binary outcome (1 = alive and 0 = dead) and changes in the relative abundance of different OTUs (abundance change = Log10 [average of normalized reads of M2 samples] – Log10 [normalized reads of M1 samples]) as an explanatory variable. Graphics (ggplot2 and dabestr packages for boxplots and Garner–Altman estimation plots, respectively) and statistics were generated in R v3.5 (https://cran.r-project.org/).
List of abbreviations
| AIC | = | Akaike information criterion |
| APACHE | = | acute physiologic assessment and chronic health evaluation |
| BMI | = | body mass index |
| CRP | = | c-reactive protein |
| DNA | = | deoxyribonucleic acid |
| FMT | = | faecal microbiota transplantation |
| ICU | = | intensive care unit |
| IM | = | intestinal microbiota |
| LMM | = | linear mixed model |
| OTU | = | operational taxonomic unit |
| OR | = | odds ratio |
| PC | = | principal coordinate |
| PCoA | = | principal coordinate analysis |
| PCR | = | polymerase chain reaction |
| RNA | = | ribonucleic acid |
| SDD | = | selective decontamination of the digestive tract |
| SOFA | = | sequential organ failure assessment. |
Authors’ contributions
GMAO, BEVD, NAGG, and ABP designed the study, conducted the research, performed the experimental analyses, and managed and obtained the resources to carry out the assessment. ABP analyzed and interpreted the molecular data. AMJR, AGV, IAC, MAYM, and JBB selected patients and coordinated sampling, storage, and transport of the biological samples. ABP wrote the manuscript. ABP and GMAO have primary responsibility for the final content. All authors helped in manuscript preparation and read and discussed the results and conclusions included. All authors read and approved the final version of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Data availability and material
The raw fastq sequences generated from the 16S amplicon sequencing of fecal DNA are publicly available at the European Nucleotide Archive (ENA)Citation56 via the project number PRJEB33360.
Ethics approval and consent to participate
This study was conducted according to the guidelines laid down in the Declaration of Helsinki and was carried out in accordance with the principles of the Belmont Report and with the approval of the ethics committees from the University of Antioquia (Act 03 of 2015), Hospital Pablo Tobón Uribe, Hospital San Vicente Fundación, and Clínica Las Américas.
Supplemental Material
Download Zip (270 KB)Acknowledgments
The authors thank all ICU nursing and laboratory staff from the health-care institutions involved in this study as well as the relatives of the recruited patients. The authors also want to thank Vivian Cepeda, Estefanía Cano, Katherine Correa, Luisa Fernanda Arroyave, and Laura Guerrero, all of which are students of the Nutrition and Dietetics programme of the Universidad de Antioquia, for helping in different stages of this multi-center work.
Supplementary material
Supplemental data for this article can be accessed publisher’s website.
Additional information
Funding
Related Research Data
References
- Jacobs MC, Haak BW, Hugenholtz F, Wiersinga WJ. Gut microbiota and host defense in critical illness. Curr Opin Crit Care. 2017;23:257–16. doi:10.1097/MCC.0000000000000424.
- Shimizu K, Ogura H, Hamasaki T, Goto M, Tasaki O, Asahara T, Nomoto K, Morotomi M, Matsushima A, Kuwagata Y, et al. Altered gut flora are associated with septic complications and death in critically ill patients with systemic inflammatory response syndrome. Dig Dis Sci. 2011;56:1171–1177. doi:10.1007/s10620-010-1418-8.
- Shimizu K, Yamada T, Ogura H, Mohri T, Kiguchi T, Fujimi S, Asahara T, Yamada T, Ojima M, Ikeda M, et al. Synbiotics modulate gut microbiota and reduce enteritis and ventilator-associated pneumonia in patients with sepsis: a randomized controlled trial. Crit Care. 2018;22:239. doi:10.1186/s13054-018-2167-x.
- D’Argenio V, Salvatore F. The role of the gut microbiome in the healthy adult status. Clin Chim Acta. 2015;451:97–102. doi:10.1016/j.cca.2015.01.003.
- Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. N Engl J Med. 2016;375:2369–2379.
- Mittal R, Coopersmith CM. Redefining the gut as the motor of critical illness. Trends Mol Med. 2014;20:214–223. doi:10.1016/j.molmed.2013.08.004.
- Cuesta JM, Singer M. The stress response and critical illness: a review. Crit Care Med. 2012;40:3283–3289. doi:10.1097/CCM.0b013e31826567eb.
- Dickson RP. The microbiome and critical illness. Lancet Respir Med. 2016;4:59–72. doi:10.1016/S2213-2600(15)00427-0.
- Haak BW, Wiersinga WJ. The role of the gut microbiota in sepsis. Lancet Gastroenterol Hepatol. 2017;2:135–143. doi:10.1016/S2468-1253(16)30119-4.
- Wolff NS, Hugenholtz F, Wiersinga WJ. The emerging role of the microbiota in the ICU. Crit Care. 2018;22:78. doi:10.1186/s13054-018-1999-8.
- Heneghan AF, Pierre JF, Tandee K, Shanmuganayagam D, Wang X, JD R. Parenteral nutrition decreases paneth cell function and intestinal bactericidal activity while increasing susceptibility to bacterial enteroinvasion. JPEN J Parenter Enteral Nutr. 2014;38:817–824.
- Ruppe E, Lisboa T, Barbier F. The gut microbiota of critically ill patients: first steps in an unexplored world. Intensive Care Med. 2018;44:1561–1564. doi:10.1007/s00134-018-5309-3.
- Shimizu K, Ogura H, Goto M, Asahara T, Nomoto K, Morotomi M, Yoshiya K, Matsushima A, Sumi Y, Kuwagata Y, et al. Altered gut flora and environment in patients with severe SIRS. J Trauma. 2006;60:126–133. doi:10.1097/01.ta.0000197374.99755.fe.
- Zaborin A, Smith D, Garfield K, Quensen J, Shakhsheer B, Kade M, Tirrell M, Tiedje J, Gilbert JA, Zaborina O, et al. Membership and behavior of ultra-low-diversity pathogen communities present in the gut of humans during prolonged critical illness. MBio. 2014;5:e01361–14. doi:10.1128/mBio.01361-14.
- Haak BW, Levi M, Wiersinga WJ. Microbiota-targeted therapies on the intensive care unit. Curr Opin Crit Care. 2017;23:167–174. doi:10.1097/MCC.0000000000000389.
- Davison JM, Wischmeyer PE. Probiotic and synbiotic therapy in the critically ill: state of the art. Nutrition. 2019;59:29–36. doi:10.1016/j.nut.2018.07.017.
- McClave SA, Patel J, Bhutiani N. Should fecal microbial transplantation be used in the ICU? Curr Opin Crit Care. 2018;24:105–111. doi:10.1097/MCC.0000000000000489.
- Fay KT, Ford ML, Coopersmith CM. The intestinal microenvironment in sepsis. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2574–2583. doi:10.1016/j.bbadis.2017.03.005.
- Jeffery IB, Lynch DB, O’Toole PW. Composition and temporal stability of the gut microbiota in older persons. Isme J. 2016;10:170–182. doi:10.1038/ismej.2015.88.
- Konturek PC, Haziri D, Brzozowski T, Hess T, Heyman S, Kwiecien S. Emerging role of fecal microbiota therapy in the treatment of gastrointestinal and extra-gastrointestinal diseases. J Physiol Pharmacol. 2015;66:483–491.
- Rampelli S, Candela M, Turroni S, Biagi E, Collino S, Franceschi C. Functional metagenomic profiling of intestinal microbiome in extreme ageing. Aging (Albany NY). 2013;5:902–912.
- Valle Gottlieb MG, Closs VE, Junges VM, Schwanke CHA. Impact of human aging and modern lifestyle on gut microbiota. Crit Rev Food Sci Nutr. 2018;58:1557–1564. doi:10.1080/10408398.2016.1269054.
- Haro C, Rangel-Zuniga OA, Alcala-Diaz JF, Gomez-Delgado F, Perez-Martinez P, Delgado-Lista J, Quintana-Navarro GM, Landa BB, Navas-Cortés JA, Tena-Sempere M, et al. Intestinal microbiota is influenced by gender and body mass index. PLoS One. 2016;11:e0154090. doi:10.1371/journal.pone.0154090.
- Gao X, Zhang M, Xue J, Huang J, Zhuang R, Zhou X. Body mass index differences in the gut microbiota are gender specific. Front Microbiol. 2018;9:1250.
- Hess AL, Benitez-Paez A, Blaedel T, LH L, JR I, Madera C. The effect of inulin and resistant maltodextrin on weight loss during energy restriction: a randomised, placebo-controlled, double-blinded intervention. Eur J Nutr. 2019;59.
- Hamid Z, Basit A, Pontis S, Piras F, Assogna F, Bossu P, Pontieri FE, Stefani A, Spalletta G, Franceschi P, et al. Gender specific decrease of a set of circulating N-acylphosphatidyl ethanolamines (NAPEs) in the plasma of parkinson’s disease patients. Metabolomics. 2019;15:74. doi:10.1007/s11306-019-1536-z.
- de la Cuesta-zuluaga J, Kelley ST, Chen Y, Escobar JS, Mueller NT, Ley RE, McDonald D, Huang S, Swafford AD, Knight R, Thackray VG. Age- and sex-dependent patterns of gut microbial diversity in human adults. mSystems. 2019;4:e00261–19.
- Wexler HM. Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev. 2007;20:593–621. doi:10.1128/CMR.00008-07.
- Kverka M, Zakostelska Z, Klimesova K, Sokol D, Hudcovic T, Hrncir T, Rossmann P, Mrazek J, Kopecny J, Verdu EF, et al. Oral administration of parabacteroides distasonis antigens attenuates experimental murine colitis through modulation of immunity and microbiota composition. Clin Exp Immunol. 2011;163:250–259. doi:10.1111/j.1365-2249.2010.04286.x.
- Brahe LK, Le Chatelier E, Prifti E, Pons N, Kennedy S, Hansen T, Pedersen O, Astrup A, Ehrlich SD, Larsen LH, et al. Specific gut microbiota features and metabolic markers in postmenopausal women with obesity. Nutr Diabetes. 2015;5:e159. doi:10.1038/nutd.2015.9.
- Schneeberger M, Everard A, AG G-V, Matamoros S, Ramirez S, Delzenne NM, Gomis R, Claret M, Cani PD. Akkermansia muciniphila inversely correlates with the onset of inflammation, altered adipose tissue metabolism and metabolic disorders during obesity in mice. Sci Rep. 2015;5:16643.
- Feng Z, Long W, Hao B, Ding D, Ma X, Zhao L, Pang X. A human stool-derived bilophila wadsworthia strain caused systemic inflammation in specific-pathogen-free mice. Gut Pathog. 2017;9:59.
- Wan YD, Zhu RX, Wu ZQ, Lyu SY, Zhao LX, Du ZJ, Pan XT. Gut microbiota disruption in septic shock patients: a pilot study. Med Sci Monit. 2018;24:8639–8646.
- Wirbel J, Pyl PT, Kartal E, Zych K, Kashani A, Milanese A, Fleck JS, Voigt AY, Palleja A, Ponnudurai R, et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat Med. 2019;25:679–689. doi:10.1038/s41591-019-0406-6.
- Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, Amiot A, Böhm J, Brunetti F, Habermann N, et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol Syst Biol. 2014;10:766. doi:10.15252/msb.20145645.
- Yazici C, Wolf PG, Kim H, Cross TWL, Vermillion K, Carroll T, Augustus GJ, Mutlu E, Tussing-Humphreys L, Braunschweig C, et al. Race-dependent association of sulfidogenic bacteria with colorectal cancer. Gut. 2017;66:1983–1994. doi:10.1136/gutjnl-2016-313321.
- Maki JJ, Looft T. Megasphaera stantonii sp. nov., a butyrate-producing bacterium isolated from the cecum of a healthy chicken. Int J Syst Evol Microbiol. 2018;68:3409–3415. doi:10.1099/ijsem.0.002991.
- McClanahan D, Yeh A, Firek B, Zettle S, Rogers M, Cheek R, Nguyen MVL, Gayer CP, Wendell SG, Mullett SJ, et al. Pilot study of the effect of plant-based enteral nutrition on the gut microbiota in chronically ill tube-fed children. JPEN J Parenter Enteral Nutr. 2019;43:899–911. doi:10.1002/jpen.v43.7.
- Hooks KB, O’Malley MA. Dysbiosis and its discontents. MBio. 2017;8:e01492–17.
- McDonald D, Ackermann G, Khailova L, Baird C, Heyland D, Kozar R, Lemieux M, Derenski K, King J, Vis-Kampen C, Knight R. Extreme dysbiosis of the microbiome in critical illness. mSphere. 2016;1:e00199–16.
- Howard BM, Kornblith LZ, Christie SA, Conroy AS, Nelson MF, Campion EM, Callcut RA, Calfee CS, Lamere BJ, Fadrosh DW, et al. Characterizing the gut microbiome in trauma: significant changes in microbial diversity occur early after severe injury. Trauma Surg Acute Care Open. 2017;2:e000108. doi:10.1136/tsaco-2017-000108.
- Wang X, Yang J, Tian F, Zhang L, Lei Q, Jiang T, Zhou J, Yuan S, Wang J, Feng Z, et al. Gut microbiota trajectory in patients with severe burn: A time series study. J Crit Care. 2017;42:310–316. doi:10.1016/j.jcrc.2017.08.020.
- Lamarche D, Johnstone J, Zytaruk N, Clarke F, Hand L, Loukov D, Szamosi JC, Rossi L, Schenck LP, Verschoor CP, et al. Microbial dysbiosis and mortality during mechanical ventilation: a prospective observational study. Respir Res. 2018;19:245. doi:10.1186/s12931-018-0950-5.
- De Filippis F, Pasolli E, Tett A, Tarallo S, Naccarati A, De Angelis M, Neviani E, Cocolin L, Gobbetti M, Segata N, et al. Distinct genetic and functional traits of human intestinal prevotella copri strains are associated with different habitual diets. Cell Host Microbe. 2019;25:444–53 e3. doi:10.1016/j.chom.2019.01.004.
- Fragiadakis GK, Smits SA, Sonnenburg ED, Van Treuren W, Reid G, Knight R, Manjurano A, Changalucha J, Dominguez-Bello MG, Leach J, et al. Links between environment, diet, and the hunter-gatherer microbiome. Gut Microbes. 2019;10:216–227. doi:10.1080/19490976.2018.1494103.
- Livanos AE, Snider EJ, Whittier S, Chong DH, Wang TC, Abrams JA, Freedberg DE. Rapid gastrointestinal loss of clostridial clusters IV and XIVa in the ICU associates with an expansion of gut pathogens. PLoS One. 2018;13:e0200322.
- Fisher K, Phillips C. The ecology, epidemiology and virulence of enterococcus. Microbiology. 2009;155:1749–1757. doi:10.1099/mic.0.026385-0.
- Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche J-D, Coopersmith CM, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315:801–810. doi:10.1001/jama.2016.0287.
- Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1. doi:10.1093/nar/gks808.
- Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi:10.1093/bioinformatics/btr507.
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi:10.1128/AEM.01541-09.
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi:10.1093/bioinformatics/btr381.
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6.
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi:10.1093/bioinformatics/btq461.
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi:10.1038/nmeth.f.303.
- Harrison PW, Alako B, Amid C, Cerdeno-Tarraga A, Cleland I, Holt S, Hussein A, Jayathilaka S, Kay S, Keane T, et al. The European nucleotide archive in 2018. Nucleic Acids Res. 2019;47:D84–D8. doi:10.1093/nar/gky1078.