
ABSTRACT
Acetyl phosphate (AcP) is generally produced from acetyl coenzyme A by phosphotransacetylase (Pta), and subsequent reaction with ADP, catalyzed by acetate kinase (Ack), produces ATP. The mechanism of ATP production in Porphyromonas gingivalis is poorly understood. The aim of this study was to explore the molecular basis of the Pta-Ack pathway in this microorganism. Pta and Ack from P. gingivalis ATCC 33277 were enzymatically and structurally characterized. Structural and mutational analyses suggest that Pta is a dimer with two substrate-binding sites in each subunit. Ack is also dimeric, with a catalytic cleft in each subunit, and structural analysis indicates a dramatic domain motion that opens and closes the cleft during catalysis. ATP formation by Ack proceeds via a sequential mechanism. Reverse transcription-PCR analysis demonstrated that the pta (PGN_1179) and ack (PGN_1178) genes, tandemly located in the genome, are cotranscribed as an operon. Inactivation of pta or ack in P. gingivalis by homologous recombination was successful only when the inactivated gene was expressed in trans. Therefore, both pta and ack genes are essential for this microorganism. Insights into the Pta-Ack pathway reported herein would be helpful to understand the energy acquisition in P. gingivalis.
Porphyromonas gingivalis, a black-pigmented Gram-negative rod-type bacterium, is a keystone pathogen linked to the onset and progression of periodontitis [Citation1,Citation2]. Although periodontitis is localized to tissues surrounding teeth, it is associated with serious systemic conditions such as cardiovascular disease [Citation3,Citation4], diabetes [Citation5], and rheumatoid arthritis [Citation6].
The main routes for ATP and energy production in P. gingivalis remain to be fully elucidated. P. gingivalis is unable to ferment carbohydrates such as glucose, although small quantities of glucose monomers can be utilized for polymer biosynthesis [Citation7]. Therefore, P. gingivalis relies on catabolism of amino acids to generate metabolic energy [Citation8,Citation9]. In many bacteria, the enzymes phosphotransacetylase (Pta, EC 2.3.1.8) and acetate kinase (Ack, EC 2.7.2.1) form a key pathway for production of ATP from excess acetyl coenzyme A (acetyl-CoA) [Citation10]. Pta produces acetyl phosphate (AcP) from acetyl-CoA, and AcP is subsequently converted to acetate by Ack, with the concomitant production of ATP through substrate-level phosphorylation.
AcP functions as a signaling molecule that is capable of serving as a phosphate donor for two-component signal transduction systems, thereby connecting central metabolism with environment sensing and signal transduction [Citation11,Citation12]. Interestingly, inactivation of the Pta-Ack pathway in Escherichia coli results in biofilm formation that is quantitatively and architecturally distinct from that in parental strains [Citation12–Citation14]. However, disruption of the Pta-Ack pathway does not affect cell growth in many bacteria, including E. coli [Citation15], Streptococcus mutans [Citation16], Streptococcus pneumoniae [Citation13], Clostridium acetobutylycum [Citation17], Clostridium tyrobutyricum [Citation18], and Bacillus subtilis [Citation19]. Indeed, the presence of alternate pathways has been demonstrated for several bacterial, including B. subtilis [Citation20], and a search of all available Ack sequences showed that more than 300 species possess multiple Ack enzymes [Citation21]. By contrast, a recent study revealed the indispensable role of the Pta-Ack pathway in Staphylococcus aureus for maintaining energy and metabolic homeostasis during overflow metabolism [Citation22].
Since P. gingivalis produces a large amount of short chain fatty acids, including acetate, propionate, and butyrate [Citation23], it is postulated that the Pta-Ack pathway, which is associated with acetate production, is conserved in P. gingivalis [Citation8,Citation9] (). Despite the importance of Pta and Ack in carbon cycling and energy metabolism in P. gingivalis, these enzymes have not been characterized in this microorganism, even though the coding genes have been assigned based on sequence homology [Citation24,Citation25]. In the current study, recombinant Pta (PgPta) and Ack (PgAck) from P. gingivalis ATCC 33277, encoded by pta (PGN_1179) and ack (PGN_1178), respectively, were prepared and enzymatically characterized. In addition, crystal structures were determined by X-ray crystallography. Based on the structures, several putative functional residues were identified and their contribution to catalytic activity was evaluated by site-directed mutagenesis. Furthermore, we examined the essentiality of pta and ack in P. gingivalis.
Figure 1. Proposed metabolic pathways for use of glutamate and aspartate in P. gingivalis based on previously described pathways [Citation8,Citation9,Citation67,6Citation8]. Broken lines indicate expected pathways that were not supported by the experimental evidence.
![Figure 1. Proposed metabolic pathways for use of glutamate and aspartate in P. gingivalis based on previously described pathways [Citation8,Citation9,Citation67,6Citation8]. Broken lines indicate expected pathways that were not supported by the experimental evidence.](/cms/asset/f39a5488-27d4-4a4b-985b-e5c6e4bbde58/zjom_a_1588086_f0001_b.gif)
Materials and methods
Bacterial strains and growth conditions
All bacterial strains used in this study are listed in . P. gingivalis ATCC 33277 and its derivatives were grown in modified GAM broth (Nissui, Tokyo, Japan), or on Brucella HK agar (Kyokuto Pharmaceutical Industrial, Tokyo, Japan) supplemented with 5% rabbit blood at 37°C in an anaerobic atmosphere (80% N2, 10% H2, 10% CO2). Ampicillin or tetracycline was added to a final concentration of 10 µg/mL and 1 µg/mL, respectively, when needed. E. coli DH5α (Invitrogen, Carlsbad, CA) and BL21 (DE3) (Novagen, Madison, WI) strains were grown aerobically at 37°C in 2 × YT or LB medium (Thermo Fisher JP, Tokyo, Japan) supplemented with 100 µg/mL ampicillin, as required.
Table 1. Bacterial strains and plasmids used.
Expression of recombinant PgPta and PgAck
Recombinant PgPta and PgAck were overproduced using E. coli after cloning each coding region between appropriate restriction sites in pGEX-6P-1 (GE Healthcare Japan, Hino, Japan) and pET28a (Merck, Darmstadt, Germany), respectively. Primers used for cloning are listed in the Supplemental Table. PgPta has a glutathione S-transferase (GST)-tag cleavable by PreScission protease, whereas PgAck possesses an N-terminal (His)6-tag followed by a thrombin cleavage site. After verification by DNA sequencing, the resulting pPR1179-Gex and pET1178-Pet constructs () were used to transform competent E. coli BL21 (DE3) cells. Transformants were cultivated at 37°C to an optical density at 600 nm of ~0.3, and isopropyl β-D-1-thiogalactopyranoside was added at a final concentration of 0.3 mM to induce expression of recombinant proteins. Culturing was continued at 37°C for ~3 h, and cells were harvested by centrifugation at 4°C.
Site-directed mutagenesis of pta and ack genes in P. gingivalis was performed using overlap extension PCR [Citation26] or a KOD-Plus-Mutagenesis Kit (Toyobo, Osaka, Japan) with primers listed in the Supplemental Table, as previously described [Citation27]. Overproduction of mutant enzymes was performed using the same procedure for wild-type (WT) enzyme described above.
Purification of recombinant PgPta and PgAck
To purify recombinant PgPta, harvested cells were resuspended in phosphate-buffered saline (PBS) and lysed by ultrasonication on ice. The lysate was centrifuged at 30,000 × g for 1 h, and the GST fusion protein in the supernatant was absorbed onto an affinity matrix glutathione-Sepharose 4B column and cleaved with PreScission protease (GE Healthcare Japan) according to the manufacturer’s protocol. The tag-digested protein solution was loaded onto a MonoQ 10/100 GL column (GE Healthcare Japan) and eluted with a six column volume linear gradient of NaCl from 100 to 500 mM. Fractions containing recombinant PgPta were pooled, concentrated, and loaded onto a HiLoad 16/60 Superdex 200 pg column (GE Healthcare Japan) pre-equilibrated with 10 mM Tris/HCl pH 7.6 containing 10 mM NaCl.
For purification of recombinant PgAck, harvested cells were resuspended in 50 mM potassium phosphate (pH 8.0) containing 250 mM NaCl, and lysed by ultrasonication on ice. The lysate was centrifuged at 30,000 × g for 1 h, and recombinant PgAck was purified using TALON Superflow resin (GE Healthcare Japan) according to the manufacturer’s protocol. The (His)6-tag was cleaved by thrombin (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s instructions, and the tag-digested protein was further purified using MonoQ 10/100 GL and HiLoad 16/60 Superdex 200 pg columns as described above for PgPta.
Purified enzymes were stored at – 20°C after adding an equal volume of 80% glycerol. Protein concentrations were determined as described previously [Citation28] and protein purity was assessed by SDS-PAGE and gel filtration chromatography using a Superdex 200 HR 10/30 column (GE Healthcare) at a flow rate of 0.3 mL/min in PBS. Molecular mass estimation of purified enzymes (240 µg/200 µL) was also performed by gel filtration chromatography using a calibrated column and protein standards (molecular weight range of 12,000–200,000) purchased from Sigma-Aldrich.
Colorimetric enzyme assays
i) PgPta activity. The ability of PgPta to produce AcP from acetyl-CoA was investigated by spectrophotometrically quantifying the consumption of acetyl-CoA during the reaction as previously described [Citation29] with several minor modifications. Briefly, each reaction (100 µL) consisted of 100 mM Tris/phosphate (pH 9.0), 20 mM KCl, varying concentrations of acetyl-CoA (50–200 µM), and 1.8 ng/mL recombinant PgPta. After reactions were initiated by addition of enzyme and precisely incubated for 3 min at 37°C, samples were spectrophotometrically examined by measuring the absorbance at 233 nm (A233). The concentration of acetyl-CoA was calculated using a standard curve, and kinetic parameters of PgPta were calculated by fitting directly to initial rate vs. substrate concentration (V vs. S) curves based on the Michaelis-Menten equation. To analyze the optimal pH, reaction mixtures consisting of 50 mM Tris/phosphate buffer were adjusted to pH 6.0, 7.0, 8.0, 9.0, or 10.0. Data were obtained from three independent experiments.
ii) PgAck activity. The activity of PgAck was determined by monitoring NADPH production as previously described [Citation30] with several modifications. Reactions (150 µL) consisted of 50 mM Tris/HCl (pH 8.0), 2 mM dithiothreitol, 3.2 mM glucose, 1.3 mM NADP, 3.7 U/mL hexokinase (Sigma-Aldrich), 0.93 U/mL glucose-6-phosphate dehydrogenase (Sigma-Aldrich), 1.3 mM MgCl2, 67 ng/mL recombinant PgAck, varying concentrations of lithium potassium AcP (0.0667, 0.133, 0.267, and 0.667 mM), and ADP (0.267, 0.667, 1.33, and 2.67 mM). Reactions were initiated by the addition of substrate and incubation for 5 min at 37°C, and the amount of ATP in the reaction mixture was determined by spectrophotometrically quantifying NADPH at A340 using standard curves. Initial velocities of PgAck were determined at four fixed ADP concentrations at different AcP concentrations, and double reciprocal (Lineweaver-Burk) plots were obtained. Secondary plots of the y intercept (velocity reciprocal) vs. the inverse concentration of ADP were used to obtain Vmax value and Km value for ADP. Secondary plots of reciprocal slopes from Lineweaver-Burk plots vs. the inverse concentration of ADP provided Km/Vmax values, which were used to calculate Km values for AcP. Assay mixtures used for divalent metal ion screening contained 1.3 mM MgCl2, 1.3 mM ZnCl2, 1.3 mM CaCl2, or 1.3 mM EDTA instead of 1.3 mM MnCl2. To analyze the optimal pH, reaction mixtures consisted of 50 mM Tris/HCl buffer with the pH adjusted to 6.0, 7.0, 8.0, 9.0, or 10.0. Data were obtained from three independent experiments.
Reverse transcription (RT)-PCR
RT-PCR analysis was carried out as previously described [Citation31]. Briefly, total RNA was isolated from P. gingivalis using ISOGEN-LS reagent (Nippon Gene, Tokyo, Japan) in accordance with the manufacturer’s instructions. Total RNA samples were treated with RNase-free DNase (TaKaRa Bio, Kusatsu, Japan) to remove traces of chromosomal DNA. Total RNA was reverse-transcribed with random hexadeoxyribonucleotide primers (TaKaRa Bio) using PrimeScript Reverse Transcriptase (TaKaRa Bio) according to the manufacturer’s instructions. Gene-specific primers used for RT-PCR are listed in the Supplemental Table. Reaction mixtures without reverse transcriptase served as negative controls to evaluate the presence of contaminating genomic DNA in samples.
Construction of gene-deficient mutants by allelic replacement
Mutant strains of P. gingivalis lacking PGN_1180, pta, ack, or PGN_1177 genes were constructed by replacing each gene with a 2,123 bp cassette containing the ampicillin resistance gene, cepA, flanked by upstream and downstream DNA sequences targeting the P. gingivalis gene of interest. Each targeting sequence was separately amplified using P. gingivalis ATCC 33277 genomic DNA as a template, and cepA was amplified from pCEPA [Citation32]. Primers used for PCR are listed in the Supplemental Table. Three amplicons were linked using an In-fusion HD Cloning kit (TaKaRa Bio) and subcloned into pMCL200 [Citation33]. The construct was sequenced to rule out unintended base changes. Each linearized plasmid was electroporated into P. gingivalis ATCC 33277 and its derivatives as previously described [Citation34]. Briefly, an overnight culture in modified GAM broth was diluted 1:5 in 5 ml of fresh modified GAM broth and incubated for 8 h at 37°C to obtain competent cells in log phase. The cells were collected, washed three times with 0.1 mM HEPES, washed once and resuspended with 10% glycerol, and then stored at – 80°C until use. The linearized plasmids (approximately 5 µg) were added to the competent cells and the mixtures were pulsed with a Gene Pulser apparatus (Bio-Rad Laboratories, Hercules, CA) at 2.5 kV with a time constant of 5 ms. The cell suspensions were immediately added to 10 ml of modified GAM broth prewarmed anaerobically, and then incubated anaerobically at 37°C for 24 h without antibiotics. Aliquots of the cell suspension were plated on Brucella HK agar with 5% rabbit blood containing appropriate antibiotics and incubated anaerobically for 10–15 days at 37°C. PCR analysis of genomic DNA from mutant strains was performed to confirm appropriate gene insertion in ampicillin-resistant mutant colonies.
Conditional mutant strains were also constructed from P. gingivalis ATCC 33277 using pComp1179 or pComp1178 (). Plasmids contained pta or ack inserted between BamHI and SalI sites in pTRAG402 [Citation27] under the control of the strong ragA promotor [Citation35].
Structure determination of recombinant PgPta and PgAck
To perform crystallization experiments, each purified protein was concentrated to 10 mg/mL. Crystallization experiments were performed at 20°C using the hanging-drop vapor diffusion method. Purified protein solution (0.8 μL) was mixed with reservoir solution (0.8 μL) and the resulting droplet was equilibrated against 200 μL reservoir solution in a well. Crystals of PgPta were obtained using reservoir solution containing 20% (w/v) polyethylene glycol (PEG) 3350 and 0.2 M ammonium tartrate dibasic. Crystals of PgPta in complex with acetyl-CoA were prepared by cocrystallization using droplets containing 5 mM acetyl-CoA equilibrated against the same reservoir. Before crystallization of PgAck, magnesium chloride was added to the protein solution at a final concentration of 10 mM. PgAck was crystallized using reservoir solution containing 0.2 M lithium sulfate monohydrate, 0.1 M Tris/HCl (pH 8.5), and 24% (w/v) PEG 3350.
X-ray diffraction data were collected using beamline NE3A at the Photon Factory Advanced Ring (Ibaraki, Japan) at 95 K. Crystals of PgPta and PgAck were cryoprotected by soaking briefly in mother liquor containing 10% (v/v) 2-methyl-2,4-pentanediol and 30% (w/v) PEG 3350, respectively. Diffraction data were indexed, integrated, and scaled using DIALS [Citation36] and SCALA [Citation37] as implemented in XIA2 [Citation38]. The structures of PgPta and PgAck were solved by molecular replacement using MOLREP [Citation39]. Search models were generated using the homology modeling server SWISS-MODEL [Citation40] based on the structure of Pta from Methanosarcina thermophila (MtPta) [Protein Data Bank (PDB) ID: 2AF4] [Citation41] and Ack from Thermotoga maritima (PDB ID: 2IIR) for PgPta and PgAck, respectively. The resultant structures were progressively refined by fitting to the electron density maps using COOT [Citation42]. Manual adjustment of structures was interspersed with refinement cycles using REFMAC5 [Citation43]. Final statistics for X-ray diffraction data and crystallographic refinement are summarized in .
Table 2. Statistics for X-ray diffraction data and crystallographic refinement of PgPta and PgAck.
Statistical analyses
Differences between groups were assessed by one-way analysis of variance and Student-Newman-Keuls multiple comparison post-tests or Student’s t-tests. Differences were considered statistically significant at p < 0.01.
Accession numbers
Atomic coordinates and structure factors for PgPta in complex with acetyl-CoA, and PgAck, have been deposited at the PDB under accession codes 6IOX and 6IOY, respectively.
Results
Purification and enzymatic characterization of recombinant PgPta and PgAck proteins
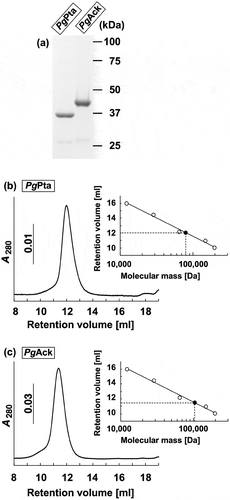
The final step in the production of ATP from acetyl-CoA in P. gingivalis is believed to be carried out by the enzymes associated with the Pta-Ack pathway (). A homology search revealed that the amino acid sequences of PgPta (PGN_1179) and PgAck (PGN_1178) from P. gingivalis share 45% and 47% identity, respectively, with MtPta and MtAck from M. thermophila (Supplemental Figures S1 and S2), which are well-characterized [Citation44–Citation46]. To enzymatically characterize PgPta and PgAck, recombinant proteins were expressed in E. coli and purified, and homogeneity was confirmed by SDS-PAGE ()). The molecular masses of denatured PgPta and PgAck were in agreement with the predicted values (35.8 kDa and 43.2 kDa, respectively). In gel filtration chromatography experiments, PgPta and PgAck eluted as single peaks at retention volumes corresponding to molecular masses of 80.5 and 108 kDa, respectively () and )), suggesting that PgPta is a homodimer in solution, and PgAck is a homodimer or homotrimer.
Figure 2. Purification and gel filtration analysis of recombinant PgPta and PgAck proteins. (a) SDS-PAGE analysis of recombinant PgPta and PgAck proteins. Samples (~5 μg) were subjected to SDS-PAGE and visualized by Coomassie Brilliant Blue staining. The positions of molecular mass markers (in kDa) are shown. (b and c) Gel filtration chromatography analysis of the molecular mass of PgPta (b) and PgAck (c). A calibration curve was constructed using protein standards (shown as open circles in the inset). Purified PgPta and PgAck are represented by closed circles.
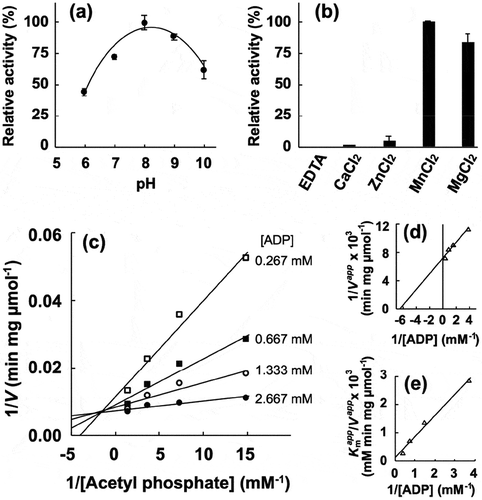
Next, the enzymatic activity of the recombinant proteins was characterized. The phosphotransacetylase activity of recombinant PgPta was evaluated by spectrophotometrically quantifying acetyl-CoA consumed during the reaction. The maximum activity of recombinant PgPta was observed at pH 9.0 ()). Kinetic parameters of PgPta were determined using the Michaelis-Menten equation ( and )), yielding kcat and Km values of 1,487 ± 63 s–1 and 63.8 ± 7.5 μM, respectively. Meanwhile, the acetate kinase activity of PgAck was determined by quantifying ATP produced from ADP and AcP. The optimal pH of PgAck was pH 8.0 ()). Among the divalent metal ions tested (Mn2+, Mg2+, Zn2+, and Ca2+), Mn2+ and Mg2+ supported the high PgAck activity ()). The activity of PgAck in the presence of Mg2+ was ~80% of that in the presence of Mn2+. No detectable activity was observed in the presence of EDTA. Initial reaction velocities of PgAck were determined at four fixed ADP concentrations at different AcP concentrations, and Lineweaver-Burk (double reciprocal) plots revealed a clear intersecting line pattern ()), indicating that PgAck acts via a sequential mechanism [Citation45]. The Vmax (145 ± 1 μmol/min/mg) and Km values for ADP (188 ± 22 μM) and AcP (22.2 ± 0.2 μM) were estimated from secondary plots () and )). The kinetic parameters are summarized in .
Table 3. Kinetic parameters for reactions catalyzed by recombinant PgPta and PgAck.
Figure 3. Enzymatic characterization of recombinant PgPta protein. (a) Effect of pH on AcP production by PgPta. The decrease in acetyl-CoA during incubation of recombinant PgPta with acetyl-CoA at pH 6.0–10.0 was spectrophotometrically quantified by measuring the absorbance at 233 nm (A233). Enzyme activities are indicated relative to the activity at pH 9.0. (b) Steady-state kinetic analysis of PgPta. Reaction mixtures contained 100 mM Tris/phosphate buffer (pH 9.0), 20 mM KCl, 1.8 ng/mL recombinant PgPta, and 50, 75, 100, 125, 150, or 200 µM acetyl-CoA. Averages from three independent experiments are presented.
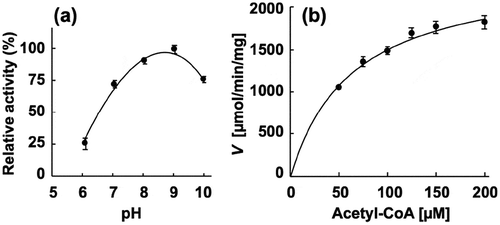
Figure 4. Enzymatic characterization of recombinant PgAck protein. (a) Effect of pH on production of ATP by PgAcK from 0.3 mM AcP and 0.3 mM ADP. Enzyme activities are indicated relative to the activity at pH 8.0. (b) Effect of divalent metal ions on ATP production by PgAcK from 0.3 mM AcP and 0.3 mM ADP. Enzyme activities at pH 8.0 are indicated relative to that in the presence of 1.3 mM MnCl2. (c) Steady-state kinetic analysis of PgAck using double reciprocal plots from initial velocity measurements determined in 50 mM Tris/HCl buffer (pH 8.0) at 37°C. Various concentrations of AcP (0.0667, 0.133, 0.267, and 0.667 mM) were tested for every fixed ADP concentration (0.267, 0.667, 1.33, and 2.67 mM). Averages from three independent experiments are presented. (d) Secondary plots of the y intercept (velocity reciprocal) vs. the inverse concentration of ADP. (e) Secondary plots of reciprocal slopes from panel (a) vs. the inverse concentration of ADP.
Structure of PgPta and enzymatic activity of its mutants
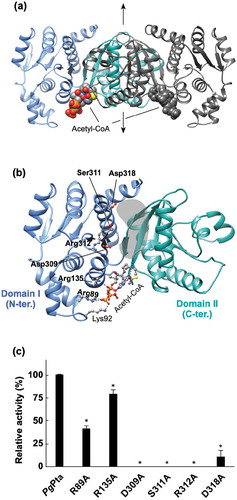
The structure of PgPta in complex with acetyl-CoA was determined at 2.0 Å resolution () and ). The asymmetric unit of the P21212 cell contains one dimer. The subunit is an α/β protein composed of two α/β domains ()); N-terminal domain I (residues 1–145 and 302–336) and C-terminal domain II (residues146–301), based on the previous definition [Citation44]. Domain II is responsible for dimerization ()). The N- and C-terminal domains are positioned side by side, and the domain arrangement results in the formation of a cleft with a putative active site at the interface.
Figure 5. Structural analysis of PgPta. (a) The dimeric structure of PgPta in complex with acetyl-CoA. The two domains of one subunit of the dimer are colored light blue (domain I) and green (domain II), and both domains of the other subunit are colored dark gray. Bound acetyl-CoA molecules are shown as spheres. The 2-fold axis is indicated by arrows. (b) The subunit structure of PgPta viewed in the same orientation as (a). Residues mutated in this study (labeled with bold letters) and bound to acetyl-CoA (Lys92) are shown in ball-and-stick representation. The gray-colored region at the domain interface is a putative active site. (c) Enzyme activity of WT and mutant PgPta proteins. Initial acetyl-CoA consumption velocities were measured, and enzyme activities are indicated relative to that of WT PgPta. Data are presented as means ± standard deviation of three independent experiments (*p < 0.01).
Three distinct conformations of the subunit structure have been reported for MtPta [Citation44]; a change in the position of domain I relative to domain II results in distinct open, closed, and partially closed conformations. Structural comparison of the subunits of PgPta in complex with acetyl-CoA with those of MtPta showed that both subunits of PgPta are in the open conformation, with a root mean square deviation (rmsd) of 0.89 Å (314 Cα atoms) and 0.90 Å (318 Cα atoms) for subunits A and B, respectively. However, the extent of the conformational change observed in PgPta is much less than that in PgAck (described below).
Structural analysis revealed an acetyl-CoA molecule bound to each subunit ()), with seven direct hydrogen bonds or electrostatic interactions between the bound acetyl-CoA and subunit B ()). The adenine base is recognized by two hydrogen bonds with the main-chain amide and carbonyl groups of Val150. The 3´-phosphate interacts with the side chains of Arg89 and Lys92 in subunit B. Arg135 forms an electrostatic interaction with the β-phosphate. The second amide nitrogen is hydrogen-bonded to the carbonyl group of Ala175. The interactions provided from Lys92 and Ala175 are missing in subunit A due to differences in χ angles of side chains and/or the bound acetyl-CoA. This results in higher B-factor values for acetyl-CoA in subunit A (74 Å2) than subunit B (52 Å2).
Six amino acid residues (Arg89, Arg135, Asp309, Ser311, Arg312, and Asp318) positioned at the cleft and the acetyl-CoA binding site were mutated and recombinant proteins were prepared (Supplemental Figure S3) to investigate their contributions to catalytic activity. All the prepared mutants except D318A gave a single peak in the gel filtration (Supplemental Figure S3), indicating the mutations did not affect the dimerization. The D318A mutation displayed a broad biphasic pattern. However, it seems that more than half of the D318A mutant is still dimer. The crystal structure clearly shows that Arg89 and Arg135 interact with the 3´-phosphate and β-phosphate of acetyl-CoA, respectively ()). Replacement of Arg89 and Arg135 resulted in a 58% and 21% decrease in specific activity, respectively ()). The other four mutated residues are located at the cleft, not in contact with Arg89 and Arg135. The polypeptide region in which Asp309, Ser311, and Arg312 are located forms part of the side of the cleft on the domain I side, with Ser311 and Arg312 positioned at the edge of the cleft, and Asp309 located near the bottom of the cleft. Mutation of these residues resulted in the complete loss of enzyme activity. Finally, Asp318 is located at the edge of the cleft on the opposite side from the observed acetyl-CoA binding site. The D318A mutant displayed only 11% of the specific activity of the WT enzyme.
Structure of PgAck and enzymatic activity of its mutants
The structure of PgAck was refined at 1.9 Å resolution (). Four subunits (A, B, C, and D) are present in the asymmetric unit of the P21 cell. Structural analysis by the PISA server [Citation47] was performed to identify potential subunit interfaces and evaluate their significance for multimer formation. Interfaces between subunits A and B, and subunits C and D (interface area of ~3000 Å2) received significantly higher scores than others (data not shown), suggesting that PgAck forms a homodimer ()), with two homodimers present in the asymmetric unit. This implies a dimeric state in solution, consistent with the ambiguous dimer or trimer predicted by gel filtration chromatography ()). The structure of the PgAck subunit is composed of two α/β domains; the N-terminal domain (residues 1–149 and 384–398) and the C-terminal domain (residues 150–383, )). The secondary structural elements of PgAck are well conserved with those of MtAck [Citation48]. The C-terminal domain of each subunit is responsible for dimerization, and the dimer is related by the 2-fold symmetry axis ()). A putative catalytic cleft is formed between the domains.
Figure 6. Structural analysis of PgAck. (a) The dimeric structure of PgAck (shown using subunits C and D). The two domains in subunit D are colored pink (N-terminal domain) and purple (C-terminal domain), and both domains of subunit C are colored dark gray. The two sulfate ions (SO42- 401 and 402) bound in the cleft in subunit D, and Pro37 at the edge of the cleft in both subunits, are shown as spheres. The 2-fold axis is indicated by arrows. (b) The subunit structure of PgAck viewed in the same orientation as (a). Residues mutated in this study (labeled with bold letters), with hydrogen-bonding interactions between the domains, and with bound sulfate ions are shown in ball-and-stick representation. Hydrogen bonds are indicated by dashed lines. An enlarged view of the active site is shown in the inset. (c) Superposition of PgAck subunits using Cα atoms of the C-terminal domain (rmsd <0.35 Å, ~234 Cα atoms). The position of the N-terminal domain relative to the C-terminal domain is different among the subunits. Subunits A, C, and D are colored yellow, light green, and blue, respectively. Although subunit B is slightly more similar in conformation to subunit A, it is not shown to simplify the figure. The two sulfate ions bound to subunit D are also illustrated. (d) Enzyme activity of WT and mutant PgAck enzymes. Initial ATP production velocities were measured, and enzyme activities are indicated relative to that of WT PgAck. Data are presented as means ± standard deviation of three independent experiments (*p < 0.01).
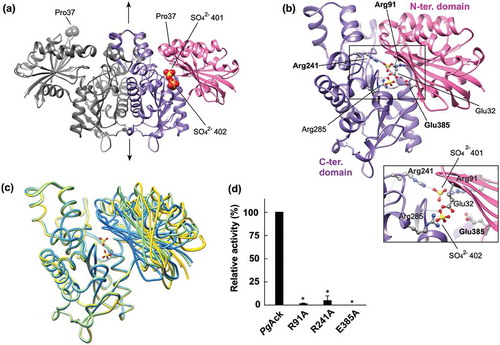
Comparison of the four subunits in the asymmetric unit revealed distinct conformations, with rmsd values ranging from 0.81 to 5.19 Å using all 398 Cα atoms ()). The structure of the C-terminal domain is almost invariant among the subunits, and the positions of the N-terminal domain relative to the C-terminal domain are different, leading to opening and closing of the cleft ()). The cleft in subunit D is the most closed, with two sulfate ions bound, presumably derived from lithium sulfate in the mother liquor. Compared with subunit D, the cleft in the other subunits is more open to varying extents and contains no bound ligands. Pro37 at the edge of the cleft is shifted 17.6, 14.0, and 11.7 Å in subunits A, B, and C, respectively, from the corresponding position in subunit D. The degree of opening is ordered subunits A, B, C, and D (highest to lowest). Enzyme activity assays revealed that Mg2+ or Mn2+ is required for catalytic activity of PgAck ()). Interestingly, although we crystallized PgAck in the presence of Mg2+ (and Mn2+, data not shown), electron density corresponding to a divalent metal cation was not observed.
We performed a mutational analysis of three residues positioned at the cleft; based on the structure of PgAck and previous mutational studies on MtAck [Citation46,Citation49–Citation51], and we selected Arg91, Arg241, and Glu385 for mutation. Arg91 and Arg241 interact with SO42- 401 in the crystal structure ()), while Glu385 is positioned ~7 Å away from both sulfate ions. Following preparation of the mutant proteins (Supplemental Figure S3), enzymatic activity was compared ()). All three mutations dramatically decreased the specific activity; R91A and R241A exhibited only 0.6% and 3.9% of the activity of the WT enzyme, and E385A displayed no detectable activity.
Gel filtration analysis of the mutants showed that each mutant protein mostly forms homodimer in common with the WT enzyme (Supplemental Figure S3). However, the peaks of E385A and R91A had obvious front shoulders with molecular mass of 217 kDa, which corresponds to almost the double of the estimated mass of the dimer (108 kDa). This result indicated that approximately 20% and 40% of the E385A and R91A mutants, respectively, appeared to be tetramer.
Transcriptional analysis of Pta and Ack genes in P. gingivalis
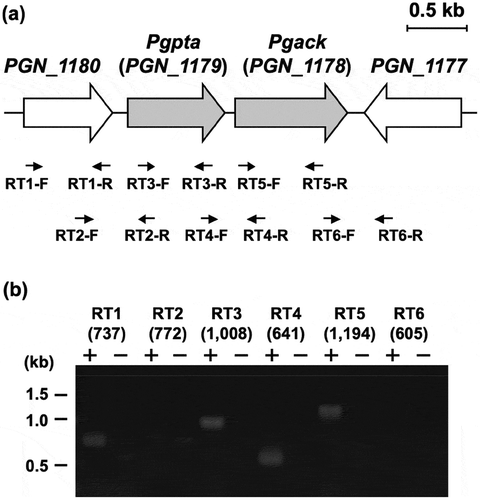
Transcriptional regulation of the pta and ack regions in P. gingivalis was characterized by RT-PCR (). DNA fragments with expected length were effectively amplified using the primers (Supplemental Table) and the genomic DNA from P. gingivalis (Supplemental Figure S4). Transcripts corresponding to the regions spanning the borders of pta and ack were detectable, demonstrating that these genes are cotranscribed in this microorganism. PCR fragments corresponding to the regions spanning the border of PGN_1180 and pta were not amplified. The function of the PGN_1180 protein is unknown. By contrast, the gene (PGN_1177) immediately downstream from ack was oppositely transcribed. Thus, only pta and ack were transcribed together as an operon. No significant products were amplified using cDNA prepared without reverse transcriptase, indicating that the RT-PCR products were not derived from contaminating chromosomal DNA ().
Figure 7. Genetic organization and transcriptional analysis of the pta-ack region of P. gingivalis ATCC 33277. (a) Gene arrangement within the pta-ack region of P. gingivalis ATCC 33277 chromosomal DNA. Large and small arrows indicate open reading frames and RT-PCR primers, respectively. (b) RT-PCR analysis of pta and ack gene expression. PCR amplicons were separated by 0.8% agarose gel electrophoresis. The predicted size (in bp) of each PCR amplicon is shown in parentheses. Lanes marked (+) or (–) indicate standard RT-PCR reactions or negative controls without cDNA, respectively. The positions of DNA size standards (in kb) are indicated on the left.
Both pta and ack genes are essential in P. gingivalis
To examine the physiological roles of pta and ack genes in P. gingivalis ATCC 33277, we attempted to construct chromosomal knockout mutants of each gene using a standard allelic replacement procedure that involves the insertion of an ampicillin resistance gene into the target gene (Supplemental Figure S5). However, attempts to obtain mutant P. gingivalis strains in which pta or ack was inactivated were unsuccessful (). Furthermore, when an erythromycin resistance cassette was used instead of an ampicillin resistance gene, no null mutants were obtainable (data not shown). By contrast, using the same procedure, it was possible to obtain null mutants of both PGN_1180 and the PGN_1177 genes, which are located upstream from pta and downstream from ack, respectively ().
Table 4. P. gingivalis transformation efficiency.
Next, we attempted to construct chromosomal knockout mutants of pta and ack genes in the P. gingivalis ATCC 33277 background using pComp1179 and pComp1178, respectively, () for expression of the respective genes in P. gingivalis under the control of the ragA promotor [Citation27]. The introduction of pta and ack genes in trans led to disruption of the corresponding genes in the chromosomal DNA of P. gingivalis, and the transformation efficiencies for pta- and ack-deficient mutants were comparable with those for PGN_1180- and PGN_1177-deficient mutants (). The growth of all the mutant strains was similar to that of the parent strain (Supplemental Figure S6),
Discussion
Knowledge of cell physiology and the conversion of nutrients to ATP, the universal molecular currency of energy transfer, is indispensable for understanding how bacteria survive, grow, reproduce, and express virulence factors. However, ATP production in P. gingivalis remains poorly understood. Indeed, the present study represents the first characterization of the Pta-Ack pathway associated with ATP production in P. gingivalis.
The kcat value of PgPta (1,487 ± 63 s–1) is comparable to that of MtPta (1,500 ± 30 s–1) [Citation52] and is more than 10-fold higher than those of other Pta orthologues [Citation53,Citation54]. By contrast, the Km of PgPta (63.8 ± 7.5 μM) for acetyl-CoA is within the range reported for other Pta orthologues (8.6 to 600 μM) [Citation53–Citation56]. The pH optimum of PgPta was 9.0, which is slightly higher than the pH 8.0 reported previously for other Pta orthologues [Citation29,Citation54].
Structural analysis of PgPta revealed two acetyl-CoA molecules bound to the dimer ()), and the acetyl-CoA binding site in each subunit is unique and identical. In MtPta, there are two CoA binding sites (1 and 2) in each subunit with different affinities [Citation41]. Replacement of Ser309 and Arg310 located at binding site 1 in MtPta greatly affected the kcat value [Citation52], whereas mutation of Arg87 and Arg133 positioned at site 2 mainly impacted the Km value [Citation57]. Given the number of interactions between the bound CoA and each site, CoA at site 1 is thought to be more tightly bound to MtPta than CoA at site 2. Therefore, in MtPta, site 1 (the high-affinity site) is suggested to be the active site [Citation41]. Site-directed mutagenesis of Arg89 and Arg135 in site 2 of PgPta (Arg87 and Arg133 in MtPta) had moderate effects on specific activity ()), indicating that they are also not essential for catalysis. By contrast, mutation of residues at site 1 (D309A, S311A, R312A, and D318A) completely or dramatically decreased the catalytic activity. These findings suggest that site 1 was also the probable active site in PgPta. Contrary to predictions, acetyl-CoA molecules were bound only to site 2 in both subunits of PgPta () and )), even though crystal packing of PgPta does not appear to interfere with binding of acetyl-CoA to site 1. Since interference of the binding of acetyl-CoA to site 1 of PgPta has not been observed in solution, such contradictory findings might be due to crystallization artifacts. Dynamic aspects, such as domain motion, can affect substrate binding and are difficult to estimate from the static picture revealed by a crystal structure. Further analyses to investigate the binding of acetyl-CoA to PgPta are therefore required.
The pH optimum for PgAck (pH 8.0) is consistent with that of most other Ack enzymes reported to date [Citation21,Citation53]. Under the conditions tested, PgAck displayed highest activity in the presence of Mn2+, and was less efficient with Mg2+. Similarly, other Ack enzymes also function optimally with Mn2+ or Mg2+ [Citation58]. The reaction velocities of PgAck exhibited conventional Michaelis-Menten kinetics, consistent with most other Ack enzymes [Citation53]. Double reciprocal plots ()) indicate that ATP formation by PgAck proceeds via a sequential mechanism.
There are four crystallographically independent PgAck subunits in the asymmetric unit with distinct conformations, corresponding to the degree of opening and closing of the catalytic cleft ()). Such conformational changes are a common feature of the acetate and sugar kinase/Hsc70/actin (ASKHA) superfamily [Citation59]. Structural analysis of MtAck with transition state analogs ADP, AlF3 (a mimic of the meta-phosphate transition state), and acetate strongly suggest that catalysis proceeds via a direct in-line phosphoryl transfer mechanism [Citation51]. ADP, AlF3, and acetate are aligned in a linear array in the cleft in MtAck, and the catalytic cleft is wide open to accommodate all three ligands. By contrast, the cleft of subunit D is almost closed in the PgAck structure determined without any ligands such as ADP, AlF3, and acetate. This structure likely represents complete cleft closure because there is a direct contact between residues Glu32 and Arg285 positioned on opposite sides of the interface of the two domains ()). Comparison of the MtAck subunit with bound transition state analogs and subunit D of PgAck reveals a shift in the N-terminal domain of 22 Å (Cα-Cα distance between residue 37 of the two enzymes) upon cleft closure. This estimated value is larger than expected (≤15 Å) for MtAck and propionate kinase, other members of the ASKHA superfamily [Citation60]. Therefore, these results strongly indicate a dramatic domain motion in PgAck during catalysis via a direct in-line phosphoryl transfer mechanism.
There are several conserved residues at the active site of Ack enzymes [Citation30], and their roles and contributions to enzymatic activity and/or substrate binding have been extensively examined for MtAck [Citation46,Citation49,Citation50,Citation60,Citation61]. Arg91 and Arg241 in MtAck interact with the carboxyl group of acetate and are catalytically essential; R91A and R241A mutants of MtAck displayed kcat values only 0.3 and 0.4% that of the WT enzyme, respectively [Citation51]. The specific activities of PgAck mutants R91A and R214A showed a similar trend to the corresponding MtAck mutants, indicating their importance in catalysis. The E385A mutant of PgAck lost all enzyme activity ()). The equivalent Glu384 in MtAck is believed to be responsible for divalent metal cation binding [Citation46,Citation60]. As shown in ), PgAck favors Mn2+ or Mg2+, either of which is indispensable for catalysis. Therefore, Glu385 in PgAck is a candidate metal-binding residue. As the result of gel filtration, the E385A and R91A mutants are like to be partly tetramer. Although it is unclear if the possible oligomeric transition affects the catalytic activity, a large proportion (60–80%) of the R91A and E385A mutants are present as dimer like the WT enzyme. Therefore, the no or little activity of the mutants would be mostly due to the single amino acid mutation.
Bacterial genes are often organized in operons with regulatory units that control genes/proteins with associated functions. The RT-PCR analysis demonstrated that ack and pta genes constitute an operon in P. gingivalis (), as has also been shown for homologous genes in several bacteria including E. coli [Citation15] and Corynebacterium glutamicum [Citation62]. Transcription of the PGN_1180 gene is independent from pta and ack. The PGN_1180 protein, the function of which has not been assigned, is therefore presumably not associated with ATP production.
Our experiments using conditional mutant strains clearly revealed that both pta and ack are essential genes in P. gingivalis, at least under the growth conditions tested (). A previous experiment using a transposon insertion mutant library demonstrated that 463 genes, including pta and ack, are essential for growth of P. gingivalis ATCC 33277 in vitro [Citation63], whereas another independent experiment using the same bacterial strain and a similar method did not identify these genes as essential [Citation64]. Of the 459 genes found to be essential by Klein et al. [Citation63], only 281 genes (61%) were predicted to be essential by Hutcherson et al. [Citation64]. It should be noted that the essentiality of genes can change depending on the growth conditions.
All living cells rapidly turn over ATP to satisfy their energy demands, and consequently ATP must be produced from ADP in a process requiring energy. In general, ATP is biosynthesized via substrate-level phosphorylation by enzymes in the cytoplasm, or through electron transfer by ATPases located on mitochondrial (eukaryotes) or cytoplasmic (prokaryotes) membranes. Since ATP production and substrate-level phosphorylation processes are directly coupled, the reactions necessarily liberate the amount of energy required to phosphorylate ADP [Citation65]. Coupling to the phosphorylation of ADP is limited to a small number of molecules for which it is thermodynamically feasible: 1,3-bisphosphoglycerate, succinyl-CoA, phosphoenolpyruvate, arginine phosphate, carbamoyl phosphate, creatine phosphate, butyryl phosphate, and AcP, catalyzed by phosphoglycerate kinase, succinyl-CoA synthetase, pyruvate kinase, arginine kinase, carbamate kinase, creatine kinase, butyrate kinase, and Ack, respectively [Citation65]. Homology searches using complete genome sequences revealed that, apart from phosphoglycerate kinase and PgAck, homologs of these enzymes are not present in P. gingivalis ATCC 33277 or W83 [Citation24,Citation25]. A homolog of phosphoglycerate kinase, one of the major enzymes in glycolysis, is present in the genome of P. gingivalis ATCC 33277 (PGN_0433). Previous experiments using radio-labeled glucose revealed only 5% uptake of glucose from the test medium by P. gingivalis, most of which contributes to cell carbohydrates and their derivatives, suggesting that glucose utilization by this organism is very poor, and carbohydrates do not appear to readily support growth [Citation7]. Therefore, the PGN_0433 protein might not necessarily function as a phosphoglycerate kinase. In addition, apart from PgAck, no other analogs were identified in the genome of P. gingivalis ATCC 33277 or W83. Further studies are therefore needed to clarify the exact mechanism of ATP production in P. gingivalis, but the essentiality of both PgPta and PgAck implies that the Pta-Ack pathway plays an important role in ATP production in this microorganism.
Supplemental Material
Download Zip (1.1 MB)Acknowledgments
We are grateful to Ms. Satoko Kumagai for assistance in sample preparation and protein crystallization. Synchrotron radiation experiments were performed at beamline NE3A in the Photon Factory Advanced Ring (Tsukuba, Japan) under proposal number 2017G675.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Lamont RJ, Koo H, Hajishengallis G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. 2018;16(12):745–14.
- Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10(10):717–725.
- Scannapieco FA. Position paper of The American academy of periodontology: periodontal disease as a potential risk factor for systemic diseases. J Periodontol. 1998;69(7):841–850.
- Dietrich T, Jimenez M, Krall Kaye EA, et al. Age-dependent associations between chronic periodontitis/edentulism and risk of coronary heart disease. Circulation. 2008;117(13):1668–1674.
- Campus G, Salem A, Uzzau S, et al. Diabetes and periodontal disease: a case-control study. J Periodontol. 2005;76(3):418–425.
- de Pablo P, Chapple IL, Buckley CD, et al. Periodontitis in systemic rheumatic diseases. Nat Rev Rheumatol. 2009;5(4):218–224.
- Shah HN, Williams RAD. Utilization of glucose and amino acids by Bacteroides intermedius and Bacteroides gingivalis. Curr Microbiol. 1987;15(5):241–246.
- Takahashi N, Sato T, Yamada T. Metabolic pathways for cytotoxic end product formation from glutamate- and aspartate-containing peptides by Porphyromonas gingivalis. J Bacteriol. 2000;182(17):4704–4710.
- Takahashi N, Sato T. Preferential utilization of dipeptides by Porphyromonas gingivalis. J Dent Res. 2001;80(5):1425–1429.
- Wolfe AJ. The acetate switch. Microbiol Mol Biol Rev. 2005;69(1):12–50.
- Klein AH, Shulla A, Reimann SA, et al. The intracellular concentration of acetyl phosphate in Escherichia coli is sufficient for direct phosphorylation of two-component response regulators. J Bacteriol. 2007;189(15):5574–5581.
- Wolfe AJ, Chang DE, Walker JD, et al. Evidence that acetyl phosphate functions as a global signal during biofilm development. Mol Microbiol. 2003;48(4):977–988.
- Ramos-Montañez S, Kazmierczak KM, Hentchel KL, et al. Instability of ackA (acetate kinase) mutations and their effects on acetyl phosphate and ATP amounts in Streptococcus pneumoniae D39. J Bacteriol. 2010;192(24):6390–6400.
- Pruss BM, Verma K, Samanta P, et al. Environmental and genetic factors that contribute to Escherichia coli K-12 biofilm formation. Arch Microbiol. 2010;192(9):715–728.
- Kakuda H, Shiroishi K, Hosono K, et al. Construction of Pta-Ack pathway deletion mutants of Escherichia coli and characteristic growth profiles of the mutants in a rich medium. Biosci Biotechnol Biochem. 1994;58(12):2232–2235.
- Kim JN, Ahn SJ, Burne RA. Genetics and physiology of acetate metabolism by the Pta-Ack pathway of Streptococcus mutans. Appl Environ Microbiol. 2015;81(15):5015–5025.
- Kuit W, Minton NP, López-Contreras AM, et al. Disruption of the acetate kinase (ack) gene of Clostridium acetobutylicum results in delayed acetate production. Appl Microbiol Biotechnol. 2012;94(3):729–741.
- Zhu Y, Liu X, Yang ST. Construction and characterization of pta gene-deleted mutant of Clostridium tyrobutyricum for enhanced butyric acid fermentation. Biotechnol Bioeng. 2005;90(2):154–166.
- Grundy FJ, Waters DA, Allen SH, et al. Regulation of the Bacillus subtilis acetate kinase gene by CcpA. J Bacteriol. 1993;175(22):7348–7355.
- Grundy FJ, Waters DA, Takova TY, et al. Identification of genes involved in utilization of acetate and acetoin in Bacillus subtilis. Mol Microbiol. 1993;10(2):259–271.
- Chan SH, Norregaard L, Solem C, et al. Acetate kinase isozymes confer robustness in acetate metabolism. PloS One. 2014;9(3):e92256.
- Sadykov MR, Thomas VC, Marshall DD, et al. Inactivation of the Pta-AckA pathway causes cell death in Staphylococcus aureus. J Bacteriol. 2013;195(13):3035–3044.
- Imai K, Ochiai K, Okamoto T. Reactivation of latent HIV-1 infection by the periodontopathic bacterium Porphyromonas gingivalis involves histone modification. J Immunol. 2009;182(6):3688–3695.
- Nelson KE, Fleischmann RD, DeBoy RT, et al. Complete genome sequence of the oral pathogenic bacterium Porphyromonas gingivalis strain W83. J Bacteriol. 2003;185(18):5591–5601.
- Naito M, Hirakawa H, Yamashita A, et al. Determination of the genome sequence of Porphyromonas gingivalis strain ATCC 33277 and genomic comparison with strain W83 revealed extensive genome rearrangements in P. gingivalis. DNA Res. 2008;15(4):215–225.
- Horton RM, Hunt HD, Ho SN, et al. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77(1):61–68.
- Yoshida Y, Sato M, Kezuka Y, et al. Acyl-CoA reductase PGN_0723 utilizes succinyl-CoA to generate succinate semialdehyde in a butyrate-producing pathway of Porphyromonas gingivalis. Arch Biochem Biophys. 2016;596:138–148.
- Pace CN, Vajdos F, Fee L, et al. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4(11):2411–2423.
- Lundie LL Jr., Ferry JG. Activation of acetate by Methanosarcina thermophila. Purification and characterization of phosphotransacetylase. J Biol Chem. 1989;264(31):18392–18396.
- Ingram-Smith C, Gorrell A, Lawrence SH, et al. Characterization of the acetate binding pocket in the Methanosarcina thermophila acetate kinase. J Bacteriol. 2005;187(7):2386–2394.
- Yoshida Y, Sasaki T, Ito S, et al. Identification and molecular characterization of tryptophanase encoded by tnaA in Porphyromonas gingivalis. Microbiology. 2009;155(Pt 3):968–978.
- Sato M, Yoshida Y, Nagano K, et al. Three CoA transferases involved in the production of short chain fatty acids in Porphyromonas gingivalis. Front Microbiol. 2016;7:1146.
- Nakano Y, Yoshida Y, Yamashita Y, et al. Construction of a series of pACYC-derived plasmid vectors. Gene. 1995;162(1):157–158.
- Nakayama K, Kadowaki T, Okamoto K, et al. Construction and characterization of arginine-specific cysteine proteinase (Arg-gingipain)-deficient mutants of Porphyromonas gingivalis. Evidence for significant contribution of Arg-gingipain to virulence. J Biol Chem. 1995;270(40):23619–23626.
- Nagano K, Murakami Y, Nishikawa K, et al. Characterization of RagA and RagB in Porphyromonas gingivalis: study using gene-deletion mutants. J Med Microbiol. 2007;56(Pt 11):1536–1548.
- Winter G, Waterman DG, Parkhurst JM, et al. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr D Struct Biol. 2018;74(Pt 2):85–97.
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 1):72–82.
- Winter G. xia2: an expert system for macromolecular crystallography data reduction. J Appl Cryst. 2010;43(1):186.
- Vagin A, Teplyakov A. Molrep: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025.
- Schwede T, Kopp J, Guex N, et al. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31(13):3381–3385.
- Lawrence SH, Luther KB, Schindelin H, et al. Structural and functional studies suggest a catalytic mechanism for the phosphotransacetylase from Methanosarcina thermophila. J Bacteriol. 2006;188(3):1143–1154.
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132.
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255.
- Iyer PP, Lawrence SH, Luther KB, et al. Crystal structure of phosphotransacetylase from the methanogenic archaeon Methanosarcina thermophila. Structure. 2004;12(4):559–567.
- Miles RD, Gorrell A, Ferry JG. Evidence for a transition state analog, MgADP-aluminum fluoride-acetate, in acetate kinase from Methanosarcina thermophila. J Biol Chem. 2002;277(25):22547–22552.
- Miles RD, Iyer PP, Ferry JG. Site-directed mutational analysis of active site residues in the acetate kinase from Methanosarcina thermophila. J Biol Chem. 2001;276(48):45059–45064.
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797.
- Buss KA, Cooper DR, Ingram-Smith C, et al. Urkinase: structure of acetate kinase, a member of the ASKHA superfamily of phosphotransferases. J Bacteriol. 2001;183(2):680–686.
- Singh-Wissmann K, Ingram-Smith C, Miles RD, et al. Identification of essential glutamates in the acetate kinase from Methanosarcina thermophila. J Bacteriol. 1998;180(5):1129–1134.
- Singh-Wissmann K, Miles RD, Ingram-Smith C, et al. Identification of essential arginines in the acetate kinase from Methanosarcina thermophila. Biochemistry (Mosc). 2000;39(13):3671–3677.
- Gorrell A, Lawrence SH, Ferry JG. Structural and kinetic analyses of arginine residues in the active site of the acetate kinase from Methanosarcina thermophila. J Biol Chem. 2005;280(11):10731–10742.
- Lawrence SH, Ferry JG. Steady-state kinetic analysis of phosphotransacetylase from Methanosarcina thermophila. J Bacteriol. 2006;188(3):1155–1158.
- Knorr R, Ehrmann MA, Vogel RF. Cloning, expression, and characterization of acetate kinase from Lactobacillus sanfranciscensis. Microbiol Res. 2001;156(3):267–277.
- Brinsmade SR, Escalante-Semerena JC. In vivo and in vitro analyses of single-amino acid variants of the Salmonella enterica phosphotransacetylase enzyme provide insights into the function of its N-terminal domain. J Biol Chem. 2007;282(17):12629–12640.
- Pelroy RA, Whiteley HR. Kinetic properties of phosphotransacetylase from Veillonella alcalescens. J Bacteriol. 1972;111(1):47–55.
- Campos-Bermudez VA, Bologna FP, Andreo CS, et al. Functional dissection of Escherichia coli phosphotransacetylase structural domains and analysis of key compounds involved in activity regulation. FEBS J. 2010;277(8):1957–1966.
- Rasche ME, Smith KS, Ferry JG. Identification of cysteine and arginine residues essential for the phosphotransacetylase from Methanosarcina thermophila. J Bacteriol. 1997;179(24):7712–7717.
- Winzer K, Lorenz K, Durre P. Acetate kinase from Clostridium acetobutylicum: a highly specific enzyme that is actively transcribed during acidogenesis and solventogenesis. Microbiology. 1997;143(Pt 10):3279–3286.
- Hurley JH. The sugar kinase/heat shock protein 70/actin superfamily: implications of conserved structure for mechanism. Annu Rev Biophys Biomol Struct. 1996;25:137–162.
- Gorrell A, Ferry JG. Investigation of the Methanosarcina thermophila acetate kinase mechanism by fluorescence quenching. Biochemistry (Mosc). 2007;46(49):14170–14176.
- Ingram-Smith C, Barber RD, Ferry JG. The role of histidines in the acetate kinase from Methanosarcina thermophila. J Biol Chem. 2000;275(43):33765–33770.
- Reinscheid DJ, Schnicke S, Rittmann D, et al. Cloning, sequence analysis, expression and inactivation of the Corynebacterium glutamicum pta-ack operon encoding phosphotransacetylase and acetate kinase. Microbiology. 1999;145(Pt 2):503–513.
- Klein BA, Tenorio EL, Lazinski DW, et al. Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics. 2012;13:578.
- Hutcherson JA, Gogeneni H, Yoder-Himes D, et al. Comparison of inherently essential genes of Porphyromonas gingivalis identified in two transposon-sequencing libraries. Mol OralMicrobiol. 2016;31(4):354–364.
- Müller V, Hess V. The minimum biological energy quantum. Front Microbiol. 2017;8:2019.
- Lovell SC, Davis IW, Arendall WB 3rd, et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50(3):437–450.
- Hendrickson EL, Xia Q, Wang T, et al. Pathway analysis for intracellular Porphyromonas gingivalis using a strain ATCC 33277 specific database. BMC Microbiol. 2009;9:185.
- Nelson KE, Zinder SH, Hance I, et al. Phylogenetic analysis of the microbial populations in the wild herbivore gastrointestinal tract: insights into an unexplored niche. Environ Microbiol. 2003;5(11):1212–1220.