
Abstract
The oral bacterium Porphyromonas gingivalis has special nutrient requirements due to its asaccharolytic nature subsisting on small peptides cleaved from host proteins. Using proteases and other virulence factors, P. gingivalis thrives as a component of a polymicrobial community in nutritionally favorable inflammatory environments. In this regard, P. gingivalis has a number of strategies that subvert the host immune response in ways that promote its colonization and facilitate the outgrowth of the surrounding microbial community. The focus of this review is to discuss at the molecular level how P. gingivalis subverts leukocytes to create a favorable environment for a select community of bacteria that, in turn, adversely affects the periodontal tissues.
Introduction
Chronic periodontitis is an oral inflammatory disease leading to the destruction of the tissues that support the teeth (periodontium) and is associated with increased risk for certain systemic disorders.Citation1,2 Recent metagenomic, metatranscriptomic, and animal model-based mechanistic studies indicate that periodontitis is characterized by polymicrobial synergy and dysbiosis.Citation3-8 Porphyromonas gingivalis is a gram-negative asaccharolytic bacterium, which expresses a variety of virulence factors () and has long been implicated in periodontitis.Citation9 In a mouse model of periodontitis, oral inoculation with P. gingivalis leads to low-level colonization of this bacterium as well as to dysbiosis, an elevation of certain populations in the oral bacterial community leading to inflammatory bone loss.Citation10 Remarkably, P. gingivalis is unable to elicit disease by itself in germ-free mice despite colonizing this host, suggesting that disease pathology requires the presence of a polymicrobial community.Citation10 This community-wide dysbiotic effect of P. gingivalis, while present at relatively low colonization levels, has led to its characterization as a keystone pathogen, that is, an organism with a disproportionately large effect on its environment relative to its abundance.Citation11,12 Contrary to the findings of some of the early culture-based microbiological studies, more recent investigations based on culture-independent molecular methods show that P. gingivalis is a quantitatively minor constituent of human periodontitis-associated biofilms.Citation3,13,14 Whether P. gingivalis can act as a keystone pathogen in human periodontitis has not been specifically addressed, although this would require an interventional study (e.g., to specifically target P. gingivalis and assess its effect on the microbiome and the disease). In this regard, in non-human primates which naturally harbor P. gingivalis in the subgingival biofilm, a gingipain-based vaccine causes a decrease both in P. gingivalis counts and in the total subgingival bacterial load (as well as inhibits bone loss),Citation15 suggesting that the presence of P. gingivalis benefits the entire microbial community.
Table 1. Virulence factors of P. gingivalis involved in immune subversion.
The pathogenicity of periodontitis and the virulence of P. gingivalis require a susceptible host. Susceptibility is influenced by host genotype (immunoregulatory defects or immunodeficiencies), stress, diet, or risk-associated behavior such as smoking.Citation16–21 Moreover, the virulence of P. gingivalis is influenced by its environment, involving host-related factors or other bacteria (such as accessory pathogens that assist P. gingivalis in terms of colonization and metabolic activities).Citation5 Consistent with this, several P. gingivalis virulence proteins including gingipains, FimA fimbriae, HtrA protease and lipid A phosphatase have been shown to be regulated by environmental factors such as temperature and hemin.Citation22-25 Moreover, the metabolic profiles of P. gingivalis (and other periodontal bacteria) are significantly altered when compared in healthy versus diseased sites from the same patient.Citation4 For instance, virulence gene expression (e.g., encoding for gingipains, collagenase, and hemagglutinin proteins) is modified, yet the precise expression pattern for the different genes varies from patient to patient suggesting that environmental factors play a role in shaping P. gingivalis virulence. Here, we review strategies utilized by P. gingivalis to compromise host immune function, which in turn can cause compositional and quantitative shifts to the oral microbiome toward a pathogenic phenotype.
P. gingivalis manipulation of leukocytes
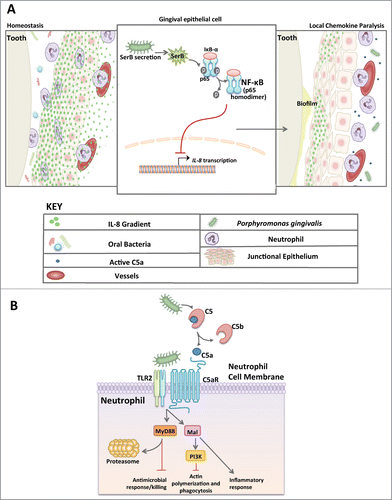
The almost exclusive niche of P. gingivalis is in the oral cavity where it hijacks leukocytes altering their migration and defense functions, and elicits inflammation to obtain nutrients from tissue breakdown.Citation26,27 The oral cavity is home to approximately 600 species of bacteria.Citation28 Unlike other mucosal tissues, the oral mucosa contains no tight junctions or mucus layer to keep microbes at bay. Instead, the gingival epithelium is charged with a steady gradient of IL-8, a cytokine that signals the migration of neutrophils through the junctional epithelium at an astonishing rate of 30,000 PMNs per minute.Citation29,30 The neutrophils create a wall-like defense system, making up the primary cellular defense in healthy oral tissues.Citation31 P. gingivalis positions itself within the sub-gingival pocket due to the strict requirements of an anaerobic lifestyle. It is here that the bacterium will necessarily encounter a neutrophil. Upon contact with the surrounding epithelium, P. gingivalis secretes a serine phosphatase (SerB) which suppresses IL-8 production by dephosphorylating serine-536 of the p65 subunit of NF-κB, thereby preventing nuclear translocation and transcription of the IL8 gene ().Citation32 Chemokine paralysis resulting from IL-8 inhibition can disrupt neutrophil migration into gingival tissues ().Citation27,33 Studies in the oral gavage model of mouse periodontitis have confirmed the capacity of P. gingivalis to inhibit the expression of neutrophil-recruiting chemokines,Citation10 as predicted by the local chemokine paralysis model.Citation27,32 Although transient (inhibited expression occurs only during the first days following P. gingivalis oral inoculation), this subversive activity can delay the recruitment of neutrophils and allow initial biofilm formation in the relative absence of neutrophil defenses. The reduction in neutrophil migration has recently been shown to disrupt an IL-17 regulatory feedback loop, thereby unleashing IL-17-mediated inflammation that drives dysbiosis.Citation34 Whether the transient inhibition of neutrophil migration leads to enhanced IL-17 expression in P. gingivalis microenvironments to contribute to dysbiosis is a distinct yet unproven possibility. Once a mature pathogenic biofilm develops that is capable of resisting neutrophil defenses, the recruitment of neutrophils can promote inflammation thereby contributing to the escalating dysbiosis.Citation26 Extensive in vivo microscopy studies have revealed hierarchical chemokines that facilitate neutrophil migration to a pinpoint locale containing bacteria or microbe-associated molecular patterns.Citation35 In gingival tissues, IL-8 directs neutrophils to the leading edge of the junctional epithelium, far away from the depths required for growth of P. gingivalis. Although transmigrating neutrophils initially follow the IL-8 gradient, they then have to move toward gradients existing in the infected or inflamed tissue. Such gradients involve chemoattractants derived from bacteria (e.g., N-formyl-methionyl-leucyl-phenylalanine) or from local activation of complement (C5a fragment).Citation36 Intriguingly, P. gingivalis expresses Arg-specific gingipains (cysteine proteases) that cleave C5 and release biologically active C5a, independently of the canonical activation of the complement cascade.Citation37 This activity enables P. gingivalis to induce a subversive crosstalk between the C5a receptor (C5aR; CD88) and Toll-like receptor (TLR)-2 ().Citation26 This C5aR-TLR2 crosstalk causes degradation of the signaling adaptor MyD88, thereby allowing decoupling of microbicidal activity from the inflammatory response which is mediated by an alternative pathway involving the MyD88-adapter-like (Mal) molecule and phosphoinositide 3-kinase (PI3K). The same Mal-PI3K pathway also causes inhibition of the small GTPase RhoA, thereby blocking actin polymerization and phagocytosis of bacteria.Citation26 Inhibition of TLR2 or C5aR counteracts P. gingivalis control of the neutrophil allowing the cell to regain effective immune clearance of the bacteria. This very precise manipulation of the neutrophil bequests P. gingivalis a safe niche rich with food, yet the subversion of the neutrophil causes the disruption of host protective mechanisms that benefits the entire oral bacterial community (more below).
Figure 1. Manipulation of neutrophil function by P. gingivalis. (A) Model of chemokine paralysis. Under homeostatic conditions, oral bacteria are kept at bay by steady recruitment of neutrophils following a gradient of IL-8 production by the gingival epithelium. P. gingivalis can manipulate the IL-8 gradient by secreting SerB, an enzyme that dephosphorylates the p65 subunit of NF-κB thereby inhibiting translocation into the nucleus and preventing IL-8 transcription. The result is chemokine paralysis that disrupts the recruitment of neutrophils into the junctional epithelium and control of the outgrowth of oral bacteria. (B) Model of Neutrophil subversion by P. gingivalis that leads to dysbiotic inflammation. Due to C5a ligand generation by Arg-specific gingipains coupled with potent TLR2 agonists (e.g., lipoproteins), P. gingivalis is able to co-activate C5aR and TLR2 resulting in Smurf1-dependent MyD88 degradation thus preventing an antimicrobial response. This signaling event also induces Mal- and PI3K-dependent inhibition of RhoA, thereby preventing phagocytosis while the same subversive pathway mediates inflammatory responses. In total, P. gingivalis can successfully decouple antimicrobial killing from a nutritionally favorable inflammatory response in neutrophils. This mechanism provides bystander support to neighboring bacteria.
The macrophage is also amenable to P. gingivalis exploitation (). While few are found in healthy gingival tissues, macrophages are most notable for their rapid response to inflammatory insult and as such are thought to play a principal role mediating an immune response as well as inflammation resolution in oral tissues.Citation38,39 The macrophage is a professional phagocyte, well known for its efficient uptake of cellular debris or invading pathogens. Normally, a bacterium is sensed by the macrophage via pattern recognition receptors and quickly phagocytosed into an acidic phagolysosome capable of killing the bacteria; during this process, an inflammasome can form leading to the induction and release of inflammatory cytokines IL-1β and IL-18.Citation40 Instead of waiting for macrophage phagocytosis to occur, P. gingivalis takes the initiative to hijack lipid rafts that form on the macrophage cellular membrane.Citation41,42 With its fimbriae and a complex of accessory proteins, P. gingivalis causes co-association of CXCR4 and TLR2; the resulting signaling crosstalk inhibits nitrogen oxide production in a manner dependent upon cAMP-dependent protein kinase A activation ().Citation43,44 Fimbriae activation of CXCR4 also leads to induction of the high-affinity conformation of complement receptor 3 that P. gingivalis exploits for safe intracellular entry.Citation45,46 Once inside the macrophage, P. gingivalis has additional methods to disrupt cellular processes that mediate bacterial killing. In this regard, it was recently shown that the lipid A moiety of the LPS structure can elicit TLR4-independent, noncanonical activation of the inflammasome.Citation47 P. gingivalis LPS is heterogeneous and contains several different lipid A moieties which can cause differential signaling through TLR4.Citation48 Remarkably, a P. gingivalis mutant fixed in its expression of a lipid A moiety that functions as a TLR4 antagonist was found to evade activation of the noncanonical inflammasome, thereby enha-ncing its intracellular survival in macrophages ().Citation49 In contrast, a P. gingivalis mutant expressing only the TLR4 agonist lipid A predictably induces noncanonical inflammasome activation cha-racterized by production of IL-1β and loses the ability to survive in the macrophage.Citation49 Noncanonical activation of the inflammasome by the LPS of gram-negative bacteria involves the participation of caspase 11.Citation50 In the noncanonical mechanism, intracellular LPS directly binds and activates caspase 11.Citation51 In contrast, TLR4 antagonist lipid A binds caspase 11 but does not induce caspase 11 oligomerization required for its activation.Citation51 This explains why P. gingivalis equipped with antagonistic or evasive lipid A interferes with the activation of both TLR4 and the caspase 11-dependent non-canonical inflammasome. Importantly, the lipid A phosphatase activity responsible for lipid A alterations was shown to be critical for the oral colonization capacity of P. gingivalis.Citation52 Moreover, the regulation of the lipid A moiety also shapes the community of oral bacteria as discussed below.Citation53
Figure 2. P. gingivalis exploitation of macrophages and dendritic cells. (A) P. gingivalis hijacking of the macrophage. P. gingivalis associates with lipid rafts on macrophages and causes the co-aggregation of CXCR4 and TLR2 with its FimA fimbriae and associated proteins. The result is an inside-out signaling event that causes complement receptor 3 (CR3) to undergo a conformational change to a ‘high affinity’ structure. P. gingivalis then utilizes CR3 for macrophage internalization. In addition to the inside-out singaling, TLR2 and CXCR4 cause activation of cAMP and subsequent PKA-dependent inhibition of inducible nitrogen oxide synthase (iNOS) ultimately preventing the bacterial killing ability of the macrophage. An additional mechanism by which P. gingivalis can increase its survival within the macrophage involves its capacity to inhibit non-canonical inflammasome activation and hence pyroptosis, a proinflammatory mechanism of lytic cell death that protects the host against infection. Since the caspase 11-dependent noncanonical mechanism of inflammasome activation is triggered by intracellular LPS, it is likely that P. gingivalis, or at least its LPS, escapes to the cytosol. (B) P. gingivalis manipulation of dendritic cell entry. P. gingivalis has a unique fimbrial protein, Mfa1, that specifically interacts with DC-SIGN on the dendritic cell surface. This binding phenomenon allows P. gingivalis to gain entry into the dendritic cell where it can survive and may be visualized within a vacuole. P. gingivalis-manipulated dendritic cells can also harbor other bacterial species as well. It is currently unclear whether P. gingivalis has to escape the vacuole in order to survive as is the case with other cell types.
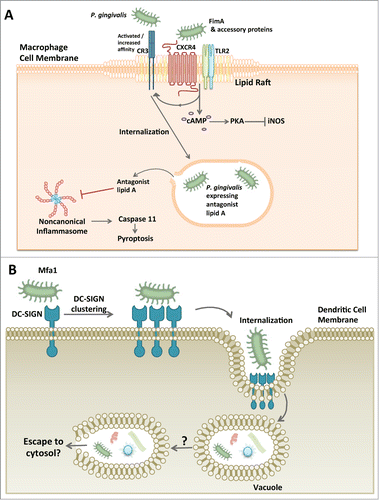
The macrophage is not the only leukocyte type which P. gingivalis may call home; the dendritic cell is also susceptible to proactive P. gingivalis internalization and manipulation. Dendritic cells in healthy gingival tissues are scarce but very effective at sampling the environment and responding rapidly to bacterial stimuli (for a review seeCitation54). Once stimulated, the dendritic cell normally becomes activated and matures into effector cell capable of eliciting a polarized T-cell response.Citation54 P. gingivalis uses its Mfa1 fimbriae to interact with a C-type lectin in dendritic cells, specifically the DC-specific ICAM-3 grabbing nonintegrin (DC-SIGN) ().Citation55 This interaction is followed by P. gingivalis internalization and survival within the dendritic cell which is further manipulated in ways that appear to contribute to an atherogenic phenotype and the systemic dissemination of P. gingivalis.Citation56 Although the association of P. gingivalis with systemic disease is beyond the scope of the present review, it is of interest to note that P. gingivalis has been detected in blood myeloid dendritic cells of patients along with several other species including Helicobacter pylori, Pseudomonas spp., Moraxella catarrhalis, Klebsiella pneumonia, and Salmonella enterica.Citation56 Although uncertain, it is tempting to hypothesize that the manipulation of dendritic cells by P. gingivalis might contribute to the intracellular survival of the other detected species. P. gingivalis causes suboptimal maturation of dendritic cells and modulates the effector response from a T helper 1 (Th1)-biased to a Th2-biased response, which may have local consequences in periodontitis. This is because the Th1 response directs effective cell-mediated immunity against periodontal bacteria.Citation57 However, this notion does not represent a consensus given the overall uncertainty regarding the precise roles of T helper subsets in periodontal disease pathogenesis.Citation58,59
As alluded to above, P. gingivalis may manipulate the adaptive immune response, although it has not been conclusively determined whether it can directly subvert lymphocytes. A microarray analysis in mice systemically exposed to P. gingivalis revealed a predominant downregulatory effect on the expression of immune response-related genes in CD4+ and CD8+ T cells.Citation60 A more recent study by an independent group provided a possible mechanism by which P. gingivalis may subvert T cell functionCitation61: Upon systemic injection in mice, P. gingivalis causes the production of high levels of IL-10 by CD11b+ cells and CD4+ as well as CD8+ T cells. IL-10, in turn, potently inhibits IFN-γ production by CD8+ T cells and CD4+ Th1 cells,Citation61 which arguably (see above) mediate protective cell-mediated immunity against periodontal bacteria.Citation62
Community-wide effects
P. gingivalis is unmistakably adept at intercepting host immune function for its own benefit, but, as alluded to above, the surrounding bacterial community can also benefit. To be precise, only those species that can both endure and exploit the inflammatory environment can really take advantage of P. gingivalis’ company. Many of these species behave as pathobionts that further exacerbate inflammatory tissue destruction.Citation16,63 Other bacterial species may be outcompeted and disappear from the escalating inflammatory environment. Consistent with the requirement of intact C5aR signaling for successful evasion of neutrophil killing by P. gingivalis in vitro, the organism fails to colonize the periodontium of C5aR-deficient mice, whereas local treatment of P. gingivalis-colonized mice with a C5aR antagonist essentially eliminates P. gingivalis, reverses dysbiosis, and inhibits development of periodontitis.Citation10,64 The C5aR-dependent evasive mechanism, as established in vitro, strictly requires a crosstalk with TLR2 and activation of downstream PI3K signaling; consistently, local inhibition of TLR2 or PI3K in the periodontium of P. gingivalis-colonized mice similarly leads to near elimination of this keystone pathogen and counteracts its earlier effect to increase the total microbial load.Citation26 It should be noted, however, that additional cell types in the periodontal environment also express the implicated molecules (C5aR, TLR2, and PI3K); their inhibition, therefore, in cells other than neutrophils might contribute to effects on P. gingivalis and the dysbiotic microbiota. For instance, P. gingivalis induces and exploits PI3K signaling also in gingival epithelial cells, where it inhibits apoptosis in a PI3K-dependent mode to promote its intracellular persistence.Citation65 In contrast to the dysbiotic effects of wild-type P. gingivalis, oral inoculation of mice with a gingipain-deficient mutant that cannot generate C5a has no influence on the microbiota.Citation26 Additional P. gingivalis virulence factors, such as the LPS lipid A moiety, can also cause alterations to the microbiota composition and bone loss in animal models of periodontal disease.Citation53 Indeed, in contrast to wild-type P. gingivalis, mutant strains that are unable to modify the lipid A moiety fail to colonize the periodontal tissue in a rabbit model of periodontitis.Citation53
Conclusion
Decades of research have identified a plethora of virulence factors of P. gingivalis, some of which are shown in . However, only recently have we started to understand how P. gingivalis integrates its virulence properties into the collective pathogenicity of the oral polymicrobial community. The emerging role of P. gingivalis involves the subversion of the host immune response in ways that enhance the fitness of the community in a nutritionally favorable inflammatory environment. It should be noted, however, that the presence of P. gingivalis does not necessarily prompt a pathological transition toward periodontal disease; rather, this bacterium signifies a risk factor for disease.Citation2,66 Indeed, periodontally healthy individuals may also harbor P. gingivalis albeit with reduced frequency relative to periodontitis patients.Citation3,67 The most likely explanations, which are not mutually exclusive, involve changes in the status of the bacterium or the host. For instance, there is considerable strain and virulence diversity within the population structure of P. gingivalis and, as alluded to above, at least some of its key virulence factors (e.g.,, gingipains and lipid A phosphatases) are regulated by local environmental conditions that are likely different among different individuals.Citation68 From a broader point of view, a susceptible host is necessary for the development of periodontitis as implied by cases of individuals who do not develop periodontitis despite considerable biofilm accumulation at dentogingival sites.Citation69,70 In this context, there might be individuals who can resist the capacity of P. gingivalis to convert a symbiotic microbiota into a dysbiotic one by virtue of their intrinsic immune status (e.g., alterations in signaling pathways required for immune subversion by P. gingivalis). Although this review focused on the manipulation of leukocytes by P. gingivalis, it should be noted that the capacity of this pathogen to subvert additional aspects of host immunity and homeostasis (e.g.,, gingival epithelial cells, complement, antimicrobial molecules) is also important and the reader is referred to other reviews.Citation71-74 Moreover, it should be noted that P. gingivalis may additionally influence the periodontal biofilm also through host-independent effects; for instance, its gingipains were shown to qualitatively and quantitatively affect the composition of polymicrobial biofilms in vitro.Citation75 The elucidation of mechanisms by which P. gingivalis promotes dysbiosis has important translational implications. In this regard, host-modulation strategies aiming to block receptors or signaling pathways by which P. gingivalis elevates the pathogenicity of the dysbiotic microbial community may offer promising options for the treatment of human periodontitis.
Disclosure of Potential Conflicts of Interest
The authors declare no potential conflicts of interest.
Funding
The authors’ research is supported by NIH/NIDCR grants; DE015254, DE017138, DE021685, and DE024716 (GH).
Related Research Data
References
- Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet 2005; 366:1809-20; PMID:16298220; http://dx.doi.org/10.1016/S0140-6736(05)67728-8
- Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol 2015; 15(1):30-44; PMID:25534621; http://dx.doi.org/10.1038/nri3785
- Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, Gamonal J, Diaz PI. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J 2013; 7:1016-25; PMID:23303375; http://dx.doi.org/10.1038/ismej.2012.174
- Jorth P, Turner KH, Gumus P, Nizam N, Buduneli N, Whiteley M. Metatranscriptomics of the human oral microbiome during health and disease. MBio 2014; 5:e01012-14; PMID:24692635; http://dx.doi.org/10.1128/mBio.01012-14
- Hajishengallis G, Lamont RJ. Beyond the red complex and into more complexity: The Polymicrobial Synergy and Dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol 2012; 27:409-19; PMID:23134607; http://dx.doi.org/10.1111/j.2041-1014.2012.00663.x
- Lamont RJ, Hajishengallis G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol Med 2014; PMID:25498392; doi:10.1016/j.molmed.2014.11.004 (Epub ahead of print)
- Rosier BT, de Jager M, Zaura E, Krom BP. Historical and contemporary hypotheses on the development of oral diseases: are we there yet? Front Cell Infect Microbiol 2014; 4:92; PMID:25077073; http://dx.doi.org/10.3389/fcimb.2014.00092
- Costalonga M, Herzberg MC. The oral microbiome and the immunobiology of periodontal disease and caries. Immunol Lett 2014; 162(2 Pt A):22-38; PMID:25447398; doi:10.1016/j.imlet.2014.08.017 (Epub ahead of print)
- Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL, Jr. Microbial complexes in subgingival plaque. J Clin Periodontol 1998; 25:134-44; PMID:9495612; http://dx.doi.org/10.1111/j.1600-051X.1998.tb02419.x
- Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 2011; 10:497-506; PMID:22036469; http://dx.doi.org/10.1016/j.chom.2011.10.006
- Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol 2012; 10:717-25; PMID:22941505; http://dx.doi.org/10.1038/nrmicro2873
- Darveau RP. The oral microbial consortium's interaction with the periodontal innate defense system. DNA Cell Biol 2009; 28:389-95; PMID:19435427; http://dx.doi.org/10.1089/dna.2009.0864
- Kumar PS, Leys EJ, Bryk JM, Martinez FJ, Moeschberger ML, Griffen AL. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing. J Clin Microbiol 2006; 44:3665-73; PMID:17021095; http://dx.doi.org/10.1128/JCM.00317-06
- Doungudomdacha S, Rawlinson A, Douglas CW. Enumeration of Porphyromonas gingivalis, Prevotella intermedia and Actinobacillus actinomycetemcomitans in subgingival plaque samples by a quantitative-competitive PCR method. J Med Microbiol 2000; 49:861-74; PMID:11023183
- Page RC, Lantz MS, Darveau R, Jeffcoat M, Mancl L, Houston L, Braham P, Persson GR. Immunization of Macaca fascicularis against experimental periodontitis using a vaccine containing cysteine proteases purified from Porphyromonas gingivalis. Oral Microbiol Immunol 2007; 22:162-8; PMID:17488441; http://dx.doi.org/10.1111/j.1399-302X.2007.00337.x
- Hajishengallis G. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol 2014; 35:3-11; PMID:24269668; http://dx.doi.org/10.1016/j.it.2013.09.001
- Kumar PS. Smoking and the subgingival ecosystem: a pathogen-enriched community. Future Microbiol 2012; 7:917-9; PMID:22913349; http://dx.doi.org/10.2217/fmb.12.71
- Stabholz A, Soskolne WA, Shapira L. Genetic and environmental risk factors for chronic periodontitis and aggressive periodontitis. Periodontol 2000 2010; 53:138-53; PMID:20403110; http://dx.doi.org/10.1111/j.1600-0757.2010.00340.x
- Zhou Q, Leeman SE, Amar S. Signaling mechanisms in the restoration of impaired immune function due to diet-induced obesity. Proc Natl Acad Sci U S A 2011; 108:2867-72; PMID:21282635; http://dx.doi.org/10.1073/pnas.1019270108
- Divaris K, Monda KL, North KE, Olshan AF, Reynolds LM, Hsueh WC, Lange EM, Moss K, Barros SP, Weyant RJ, et al. Exploring the genetic basis of chronic periodontitis: a genome-wide association study. Hum Mol Genet 2013; 22:2312-24; PMID:23459936; http://dx.doi.org/10.1093/hmg/ddt065
- Bagaitkar J, Williams LR, Renaud DE, Bemakanakere MR, Martin M, Scott DA, Demuth DR. Tobacco-induced alterations to Porphyromonas gingivalis-host interactions. Environ Microbiol 2009; 11:1242-53; PMID:19175666; http://dx.doi.org/10.1111/j.1462-2920.2008.01852.x
- Xie H, Cai S, Lamont RJ. Environmental regulation of fimbrial gene expression in Porphyromonas gingivalis. Infect Immun 1997; 65:2265-71; PMID:9169762
- Al-Qutub MN, Braham PH, Karimi-Naser LM, Liu X, Genco CA, Darveau RP. Hemin-dependent modulation of the lipid A structure of Porphyromonas gingivalis lipopolysaccharide. Infect Immun 2006; 74:4474-85; PMID:16861633; http://dx.doi.org/10.1128/IAI.01924-05
- Curtis MA, Percival RS, Devine D, Darveau RP, Coats SR, Rangarajan M, Tarelli E, Marsh PD. Temperature-dependent modulation of Porphyromonas gingivalis lipid A structure and interaction with the innate host defenses. Infect Immun 2011; 79:1187-93; PMID:21220483; http://dx.doi.org/10.1128/IAI.00900-10
- Roy F, Vanterpool E, Fletcher HM. HtrA in Porphyromonas gingivalis can regulate growth and gingipain activity under stressful environmental conditions. Microbiology 2006; 152:3391-8; PMID:17074908; http://dx.doi.org/10.1099/mic.0.29147-0
- Maekawa T, Krauss JL, Abe T, Jotwani R, Triantafilou M, Triantafilou K, Hashim A, Hoch S, Curtis MA, Nussbaum G, et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 2014; 15:768-78; PMID:24922578; http://dx.doi.org/10.1016/j.chom.2014.05.012
- Darveau RP, Belton CM, Reife RA, Lamont RJ. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun 1998; 66:1660-5; PMID:9529095
- Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG. The human oral microbiome. J Bacteriol 2010; 192:5002-17; PMID:20656903; http://dx.doi.org/10.1128/JB.00542-10
- Tonetti MS, Imboden MA, Lang NP. Neutrophil migration into the gingival sulcus is associated with transepithelial gradients of interleukin-8 and ICAM-1. J Periodontol 1998; 69:1139-47; PMID:9802714; http://dx.doi.org/10.1902/jop.1998.69.10.1139
- Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol 2010; 8:481-90; PMID:20514045; http://dx.doi.org/10.1038/nrmicro2337
- Ryder MI. Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontol 2000 2010; 53:124-37; PMID:20403109; http://dx.doi.org/10.1111/j.1600-0757.2009.00327.x
- Takeuchi H, Hirano T, Whitmore SE, Morisaki I, Amano A, Lamont RJ. The serine phosphatase SerB of Porphyromonas gingivalis suppresses IL-8 production by dephosphorylation of NF-κB RelA/p65. PLoS Pathog 2013; 9:e1003326; PMID:23637609; http://dx.doi.org/10.1371/journal.ppat.1003326
- Bainbridge B, Verma RK, Eastman C, Yehia B, Rivera M, Moffatt C, Bhattacharyya I, Lamont RJ, Kesavalu L. Role of Porphyromonas gingivalis phosphoserine phosphatase enzyme SerB in inflammation, immune response, and induction of alveolar bone resorption in rats. Infect Immun 2010; 78:4560-9; PMID:20805334; http://dx.doi.org/10.1128/IAI.00703-10
- Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, Abusleme L, Zenobia C, Hosur KB, Abe T, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med 2014; 6:229ra40; PMID:24670684
- Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med 2011; 17:1381-90; PMID:22064428; http://dx.doi.org/10.1038/nm.2514
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 2013; 13:159-75; PMID:23435331; http://dx.doi.org/10.1038/nri3399
- Wingrove JA, DiScipio RG, Chen Z, Potempa J, Travis J, Hugli TE. Activation of complement components C3 and C5 by a cysteine proteinase (gingipain-1) from Porphyromonas (Bacteroides) gingivalis. J Biol Chem 1992; 267:18902-7; PMID:1527018
- Chapple CC, Srivastava M, Hunter N. Failure of macrophage activation in destructive periodontal disease. J Pathol 1998; 186:281-6; PMID:10211117; http://dx.doi.org/10.1002/(SICI)1096-9896(1998110)186:3%3c281::AID-PATH200%3e3.0.CO;2-7
- Hasturk H, Kantarci A, Van Dyke TE. Oral inflammatory diseases and systemic inflammation: role of the macrophage. Front Immunol 2012; 3:118; PMID:22623923; http://dx.doi.org/10.3389/fimmu.2012.00118
- Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 2009; 10:241-7; PMID:19221555; http://dx.doi.org/10.1038/ni.1703
- Hajishengallis G, Wang M, Harokopakis E, Triantafilou M, Triantafilou K. Porphyromonas gingivalis fimbriae proactively modulate β2 integrin adhesive activity and promote binding to and internalization by macrophages. Infect Immun 2006; 74:5658-66; PMID:16988241; http://dx.doi.org/10.1128/IAI.00784-06
- Wang M, Hajishengallis G. Lipid raft-dependent uptake, signalling and intracellular fate of Porphyromonas gingivalis in mouse macrophages. Cell Microbiol 2008; 10:2029-42; PMID:18547335; http://dx.doi.org/10.1111/j.1462-5822.2008.01185.x
- Hajishengallis G, McIntosh ML, Nishiyama SI, Yoshimura F. Mechanism and implications of CXCR4-mediated integrin activation by Porphyromonas gingivalis. Mol Oral Microbiol 2013; 28:239-249; http://dx.doi.org/10.1111/omi.12021
- Hajishengallis G, Wang M, Liang S, Triantafilou M, Triantafilou K. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc Natl Acad Sci U S A 2008; 105:13532-7; PMID:18765807; http://dx.doi.org/10.1073/pnas.0803852105
- Hajishengallis G, McIntosh ML, Nishiyama SI, Yoshimura F. Mechanism and implications of CXCR4-mediated integrin activation by Porphyromonas gingivalis. Mol Oral Microbiol 2013; 28:239-49; PMID:23331495; http://dx.doi.org/10.1111/omi.12021
- Hajishengallis G, Harokopakis E. Porphyromonas gingivalis interactions with complement receptor 3 (CR3): innate immunity or immune evasion? Front Biosci 2007; 12:4547-57; PMID:17485396; http://dx.doi.org/10.2741/2409
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013; 341:1246-9; PMID:23887873; http://dx.doi.org/10.1126/science.1240248
- Coats SR, Jones JW, Do CT, Braham PH, Bainbridge BW, To TT, Goodlett DR, Ernst RK, Darveau RP. Human Toll-like receptor 4 responses to P. gingivalis are regulated by lipid A 1- and 4'-phosphatase activities. Cell Microbiol 2009; 11:1587-99; PMID:19552698; http://dx.doi.org/10.1111/j.1462-5822.2009.01349.x
- Slocum C, Coats SR, Hua N, Kramer C, Papadopoulos G, Weinberg EO, Gudino CV, Hamilton JA, Darveau RP, Genco CA. Distinct lipid A moieties contribute to pathogen-induced site-specific vascular inflammation. PLoS Pathog 2014; 10:e1004215; PMID:25010102; http://dx.doi.org/10.1371/journal.ppat.1004215
- Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 2014; 157:1013-22; PMID:24855941; http://dx.doi.org/10.1016/j.cell.2014.04.007
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014; 514:187-92; PMID:25119034; http://dx.doi.org/10.1038/nature13820
- Zenobia C, Luo XL, Hashim A, Abe T, Jin L, Chang Y, Jin ZC, Sun JX, Hajishengallis G, Curtis MA, et al. Commensal bacteria-dependent select expression of CXCL2 contributes to periodontal tissue homeostasis. Cell Microbiol 2013; 15:1419-26; PMID:23433011; http://dx.doi.org/10.1111/cmi.12127
- Zenobia C, Hasturk H, Nguyen D, Van Dyke TE, Kantarci A, Darveau RP. Porphyromonas gingivalis lipid A phosphatase activity is critical for colonization and increasing the commensal load in the rabbit ligature model. Infect Immun 2014; 82:650-9; PMID:24478080; http://dx.doi.org/10.1128/IAI.01136-13
- Cutler CW, Jotwani R. Dendritic cells at the oral mucosal interface. J Dent Res 2006; 85:678-89; PMID:16861283; http://dx.doi.org/10.1177/154405910608500801
- Zeituni AE, Jotwani R, Carrion J, Cutler CW. Targeting of DC-SIGN on human dendritic cells by minor fimbriated Porphyromonas gingivalis strains elicits a distinct effector T cell response. J Immunol 2009; 183:5694-704; PMID:19828628; http://dx.doi.org/10.4049/jimmunol.0901030
- Carrion J, Scisci E, Miles B, Sabino GJ, Zeituni AE, Gu Y, Bear A, Genco CA, Brown DL, Cutler CW. Microbial carriage state of peripheral blood dendritic cells (DCs) in chronic periodontitis influences DC differentiation, atherogenic potential. J Immunol 2012; 189:3178-87; PMID:22891282; http://dx.doi.org/10.4049/jimmunol.1201053
- Gemmell E, Yamazaki K, Seymour GJ. The role of T cells in periodontal disease: homeostasis and autoimmunity. Periodontol 2000 2007; 43:14-40; PMID:17214833; http://dx.doi.org/10.1111/j.1600-0757.2006.00173.x
- Garlet GP. Destructive and protective roles of cytokines in periodontitis: A re-appraisal from host defense and tissue destruction viewpoints. J Dent Res 2010; 89:1349-63; PMID:20739705; http://dx.doi.org/10.1177/0022034510376402
- Gaffen SL, Hajishengallis G. A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J Dent Res 2008; 87:817-28; PMID:18719207; http://dx.doi.org/10.1177/154405910808700908
- Gemmell E, Drysdale KE, Seymour GJ. Gene expression in splenic CD4 and CD8 cells from BALB/c mice immunized with Porphyromonas gingivalis. J Periodontol 2006; 77:622-33; PMID:16584343; http://dx.doi.org/10.1902/jop.2006.050211
- Gaddis DE, Maynard CL, Weaver CT, Michalek SM, Katz J. Role of TLR2 dependent-IL-10 production in the inhibition of the initial IFN-γ T cell response to Porphyromonas gingivalis. J Leukoc Biol 2012; 93:21-31; PMID:23077245; http://dx.doi.org/10.1189/jlb.0512220
- Gemmell E, Yamazaki K, Seymour GJ. The role of T cells in periodontal disease: homeostasis and autoimmunity. Periodontology 2000 2007; 43:14-40; PMID:NOT_FOUND; http://dx.doi.org/10.1111/j.1600-0757.2006.00173.x
- Jiao Y, Hasegawa M, Inohara N. The Role of Oral Pathobionts in Dysbiosis during Periodontitis Development. J Dent Res 2014; 93:539-46; PMID:24646638; http://dx.doi.org/10.1177/0022034514528212
- Abe T, Hosur KB, Hajishengallis E, Reis ES, Ricklin D, Lambris JD, Hajishengallis G. Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (CD88) antagonist. J Immunol 2012; 189:5442-8; PMID:23089394; http://dx.doi.org/10.4049/jimmunol.1202339
- Yilmaz O, Jungas T, Verbeke P, Ojcius DM. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun 2004; 72:3743-51; PMID:15213114; http://dx.doi.org/10.1128/IAI.72.7.3743-3751.2004
- Darveau RP. Porphyromonas gingivalis neutrophil manipulation: risk factor for periodontitis? Trends Microbiol 2014; PMID:25001854; http://dx.doi.org/10.1016/j.tim.2014.06.006 (Epub ahead of print)
- Haffajee AD, Cugini MA, Tanner A, Pollack RP, Smith C, Kent RL, Jr., Socransky SS. Subgingival microbiota in healthy, well-maintained elder and periodontitis subjects. J Clin Periodontol 1998; 25:346-53; PMID:9650869; http://dx.doi.org/10.1111/j.1600-051X.1998.tb02454.x
- Darveau RP, Hajishengallis G, Curtis MA. Porphyromonas gingivalis as a potential community activist for disease. J Dent Res 2012; 91:816-20; PMID:22772362; http://dx.doi.org/10.1177/0022034512453589
- Laine ML, Crielaard W, Loos BG. Genetic susceptibility to periodontitis. Periodontol 2000 2012; 58:37-68; PMID:22133366; http://dx.doi.org/10.1111/j.1600-0757.2011.00415.x
- Socransky SS, Haffajee AD. Evidence of bacterial etiology: a historical perspective. Periodontol 2000 1994; 5:7-25; PMID:9673160; http://dx.doi.org/10.1111/j.1600-0757.1994.tb00016.x
- Bostanci N, Belibasakis GN. Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol Lett 2012; 333:1-9; PMID:22530835; http://dx.doi.org/10.1111/j.1574-6968.2012.02579.x
- Barth K, Remick DG, Genco CA. Disruption of immune regulation by microbial pathogens and resulting chronic inflammation. J Cell Physiol 2013; 228:1413-22; PMID:23255141; http://dx.doi.org/10.1002/jcp.24299
- Yilmaz O. The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology 2008; 154:2897-903; PMID:18832296; http://dx.doi.org/10.1099/mic.0.2008/021220-0
- Guo Y, Nguyen KA, Potempa J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon's knife to a meat chopper-like brutal degradation of proteins. Periodontol 2000 2010; 54:15-44; PMID:20712631; http://dx.doi.org/10.1111/j.1600-0757.2010.00377.x
- Bao K, Belibasakis GN, Thurnheer T, Aduse-Opoku J, Curtis MA, Bostanci N. Role of Porphyromonas gingivalis gingipains in multi-species biofilm formation. BMC Microbiol 2014; 14:258; PMID:25270662; http://dx.doi.org/10.1186/s12866-014-0258-7
- Popadiak K, Potempa J, Riesbeck K, Blom AM. Biphasic effect of gingipains from Porphyromonas gingivalis on the human complement system. J Immunol 2007; 178:7242-50; PMID:17513773; http://dx.doi.org/10.4049/jimmunol.178.11.7242
- Potempa M, Potempa J, Kantyka T, Nguyen KA, Wawrzonek K, Manandhar SP, Popadiak K, Riesbeck K, Eick S, Blom AM. Interpain A, a cysteine proteinase from Prevotella intermedia, inhibits complement by degrading complement factor C3. PLoS Pathog 2009; 5:e1000316; PMID:19247445; http://dx.doi.org/10.1371/journal.ppat.1000316
- Nussbaum G, Shapira L. How has neutrophil research improved our understanding of periodontal pathogenesis? J Clin Periodontol 2011; 38:49-59; PMID:21323704; http://dx.doi.org/10.1111/j.1600-051X.2010.01678.x
- Wang M, Krauss JL, Domon H, Hosur KB, Liang S, Magotti P, Triantafilou M, Triantafilou K, Lambris JD, Hajishengallis G. Microbial hijacking of complement-toll-like receptor crosstalk. Sci Signal 2010; 3:ra11; PMID:20159852
- Potempa M, Potempa J, Okroj M, Popadiak K, Eick S, Nguyen KA, Riesbeck K, Blom AM. Binding of complement inhibitor C4b-binding protein contributes to serum resistance of Porphyromonas gingivalis. J Immunol 2008; 181:5537-44; PMID:18832711; http://dx.doi.org/10.4049/jimmunol.181.8.5537
- Mahtout H, Chandad F, Rojo JM, Grenier D. Porphyromonas gingivalis mediates the shedding and proteolysis of complement regulatory protein CD46 expressed by oral epithelial cells. Oral Microbiol Immunol 2009; 24:396-400; PMID:19702953; http://dx.doi.org/10.1111/j.1399-302X.2009.00532.x
- Coats SR, Jones JW, Do CT, Braham PH, Bainbridge BW, To TT, Goodlett DR, Ernst RK, Darveau RP. Human Toll-like receptor 4 responses to P. gingivalis are regulated by lipid A 1- and 4'- phosphatase activities. Cell Microbiol 2009; 11:1587-99; PMID:19552698; http://dx.doi.org/10.1111/j.1462-5822.2009.01349.x
- Maresz KJ, Hellvard A, Sroka A, Adamowicz K, Bielecka E, Koziel J, Gawron K, Mizgalska D, Marcinska KA, Benedyk M, et al. Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PLoS Pathog 2013; 9:e1003627; PMID:24068934; http://dx.doi.org/10.1371/journal.ppat.1003627
- Wegner N, Wait R, Sroka A, Eick S, Nguyen KA, Lundberg K, Kinloch A, Culshaw S, Potempa J, Venables PJ. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheumatol 2010; 62:2662-72; PMID:NOT_FOUND; http://dx.doi.org/10.1002/art.27552
- Yilmaz O, Yao L, Maeda K, Rose TM, Lewis EL, Duman M, Lamont RJ, Ojcius DM. ATP scavenging by the intracellular pathogen Porphyromonas gingivalis inhibits P2X7-mediated host-cell apoptosis. Cell Microbiol 2008; 10:863-75; PMID:18005240; http://dx.doi.org/10.1111/j.1462-5822.2007.01089.x
- Choi CH, Spooner R, DeGuzman J, Koutouzis T, Ojcius DM, Yilmaz O. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell Microbiol 2013; 15:961-76; PMID:23241000; http://dx.doi.org/10.1111/cmi.12089
- Hajishengallis G, Lambris JD. Microbial manipulation of receptor crosstalk in innate immunity. Nat Rev Immunol 2011; 11:187-200; PMID:21350579; http://dx.doi.org/10.1038/nri2918
- Hajishengallis G, Lamont RJ. Breaking bad: Manipulation of the host response by Porphyromonas gingivalis. Eur J Immunol 2014; 44:328-38; PMID:24338806; http://dx.doi.org/10.1002/eji.201344202
- Zeituni AE, McCaig W, Scisci E, Thanassi DG, Cutler CW. The native 67-kilodalton minor fimbria of Porphyromonas gingivalis is a novel glycoprotein with DC-SIGN-targeting motifs. J Bacteriol 2010; 192:4103-10; PMID:20562309; http://dx.doi.org/10.1128/JB.00275-10
- Nakayama K. Porphyromonas gingivalis cell-induced hemagglutination and platelet aggregation. Periodontol 2000 2010; 54:45-52; PMID:20712632; http://dx.doi.org/10.1111/j.1600-0757.2010.00351.x