
ABSTRACT
Gestational diabetes mellitus (GDM) is a metabolic complication that manifests as hyperglycemia during the later stages of pregnancy. In high resource settings, careful management of GDM limits risk to the pregnancy, and hyperglycemia typically resolves after birth. At the same time, previous studies have revealed that the gut microbiome of infants born to mothers who experienced GDM exhibit reduced diversity and reduction in the abundance of several key taxa, including Lactobacillus. What is not known is what the functional consequences of these changes might be. In this case control study, we applied 16S rRNA sequence surveys and metatranscriptomics to profile the gut microbiome of 30 twelve-month-old infants − 16 from mothers with GDM, 14 from mothers without – to examine the impact of GDM during pregnancy. Relative to the mode of delivery and sex of the infant, maternal GDM status had a limited impact on the structure and function of the developing microbiome. While GDM samples were associated with a decrease in alpha diversity, we observed no effect on beta diversity and no differentially abundant taxa. Further, while the mode of delivery and sex of infant affected the expression of multiple bacterial pathways, much of the impact of GDM status on the function of the infant microbiome appears to be lost by twelve months of age. These data may indicate that, while mode of delivery appears to impact function and diversity for longer than anticipated, GDM may not have persistent effects on the function nor composition of the infant gut microbiome.
Introduction
The human gut microbiome is increasingly viewed as a key determinant of health, with evidence supporting links to an ever-increasing number of diseases from diabetes and obesity to depression.Citation1,Citation2 While the microbiome can exhibit dramatic changes over the course of an individual’s life, how it develops over the first three years has a critical impact on determining its future contributions to health, disease, and immune system maturation.Citation3–5 Among the most impactful factors that contribute to this development are mode of delivery, use of antenatal or postpartum antibiotics, and diet. Initial colonization is largely driven by mode of delivery, with vaginal deliveries associated with the dominance of Bifidobacterium, Lactobacillus and Bacteroides ,Citation6 species that experience reduced abundance in infants delivered through Caesarean-section (C-section). As an infant’s diet changes from primarily breast or formula milk feeding with the introduction of solids, the microbiome increases in complexity.Citation7 While most studies have focused on the dynamics of community composition using 16S rDNA sequence surveys, our knowledge of functional changes associated with the microbiome at this critical stage of development is limited.
In addition to these impactful postnatal factors, the metabolic status of the mother during pregnancy has also been found to contribute to the formation of the infant microbiomeCitation8 and future health status.Citation9 In utero exposure to maternal type 1 diabetes (T1D), and in particular poorly regulated T1D, has recently been associated with a distinct infant rectal and skin microbiomes as early as the first day of life.Citation10 Gestational diabetes mellitus (GDM) is a relatively common metabolic derangement of pregnancy, occurring in 3–20% of pregnanciesCitation11 in which mothers exhibit hyperglycemia during the later stages of gestation.Citation12 While maternal hyperglycemia is generally manageable with dietary changes, and in fewer cases exogenous insulin or metformin, and often resolves after birth, it can increase the risk of type 2 diabetes and other metabolic disorders in both the mother and offspring in later life.Citation13–15 16S rRNA gene surveys have shown that the gut microbiota of infants born to mothers with GDM exhibit a significant decrease in alpha diversity,Citation16,Citation17 together with a loss in abundance of several taxa, including Lactobacillus and Flavonifractor.Citation18 A recent study evaluating the neonatal ear-skin microbiome following delivery found differences in infants born to women with type 1 diabetes compared to neonates born to control mothers. The composition of the neonatal ear-skin microbiome was related to maternal levels of HbA1c in first trimester in a beta-diversity analysis.Citation10 While such dysbioses in infants have been postulated as contributory to the increased risk of cardiometabolic diseases, little is known concerning the functional implications of such microbiome shifts.
Whole microbiome RNA sequencing, or metatranscriptomics, is a method of surveying the function of a community of microbes. Previous metatranscriptomics studies focusing on the developing infant gut microbiome have identified characteristics of gene expression of major taxa in the infant microbiome.Citation19–21 Observations regarding function and expression help to form a more complete picture of the early microbiome and how important factors may shape it. In this study, we used metatranscriptomics to functionally profile the microbial communities associated with stool samples from the 12-month-old infant gut. In addition to examining how the function of these communities respond to factors such as mode of delivery, breast feeding status and sex of the infant, we also examined how exposure to GDM impacts both microbial community structure and function. To our knowledge, this is the first study to perform metatranscriptomic community functional profiling on the infant gut microbiome in the context of GDM.
Materials and methods
Detailed methodology can be found in supplemental files.
Study design
Pregnant women with no prior diagnosis of diabetes were recruited during routine visits in late 2nd to early 3rd trimester at Mount Sinai Hospital, Toronto, Canada. The diagnosis of GDM was made using a sequential 2-step process as per the Diabetes Canada Clinical Guidelines.Citation11 In brief, between 24 and 28 weeks of gestation, women undergo a 1 h 50 g glucose challenge test (GCT). Women were recruited both before and after the 1-h 50 g glucose challenge test. The cohort was enriched for mothers that failed the GCT (1 h glucose >7.8 mmol/L) who are more likely to be diagnosed with GDM than those with a passing GCT result. Study participants were thereafter stratified by GDM status as measured through 2 h, 75 g oral glucose tolerance test (OGTT) performed within 1–2 weeks following the GCT, and defined according to established criteria.Citation11 The glucose levels sampled during OGTT were used to estimate the area-under-the-glucose-curve (AUCglucose) according to the trapezoid rule.
Data collection
Participating women attended study visits that included questionnaires in late pregnancy and at 3-months and 12-months postpartum. Offspring were assessed at birth, 3, and 12 months. Infants were assessed for anthropometric measurements, and the parent completed questionnaires related to their infant’s health and nutrition. Participating mothers were provided with a sample collection kit to collect their child’s stool sample just prior to the study visit at 3 and 12 months of age. Each kit contained diaper liners, two sterile polypropylene containers, a set of gloves, and plastic collection spoons. Stool samples were collected at home and placed into a sterile polypropylene container. Each stool sample was separated into two containers for 16S rDNA and metatranscriptomic profiling. The samples were kept in the freezer at home and brought with an ice pack at the scheduled visit and were then stored at −80°C. Stool samples were collected and sent for 16S rDNA profiling (3 months and 1 year) and metatranscriptomics profiling (1 year). Additional clinical measurements included: sex, exclusive breastfeeding status, mode of delivery, maternal BMI, and infant birth BMI. Medications and supplements taken by participants during pregnancy include prenatal vitamins, antacids, antihistamines, and medications for hypothyroidism and asthma. Antibiotic and probiotic use, as well as use of other medications was collected postnatally for infants.
16S sample preparation and analysis
Prior to nucleotide extraction, stool samples were homogenized. DNA extraction was completed using Omega E.Z.N.A.TM Stool DNA Isolation Kit. Amplification of the V4 region was performed with uniquely barcoded 515F (forward) and 806 R (reverse) sequencing primers to allow for multiplexing.Citation22 High-throughput sequencing targeted the hypervariable V4 region of the 16S rRNA gene, using 150 bp x 2 read length. Sequencing was performed at The Centre for the Analysis of Genome Evolution and Function (CAGEF) at the University of Toronto. Primers were adapted to incorporate Illumina adapters with indexing barcodes and sequenced using the Illumina MiSeq platform. QIIME 2 v2023.2 was used to process raw FASTQ files and cluster reads into amplicon sequence variants (ASVs).Citation23 Adonis2 from the vegan 2.6–4 R package was used to conduct permutational analysis of variance (PERMANOVA) statistical tests on beta diversity, and ANCOM-BC 2.1.2 was used to test for differential abundance.Citation24–26 Analysis of variance (ANOVA) was used to assess for differences in alpha diversity.
Metatranscriptomics sequencing
Stool collected at 12-months was processed using the RNeasy PowerSoil Total RNA kit, using 400–500 mg of each sample. The extracted RNA was cleaned using the Illumina Ribo-Zero Gold rRNA Removal Kit (Epidemiology), followed by the Zymo RNA Clean and Concentrator kit. The cleaned RNA was then converted into cDNA using the NEBNext Ultra II Directional RNA Library prep kit. Libraries were sequenced using the Illumina NextSeq platform to generate ~ 20,000,000 150bp paired end reads per sample, with 1% PhiX spike-in as standard.
Metatranscriptomics reads processing
Reads were processed to remove low-quality reads, host reads, and other unnecessary reads using the MetaPro pipeline, which also annotates gene reads using the ChocoPhlAn pangenome database from HUMAnN 2.0 and NCBI non-redundant (NR) protein databases.Citation27–29 Human reads were removed through sequence similarity searches against the GRCh38 human reference genome. Enzyme annotations are assigned according to the Swiss-Prot database and taxonomic annotations also utilize ChocoPhlAn and NCBI NR databases, in addition to Kaiju 1.9.0 and Centrifuge 1.0.4.Citation30–32
Metatranscriptomics analyses
adonis from the vegan 2.3–5 R package was used to perform PERMANOVA to compare Bray-Curtis beta diversity between samples.Citation33 Data for differential expression was filtered to remove genes with fewer than 6 counts in 6 samples, resulting in 21,563 genes for differential expression and set enrichment analysis. DESeq2 3.16 was used to identify differentially abundant taxa and differentially expressed genes.Citation34 Gene set enrichment analysis was done using fgsea 1.26.0.Citation35
Statistical analyses
Maternal and infant descriptive data are presented as the mean ± SE. For comparison between groups, Student’s t test was used for continuous variables and Chi-square for categorical variables. Statistical significance was considered at p < .05.
30 total infant gut samples were included in 16S and metatranscriptomics analysis. At the ASV level, 16S alpha diversity comparisons were calculated using ANOVA, and beta diversity comparisons were made using PERMANOVA. ANCOM-BC was used to perform differential taxon abundance analysis.Citation36 Bray-CurtisCitation37 distance comparisons of gene expression was similarly made using PERMANOVA. Differential gene expression was determined using DESeq2,Citation34 and downstream gene set enrichment analysis was performed using the resultant log2 fold change values with fgsea.Citation35
Results
Description of cohort
Participants were recruited (n = 44) prior to the 2 h OGTT, performed at a mean gestation of 29.9 ± 2.9 weeks. Maternal medication use during pregnancy included prenatal vitamins (n = 16), L-thyroxine (n = 8), Diclectin (n = 3), Iron supplements (n = 2), Omega 3 supplements (n = 2) and antacids (n = 2). Mean gestational age at birth was 39.1 ± 1.2 weeks and mean infant birthweight 3.4 ± 0.5 g. Mothers in this study diagnosed with GDM were either treated with insulin or through diet. None of the women in this study experienced hypertension during pregnancy with the exception of 1 woman with preeclampsia. Of the 44 infants recruited, 30 (n = 14 GDM, n = 16 nonGDM) had available samples to conduct both 16S rDNA amplicon sequencing and metatranscriptomics at 12 months. Twenty-eight (n = 14 GDM, n = 14 nonGDM) of these samples were available at 3 months for 16S analysis. The remaining 14 infants who had stool available for only 16S rDNA sequencing and were not included. There was no change in our findings from 16S rDNA sequencing results after exclusion of the 14 infants who did not have stool for metatranscriptome analysis. Postnatally, all infants were exclusively or partially breastfed. Prior antibiotic intake at 3 months of age occurred in one infant (GDM) while probiotics were given to 5 infants (3 nonGDM, 2 GDM). Between 3 and 12 months of age, 3 infants had taken a course of antibiotics (2 nonGDM, 1 GDM) and 3 infants had received probiotics (2 nonGDM, 1 GDM). Participant characteristics are summarized in and Supplemental . Study design is outlined in .
Figure 1. Overview of clinical study design. Patient info was collected at birth, 3 months of infant age, and 12 months of infant age. 16S sequencing was performed on 42 stool samples collected at 3 months and 44 stool samples collected at 12 months. Metatranscriptomics sequencing was performed on 30 of the stool samples collected at 12 months. 28 samples were included for 16S analysis at 3 months. 30 samples were included for 16S and metatranscriptomics analysis at 12 months. See for demographic information.
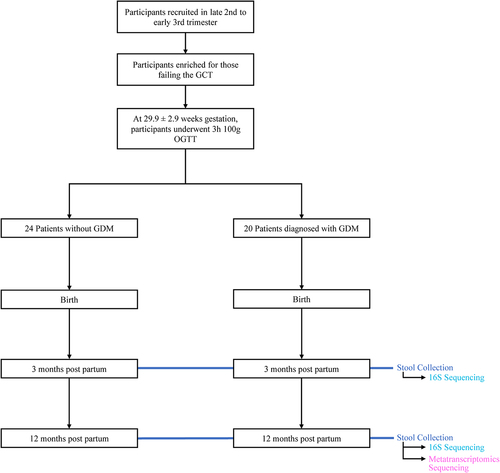
Table 1. Study demographic table.
GDM has limited impact on the microbial community in the developing infant gut
To examine the impact of GDM on the developing infant gut community, we performed 16S rRNA amplicon sequencing to profile stool samples collected at 3-month (n = 28) and 12-month (n = 30) timepoints after birth. From the 6,124,664 sequenced reads (median 83,769) we identified 1946 bacterial OTUs across all 58 samples. Shannon Index analysis revealed an increase in alpha diversity for the 12-month samples relative to the 3-month samples (p < .001, ANOVA) and that vaginal delivery decreases community diversity at 3 months postpartum (p < .001, ANOVA) and increases at 12 months (p < .001, ANOVA; ). Similarly, nonGDM maternal status infants (p = .038; ) and exclusive breastfeeding status (EBF; p < .001) were associated with an increase in alpha diversity at 3 months, but not at 12 months, in the infant cohort.
Figure 2. Diversity analyses of 16S data by mode of delivery. (ab) Shannon Alpha diversity analysis. Analysis of Variance (ANOVA) was used to test for statistical significance. (cd) Bray-curtis principal coordinate analysis beta diversity analysis. Ellipses represent 95% confidence interval for a multivariate normal distribution. n = 19, Cesarean-section; n = 39, Vaginal. n = 28, 3 months; n = 30, 12 months. *p ≤ .05, **p ≤ .01, ***p ≤ .001.
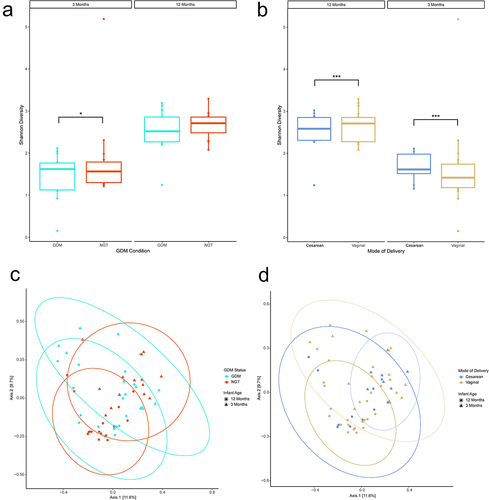
Evaluation of beta-diversity (Supplementary Table S2) at the genus level found that infant age (p < .001, PERMANOVA), mode of delivery (p = .045, PERMANOVA, ), and exclusive breastfeeding (p = .004, PERMANOVA) had a significant effect on sample clustering. When assessing beta diversity within 3-month samples, only exclusive breastfeeding status had a significant effect on clustering (p = .048, PERMANOVA). Neither GDM status nor GDM treatment were associated with any significant clustering effect (, Supplemental Figure 1). Within 12-month samples, none of the comparisons tested had a significant clustering effect on beta diversity. These findings were repeated at the family level. No significant effects were observed at the phylum level.
In order to uncover any taxa-level effects of GDM on gut composition, we examined individual changes in abundance of taxa (Supplementary Table S3). Using ANCOM-BC to test for differential abundance at a genus level, we found that 23 taxa were differentially abundant between the two infant ages, with 17 of these genera decreased in 12-month infants. At 12 months, mode of delivery was associated with 2 differentially abundant genera: Fusobacterium (p = 3.36e-2, ANCOM-BC), which was more abundant in c-section delivered infants, and Streptococcus (p = 9.85e-2, ANCOM-BC), more abundant in vaginally delivered infants. At a family level (Supplementary Table S4), Bacteroidaceae was decreased in C-section infants, across all infant ages (p = 3.23e-2, ANCOM-BC) and at 3 months (p = 4.65e-2, ANCOM-BC).
Metatranscriptomics taxonomic expression differed significantly from 16S taxonomic composition
To functionally interrogate the gut microbiome of the 12-month-old infants, we performed metatranscriptomics on 30 unique samples, resulting in the generation of an average of 21,154,913.4 150 bp paired-end sequence reads per sample (Supplementary Table S5). After filtering low quality, host, and rRNA, putative mRNA reads were annotated to genes using the MetaPro pipeline.Citation38 An average of 25.47% of reads were retained per sample, representing an average of 5,366,805 annotated putative mRNA reads. After gene-level filtering 21,563 genes were retained for analysis, mapping to 607 unique Enzyme Commision (EC) identifiers. This, along with the number of annotated mRNA reads, falls within numbers previously found in metatranscriptomics studies, suggesting adequate gene representation.Citation26,Citation39–43 The percentage of putative mRNA reads out of total reads (44.17%) is similar to that established by previous studies.Citation41,Citation44,Citation45
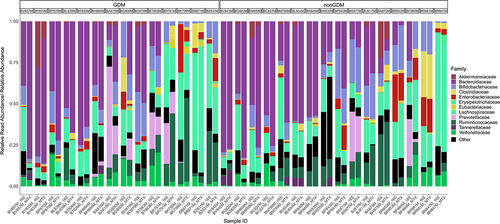
The phylum rank was dominated by Bacteroidetes and Firmicutes in both the GDM and nonGDM samples (Supplemental ). In both GDM and nonGDM samples, the Bacteroidaceae family was most prevalent (). We then used DESeq2Citation34 to find families that exhibited a significantly different level of expression in metatranscriptomics samples. Of the 404 families detected here, 11 differentially expressing taxa were identified as differentially expressing between GDM and nonGDM infants (Supplemental Table S6), and 6 between vaginally and cesarean-section-delivered infants (Supplemental Table S7). Additionally, Bray-Curtis clustering of gene expression was found to differ significantly from 16S taxonomic composition (p < .001) as found by PERMANOVA, further highlighting the difference between expression and composition. This discordance applied to the phylum rank (p < .001), as well as the family rank (p < .001).
Figure 3. Stacked bar chart illustrating the relative abundance of families in the 16S samples compared to the relative read abundance expressed by each family in metatranscriptomics (MTX) samples. Samples are stratified by gestational diabetes (GDM) condition of the mother. Sorted by decreasing relative abundance of Bacteroidetes phylum in MTX samples. Limited to the top 12 most abundant families. Colours of each family correspond to color of the parent phyla in Supplemental Figure 2 (e.g., Lachnospiraceae is color coded to the Firmicutes phylum). n = 30.
Mode of delivery has the largest impact on individual gene expression in the 12-month infant gut
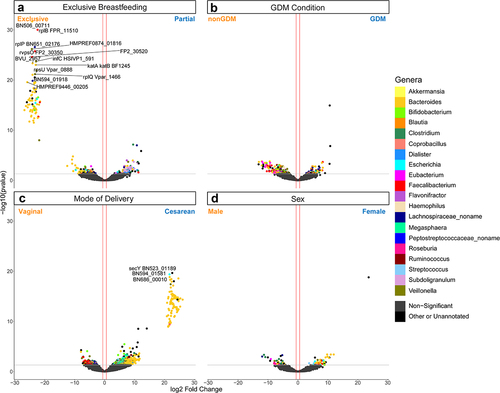
We next examined gene-level expression in our data. DESeq2 identified 980 DEGs associated mode of delivery, nearly thrice the number of DEGs found in any other comparison (, Supplemental Figure S3a-d, Supplemental Table S8). Considering the larger impact of mode of delivery on gene expression, each comparison was then reexamined after controlling for the impact of mode of delivery on function. Here, 226 genes were differentially expressed in the GDM status condition, 90 DEGs associated with sex, and 269 DEGs linked to EBF (). Examination of the genera that express these DEGs reveals that Bacteroides are usually the most abundant taxa, with the exception of GDM condition-associated DEGs, where Veillonella expressed 20% of the genes. Faecalibacterium-annotated genes are also common across these DEGs. In EBF-associated DEGs, a significantly higher number of DEGs were annotated to Subdoligranulum than might be expected in the population (p < .001, hypergeometric test).
Figure 4. Volcano plot depicting differentially expressed gene (DEG) results for mode of delivery and mode of delivery-controlled factors. Log2 fold change and -log10(p value) are both from results of poscounts DESeq2. Colour of dot corresponds to taxonomic annotation. Grey points are non-DEGs. (a) nonGDM vs GDM. (b) Vaginal birth vs cesarean section birth. (c) Male vs female. (d) Exclusive breastfeeding vs. partial breastfeeding. Upregulation and downregulation are relative to the labels. n = 21563 genes.
Gestational diabetes condition was not associated with any enriched gene ontology terms
In order to examine which broader categories of function were represented by these DEGs, we performed a gene set enrichment analysis using the fgsea R package.Citation35 Here, we examined enrichment using three different databases: the biological process ontology of the gene ontology resource (GO; ), the comprehensive antibiotic resistance database (CARD; Supplemental ), and the carbohydrate-active enzymes database (CAZy/CAZyme; ).Citation46–48 Mode of delivery was associated with an increase of tricarboxylic acid cycle (TCA; P 2.46e-2, fgsea), glycolytic process (p = 2.46e-2, fgsea), and translation (p < 0.001, fgsea) GO terms in C-section infants. Similarly, in the CAZy enrichment, mode of delivery was associated with 5 CAZy families, all of which upregulated in C-section infants as well.
Figure 5. Bubble charts detailing each Gene Ontology (GO) biological process term and carbohydrate-active enzyme (CAZy) family that was significantly enriched in our differential expression results. Enriched terms were identified using the fgsea R package.Citation35 Size of bubble indicates the number of DEGs from a given functional category associated with the factor. Enrichment score indicates degree and direction of enrichment for a given term. GO terms and CAZy families are sorted by adjusted p-value. Mode of delivery (MoD) controlled terms are indicated. (a) Bubble chart of GO term enrichment. MoD-controlled GDM condition is omitted as there were no significant terms. (b) Bubble chart of CAZy enzymes. CBM = Carbohydrate-Binding Modules, GH = Glycoside Hydrolases, GT = GlycosylTransferases.
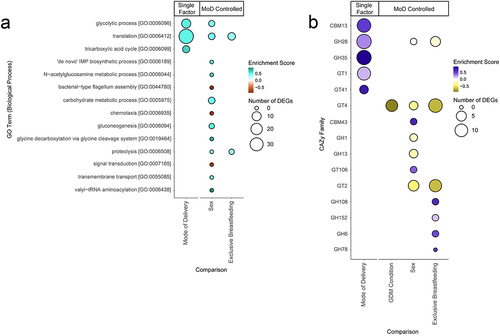
After controlling for mode of delivery, GDM DEGs were enriched for the glycosyl transferase (GT) family 4 CAZyme (p = 9.03e-4, fgsea) within GDM infants. EBF DEGs were enriched for 7 CAZy families, including 5 glycoside hydrolase (GH) families, and two CARD sets, all of which are increased in enrichment in partially breastfed infants, apart from an isoniazid resistance term (p = 4.26e-2, fgsea). Significant GO terms include translation (p = 5.88e-3, fgsea) and proteolysis (p = 3.58e-7, fgsea), both of which are enriched in partially breastfed infants. Sex was associated with the highest number of GO terms and CAZy families, though direction of enrichment was not consistent. These include 5 carbohydrate metabolism terms enriched in female infants, and 2 motility terms enriched in male infants. Terms relating to microbiome product usage or generation such as short-chain fatty acids and LPS were not found to be enriched in any comparison.
Discussion
This case control study sought to profile the changes experienced by the infant gut microbiome in response to a pregnancy complicated by GDM, while concurrently exploring the effect of mode of delivery, breastfeeding, and sex. While previous studies have examined the impact of GDM on the composition of the infant gut microbiome, we believe this is the first study that incorporates a functional metatranscriptomics approach. Our results indicate that maternal GDM is associated with a decrease in alpha diversity across all infant ages and in the 3-month infants specifically. This corroborates existing abundance-based microbiome research, in which alpha diversity has been shown to be significantly lower in neonates of mothers with GDM.Citation49–51 This is not always consistent in literature; studies involving infants ranging from 1-week to 4-years of age have found no differences in alpha diversity between GDM and nonGDM infants.Citation17,Citation52 As GDM did not have a significant impact on alpha diversity in the 12-month infants, it is possible that alpha diversity in GDM infants increases to meet the levels of nonGDM infants sometime before 12 months of age due to changing diet and environmental exposure.Citation3 This may also apply to the non-significant difference in beta diversity between GDM and nonGDM infants in our data. Previous literature indeed have found differences in beta diversity in GDM infants.Citation16,Citation17,Citation51,Citation52 In our data, it is also possible that the nonGDM mothers having already failed the GCT indicating some degree of dysglycemia resulted in the interindividual differences between GDM and nonGDM infant guts failing to be distinct enough. Furthermore, the women with GDM participating in our study were extremely well controlled with diet and/or insulin, and this may further have diminished differences in infant microbiota, as evidenced by similar birth weights between GDM and nonGDM infants ().
Mode of delivery was linked with a significant difference in alpha diversity in 3-month and 12-month infants. Mode of delivery also significantly associated with beta diversity when examining samples of all ages. Existing literature offers somewhat conflicting results in regards to how mode of delivery induces compositional changes to the microbiome over the first year of life.Citation53–57 While mode of delivery was associated with changes in the gut microbiome early on, some studies suggest its influence wanes by 6 months of age, possibly as early as 8 weeks.Citation57–61 Research centered on the gut microbiome of older children (≥3 years) have found few differences in overall diversity associated with birthing method, though individual taxa had changed.Citation62,Citation63 Our results support the notion that perturbations to alpha and beta diversity by delivery remain detectable within the first year.
In our 16S samples, we found a reduction of Bacteroidaceae abundance in C-section infants across both infant ages and at 3 months specifically. This has previously been shown to be a common result of C-section infants receiving less exposure to the maternal microbiome.Citation60,Citation64 In comparison, 6 families were differentially expressive in the metatranscriptomics samples, which also include a reduction in Lactobacillaceae- and Ruminococcaceae-derived transcripts in C-section delivered infants. Both of these families contains species considered beneficial to the infant gut, such as Lactobacillus acidophilus and Faecalibacterium prausnitzii.Citation65–69 These taxa were likely passed down to the vaginally delivered infants,Citation42,Citation70 and the reduction in abundance of these species could represent a long-term consequence of C-section delivery.
Since there were no mothers in this study that exclusively used infant formula milk, groups compared between exclusive and partial breastfeeding diets. Therefore, the effect being measured is likely more subtle than comparison between exclusively breastfed and exclusively formula-fed infant gut microbiota. Breast milk aids in nurturing infant gut microbes through a supply of human milk oligosaccharides (HMOs) among other metabolites, vitamins and immunoglobulins.Citation66,Citation71 These HMOs are utilized by Bifidobacterium and Bacteroides species in the gut,Citation72 the latter of which expressed a large proportion of transcripts in our data. A meta-analysis of 7 studies conducted by Ho et al. found that non-exclusively breastfed infants under 6 months of age displayed higher gut Shannon alpha diversity.Citation73 Breastfeeding status also had a strong effect on our 16S data, with partially breastfed infants experiencing a decrease in alpha diversity across all infant ages and at 3 months. Similarly, breastfeeding status was associated with significant clustering by beta-diversity at all infant ages and at 3 months; it is possible these differences in diversity are eventually lost by 12 months. Increasing variety in diet, such as introduction of solid foods, or changes in milk feeding frequency may contribute to this increase in diversity.Citation74,Citation75
Few studies on the infant gut microbiome have applied metatranscriptomics to study the infant gut microbiome, and they have tended to focus on preterm births.Citation26,Citation76,Citation77 Beyond these, one study reported pathway changes in a longitudinal study over the first year of life,Citation19 while another study profiled changes in microbiome activity alongside solid food adaptation, outlining steps toward an adult-like composition.Citation21 In comparison, our metatranscriptomics results focused on twelve-month old infants, aiming to reveal factors that may have sustained effects on microbiome activity. Our results demonstrate that by 12 months, the persisting effect of maternal GDM status and EBF is modest, while mode of delivery and sex both influence the function of the microbiome, based on functional enrichment analysis.
As there have been no metatranscriptomics studies on GDM, most predictions on the functional changes GDM induce are derived from correlating taxonomic abundance changes to factors in health.Citation50,Citation78 We found no significantly enriched functions, as defined by GO terms, even after controlling for the effects of mode of delivery. We found only one significantly enriched CAZy family (GT4) upregulated in GDM infants after controlling for mode of delivery, despite GDM being primarily an issue of carbohydrate metabolism in the host. Notably, GTs have previously been implicated in diabetes as a potential therapeutic targetCitation79 and have been observed to change in activity in response to hyperglycemia.Citation80 However, the higher number of CAZymes enriched in our other analyses suggest a marginal influence on carbohydrate metabolism. While there were a higher number of DEGs associated with GDM than sex, our results suggest that, by 12 months of age, there is no significant persistent perturbation of functional categories in the infant gut resulting from maternal GDM status. As highlighted below, we cannot rule out the possibility that the diet and/or treatment with insulin of the mothers diagnosed with GDM in this study were able to overcome any potential negative effects of GDM on the infant gut, which has previously been shown through metagenomics by Sugino et al.Citation81 The study compared a conventional diet with a diet that was higher in complex carbohydrates and lower in fat in women diagnosed with GDM. As a result, infants of the mothers fed the treatment diet displayed increased gut alpha diversity over time. Indeed, previous work from our cohort has consistently demonstrated lower birthweight of infants born to women with GDM, compared to those with normal or milder dysglycemia during pregnancy, which indicates excellent glycemic control in this GDM cohort during pregnancy.Citation82
Three terms were significantly enriched in infants delivered through C-section: translation, glycolytic process, and tricarboxylic acid cycle. The enrichment of two carbohydrate metabolism-related terms together with translation, which correlates with cell growth,Citation83 suggest that there may be a relative increase in microbial cell proliferation in infants delivered through C-section compared to vaginally delivered infants. This is supported by the overall direction of expression for most mode of delivery DEGs () and the direction of the significantly enriched CAZy families (), all of which are enriched in samples associated with C-sections. Along with the high number of DEGs and the observed changes in diversity, our results point to the mode of delivery as having a clear effect on both the structure and function of the 12-month-old infant gut microbiome.
Post mode of delivery-control, sex was associated with 13 enriched GO terms, the most of any comparison tested. Five terms were directly related to nutrient metabolism, primarily carbohydrate metabolism, all of which had been enriched within female infant gut microbiomes. Similarly, seven CAZy families were significantly enriched, though these were not conformally unidirectional. PICRUSt analysis of 16S data has previously identified some sex-specific differences between pre-term, dizygotic twin male and female gut function; though there were carbohydrate metabolism pathways differentially regulated in these results, many other unrelated pathways were differentially regulated as well.Citation24 Hormones appear to affect the composition of the infant gut since birth,Citation25 and our results point to some early microbiome differences in function as well.
Only two GO terms – proteolysis and translation – were enriched in the mode of delivery-controlled EBF. A previous meta-analysis by Ho et al.Citation73 involving infants less than six months of age found that most KEGG pathways that differed between partially and exclusively breastfed infants were involved with metabolism. One previous metatranscriptomics study in preterm infants found that breastfed infants and formula-fed infants were enriched for different metabolic functions.Citation26 Though in our data, there were seven CAZy families significantly enriched between infants of varying EBF status, they were not consistently enriched in one direction.
Several additional methodological considerations in this study warrant further discussion. Firstly, the infants of mothers with GDM were monitored during pregnancy in a tertiary care center, and their infants demonstrate lower birthweight than the control group, suggesting effective glucose management during pregnancy.Citation82 As a result, this may have acted to mitigate the potential microbiome-modifying effects of the condition. Secondly, the reference control group was composed of mothers who failed the initial GCT but did not meet criteria for GDM on glucose tolerance testing. While glycemic management intervention is therefore not indicated for this group of mothers, they are experiencing subclinical dysglycemia. The two groups of mothers were not significant dissimilar in terms of BMI, nor did their infants separate based on birth weight. While there have been several similar cohort studies in which this lack of group body habitus divergence was observed,Citation16,Citation51,Citation84 we must consider the alternate possibility that even exposure to subclinical dysglycemia may alter the infant gut microbiome in a manner which is similar to GDM. Although prenatal medication was recorded, perinatal and postnatal medications were not, both of which have been observed to impact the infant microbiome.Citation85 Furthermore, the relatively small sample size together with the relatively late (12 months) timepoint used in this study may have limited our ability to identify functional correlates of GDM status in the mother, which might be expected to be more prominent at timepoints closer to birth. Previous benchmarking studies have found that different methods applied at each step of sample collection, processing (e.g. choice of method for nucleic acid extractionCitation86,Citation87 and analysis can bias the recovery of nucleotides from individual taxa. As a result, certain microbes may be underrepresented in this study, although a previous study suggests that biases may be reduced for RNA extraction protocols.Citation88 Lastly, as this study was exploratory and metatranscriptomics has not been previously applied to studying GDM to our knowledge, there were no prior studies for sample size calculations. However, previous studies involving 16S analysis of GDM newborns and metatranscriptomics analysis of infants examining breastfeeding modes have similar sample sizes,Citation16,Citation26 and the Bray-Curtis analysis utilized in this study has been shown to be the most sensitive indicator of beta diversity.Citation89
While GDM has been observed to affect the composition of the early infant microbiome, in this study we were able to elucidate the functional consequences of GDM further into the development of the infant gut. Though our results support some changes in alpha diversity, GDM exerted little effect on function in the 12-month-old infant gut. As the gut develops with the introduction of solid foods and weaning from breastfeeding, the effect of GDM exposure becomes less prominent against an increasingly complex gut community. Similarly, while there may be differences in diversity remaining, our results suggest that the impact of EBF wanes by one year. Some functional differences are visible at 12 months in sex and mode of delivery, with the latter likely having the greatest influence. These data may indicate that, while mode of delivery appears to impact function and diversity for longer than anticipated, GDM may not have persistent effects on the function nor composition of the infant gut microbiome, at least when compared to mothers who failed OCT but did not experience GDM. Regulation of diet and glycemic control may alleviate the potential impact of GDM on the infant gut microbiome. Future studies in this area could explore correlations between the maternal and infant gut, the mitigating effects of diet, peripartum antibiotics, and other interventions on the impact of GDM on the infant gut.
Author contributions
JH and JP conceived and designed the study. KH organized patient recruitment and collection of samples and metadata. PWW and DG supervised extraction of nucleic material for high throughput sequencing. RVC and JC performed sequence data analyses. RVC, JP, JH, and PR wrote the manuscript. All authors reviewed and/or edited the paper.
Ethics statement
The research ethics board at The Hospital for Sick Children and Mount Sinai Hospital approved the protocol. All mothers provided written informed consent for their child. The work described was carried out in accordance with the code of ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans. Informed consent was obtained from all research participants according to the protocol approved by the Research Ethics Board of the Hospital for Sick Children and Mount Sinai Hospital (REB# 1000007561).
Supplemental Material
Download Zip (3.5 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Sequence data is deposited at the NCBI under BioProject repository identifier PRJNA1013505.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2024.2356277
Additional information
Funding
References
- Iatcu CO, Steen A, Covasa M. Gut microbiota and complications of type-2 diabetes. Nutrients. 2021;14(1):166. doi:10.3390/nu14010166.
- Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28:203–17.
- Moore RE, Townsend SD. Temporal development of the infant gut microbiome. Open Biol. 2019;9(9):190128. doi:10.1098/rsob.190128.
- Gensollen T, Blumberg RS. Correlation between early life regulation of immune system by microbiota and allergy development. J Allergy Clin Immunol. 2017;139(4):1084–1091. doi:10.1016/j.jaci.2017.02.011.
- Zhang H, Zhang Z, Liao Y, Zhang W, Tang D. The complex link and disease between the gut microbiome and the immune system in infants. Front Cell Infect Microbiol. 2022;12:924119. [accessed 2023 Oct 20]. doi:10.3389/fcimb.2022.924119.
- Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H. et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17(5):690–703. doi:10.1016/j.chom.2015.04.004.
- Homann CM, Rossel CAJ, Dizzell S, Bervoets L, Simioni J, Li J, Gunn E, Surette MG, de Souza RJ, Mommers M. et al. Infants’ first solid foods: impact on gut microbiota development in two intercontinental cohorts. Nutrients. 2021;13(8):2639. doi:10.3390/nu13082639.
- Hu J, Nomura Y, Bashir A, Fernandez-Hernandez H, Itzkowitz S, Pei Z, Stone J, Loudon H, Peter I. Diversified microbiota of meconium is affected by maternal diabetes status. PLOS ONE. 2013;8(11):e78257. doi:10.1371/journal.pone.0078257.
- Blotsky AL, Rahme E, Dahhou M, Nakhla M, Dasgupta K. Gestational diabetes associated with incident diabetes in childhood and youth: a retrospective cohort study. CMAJ. 2019;191(15):E410–E417. doi:10.1503/cmaj.181001.
- Gajecka M, Gutaj P, Jaskiewicz K, Rydzanicz M, Szczapa T, Kaminska D, Kosewski G, Przyslawski J, Ploski R, Wender-Ozegowska E. et al. Effects of maternal type 1 diabetes and confounding factors on neonatal microbiomes. Diabetologia. 2024;67(2):312–326. doi:10.1007/s00125-023-06047-7.
- Feig DS, Berger H, Donovan L. “Diabetes and pregnancy”. Diabetes Canada clinical practice guidelines expert committee. Can J Diabetes. 2018;42:S6–9.
- Plows JF, Stanley JL, Baker PN, Reynolds CM, Vickers MH. The pathophysiology of gestational diabetes mellitus. Int J Mol Sci. 2018;19(11):3342. doi:10.3390/ijms19113342.
- Scholtens DM, Kuang A, Lowe LP, Hamilton J, Lawrence JM, Lebenthal Y, Brickman WJ, Clayton P, Ma RC, McCance D. et al. Hyperglycemia and Adverse Pregnancy Outcome Follow-up Study (HAPO FUS): maternal glycemia and childhood glucose metabolism. Diabetes Care. 2019;42(3):381–392. doi:10.2337/dc18-2021.
- Ponzo V, Fedele D, Goitre I, Leone F, Lezo A, Monzeglio C, Finocchiaro C, Ghigo E, Bo S. Diet-gut microbiota interactions and Gestational Diabetes Mellitus (GDM). Nutrients. 2019;11(2):E330. doi:10.3390/nu11020330.
- Kc K, Shakya S, Zhang H. Gestational diabetes mellitus and Macrosomia: a literature review. Ann Nutr Metab. 2015;66(Suppl. 2):14–20. doi:10.1159/000371628.
- Su M, Nie Y, Shao R, Duan S, Jiang Y, Wang M, Xing Z, Sun Q, Liu X, Xu W. et al. Diversified gut microbiota in newborns of mothers with gestational diabetes mellitus. PLOS ONE. 2018;13(10):e0205695. doi:10.1371/journal.pone.0205695.
- Crusell MKW, Hansen TH, Nielsen T, Allin KH, Rühlemann MC, Damm P, Vestergaard H, Rørbye C, Jørgensen NR, Christiansen OB. et al. Comparative studies of the gut microbiota in the offspring of mothers with and without gestational diabetes. Front Cell Infect Microbiol. 2020;10:536282. doi:10.3389/fcimb.2020.536282.
- Soderborg TK, Carpenter CM, Janssen RC, Weir TL, Robertson CE, Ir D, Young BE, Krebs NF, Hernandez TL, Barbour LA. et al. Gestational diabetes is uniquely associated with altered early seeding of the infant gut microbiota. Front Endocrinol (Lausanne). 2020;11:603021. doi:10.3389/fendo.2020.603021.
- Gosalbes MJ, Compte J, Moriano-Gutierrez S, Vallès Y, Jiménez-Hernández N, Pons X, Artacho A, Francino MP. Metabolic adaptation in the human gut microbiota during pregnancy and the first year of life. EBioMedicine. 2019;39:497–509. doi:10.1016/j.ebiom.2018.10.071.
- Vatanen T, Sakwinska O, Wilson B, Combremont S, Cutfield WS, Chan SY, Godfrey KM, Sakwinska O, Cutfield WS, Chan SY. et al. Transcription shifts in gut bacteria shared between mothers and their infants. Sci Rep. 2022;12(1):1276. doi:10.1038/s41598-022-04848-1.
- Hugenholtz F, Ritari J, Nylund L, Davids M, Satokari R, de Vos WM. Feasibility of metatranscriptome analysis from infant gut microbiota: adaptation to solid foods results in increased activity of firmicutes at six months. Iran J Pediatr Hematol Oncol. 2017;2017:9547063. doi:10.1155/2017/9547063.
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. Isme J. 2012;6(8):1621–1624. doi:10.1038/ismej.2012.8.
- Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2 | Nature Biotechnology. [accessed 2023 Sep 12]. https://www.nature.com/articles/s41587-019-0209-9.
- Chen J, Li H, Hird SM, Chen M-H, Xu W, Maas K, Cong X. Sex differences in gut microbial development of preterm infant twins in early life: a longitudinal analysis. Front Cell Infect Microbiol. 2021;11:671074. doi:10.3389/fcimb.2021.671074.
- Valeri F, Endres K. How biological sex of the host shapes its gut microbiota. Front Neuroendocrinol. 2021;61:100912. doi:10.1016/j.yfrne.2021.100912.
- Wang Z, Neupane A, Vo R, White J, Wang X, Marzano SYL. Comparing gut microbiome in mothers’ own breast milk- and formula-fed moderate-late preterm infants. Front Microbiol. 2020;11:891. doi:10.3389/fmicb.2020.00891.
- Taj B, Adeolu M, Xiong X, Ang J, Nursimulu N, Parkinson J. MetaPro: a scalable and reproducible data processing and analysis pipeline for metatranscriptomic investigation of microbial communities. Bioinformatics. 2021; doi:10.1101/2021.02.23.432558.
- Beghini F, McIver LJ, Blanco-Míguez A, Dubois L, Asnicar F, Maharjan S, Mailyan A, Manghi P, Scholz M, Thomas AM. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife. 2021;10:e65088. doi:10.7554/eLife.65088.
- NCBI Resource Coordinators Agarwala R, Barrett T, Beck J, Benson DA, Bollin C, Bolton E, Bourexis D, Brister JR, Bryant SH, Canese K. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2018;46(D1):D8–D13. doi:10.1093/nar/gkx1095.
- The UniProt Consortium Bateman A, Martin M-J, Orchard MJ, Magrane M, Agivetova R, Ahmad S, Alpi E, Bowler-Barnett EH, Britto R, Bursteinas B. et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49(D1):D480–D489. doi:10.1093/nar/gkaa1100.
- Menzel P, Ng KL, Krogh A. Fast and sensitive taxonomic classification for metagenomics with kaiju. Nat Commun. 2016;7(1):11257. doi:10.1038/ncomms11257.
- Kim D, Song L, Breitwieser FP, Salzberg SL. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016;26(12):1721–1729. doi:10.1101/gr.210641.116.
- Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, Minchin PR, O’hara RB, Simpson GL, Solymos P, Stevens MHH. et al. Vegan: community ecology package. 2020. https://CRAN.R-project.org/package=vegan.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8.
- Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A. Fast gene set enrichment analysis. bioRxiv. [accessed 2021 Feb 1]. 060012. doi:10.1101/060012.
- Lin H, Peddada SD. Analysis of compositions of microbiomes with bias correction. Nat Commun. 2020;11(1):3514. doi:10.1038/s41467-020-17041-7.
- Arbizu M. pairwiseAdonis: Pairwise multilevel comparison using adonis. Published online. 2020.
- Andrews S. FastQC. 2019. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Damon C, Lehembre F, Oger-Desfeux C, Luis P, Ranger J, Fraissinet-Tachet L, Marmeisse R. Metatranscriptomics reveals the diversity of genes expressed by eukaryotes in forest soils. PLOS ONE. 2012;7(1):e28967. doi:10.1371/journal.pone.0028967.
- Jorth P, Turner KH, Gumus P, Nizam N, Buduneli N, Whiteley M, Kolter R. Metatranscriptomics of the human oral microbiome during health and disease. mBio. 2014;5(2):10–128. doi:10.1128/mbio.01012-14.
- Jiang Y, Xiong X, Danska J, Parkinson J. Metatranscriptomic analysis of diverse microbial communities reveals core metabolic pathways and microbiome-specific functionality. Microbiome. 2016;4(1):2. doi:10.1186/s40168-015-0146-x.
- Asnicar F, Manara S, Zolfo M, Truong DT, Scholz M, Armanini F, Ferretti P, Gorfer V, Pedrotti A, Tett A, Segata N. et al. Studying vertical microbiome transmission from mothers to infants by strain-level metagenomic profiling. mSystems. 2017;2(1):081828. accessed 2016 Oct 21. doi:10.1128/mSystems.00164-16.
- Lay C, Chu CW, Purbojati RW, Acerbi E, Drautz-Moses DI, de Sessions PF, Jie S, Ho E, Kok YJ, Bi X. et al. A synbiotic intervention modulates meta-omics signatures of gut redox potential and acidity in elective caesarean born infants. BMC Microbiol. 2021;21(1):191. doi:10.1186/s12866-021-02230-1.
- Zou A, Nadeau K, Xiong X, Wang PW, Copeland JK, Lee JY, Pierre JS, Ty M, Taj B, Brumell JH. et al. Systematic profiling of the chicken gut microbiome reveals dietary supplementation with antibiotics alters expression of multiple microbial pathways with minimal impact on community structure. Microbiome. 2022;10(1):127. doi:10.1186/s40168-022-01319-7.
- Xiong X, Bales ES, Ir D, Robertson CE, McManaman JL, Frank DN, Parkinson J. Perilipin-2 modulates dietary fat-induced microbial global gene expression profiles in the mouse intestine. Microbiome. 2017;5(1):117. doi:10.1186/s40168-017-0327-x.
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47(D1):D330–D338. doi:10.1093/nar/gky1055.
- Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, Huynh W, Nguyen ALV, Cheng AA, Liu S. et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020;48(D1):D517–D525. doi:10.1093/nar/gkz935.
- Drula E, Garron ML, Dogan S, Lombard V, Henrissat B, Terrapon N. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 2022;50(D1):D571–D577. doi:10.1093/nar/gkab1045.
- Wang J, Zheng J, Shi W, Du N, Xu X, Zhang Y, Ji P, Zhang F, Jia Z, Wang Y. et al. Dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus. Gut. 2018;67(9):1614–1625. doi:10.1136/gutjnl-2018-315988.
- Ponzo V, Ferrocino I, Zarovska A, Amenta MB, Leone F, Monzeglio C, Rosato R, Pellegrini M, Gambino R, Cassader M. et al. The microbiota composition of the offspring of patients with gestational diabetes mellitus (GDM). Loor JJ, ed. PLOS ONE. 2019;14(12):e0226545. doi:10.1371/journal.pone.0226545.
- Chen T, Qin Y, Chen M, Zhang Y, Wang X, Dong T, Chen G, Sun X, Lu T, White RA. et al. Gestational diabetes mellitus is associated with the neonatal gut microbiota and metabolome. BMC Med. 2021;19(1):120. doi:10.1186/s12916-021-01991-w.
- Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Kling Bäckhed H, Gonzalez A, Werner J, Angenent L, Knight R. et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150(3):470–480. doi:10.1016/j.cell.2012.07.008.
- Stewart CJ, Embleton ND, Clements E, Luna PN, Smith DP, Fofanova TY, Nelson A, Taylor G, Orr CH, Petrosino JF. et al. Cesarean or vaginal birth does not impact the longitudinal development of the gut microbiome in a Cohort of exclusively preterm infants. Front Microbiol. 2017;8:1008. doi:10.3389/fmicb.2017.01008.
- Bokulich NA, Chung J, Battaglia T, Henderson N, Jay M, Li H, Lieber DA, Wu F, Perez-Perez GI, Chen Y. et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med. 2016;8(343):ra34382–ra34382. doi:10.1126/scitranslmed.aad7121.
- Lyu L, Zhou X, Zhang M, Liu L, Niu H, Zhang J, Chen S, Gong P, Jiang S, Pan J. et al. Delivery mode affects intestinal microbial composition and the development of intestinal epithelial cells. Front Microbiol. 2021;12:626144. doi:10.3389/fmicb.2021.626144.
- Stinson LF, Payne MS, Keelan JA. A critical review of the bacterial baptism hypothesis and the impact of cesarean delivery on the infant microbiome. Front Med. 2018;5:135. doi:10.3389/fmed.2018.00135.
- Reyman M, van Houten MA, van Baarle D, Bosch AATM, Man WH, Chu MLJN, Arp K, Watson RL, Sanders EAM, Fuentes S. et al. Impact of delivery mode-associated gut microbiota dynamics on health in the first year of life. Nat Commun. 2019;10(1):4997. doi:10.1038/s41467-019-13014-7.
- Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, Aagaard KM. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat Med. 2017;23(3):314–326. doi:10.1038/nm.4272.
- Mitchell CM, Mazzoni C, Hogstrom L, Bryant A, Bergerat A, Cher A, Pochan S, Herman P, Carrigan M, Sharp K. et al. Delivery mode affects stability of early infant gut microbiota. Cell Rep Med. 2020;1(9):100156. doi:10.1016/j.xcrm.2020.100156.
- Rutayisire E, Huang K, Liu Y, Tao F. The mode of delivery affects the diversity and colonization pattern of the gut microbiota during the first year of infants’ life: a systematic review. BMC Gastroenterol. 2016;16(1):86. doi:10.1186/s12876-016-0498-0.
- Hill CJ, Lynch DB, Murphy K, Ulaszewska M, Jeffery IB, O’Shea CA, Watkins C, Dempsey E, Mattivi F, Tuohy K. et al. Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET Cohort. Microbiome. 2017;5(1):4. doi:10.1186/s40168-016-0213-y.
- Cioffi CC, Tavalire HF, Neiderhiser JM, Bohannan B, Leve LD, Whisner C. History of breastfeeding but not mode of delivery shapes the gut microbiome in childhood. Whisner C, ed. PLOS ONE. 2020;15(7):e0235223. doi:10.1371/journal.pone.0235223.
- Salminen S, Gibson GR, McCartney AL, Isolauri E. Influence of mode of delivery on gut microbiota composition in seven year old children. Gut. 2004;53(9):1388–1389. doi:10.1136/gut.2004.041640.
- Matharu D, Ponsero AJ, Dikareva E, Korpela K, Kolho K-L, de Vos WM, Salonen A. Bacteroides abundance drives birth mode dependent infant gut microbiota developmental trajectories. Front Microbiol. 2022;13:953475. doi:10.3389/fmicb.2022.953475.
- Azad MAK, Sarker M, Li T, Yin J. Probiotic species in the modulation of gut microbiota: an overview. Biomed Res Int. 2018;2018:9478630. doi:10.1155/2018/9478630.
- Zhang X, Mushajiang S, Luo B, Tian F, Ni Y, Yan W. The composition and concordance of lactobacillus populations of infant gut and the corresponding breast-milk and maternal gut. Front Microbiol. 2020;11:11. [accessed 2023 Feb 14]. doi:10.3389/fmicb.2020.597911.
- Härtel C, Pagel J, Spiegler J, Buma J, Henneke P, Zemlin M, Viemann D, Gille C, Gehring S, Frommhold D. et al. Lactobacillus acidophilus/Bifidobacterium infantis probiotics are associated with increased growth of VLBWI among those exposed to antibiotics. Sci Rep. 2017;7(1):5633. doi:10.1038/s41598-017-06161-8.
- Fortmann I, Marißen J, Siller B, Spiegler J, Humberg A, Hanke K, Faust K, Pagel J, Eyvazzadeh L, Brenner K. et al. Lactobacillus Acidophilus/Bifidobacterium infantis probiotics are beneficial to extremely low gestational age infants fed human milk. Nutrients. 2020;12(3):850. doi:10.3390/nu12030850.
- Lopez-Siles M, Duncan SH, Garcia-Gil LJ, Martinez-Medina M. Faecalibacterium prausnitzii: from microbiology to diagnostics and prognostics. ISME J. 2017;11(4):841–852. doi:10.1038/ismej.2016.176.
- Zhang C, Li L, Jin B, Xu X, Zuo X, Li Y, Li Z. The effects of delivery mode on the gut microbiota and health: state of art. Front Microbiol. 2021;12:724449. [accessed 2023 Aug 21]. https://www.frontiersin.org/articles/10.3389/fmicb.2021.724449.
- Masi AC, Stewart CJ. Untangling human milk oligosaccharides and infant gut microbiome. iScience. 2021;25(1):103542. doi:10.1016/j.isci.2021.103542.
- Kijner S, Cher A, Yassour M. The infant gut commensal bacteroides dorei presents a generalized transcriptional response to various human milk oligosaccharides. Front Cell Infect Microbiol. 2022;12:854122. [accessed 2023 Feb 14]. doi:10.3389/fcimb.2022.854122.
- Ho NT, Li F, Lee-Sarwar KA, Tun HM, Brown BP, Pannaraj PS, Bender JM, Azad MB, Thompson AL, Weiss ST. et al. Meta-analysis of effects of exclusive breastfeeding on infant gut microbiota across populations. Nat Commun. 2018;9(1):4169. doi:10.1038/s41467-018-06473-x.
- Ma J, Li Z, Zhang W, Zhang C, Zhang Y, Mei H, Zhuo N, Wang H, Wang L, Wu D. et al. Comparison of gut microbiota in exclusively breast-fed and formula-fed babies: a study of 91 term infants. Sci Rep. 2020;10(1):15792. doi:10.1038/s41598-020-72635-x.
- Moossavi S, Sepehri S, Robertson B, Bode L, Goruk S, Field CJ, Lix LM, de Souza RJ, Becker AB, Mandhane PJ. et al. Composition and variation of the human milk microbiota are influenced by maternal and early-life factors. Cell Host Microbe. 2019;25(2):324–335.e4. doi:10.1016/j.chom.2019.01.011.
- Fettweis JM, Serrano MG, Brooks JP, Edwards DJ, Girerd PH, Parikh HI, Huang B, Arodz TJ, Edupuganti L, Glascock AL. et al. The vaginal microbiome and preterm birth. Nat Med. 2019;25(6):1012–1021. doi:10.1038/s41591-019-0450-2.
- Sher Y, Olm MR, Raveh-Sadka T, Brown CT, Sher R, Firek B, Baker R, Morowitz MJ, Banfield JF. et al. Combined analysis of microbial metagenomic and metatranscriptomic sequencing data to assess in situ physiological conditions in the premature infant gut. PLOS ONE. 2020;15(3):e0229537. doi:10.1371/journal.pone.0229537.
- Man B, Koenig MD, Bernabe BP, Nagelli U, Tussing-Humphreys L. The role of the gut microbiota in the prevention and management of gestational diabetes mellitus: are we there yet? J Perinat Neonatal Nurs. 2020;34(3):195–198. doi:10.1097/JPN.0000000000000497.
- Rudman N, Gornik O, Lauc G. Altered N-glycosylation profiles as potential biomarkers and drug targets in diabetes. FEBS Lett. 2019;593(13):1598–1615. doi:10.1002/1873-3468.13495.
- Lee CL, Chiu PCN, Pang PC, Chu IK, Lee K-F, Koistinen R, Koistinen H, Seppälä M, Morris HR, Tissot B. et al. Glycosylation failure extends to glycoproteins in gestational diabetes mellitus. Diabetes. 2011;60(3):909–917. doi:10.2337/db10-1186.
- Sugino KY, Hernandez TL, Barbour LA, Kofonow JM, Frank DN, Friedman JE. A maternal higher-complex carbohydrate diet increases bifidobacteria and alters early life acquisition of the infant microbiome in women with gestational diabetes mellitus. Front Endocrinol. 2022;13:921464. [accessed 2023 Aug 21]. doi:10.3389/fendo.2022.921464.
- Retnakaran R, Ye C, Hanley AJ, Connelly PW, Sermer M, Zinman B. Treatment of gestational diabetes mellitus and maternal risk of diabetes after pregnancy. Diabetes Care. 2023;46(3):587–592. doi:10.2337/dc22-1786.
- Dong H, Nilsson L, Kurland CG. Co-variation of tRNA abundance and codon usage inEscherichia coliat different growth rates. J Mol Biol. 1996;260(5):649–663. doi:10.1006/jmbi.1996.0428.
- Zhu Q, Yang X, Zhang Y, Shan C, Shi Z, Byndloss MX. Role of the gut microbiota in the increased infant body mass index induced by gestational diabetes mellitus. Byndloss MX, ed. mSystems. 2022;7(5):e00465–22. doi:10.1128/msystems.00465-22.
- Huang H, Jiang J, Wang X, Jiang K, Cao H. Exposure to prescribed medication in early life and impacts on gut microbiota and disease development. EClinicalMedicine. 2024;68:68. doi:10.1016/j.eclinm.2024.102428.
- Costea PI, Zeller G, Sunagawa S, Pelletier E, Alberti A, Levenez F, Tramontano M, Driessen M, Hercog R, Jung F-E. et al. Towards standards for human fecal sample processing in metagenomic studies. Nat Biotechnol. 2017;35(11):1069–1076. doi:10.1038/nbt.3960.
- Han Z, Sun J, Lv A, Wang A. Biases from different DNA extraction methods in intestine microbiome research based on 16S rDNA sequencing: a case in the koi carp, cyprinus carpio var. Koi. Microbiologyopen. 2019;8(1):e00626. doi:10.1002/mbo3.626.
- Scholes AN, Lewis JA. Comparison of RNA isolation methods on RNA-Seq: implications for differential expression and meta-analyses. BMC Genomics. 2020;21(1):249. doi:10.1186/s12864-020-6673-2.
- Kers JG, Saccenti E. The power of microbiome studies: some considerations on which alpha and beta metrics to use and how to report results. Front Microbiol. 2022;12:796025. [accessed 2024 Feb 29]. doi:10.3389/fmicb.2021.796025.